

1. THE BEGINNING OF THE STORY
My interest in the biological sciences began during my third year of graduate school at Caltech. I had just completed a detailed study of the thermal and photochemistry of 1,1‐diazenes in the Dervan lab. However, rather than write my thesis, I decided to remain at Caltech and take on a project focused on generating sequence‐specific DNA cleaving molecules. Having never taken courses in biology and having no experience in DNA chemistry, this challenge was a bit daunting, but after a number of wrong turns, we managed to generate a series of poly‐pyrrole‐based amides that had quite impressive selectivity. At the time we had to write five proposals for our thesis at Caltech (a good thing) so in addition to the proposals I had formulated around reactive intermediates (which I would still like to do), I began to explore the chemistry–biology interface. I turned to proteins which had a bigger set of building blocks then DNA, and thus were more attractive to a young physical organic chemist. And as a chemist, the first question that came to mind was why God chose these particular 20 amino acids as the building blocks for life, many of which are devoid of interesting functional groups. It struck me that if one could rationally expand the set of amino acid building blocks, one could add new chemistries to proteins—both as biological probes, as well as to create proteins with novel functions. So, I began to teach myself the key elements of the central dogma that translated the genetic code to protein sequence and formulated an in vitro plan to site‐specifically introduce novel amino acids into proteins. Toward this end, I thought it might be useful to learn some protein chemistry and therefore applied to the Walsh lab (then at MIT) for a postdoc. However, while I was writing my thesis opportunity knocked and I accepted a position at Berkeley. Nonetheless, I decided to spend a year or so in the Walsh lab to learn basic molecular biology and protein chemistry.
2. ADDING BUILDING BLOCKS TO THE GENETIC CODE IN VITRO
I started at Berkeley in 1985 with a group of four (very brave) students (Chris Noren, Spencer Anthony‐Cahill, Jeff Jacobs, and Ron Zuckermann) who had joined the lab while I was a postdoc at MIT. We took on a lot for a young, inexperienced group‐working on an expanded genetic code, catalytic antibodies, and engineering sequence‐specific DNases and RNases. Our approach to adding new building blocks to the code was based on a number of key design considerations. It was clear that the ribosome could accept a wide array of amino acid side chains so we anticipated one could make significant alterations in protein side chain and possibly backbone structures. To encode a noncanonical amino acid (ncAA) one needed a “blank” codon and it was apparent that termination codons could be used for this purpose based on naturally occurring amber (TAG) and ochre (TAA) nonsense suppressor tRNAs. One needed a method to load the suppressor tRNA with the noncanonical amino acid of choice, and once delivered to the ribosome, the tRNA could not be a substrate for its cognate (or any other) aminoacyl‐tRNA synthetase (aaRS), to avoid being re‐acylated with a canonical amino acid which would result in heterogeneous mixtures of mutant proteins. An analysis of the literature suggested that the anticodon loop of yeast Phe tRNA was a key recognition feature of its cognate phenylalanyl‐tRNA synthetase (yPheRS), such that mutation of the anticodon to recognize the amber termination codon TAG would eliminate recognition by yPheRS. At the same time, Hecht had developed methods for chemically acylating tRNAs with amino acids, which although stoichiometric would provide enough of the aminoacylated tRNA to make useful amounts of protein in an in vitro S‐30‐based transcription/translation system. However, it took some work to develop a robust system, including a simplified RNA ligase‐based method for aminoacylating tRNAs with nitroveratryloxy‐(NVOC)‐N‐protected amino acid esters that could be easily deprotected with light. We also managed to produce the tRNA substrate at scale using T7 RNA polymerase runoff transcription.
After a number of missteps and a lot of hard work, we did manage to develop a robust system that allowed us and others to begin to introduce a variety of noncanonical amino acids site‐specifically into proteins, initially on milligram scale, but later with improvements in in vitro protein synthesis systems, in gram quantities. We used this system to introduce a series of noncanonical amino acids into proteins as probes of protein structure and function. For example, we were able to introduce biophysical probes including fluorescent, photocrosslinking, and spin‐labeled amino acids into proteins. We also introduced photocaged amino acids at specific sites in proteins to control their activity with light. To determine the strength of side‐chain hydrogen bonds in proteins, a series of isosteric tyrosine analogues were used to establish a free energy correlation between protein stability and hydroxyl group pKa. Indeed, many of the key elements of this technology are still being used today. However, one day I gave a seminar on this work at Berkeley and one colleague asked the question could the same thing be done in a living organism. Although mutant proteins had been made by stoichiometric microinjection of chemically aminoacylated tRNAs into oocytes, this was a much more challenging problem and my off the cuff answer was that I thought it impossible…but it got me thinking.
3. ADDING NEW BUILDING BLOCKS TO THE GENETIC CODES OF LIVING ORGANISMS
Expansion of the genetic code of a living organism requires adding new components to the translational machinery which have exquisite bioorthogonality to the endogenous components (do not cross‐react with endogenous tRNAs and aaRS's). Today, this notion of bioorthogonality is a general theme in chemical biology, but at the time it was just an emerging concept. We had already determined one could use the amber termination codon to efficiently and site‐specifically encode noncanonical amino acids in vitro and the same strategy would likely work in living cells. It was unclear whether readthrough of the natural stop codons would impair cell survival, but retrospectively, this appears not to be an issue since grams per liter of proteins containing ncAAs have been made in both bacterial and mammalian expression systems. In addition, one needed a tRNA that specifically translated the nonsense codon but was not a substrate for any of the endogenous aaRSs in the host cell to ensure high translational fidelity. One next had to generate an aaRS that aminoacylated this new tRNA and no other tRNA in the host cell (>80 in the case of Escherichia coli). Even more challenging was to engineer/evolve this aaRS to recognize the noncanonical amino acid of interest and no other amino acids in the host cell. Finally, the noncanonical amino acid had to get into the cytoplasm of the host in reasonable concentrations and not be cytotoxic. We initially set out to solve this problem in bacteria by evolving a bioorthogonal tRNA–aaRS pair from an existing bacterial pair, but this proved to be problematic. Then in a discussion with Paul Schimmel about his work in aaRS‐tRNA recognition, I realized that the recognition of tRNAs by their cognate synthetases involved interactions in the acceptor stem that were conserved in bacteria and distinct from those in archaea bacteria and eukaryotes. With this in mind, we chose the Methanococcus jannaschii (Mj) tRNA(Tyr)/MjTyrRS pair as our first candidate orthogonal bacterial pair based on the following criteria: the anticodon could likely be converted to CUA without impairing aaRS recognition, there is no editing domain, and the active site is made up largely of side chains and not protein backbone suggesting that it could be reconfigured to bind ncAAs. However, in the end, it was necessary to improve the biorthogonality of this tRNA using a series of positive and negative selections.
The next challenge was to alter the specificity of the aaRS to recognize the ncAA of interest and no endogenous amino acid in the host. We wanted a general method that was relatively rapid and could be applied to a large array of structurally diverse ncAAs. To accomplish this, we developed a two‐step selection scheme that could select from a library of aaRS active site mutants synthetases that could incorporate the ncAA at a promiscuous site in response to UAG in an essential protein. The negative selection involved suppression of the nonsense codon in a lethal gene product in the absence of the ncAA (Figure 1). This positive/negative selection scheme proved very powerful and together with variant schemes has allowed our lab and many others to genetically encode over 200 distinct ncAAs with excellent efficiencies and fidelities. Similar schemes have been used to create biorthogonal pairs to genetically encode ncAAs in higher organisms including yeast, worms, flies mammalian cells, and most recently in human hematopoietic and embryonic stem cells. Finally, although many ncAAs are taken up by cells, we have shown that Lys‐based dipeptides allow the transport and subsequent release of highly polar amino acids that have poor cellular bioavailability. In addition, biosynthetic pathways have been engineered into organisms that allow bacteria to both biosynthesize and genetically encode unnatural amino acids.
FIGURE 1.
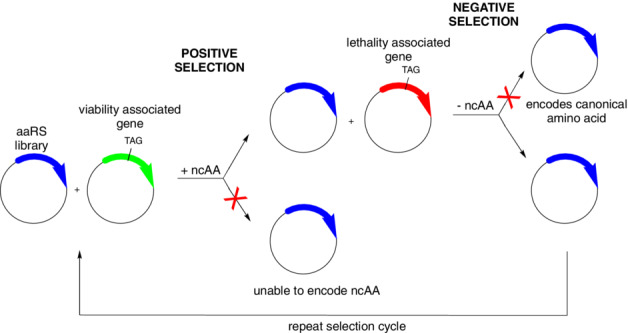
Standard protocol for generation of an aminoacyl‐tRNA synthetase (aaRS) to encode noncanonical amino acids (ncAAs).
4. A DIVERSE ARRAY OF NEW BUILDING BLOCKS
The above technology has allowed my lab and many others to genetically encode a remarkable array of structurally diverse noncanonical amino acids (these are nicely reviewed in References 1 and 2; Figure 2). Some of the earliest ncAAs to be encoded were photocrosslinkers which are now a widely used tool to covalently fix protein–protein interactions in living cells, allowing the isolation of interactors that may not be stable to other methods of isolation. We and others have also encoded ncAAs that can be used to control protein activity with light including catalytic activity, transcription, cell signaling, and protein localization. Indeed, we are now using a tetrazine analogue to photocage protein folding/unfolding on a nanosecond time scale. A series of biophysical probes including fluorescent amino acids, IR active probes, isotopically and spin‐labelled amino acids have also been genetically encoded, as have a number of postranslationally modified amino acids including phospho‐Tyr and phosphono‐Tyr and acylated lysines that are found in histones. We and others also showed that in addition to amino acid side chains, one could also make changes to the protein backbone, for example, introduce alpha hydroxy acids into proteins. We were somewhat surprised by the number of structurally distinct ncAAs we were able to encode but solution of the X‐ray crystal structures of several evolved synthetases showed the active sites to have a high degree of structural plasticity. It also became clear that some of the evolved synthetases as well as those that have naturally evolved to encode pyrrolysine in certain methanogens are polyspecific, that is they can aminoacylate their cognate tRNA with a number of different ncAAs but in general do not use the common 20 amino acids as substrates. Other useful ncAAs that have been encoded are metal ion binding amino acids, amino acids with altered pKas and steric properties, and redox‐active ncAAs. We are currently encoding a series of cofactors which should simplify the generation of proteins with both natural and synthetic cofactors. Recently, ncAAs have been used to create biorthogonal protein interfaces in essential proteins to make organisms dependent on an ncAA for survival, an approach that can be applied to the creation of conditional vaccines and biological containment. Another novel application of ncAAs was the demonstration that immunogenic amino acids can be used to break tolerance to host proteins.
FIGURE 2.
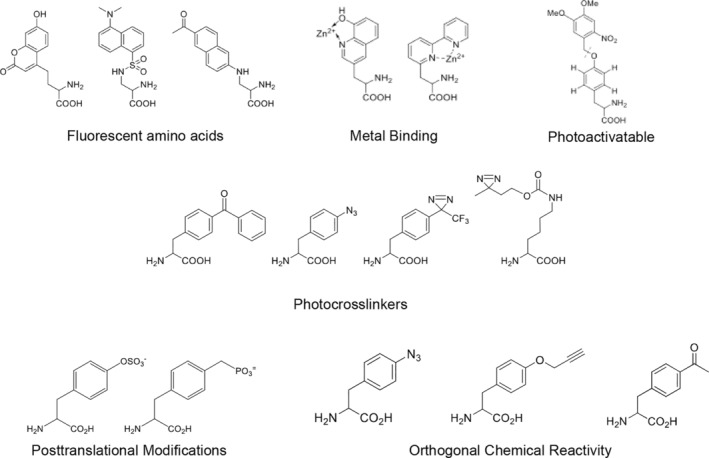
Examples of genetically encoded noncanonical amino acids with novel functions
One of the most useful classes of genetically encoded amino acids are ncAAs that have biorthogonal chemical reactivity both in vitro and in living cells. These genetically encoded chemistries include oxime formation, 1,3‐dipolar addition reactions, isothiocyanate couplings, Diels‐Alder and Michael addition reactions, cross‐coupling, metathesis reactions and fluorosulfate addition reactions. One of the first and most robust biorthogonal reactions for selective protein modification was based on a genetically encoded p‐acetylphenylalanine. This amino acid reacts selectively with alkoxy‐amines and has been used to modify proteins site‐specifically with biophysical probes, long‐chain polyethylene glycols, beads and resins, polypeptides, oligonucleotides, and drugs. In fact, there are eight such candidates that have been advanced into human clinical trials by Ambrx and others. This ability to precisely control and optimize the pharmacological properties of such conjugates enables optimization of the half‐life, efficacy, and physical properties of biologics, similar to what is routinely done with small molecules. Ambrx has optimized the platform to the degree that they can make up to 10 g/L mutant protein in bacteria and 3–4 g/L in mammalian cells, which underscores our ability to reprogram the translational machinery to encode ncAAs without compromising normal cellular processes. And recently, reactive amino acids have been used for proximity‐induced modification of protein targets to irreversibly inhibit cell signaling. Collectively, these evolved aaRS's have been distributed broadly throughout the scientific community (>2,000 distributions from my lab alone) as probes of protein structure and function and to engineer proteins with new activities.
5. EVOLVING PROTEINS WITH AN EXPANDED GENETIC CODE
The above technology allowed us to genetically encode new chemistries with relative ease, despite the fact that the genetic code itself has been conserved throughout evolution and through all kingdoms of life. This then begs the questions—why these 20 amino acids and not others, for example, why not a carbonyl group, and why stop at 20. In other words, what would life look like if God had worked rather than rested on the seventh day. We began to address this question by putting bacteria under various forms of selective pressure, providing the bacteria with additional genetically encoded ncAAs, and examining how the bacteria evolved to these challenges. In an early experiment, we created a selection in which active HIV protease expressed in E. coli led to cell death. We then screened for inhibitors of the protease from a library of expressed cyclic peptides in bacteria encoding distinct 21st amino acids. What we found in the survivors were cyclic peptides with a chemically reactive biorthogonal keto amino acid that formed a Schiff base with a Lys on the surface of the protease, likely blocking dimerization‐ a very novel mechanism for inhibiting an enzyme. In another experiment we evolved a protein in which a single ncAA mutation increased the thermal stability of the protein by a remarkable 17°C, in this case by covalently stabilizing the protein homodimer interface, again with a keto amino acid. In other experiments ncAAs have been shown to confer increased catalytic activity to enzymes and growth advantages to phage. But, the question remains—why these 20 amino acids since noncanonical amino acids can in fact provide unique solutions to evolutionary challenges, and it is relatively easy to add more building blocks to the code.
6. MORE “BLANK” CODONS
There is also the question of why the 20 common amino acids versus 21 or more. To address this question, we explored ways to generate an expanded set of orthogonal codons. One obvious approach was to use two of the three stop codons, and indeed, we were able to use amber and ochre nonsense codons together to efficiently introduce two distinct ncAAs into proteins using a pair of mutually orthogonal tRNA/aaRS pairs. To date, a number of such mutually orthogonal tRNA/aaRS pairs have been generated in our lab and in other labs that allow one to encode two or more structurally distinct ncAAs in a single protein. To obtain additional codons beyond the natural termination codons we next focused our efforts on a four‐base anticodon–codon pair. This required some in vitro evolution of the tRNA sequence to obtain good suppression efficiencies but worked quite well. This strategy was further improved when the four base codon was based on TAGX (X = G, C, T, A) due to the fact that the Church lab substituted all the TAG termination signals in the E. coli genome with TAA so that the termination factor (RF1) which competes with four base frameshift translation of TAGX could be removed. More recently, the Chin lab has removed two Ser codons from the bacterial genetic code and reassigned these codons to ncAAs. They have also developed an orthogonal ribosome translation system for the selective translation of proteins containing ncAAs. Today work in other labs is focused on encoding all 20 common amino acids with four base codons, and my own lab is attempting to make an entire four base mitochondrial genome in yeast. One interesting idea is whether one can begin to make templated synthetic polymers of defined length and sequence in bacteria using a combination of an expanded codon and amino acid set.
Another strategy we pursued upon joining Scripps in 1999 (first part‐time, then full‐time both at Scripps and GNF) was to create altogether new codons by expansion of the genetic alphabet itself, an approach that was inspired by Benner's work on new base pairs with biorthogonal hydrogen bonding patterns. I recruited Floyd Romesberg to my early Scripps lab, first as a research assistant professor then as independent tenure track faculty, to join in this effort. Together we pursued a strategy to create a third selective, stable base pair with orthogonality to the Watson–Crick G‐C and A‐T base pairs based on hydrophobicity (i.e., hydrophobic base pairs will form a stable self‐pair in water but will not pair with bases containing hydrogen bonding groups). Through a large synthetic and sometimes empirical effort we were able to generate orthogonal hydrophobic base pairs with stability comparable to the Watson–Crick base pairs and that could be selectively incorporated by DNA polymerase in vitro. Impressively Floyd was able to independently extend this work to a hydrophobic base pair that could be incorporated into plasmids in bacteria to encode an ncAA. Indeed, this methodology has been used to make a clinical stage PEG‐modified cytokine. Our own attention turned to asking whether we could replace every cytidine in the E. coli genome with a modified C, specifically 5′ hydroxymethyl C. While we made very good headway on this project, we serendipitously created a strain of E. coli where approximately half the genome is composed of ribonucleotides, incorporated through covalent deoxy‐ and ribonucleotide phosphodiester linkages. We are currently exploring the basis for formation of these chimeric structures in orthogonal bacterial replication systems in which defined changes can be made to the bacterial genome to assess the effects on ribonucleotide incorporation into plasmid DNA.
7. CONCLUSION
In conclusion, we had no idea that our initial work focused on the in vitro introduction of ncAAs into proteins with chemically misacylated tRNAs would ultimately allow us to add new chemistries to the genetic codes of living organisms. Looking back, it is a testament to our collective synthetic prowess that we were ultimately able to manipulate one of the most central aspects of life itself—the genetic code—removing a billion plus year constraint on the chemical building blocks of life. The ability to reprogram the biological machinery using concepts such as bio‐orthogonality together with in vitro evolution methods and other chemical and biological technologies has created a whole new opportunity for chemists and biologists alike in synthetic biology, one which is focused not on small molecules and natural products but rather on altering the structures and functions of complex biomolecules and biological systems. Synthetic biology, like chemical synthesis, allows us to modify molecules to create new chemical, biological, and materials properties, but has significantly expanded the complexity of molecules and systems of molecules of interest. Although this Recollection has largely focused on our own work, many labs have and continue to make major contributions to the field, and I am especially grateful to an incredible group of coworkers who made all this possible.
AUTHOR CONTRIBUTIONS
Peter Schultz: Writing – original draft (equal).
Biography
Peter Schultz is the CEO, President, and the Skaggs Presidential Chair Professor at Scripps Research. Schultz is a pioneer in the fields of chemical and synthetic biology—his work includes expanding the genetic code, catalytic antibodies, regenerative medicine, and the application of molecular diversity technologies to challenges in energy, materials, and human health. Schultz has founded nine companies, established the Genomics Institute of the Novartis Research Foundation in 1999 and served as its Director until 2010, and in 2012, he established Calibr, a nonprofit drug discovery institute. Schultz is the author of more than 600 scientific publications and has trained over 300 coworkers, many of whom are on the faculties of major institutions. He is a member of the National Academy of Sciences, USA and Institute of Medicine, and has won many awards including the Wolf Prize in Chemistry, the Paul Erhlich and Ludwig Darmstaedter Award, The Tetrahedron Prize, the Arthur C. Cope Award, the NAS Award in Chemistry and the Solvay Prize.
Schultz P. Expanding the genetic code. Protein Science. 2023;32(1):e4488. 10.1002/pro.4488
Review Editor: John Kuriyan
REFERENCES
- 1. Young DD, Schultz PG. Playing with the molecules of life. ACS Chem Biol. 2018;13(4):854–870. 10.1021/acschembio.7b00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Diercks CS, Dik D, Schultz PG. Adding new chemistries to the central dogma of molecular biology. Chem. 2021;7(11):2883–2895. 10.1016/j.chempr.2021.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]