

Abstract
Intrinsically disordered proteins (IDPs) play important roles in a wide range of biological processes and have been associated with various diseases, including cancer. In the last few years, cancer genome projects have systematically collected genetic variations underlying multiple cancer types. In parallel, the number and different types of disordered proteins characterized by experimental methods have also significantly increased. Nevertheless, the role of IDPs in various types of cancer is still not well understood. In this work, we present DisCanVis, a novel visualization tool for cancer mutations with a special focus on IDPs. In order to aid the interpretation of observed mutations, genome level information is combined with information about the structural and functional properties of proteins. The web server enables users to inspect individual proteins, collect examples with existing annotations of protein disorder and associated function or to discover currently uncharacterized examples with likely disease relevance. Through a REST API interface and precompiled tables the analysis can be extended to a group of proteins.
Keywords: cancer genome projects, intrinsically disordered proteins, mutations, sequence predictions, sequence variations, visualization, web server
1. INTRODUCTION
Large‐scale sequencing technologies have transformed biological sciences and enabled the systematic cataloging of genetic variations underlying cancer and various genetic diseases. One of the largest projects, The Cancer Genome Atlas (TCGA) was established to provide a comprehensive catalog of cancer genome profiles through next‐generation sequencing methods and other high‐throughput technologies (Tomczak et al., 2015). The results of these efforts are publicly available through various databases, such as COSMIC (Tate et al., 2019). The interpretation of the data, however, is far from straightforward, as many mutations are randomly occurring, so‐called passenger mutations and only a small subset of mutations have a direct role in driving disease development. While some of the cancer drivers genes are well‐characterized, many cancer drivers that occur less frequently or in less frequent types of cancers are still emerging. Various visualization tools can help researchers and clinicians to explore and analyze the vast collection of genetic variations. These tools include genome browsers, such as the UCSC (Kent et al., 2002) or ENSEMBL Genome Browser (Cunningham et al., 2022), which focus on the genomic context. The SwissProt (Bairoch & Apweiler, 2000) database provides a rich source of functional and structural annotations at the protein level. Its own visualization tool, similarly to ProViz (Jehl et al., 2016), can present various protein features but is not suitable for the interpretation of genetic mutations. In order to understand the effect of mutations and to develop treatment strategies it is often essential to combine both genome and protein level information. This is especially important for the class of intrinsically disordered proteins (IDPs) that has recently emerged to play important roles in various diseases—cancer in particular—and represent a currently untapped pool of potential drug targets.
Intrinsically disordered proteins represent roughly a third of human protein residues (Ward et al., 2004). These proteins and protein regions can be characterized by an ensemble of rapidly interconverting conformations instead of a single well‐defined three‐dimensional structure (Dyson & Wright, 1998; Gong et al., 2016). Their function is largely complementary to that of globular proteins and are mostly involved in regulatory and signaling processes (Uversky, 2013). Many IDRs participate in protein–protein interactions which can undergo coupled folding and binding or can form highly dynamic, fuzzy interactions which can drive phase separation (Fuxreiter & Tompa, 2012; Feng et al., 2019; Chiu et al., 2022). Sites of PTMs are often located within intrinsically disordered regions, providing means to regulate the structural and functional states of these proteins. IDRs have been associated with many diseases, including cancer. Although many known cancer drivers contain IDRs, the relationship between protein disorder and cancer can be indirect (Pajkos et al., 2012). Cancer mutations targeting IDRs have been systematically analyzed in only a few studies (Mészáros et al., 2021; Zou et al., 2022). Currently, the number of cases where the direct connection between mutations altering IDRs and cancer is established is limited (Mészáros et al., 2021). However, with the growing number of experimentally characterized disordered proteins and their widening roles in various biological processes, their importance for cancer is expected to increase.
To help further research in this area, we present a novel web server that can be used to explore, visualize, and analyze cancer mutations with a special focus on IDPs. In order to aid the interpretation of observed mutations, we combine both genome and protein level information. For the reliable assessment of protein disorder, we collect experimental annotations which are complemented by state‐of‐the art prediction methods. Functional information collected from the SwissProt databases is enhanced by annotations from additional resources related to protein disorder. The web server also enables users to carry out analysis, focus on specific subsets to analyze existing annotations or to discover novel candidate genes. The usability of the web server is demonstrated through specific examples.
2. RESULTS
2.1. Server description
2.1.1. Overview of the server
DisCanVis uses the second main version of DJANGO (v. 2.0.4) as a back‐end kernel. Information displayed is stored in a standardized relational database provided by the MySQL framework. Queried data are either accessed directly from the database or calculated on‐the‐fly from available data: no third party APIs are utilized. The user front‐end was built using a combination of DJANGO template language, jQuery (v. 3.6.0) and the latest version of Bootstrap (v. 5.0). Despite the intensive use of cutting‐edge web technologies, DisCanViz supports all HTML5 and WebP compatible browsers.
DisCanVis is available at https://discanvis.elte.hu/. Its interface offers multiple convenient approaches for users to find relevant data. The homepage contains a general description of the web server with direct links to the Getting started, Examples and API pages, where more detailed information is available. There are four main tabs that provide access to the search, browse, help, and statistics pages. The Search page allows users to query the database using different terms derived from the UniProt and COSMIC databases: Accession, Entry, full and partial name from the UniProt and identifiers from the COSMIC database are all accepted. An example is provided to help novice users. When multiple entries are found, the user can further refine the search.
2.1.2. Entry page
The main goal of our visualization tool is to integrate mutational data with genome and protein level information, as both levels are important to interpret the functional and structural impact of the observed genetic variations. We chose the human proteome sequences as the base set, as these entries contain the most complete protein level annotations (UniProt Consortium, 2021). The sequences were mapped to COSMIC transcripts, as well as the human genome through UCSC tools (see Methods). This enabled us to pull genome and transcriptome level annotations for the protein sequences. The final dataset currently contains 18,965 sequences.
Each entry has a header section. The header shows the protein name according to UniProt. By clicking on the icon left of the protein name, additional details are shown about the entry, such as the gene name, the chromosome, and length of the protein sequence. In addition, the UniProt accession and ENSEMBL transcript ID are shown with links to the corresponding databases. On the right hand side of the header section, the cancer driver status is given. By clicking on the icon next to this information, the source of this categorization can be accessed. The next few icons enable the user to select features that are shown in more detail, to select cancer types in the mutation profile and to present a brief summary table about the mutations. There is an additional download icon, enabling users to access all protein specific data, including mutations and annotations. The final icon returns to the homepage of the database.
The top of the page contains an overview of the whole protein, which shows the cancer mutations, domains and the combined disorder information along the sequence. There is a slider that can be moved along the sequence, indicating the region shown in more details below.
The detailed information for the selected region can be divided into four main sections. The first section presents the position indicators, the sequence and additional information that can be helpful to assess the relevance of variations. These include for example exon boundaries which can be useful to identify mutations located at splicing sites. Additional information include repeat regions identified by the tandem repeats finder (TRF) method (Benson, 1999). Such regions often show increased mutational rates. In our previous work (Mészáros et al., 2021), we found that low genomic conservation calculated by the PhastCons method (Siepel et al., 2005) is a good indicator of regions that contain more mutations without likely disease relevance. We also indicate polymorphisms that commonly occur in the human population. In general, mutations that occur in repeat regions, have low genomic conservation or coincide with common polymorphisms are likely to correspond to passenger mutations.
The next section presents genetic variations: both general disease mutations and specific cancer mutations. We collected pathogenic “Disease” variants from The UniProt Humsavar database (version 2022.02.) (Richards et al., 2015) with links to the OMIM database (Hamosh et al., 2005). We incorporated ClinVar variants labeled as pathogenic/likely pathogenic (Landrum et al., 2020). Currently, cancer mutations generated by the TCGA projects are shown. Single amino acid change variations are shown as bars, with the height of the bar proportional to the number of mutations in the given position. In‐frame indels and truncating (frameshift and nonsense) mutations are shown separately. In these cases, color intensity is proportional to the number of observed variations. On the top of the page, cancer types can be restricted and this filtering is also reflected in the header section. Mutations collected from the COSMIC database (Tate et al., 2019), which includes both large‐scale and targeted studies, can also be accessed, but are not shown by default. We also highlight significantly mutated regions. Driver mutations often accumulate in specific regions, especially when a large number of samples are analyzed together, while passenger mutations are expected to be distributed evenly along the sequence. To identify such regions, we used the iSiMPre method (Mészáros et al., 2016). The main advantage of iSiMPre is that it can automatically find boundaries of regions that are enriched in mutations, without prior definition of region of interest.
We gathered various information about the structural state of proteins. We show known domains according to the PFAM database (Mistry et al., 2021). We indicate structures from the PDB (Berman et al., 2000) corresponding to the given entry with red lines indicating missing residues in case of X‐ray structures and mobile regions in case of NMR structures. In most cases, such regions indicate protein disorder. Information about experimentally verified disordered regions is transferred from the MobiDB database (Piovesan et al., 2021). However, even incorporating annotations based on homology transfer, the number of entries with experimental evidence is still limited. Therefore, we included disorder prediction methods, such as IUPred (Erdős et al., 2021), the pLDDT scores of the AlphaFold2 (Varadi et al., 2022) method, and the ANCHOR (Mészáros et al., 2018) prediction to highlight disordered binding regions. In general, disordered regions are indicated by red, while ordered regions with blue color. Even experimental annotations can be wrong and predictions obtained with different methods often contradict each other. To help users to reconcile such cases, we developed a combined disorder approach. This method is based on a simple decision tree and favors experimental methods, highly confident pLDDT predictions, and IUPred predictions in this order (Figure 1).
FIGURE 1.
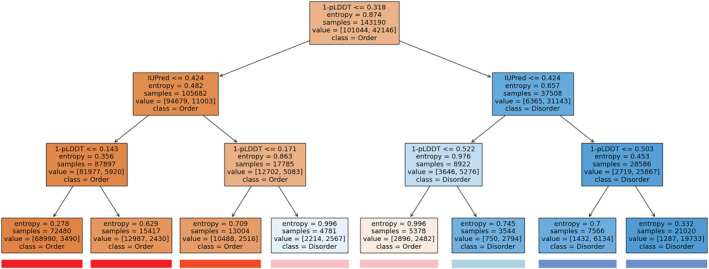
Decision tree calculated from the prediction of IUPred3 and the pLDDT scores of AlphaFold2 on the CAID‐PDB dataset using Shannon entropy as the base for information gain. Color boxes underneath each leaf node represent the coloring of sequential features on DisCanVis.
One of the primary sources of functional annotation is the SwissProt database. We collect regions of interest and binding region annotations directly from there. We added short linear motifs and SLiM switches from the ELM databases (Kumar et al., 2022). Disordered regions that undergo coupled folding and binding by interacting with globular proteins or another disordered protein are collected from the DIBS and MFIB databases, respectively (Schad et al., 2018; Fichó et al., 2017). All these regions can be selected and are linked to their source databases. Post‐translational modifications from dbPTM and PhosphositePlus (Li et al., 2022; Hornbeck et al., 2012) are also indicated, using different representations for phosphorylation, acetylation, methylation and ubiquitination. In addition, we show regions that are involved in driving the formation of membraneless organelles through phase separation based on annotations in the PhaSePro database (Mészáros et al., 2020).
The final section presents position specific sequence conservation calculated from multiple sequence alignment of orthologs (see Materials and Methods, Section 4). To enable the mapping of the conservation scores to the query protein sequence, deletion‐free alignments were used (with respect to the query protein). Since functional disorder regions—mainly linear motifs—are often conserved only in certain lineages, conservation information is shown separately for the main evolutionary levels, allowing the detection of lineage‐specific conservation patterns.
Highlighting a single position in the sequence brings up a side panel which presents the detailed information about the various features corresponding to the selected position, including the list of samples the position was mutated. This table can be downloaded in JSON and text format.
2.1.3. Browse tables
In order to ease the access to the data presented in DisCanVis, we introduced multiple tables that group the data based on certain properties. All tables contain direct links to the selected entry as well as additional information about the protein derived from the UniProt database and some basic statistics about the mutations collected with respect to either a selected region or the whole protein, depending on the table. Each column in each table is presented with a respective search entry on the top of the column to make the filtering of the data easier. Besides keyword based searches, the entry fields accept general regular expression based searches as well.
The first table labeled “Drivers” contains known cancer drivers (Tate et al., 2019; Martínez‐Jiménez et al., 2020). We included a specific table for proteins containing significantly mutated regions according to the iSiMPRe method (Mészáros et al., 2016). “Experimental disorder” table contains proteins with experimentally verified disordered regions derived from the MobiDB (Piovesan et al., 2021) database with either “curated” or “homology” level evidence. The “ELM” and “ELM Switches” tables contain proteins with regions obtained from the Eukaryotic Linear Motif Database Instances and Switches section, respectively. The sixth option called “Binding domain” contains proteins with regions that correspond to a linear peptide‐binding domain. The set of binding domains used for this table were obtained from the PixelDB database (Frappier et al., 2018). The last two tables offer user defined filtering. In the table labeled as “Chromosome” users can filter proteins by the label of the chromosome they are located in using a selection tool, while in the “GO term” table we offer an input field where users can input a Gene Ontology (The Gene Ontology Consortium, 2019) term and query proteins with the given label.
2.2. Examples
The examples described here present different use cases highlighting various features of our visualization tool.
Our first example corresponds to a fully disordered protein, POU domain class 2‐associating factor 1 (POU2AF1, UniProt: Q16633). This gene corresponds to a transcriptional coactivator involved in B‐cell differentiation, however, in addition to lymphoid tissues, it is also expressed in intestine and stomach tissue, according to the Human Protein Atlas (Uhlén et al., 2015). POU2AF1 contains a PFAM domain which indicates strong sequence conservation. Furthermore, the N‐terminal region of the protein forms a complex with the OCT1 or OCT2 transcription factors and an octamer DNA sequence. However, this region was shown to be disordered in isolation by experimental methods (Lee et al., 2001). According to the IDEAL database, the complete human protein is annotated as disordered while its mouse ortholog—which shares 89.5% sequence identity with the human protein—is annotated in the DisProt database as fully disordered. It is a known cancer gene according to the Cancer Census database. It shows low cancer specificity and the mutations from TCGA are distributed largely evenly along the protein, showing a slight preference for the N‐terminal region involved in binding to OCT2. It was suggested that mutations can subtly alter the DNA‐binding preference of OCT2, leading to the transactivation of noncanonical target genes (Hodson et al., 2016). As shown on Figure 2, the overview indicates that it is a known cancer driver. Clicking on the corresponding icon brings up further information about the cancer driver status. The overview shows that basically the whole protein is disordered with the exception of a short segment in the middle which has ambiguous characteristics. Interestingly, the AlphaFold2 method predicts a beta hairpin structure for this region. While the whole protein is annotated as disordered, the experimental method used for the characterization (SAXS) does not have the resolution to exclude the possibility of small compact structure within the generally disordered protein.
FIGURE 2.

Summary mutational and structural profile of the POU2AF1. Mutations are depicted with lollipops. Sites with larger lollipops contain more mutations. The PD‐C2‐AF1 PFAM domain is presented by a green box. At the bottom of the profile, disordered and ambiguous regions are indicated with red and pink colors, respectively.
Another potential application of our visualization is to identify known linear motifs that can be involved in disease development. The RAF proto‐oncogene serine/threonine‐protein kinase (RAF1 UniProt: P04049) is involved in the regulation of cell proliferation and differentiation by playing an important role in the RAS/RAF/MEK/ERK signaling pathways (Cobb et al., 1994). Dysregulation of the RAS/RAF/MEK/ERK pathway causes Noonan syndrome, a common developmental disorder. Noonan syndrome associated mutations in RAF1 are enriched within an conserved 14‐3‐3 binding phosphopeptide motif which is located in the middle of the sequence (254–262). These mutations impair the phosphorylation of S259 inhibiting the binding of RAF1 to 14‐3‐3 and resulting abnormal activation of ERK, hence leading to Noonan syndrome development (Kobayashi et al., 2010). Ancient evolutionary conservation of the motif can be observed clearly by the taxonomic level based conservation viewer, which highlights a universal role of the motif in evolutionary terms (Figure 3). In addition to Noonan syndrome, RAF1 mutations have been linked to several types of cancer, such as bladder cancer (Bekele et al., 2021). Observing the mutational landscape of RAF1, a cluster of cancer‐related mutations are found in the known 14‐3‐3 binding phosphopeptide motif, indicating a potential role of the motif in cancer development (Figure 3). These results show the importance of analyzing known functional disordered regions as disease‐risk factors, for which our database is a great starting point.
FIGURE 3.
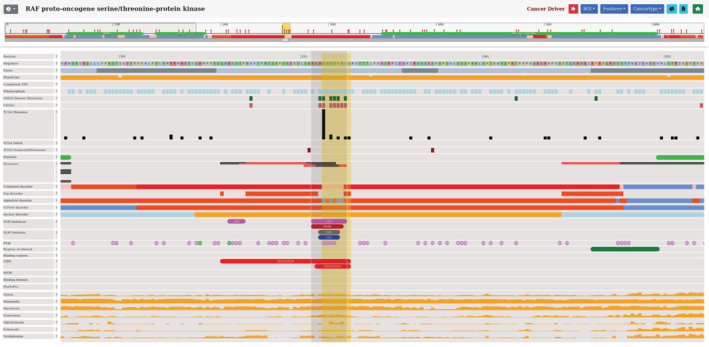
Visualization of RAF1 showing the 14‐3‐3 binding phosphopeptide motif region. Features with no data within this region are turned off. The mutation hotspots can be observed at the top of the figure in the genetic variation section. OMIM disease and cancer‐associated mutations are depicted by green and black boxes, respectively. Functional annotations and structural information are presented in the middle of the figure. Evolutionary conservation scores are shown at the bottom of the figure.
Previous results highlighted that cancer mutations often target the proteasomal degradation process and preferentially through disordered regions (Mészáros et al., 2017). In most cases, the degradation of proteins is mediated by degrons, short linear motifs that are recognized by specific E3 ligases. Known degron motifs are collected in the ELM database and the number of mutations within these motifs can be explored by the ELM table and specifying “DEG” as the ELM type. The most cancer‐mutated degrons are CTNNB1, NFE2L2, and MYC (Figure 4), which are all known cancer drivers. However, the next entry with the most mutations is WNK3, which is currently not characterized as a cancer driver. WNK3 is a member of the WNK family containing three additional paralogs. Searching for the WNK term in the identifier box shows that WNK1 and WNK4 also have a degron motif annotated. Interestingly, while WNK4 has a single cancer mutation, it has multiple disease mutations associated with Gordon's hypertension syndrome (pseudohypoaldosteronism).
FIGURE 4.
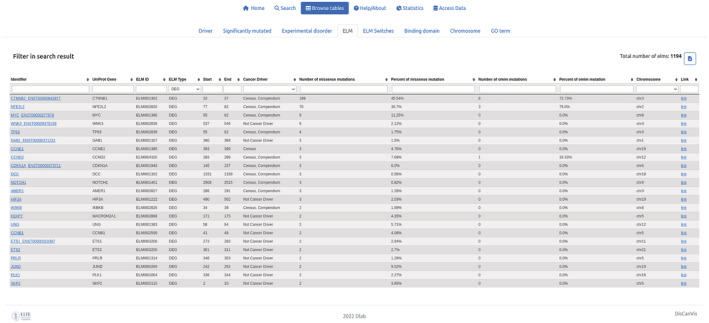
The output of searching for degrons in the ELM table using the “Browse tables” function of the database. Searching is carried out by typing the “DEG” expression into the “ELM type” text box. List of proteins with known degron motifs are sorted according to the number of missense mutations annotated within the motif region. The WNK3 degron is the 4th most mutated motif in the list with six mutations.
The degron motifs in the WNK family are recognized by the KLHL3 protein of the Cullin‐RING E3 ubiquitin ligase complex. KLHL3 binds to a conserved acidic degron motif located within the WNK isoforms, inducing their ubiquitylation and proteasomal degradation of the substrates (Gong et al., 2015; Takahashi et al., 2013). The same type of disease‐causing mutations target the binding domains as well. The effect of the missense mutations in this system causing Gordon's hypertension syndrome (pseudohypoaldosteronism) (Schumacher et al., 2014; Sohara & Uchida, 2016). In the visualization of WNK4, the positions of the degron that are affected in Gordon's syndrome can be easily detected. Moreover, the database shows that PDB structures of this region are available, allowing more detailed analysis of the mutations. While WNK2 and WNK4 are classified as cancer drivers, there is no study about the cancer associated mutations of WNK3 degron. These results shed new light on the WNK3 degron from a disease perspective. For a more general analysis of the involvement of the protein ubiquitination in cancer, the GO term could be a good starting point. Using the corresponding term GO:0016567, we can see that many proteins associated with this process contain a large number of cancer or other disease mutations, in addition to KLHL3.
Our visualization tool can also be used to explore regions that contain a significant number of mutations, but are not yet well‐characterized. One such example is Ataxin‐1 (ATXN1 UniProt: P54253). ATXN1 is a chromatin‐binding factor gene involved in regulating gene expression. The protein contains polyglutamine (polyQ) region, which is expanded in the neurodegenerative disease spinocerebellar ataxia 1 (SCA1). The polyQ region has contradicting structural annotations. Although it is localized within a conserved sequence family the domain is labeled disordered according to PFAM. AlphaFold2 method predicts the polyQ tract as helical, however, IUPred together with the combined disorder method predict this region as disordered. Although ATXN1 was shown to interact with multiple proteins (Zhang et al., 2020a), its function is still not well‐characterized. It was known to form distinctive intranuclear bodies and was recently shown to undergo phase separation via the formation of a liquid droplet state (Zhang et al., 2020b). This transition is enhanced by various factors and over time. Interestingly, cancer mutations are accumulated in the specific polyQ region (Figure 5). ATXN1 is not a known cancer gene, although it was connected to cervical cancer (Kang et al., 2017). Nevertheless, the large number of cancer mutations within the polyQ region suggest that this gene can play an important role not only in neurodegenerative disease but also in cancer.
FIGURE 5.
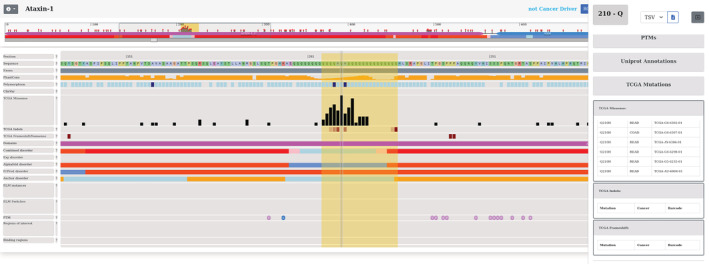
Entry page of ATXN1 positioned on the mutational hotspot region. Missense mutations and indels are depicted by black and red boxes, respectively. The specific region contains common polymorphisms (indicated by dark blue boxes). Protein disorder information is presented in the middle of the plot. At the bottom of the figure, in the functional annotation section there is no data displayed about the region of interest. Sequence conservation viewer is turned off. The panel on the right side presents more detailed information about the selected position (Q210), including the sample IDs in which the position was mutated. The region highlighted by a yellow box represents the significantly mutated region.
3. CONCLUSION
In this work, we present a novel visualization tool that can help the interpretation of cancer and other disease mutations at the level of protein sequences. With detailed annotations for structural states and functional roles, the web server is particularly useful to explore the role of IDPs in cancer. However, it can also be used to study other proteins, including globular proteins, for example those that bind disordered regions. In addition to inspecting individual proteins, users can explore the collection of known cancer drivers, proteins containing experimentally verified disordered regions or known short linear motifs, based on chromosome or shared GO ontology terms. The catalog of cancer and disease mutations is expanding rapidly. In the future, we plan to regularly update DisCanVis with additional cancer mutations from COSMIC and from large‐scale cancer genome projects and incorporate further disease mutations. It would be interesting to add targetability options as well. Nevertheless, the presented examples demonstrate the usability of DisCanVis to explore disease mutations in intrinsically disordered regions.
4. MATERIALS AND METHODS
We downloaded the reviewed proteins of the human proteome from the Uniprot database (08.2022) (UniProt Consortium, 2021). We also downloaded COSMIC data files (v96) (Tate et al., 2019). We used the COSMIC Mutation Data, a tab separated table of all COSMIC coding point mutations from targeted and genome wide screens from the current release as well as the CDS sequence for all the genes in COSMIC. The transcribed CDS sequences were mapped against the UniProt sequences using BLAST (Boratyn et al., 2013). The aim of the mapping was to find the closest transcript corresponding to the UniProt sequences. We could map 18,525 SwissProt sequences to COSMIC transcripts. In a few cases, the best hit was provided by Trembl (270) or a SwissProt isoform sequence (170). Altogether, our canonical set contained 18,965 entries. As an identifier, we kept the COSMIC names, which either corresponded to the gene name, or the gene name together with the ENSEMBL transcript ID.
The transcript sequences were then mapped to the human genome (hg38) using UCSC Genome Browser command line tools (Kuhn et al., 2013). The resulting mapping was used to find exon boundaries and to lift annotations from the UCSC Genome Browser site regarding SNPs and ClinVar mutations (Holmes et al., 2020; Sherry et al., 2001) and the genome conservation values according to the phastCons method (Siepel et al., 2005). Annotations of the corresponding UniProt entries were collected and the UniProt accession was used to find corresponding information in the ELM and ELM switches databases and post‐translational modifications from the PhosphositePlus and dbPTM databases.
Cancer mutations from the COSMIC database we mapped to UniProt sequences. We only considered single mutations, in‐frame insertions and deletions and nonsense mutations. Variations from the TCGA project were treated separately. Significantly mutated regions were determined by the iSiMPre method (Mészáros et al., 2016). Cancer drivers were collected from two sources: IntOGen and COSMIC (Tate et al., 2019; Martínez‐Jiménez et al., 2020).
We calculated various features based on the sequence. We run the SEG and DUST methods on the protein sequence to identify low complexity regions and the TRF method on the transcript sequence to identify repeat regions as these methods could help to filter out passenger mutations (Mészáros et al., 2021). Experimentally verified disordered regions were collected from the MobiDB database (Piovesan et al., 2021) using the tag “curated‐disorder‐merge” which included disorder annotation obtained by homology transfer. For disorder prediction we used the IUPred3 method (using default parameters) (providing IUPred and ANCHOR scores for disordered regions and disordered binding regions, respectively). We also collected the pLDDT scores from the AlphaFold2 database (Varadi et al., 2022). We added PFAM annotations (Mistry et al., 2021) to the UniProt entries as well. To reconcile the different classifications, a combined disorder profile was generated for each protein which combined the prediction of IUPred3 with the pLDDT scores of AlphaFold using a shallow decision tree. The decision tree was trained on the CAID‐PDB dataset (Necci et al., 2021) using Shanon entropy as a base for information gain. Experimentally verified information from MobiDB overwrites the results of the decision tree except when the two are in complete disagreement, for which we introduce a lighter colored disorder annotation.
Evolutionary conservation was calculated using a dataset of orthologous sequences, which was generated by running the GOPHER prediction algorithm (default settings) against the UniProt reference proteomes (Davey et al., 2007). In order to calculate position specific conservation scores, multiple sequence alignments of orthologs for each protein in our database were constructed using the MAFFT algorithm (default parameters) (Finn et al., 2017). Global conservation values were calculated for each position using the trident scoring approach (recommended settings) (Valdar, 2002). To provide a more informative sequence conservation viewer, the orthologous sequences were classified into the major nested evolutionary levels (Mammalia, Vertebrata, Eumetazoa, Opisthokonta, and Eukaryota) and the calculation was carried out at each level separately. For the evolutionary analysis at least three predicted orthologs were required at each taxonomy level.
AUTHOR CONTRIBUTIONS
Norbert Deutsch: Data curation (equal); formal analysis (equal); methodology (equal); resources (equal); software (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Gábor Erdős: Conceptualization (equal); data curation (equal); methodology (equal); resources (equal); software (equal); supervision (equal); writing – original draft (equal); writing – review and editing (equal). Mátyás Pajkos: Conceptualization (equal); data curation (equal); formal analysis (equal); methodology (equal); writing – original draft (equal); writing – review and editing (equal). Zsuzsanna Dosztányi: Conceptualization (equal); formal analysis (equal); funding acquisition (equal); methodology (equal); project administration (equal); supervision (equal); writing – original draft (equal); writing – review and editing (equal).
CONFLICT OF INTEREST
Zsuzsanna Dosztányi is a member of the Scientific Advisory Board of New Equilibrium Biosciences.
ACKNOWLEDGMENTS
This study was supported by the Hungarian Scientific Research Fund OTKA K129164 (Zsuzsanna Dosztányi) and by the European Union's Horizon 2020 research and innovation programme under grant agreement no. 778247. Support from ELIXIR Hungary (www.elixir‐hungary.org) and ELIXIR Implementations Studies is also acknowledged. The results shown here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga
Deutsch N, Pajkos M, Erdős G, Dosztányi Z. DisCanVis: Visualizing integrated structural and functional annotations to better understand the effect of cancer mutations located within disordered proteins. Protein Science. 2023;32(1):e4522. 10.1002/pro.4522
Review Editor: Nir Ben‐Tal
Funding information H2020 Marie Skłodowska‐Curie Actions, Grant/Award Number: 778247; Hungarian Scientific Research Fund (Országos Tudományos Kutatási Alapprogramok), Grant/Award Number: K129164
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study. The web server is freely available for academic researchers at https://discanvis.elte.hu/.
REFERENCES
- Bairoch A, Apweiler R. The SWISS‐PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekele RT, Samant AS, Nassar AH, So J, Garcia EP, Curran CR, et al. RAF1 amplification drives a subset of bladder tumors and confers sensitivity to MAPK‐directed therapeutics. J Clin Invest. 2021;131:e147849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boratyn GM, Camacho C, Cooper PS, Coulouris G, Fong A, Ma N, et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013;41:W29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S‐H, Ho W‐L, Sun Y‐C, Kuo J‐C, Huang J‐R. Phase separation driven by interchangeable properties in the intrinsically disordered regions of protein paralogs. Commun Biol. 2022;5:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb MH, Hepler JE, Cheng M, Robbins D. The mitogen‐activated protein kinases, ERK1 and ERK2. Semin Cancer Biol. 1994;5:261–8. [PubMed] [Google Scholar]
- Cunningham F, Allen JE, Allen J, Alvarez‐Jarreta J, Amode MR, Armean IM, et al. Ensembl 2022. Nucleic Acids Res. 2022;50:D988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey NE, Edwards RJ, Shields DC. The SLiMDisc server: short, linear motif discovery in proteins. Nucleic Acids Res. 2007;35:W455–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Equilibrium NMR studies of unfolded and partially folded proteins. Nat Struct Biol. 1998;5:499–503. [DOI] [PubMed] [Google Scholar]
- Erdős G, Pajkos M, Dosztányi Z. IUPred3: prediction of protein disorder enhanced with unambiguous experimental annotation and visualization of evolutionary conservation. Nucleic Acids Res. 2021;49:W297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Chen X, Wu X, Zhang M. Formation of biological condensates via phase separation: characteristics, analytical methods, and physiological implications. J Biol Chem. 2019;294:14823–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichó E, Reményi I, Simon I, Mészáros B. MFIB: a repository of protein complexes with mutual folding induced by binding. Bioinformatics. 2017;33:3682–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ, et al. InterPro in 2017‐beyond protein family and domain annotations. Nucleic Acids Res. 2017;45:D190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappier V, Duran M, Keating AE. PixelDB: protein‐peptide complexes annotated with structural conservation of the peptide binding mode. Protein Sci. 2018;27:276–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxreiter M, Tompa P. Fuzzy complexes: a more stochastic view of protein function. Adv Exp Med Biol. 2012;725:1–14. [DOI] [PubMed] [Google Scholar]
- Gong H, Zhang S, Wang J, Gong H, Zeng J. Constructing structure ensembles of intrinsically disordered proteins from chemical shift data. J Comput Biol. 2016;23:300–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Wang J, Yang J, Gonzales E, Perez R, Hou J. KLHL3 regulates paracellular chloride transport in the kidney by ubiquitination of claudin‐8. Proc Natl Acad Sci U S A. 2015;112:4340–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson DJ, Shaffer AL, Xiao W, Wright GW, Schmitz R, Phelan JD, et al. Regulation of normal B‐cell differentiation and malignant B‐cell survival by OCT2. Proc Natl Acad Sci U S A. 2016;113:E2039–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes JB, Moyer E, Phan L, Maglott D, Kattman B. SPDI: data model for variants and applications at NCBI. Bioinformatics. 2020;36:1902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post‐translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehl P, Manguy J, Shields DC, Higgins DG, Davey NE. ProViz‐a web‐based visualization tool to investigate the functional and evolutionary features of protein sequences. Nucleic Acids Res. 2016;44:W11–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang A‐R, An H‐T, Ko J, Choi E‐J, Kang S. Ataxin‐1 is involved in tumorigenesis of cervical cancer cells via the EGFR‐RAS‐MAPK signaling pathway. Oncotarget. 2017;8:94606–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Aoki Y, Niihori T, Cavé H, Verloes A, Okamoto N, et al. Molecular and clinical analysis of RAF1 in Noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum Mutat. 2010;31:284–94. [DOI] [PubMed] [Google Scholar]
- Kuhn RM, Haussler D, Kent WJ. The UCSC genome browser and associated tools. Brief Bioinform. 2013;14:144–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Michael S, Alvarado‐Valverde J, Mészáros B, Sámano‐Sánchez H, Zeke A, et al. The eukaryotic linear motif resource: 2022 release. Nucleic Acids Res. 2022;50:D497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48:D835–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L, Stollar E, Chang J, Grossmann JG, O'Brien R, Ladbury J, et al. Expression of the Oct‐1 transcription factor and characterization of its interactions with the Bob1 coactivator. Biochemistry. 2001;40:6580–8. [DOI] [PubMed] [Google Scholar]
- Li Z, Li S, Luo M, Jhong J‐H, Li W, Yao L, et al. dbPTM in 2022: an updated database for exploring regulatory networks and functional associations of protein post‐translational modifications. Nucleic Acids Res. 2022;50:D471–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐Jiménez F, Muiños F, Sentís I, Deu‐Pons J, Reyes‐Salazar I, Arnedo‐Pac C, et al. A compendium of mutational cancer driver genes. Nat Rev Cancer. 2020;20:555–72. [DOI] [PubMed] [Google Scholar]
- Mészáros B, Erdos G, Dosztányi Z. IUPred2A: context‐dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018;46:W329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros B, Erdős G, Szabó B, Schád É, Tantos Á, Abukhairan R, et al. PhaSePro: the database of proteins driving liquid‐liquid phase separation. Nucleic Acids Res. 2020;48:D360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros B, Hajdu‐Soltész B, Zeke A, Dosztányi Z. Mutations of intrinsically disordered protein regions can drive cancer but lack therapeutic strategies. Biomolecules. 2021;11:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros B, Kumar M, Gibson TJ, Uyar B, Dosztányi Z. Degrons in cancer. Sci Signal. 2017;10:eaak9982. [DOI] [PubMed] [Google Scholar]
- Mészáros B, Zeke A, Reményi A, Simon I, Dosztányi Z. Systematic analysis of somatic mutations driving cancer: uncovering functional protein regions in disease development. Biol Direct. 2016;11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021;49:D412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necci M, Piovesan D, Predictors CAID, Curators DP, Tosatto SCE. Critical assessment of protein intrinsic disorder prediction. Nat Methods. 2021;18:472–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajkos M, Mészáros B, Simon I, Dosztányi Z. Is there a biological cost of protein disorder? Analysis of cancer‐associated mutations. Mol Biosyst. 2012;8:296–307. [DOI] [PubMed] [Google Scholar]
- Piovesan D, Necci M, Escobedo N, Monzon AM, Hatos A, Mičetić I, et al. MobiDB: intrinsically disordered proteins in 2021. Nucleic Acids Res. 2021;49:D361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schad E, Fichó E, Pancsa R, Simon I, Dosztányi Z, Mészáros B. DIBS: a repository of disordered binding sites mediating interactions with ordered proteins. Bioinformatics. 2018;34:535–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher F‐R, Sorrell FJ, Alessi DR, Bullock AN, Kurz T. Structural and biochemical characterization of the KLHL3‐WNK kinase interaction important in blood pressure regulation. Biochem J. 2014;460:237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohara E, Uchida S. Kelch‐like 3/Cullin 3 ubiquitin ligase complex and WNK signaling in salt‐sensitive hypertension and electrolyte disorder. Nephrol Dial Transplant. 2016;31:1417–24. [DOI] [PubMed] [Google Scholar]
- Takahashi D, Mori T, Wakabayashi M, Mori Y, Susa K, Zeniya M, et al. KLHL2 interacts with and ubiquitinates WNK kinases. Biochem Biophys Res Commun. 2013;437:457–62. [DOI] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium . The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47:D330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomczak K, Czerwińska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol. 2015;19:A68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue‐based map of the human proteome. Science. 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium . UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN. The most important thing is the tail: multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 2013;587:1891–901. [DOI] [PubMed] [Google Scholar]
- Valdar WSJ. Scoring residue conservation. Proteins. 2002;48:227–41. [DOI] [PubMed] [Google Scholar]
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res. 2022;50:D439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–45. [DOI] [PubMed] [Google Scholar]
- Zhang S, Hinde E, Parkyn Schneider M, Jans DA, Bogoyevitch MA. Nuclear bodies formed by polyQ‐ataxin‐1 protein are liquid RNA/protein droplets with tunable dynamics. Sci Rep. 2020;10:1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Williamson NA, Duvick L, Lee A, Orr HT, Korlin‐Downs A, et al. The ataxin‐1 interactome reveals direct connection with multiple disrupted nuclear transport pathways. Nat Commun. 2020;11:3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Pan T, Gao Y, Chen R, Li S, Guo J, et al. Pan‐cancer assessment of mutational landscape in intrinsically disordered hotspots reveals potential driver genes. Nucleic Acids Res. 2022;50:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study. The web server is freely available for academic researchers at https://discanvis.elte.hu/.