

Abstract
Metal ions bound to macromolecules play an integral role in many cellular processes. They can directly participate in catalytic mechanisms or be essential for the structural integrity of proteins and nucleic acids. However, their unique nature in macromolecules can make them difficult to model and refine, and a substantial portion of metal ions in the PDB are misidentified or poorly refined. CheckMyMetal (CMM) is a validation tool that has gained widespread acceptance as an essential tool for researchers working on metal‐macromolecule complexes. CMM can be used during structure determination or to validate metal binding sites in structural models within the PDB. The functionalities of CMM have recently been greatly enhanced and provide researchers with additional information that can guide modeling decisions. The new version of CMM shows metals in the context of electron density maps and allows for on‐the‐fly refinement of metal binding sites. The improvements should increase the reproducibility of biomedical research. The web server is available at https://cmm.minorlab.org.
Keywords: drug discovery, metal binding sites validation, reproducibility, structure refinement, metalloprotein
1. INTRODUCTION
Reproducibility in biomedical research has become a hot issue. Reports indicate that academic results cannot be reproduced in a commercial environment in >50% of the cases (Prinz et al., 2011), mainly due to incomplete or inaccurate descriptions of experimental procedures and results (Errington, Denis, et al., 2021; Errington, Mathur, et al., 2021; Nosek & Errington, 2020). Some reproducibility issues arise from variations caused by the experience, bias, poor experimental protocols, and idiosyncrasies of the researcher. Consequently, the availability of validation tools specific to each area of research is highly desirable.
Structural biology is not immune to reproducibility issues. Ensuring the quality and reliability of structural data is particularly important, given its central role in modern biomedicine (Wlodawer et al., 2008, 2013). On average, every structure in the Protein Data Bank (PDB) is downloaded >30,000 times; therefore, any PDB deposit inaccuracies are proliferated and may impair subsequent research areas (Zheng, Hou, et al., 2014).
The macromolecular structures deposited in the PDB contain fundamental information necessary for drug discovery and characterization of proteins (Burley et al., 2019). However, the quality of macromolecular structures is inconsistent (Chruszcz et al., 2010; Cooper et al., 2011; Domagalski et al., 2014). Modeling errors arise from several sources, including the subjectivity of building models in poor electron density that results from flexibility of some regions; however, unaccounted density should be noted in a publication or PDB comments.
Metal ions such as iron, magnesium, and zinc are crucial to life. Around 34% of all PDB deposits and 40% of all enzymes deposits in the PDB contain at least one metal ion. Metals play vital roles in both the function and architecture of biological macromolecules, including the catalysis of biochemical reactions, electron transport, binding of gases, and stabilization of nucleotides and other polyphosphate compounds. While tools for the validation of protein structures are common and well‐established (Vaguine et al., 1999), the metal coordinating environments in protein structures are not easy to validate, resulting in an abundance of misidentified and/or suboptimally modeled metal ions in the PDB as it is presented in Figure 1. The validation of small molecular components of macromolecular structures is not highly advanced (Pozharski et al., 2013; Weichenberger et al., 2013), and metal binding sites are not an exception (Handing et al., 2018; Zheng, Chordia, et al., 2014).
FIGURE 1.
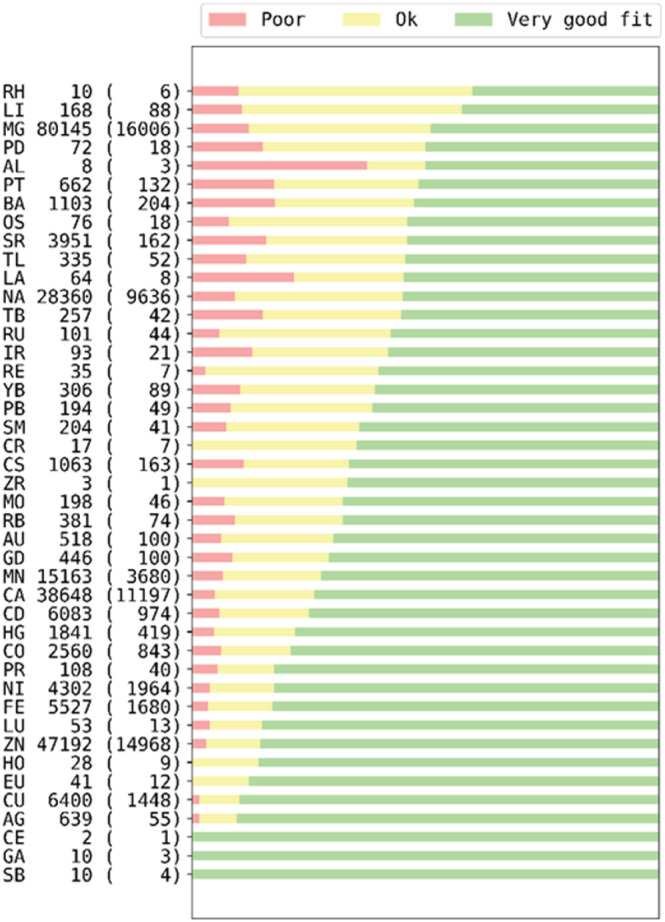
The metal site quality as judged by the RSCC parameter (Smart et al., 2018). The descriptors of the vertical axis are metal site, number of metal sites, and the number of deposits in the PDB.
Validating metal ions in macromolecular structural models is a multidisciplinary problem involving parallel crystallographic, chemical, experimental, and biological considerations. Modeling metal binding requires analysis of chemical properties and examination of geometric distortions that can be introduced by the macromolecule (Kuppuraj et al., 2009; Zheng et al., 2008). In addition, it is necessary to consider not only the resolution of the experimental data but also the overall structure quality and sample preparation (Majorek et al., 2014; Niedzialkowska et al., 2016). The data mining tools we have developed show that many metal ions are dubiously modeled (Zheng et al., 2008).
1.1. CMM WEB resource
CheckMyMetal (CMM) is a sophisticated but easy‐to‐use metal binding site validation server that addresses this previously neglected aspect of macromolecular validation and is freely available at https://cmm.minorlab.org. Initially launched in 2012, CMM (Zheng, Chordia, et al., 2014) has proved very successful in validating metal binding sites in macromolecular structures and detecting many metal assignment misidentifications. The system is widely used—researchers have submitted around 130,000 protein structures from over 6900 distinctive computer addresses in 54 countries. Over the years, scientists from this large and diverse group of users have suggested many ways to improve CMM. Herein we describe some of the significant enhancements to this validation resource.
CMM identifies validation problems in three distinct yet related aspects: coordination chemistry, agreement of experimental B‐factors, or occupancy, and the composition and motif of the metal binding environment. The new version reported herein (1) uses and shows electron density maps, (2) gives the possibility to refine the structures, (3) displays information about the crystallization conditions if present in the PDB deposit, and (4) significantly improves guidance for alternative metal identification. These combined functionalities allow for interactive modeling and refinement of metal binding sites. A new “Help and examples” section has been added to the website.
1.2. CMM validation parameters
CMM uses six parameters independent of the structure determination method and two diffraction‐specific parameters to assess the quality of each metal‐binding site. As before, CMM uses a red‐yellow‐green color scheme to classify each parameter into one of the three categories: Dubious (previously called outlier), Borderline, or Acceptable. Classification is based on statistics described in (Zheng et al., 2008), slightly modified by the experience gained during CMM's operation. Table 1 lists the current classification criteria. An example of validation parameter evaluation is presented in Figure 2, showing the distribution of zinc valency.
TABLE 1.
CMM validation parameters
| Parameters | Acceptable | Borderline | Dubious | ||
|---|---|---|---|---|---|
| Low | High | Low | High | ||
| Occupancy | [0.9, 1.0] | (0.1, 0.9) | [0.0, 0.1] | ||
| B‐factor ratio | [0.86,1.0] | [0.54, 0.86) | [0.0, 0.54) | ||
| Atomic contacts | Usual donor atoms# | Occasionally found donors# | Unusual# | ||
| Coordination geometry | Preferred coordination geometry# | Other coordination numbers# | Unusual# | ||
| Valence | |||||
| I | |||||
| Na, K, Cu | [0.7, 1.3] | [0.4, 0.7) | (1.3, 1.6] | [0, 0.4) | >1.6 |
| Mg, Ca | [1.7, 2.3] | [1.4, 1.7) | (2.3, 2.6] | [0, 1.4) | >2.6 |
| II | |||||
| Mn, Fe*, Co | [1.6, 2.4] | [1.2, 1.6) | (2.4, 2.8] | [0, 1.2) | >2.8 |
| Ni, Cu*, Zn | [1.7, 2.3] | [1.3, 1.7) | (2.3, 2.7] | [0, 1.3) | >2.7 |
| III | |||||
| Fe, Co*, Ni* | [2.7, 3.3] | [2.5, 2.7) | (3.3, 3.5] | [0, 2.5) | >3.5 |
| nVECSUM | [0, 0.10] | (0.10, 0.23] | (0.23, 1.0] | ||
| gRMSD | [0, 13.5] | (13.5, 21.5] | (21.5, 180] | ||
| Vacancy | [0, 10%] | (10%, 25%] | (25%, 100%] | ||
Note: The thresholds for the occupancy parameter are updated in the present CMM version. The parameters included in scoring metal ions for the approximated ranking are in bold. For the parameters with different thresholds for low‐resolution structures, both sets of thresholds are shown in the low and high columns. An asterisk (*) after a metal in the valence cell indicates insufficient data to set the threshold values for that metal. A hash symbol (#) indicates that these threshold values were taken or derived from previous studies (Harding et al., 2010; Kuppuraj et al., 2009). The square and round brackets indicate a closed and open interval.
FIGURE 2.
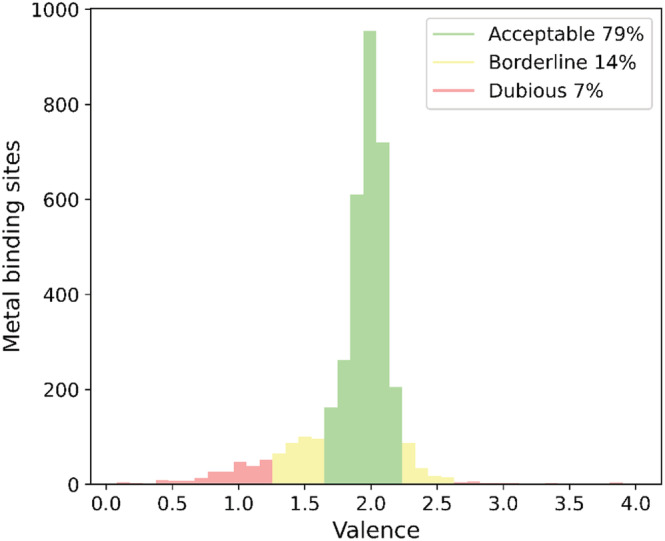
Distribution of the valence parameter for Zn binding sites in PDB deposits of proteins with resolution better than 1.5 Å derived from x‐ray and neutron diffraction only. The acceptable (79%), borderline (14%), and dubious (7%) cases are shown in green, yellow, and red, respectively. The intervals are defined according to data in Table 1.
2. RESULTS
Analyzing a structural model from the PDB or uploaded through the interface opens the validation module of CMM (Figure 3a). In addition to color‐coded validation parameters, a user can view the selected metal binding site with electron density maps, if available. On the right of this view, the distribution of metal‐electron donor atoms distances from the Cambridge Structural Database (CSD) and the actual metal‐electron donor atoms distances are shown in a histogram. Figure 3 shows the sodium‐binding site from the PDB deposit 3EEF. The six sodium‐oxygen distances fall below the most likely distance between such atoms, as observed in the CSD. In cases when the CMM validation parameters indicate that the metal binding site may require a detailed visual inspection, a scientist can switch to the model tab that provides the ability to change the metal ion. The new validation parameters characterizing the metal binding site are calculated and instantly displayed after completion of the structure refinement.
FIGURE 3.
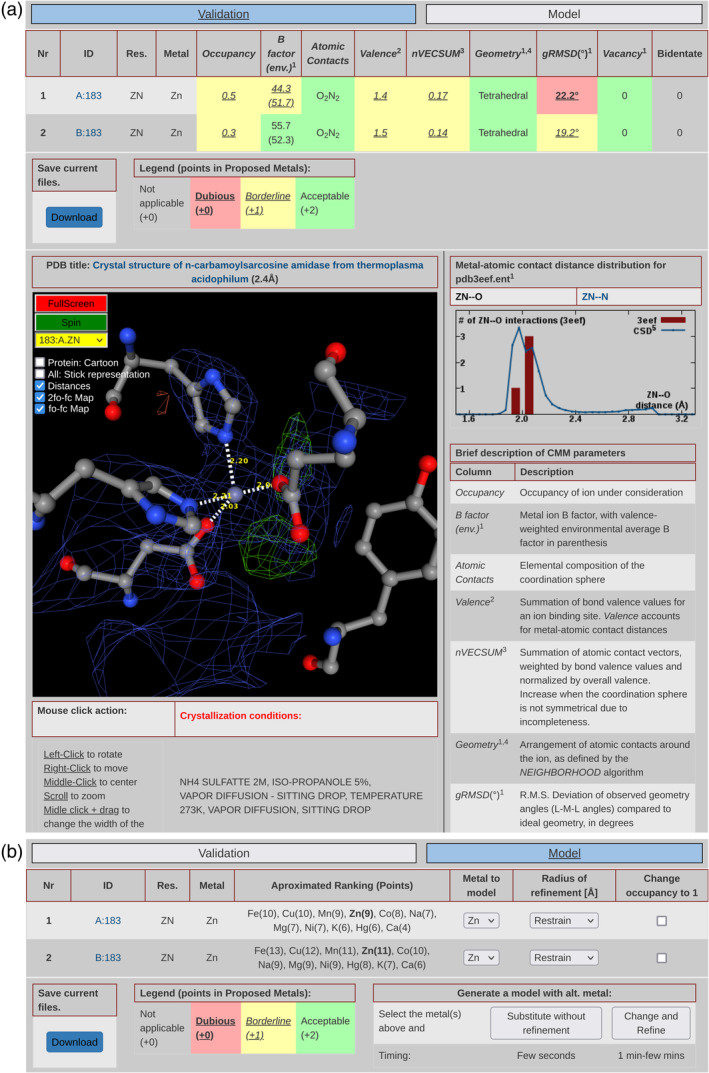
New functionalities of CMM. (a) The CMM validation analysis after uploading a structural model. On the top, a user can switch between the validation and model modules. Below is a table with parameters per each metal binding site. The NGL viewer renders the view of the selected metal binding site. The distribution of distances between metal and binding atoms is shown on the right side. Under the viewer window, the crystallization conditions are displayed, if available. (b) In the model tab, the ranking of alternative metal ions is presented for each metal binding site. Metal ions reported in crystallization conditions are underlined, and the currently modeled ion is in bold. For each metal binding site, a user can change the metal ion, adjust the radius of refinement, change the occupancy, and apply all these changes with or without structure refinement.
The current version of CMM provides many new functionalities. Visually, the most notable change is the use of the modern NGL viewer (Rose et al., 2018; Rose & Hildebrand, 2015), which displays electron density maps, including anomalous density maps when an anomalous signal is present in the data. The view is centered on the metal and shows the surrounding environment. It is now possible to examine details in full‐screen mode. Atomic contacts with the metal less than 3.2 Å are shown with a dotted line with the interatomic distances. By default, only residues close to the metal are shown, but the rest of the protein can be displayed in a cartoon or stick representations to help contextualize the metal binding site. All of the NGL viewer controls are available, that is, rotation, translation, zooming, panning, etc. To switch between the different metal binding sites of the protein, one may either click on the site ID in the site list or select the site in the in‐display menu.
2.1. CMM input data
Structures that the PDB has released can be analyzed by providing their PDB ID. All the relevant files, such as structure factors and electron density maps, are automatically loaded if available. As previously, structures with metals can be uploaded, but the present CMM version allows electron density maps to be uploaded as well, including anomalous difference maps. CMM can now model and refine x‐ray structures using uploaded structure factor data. Some structural models deposited before 2007 and structures determined with methods other than x‐ray/neutron diffraction do not have structure factor files; thus, maps and refinement are not available.
2.2. Modeling and refinement
There are many reasons a metal binding site may be suboptimal, and some, such as poor density of the map, cannot be remedied. However, previous experience shows that cognitive bias frequently influences modeling decisions. One result of cognitive bias influencing modeling decisions was exemplified when we encountered examples where another metal ion in the crystallization solution should be clearly placed into the model. When validation parameters or the difference map for a particular metal‐binding site are questionable, then the assignment of the metal should be considered in detail. CMM provides a mechanism to check a model containing an alternative metal for one or more metal binding sites.
One of the most significant improvements to CMM is the introduction of modeling and on‐the‐fly refinement functionality in REFMAC5 (Kovalevskiy et al., 2016; Murshudov et al., 2011). Switching to the modeling module of CMM changes the central table (Figure 3b) functionality. Within the modeling module, each metal binding site is listed as a table row, but the validation information is replaced with information and possible options for altering the model. A ranked list of suggested metals is provided for each metal site, along with the score calculated for each alternative metal. These rankings are a simple sum of the score for each parameter, with Acceptable parameters having weight two times higher than Borderline parameters. Obviously, this should be considered as rough approximations to guide the order of a more thorough analysis.
x‐Ray PDB deposits contain a free‐form text field with author‐specified crystallization conditions, but this information is missing for 12% of all structures containing metals. Metals present in the crystallization conditions will be underlined in the list of alternative metals. CMM looks for keywords describing metals (i.e., Na+, sodium, Na2SO4) in “REMARK 280” records to get information about possible metals. However, the conditions listed in the deposit may not list all metal ions. Similarly, ions introduced before the crystallization experiment may be bound to the macromolecule. For example, many structures do not mention the solution the protein is stored in before setting up drops, and the counter ion used to adjust the pH of buffers is often omitted. Suppose that a protein was stored in a solution containing sodium chloride. In that case, sodium may be bound to the protein even if it is not explicitly declared to be in the crystallization solution. Some of the missing crystallization information may be present in the publication. Thus, the ions CMM underlines should have priority, but other ions should also be considered. Similarly, some metals from the endogenous source may be tightly bound to the protein and thus present in the structure.
Each metal binding site in the table has options for model generation with alternative metals. An option to set the occupancy of the metal ion to one is present and selected by default. A new model can be generated using a simple metal substitution. For simple substitutions, some parameters, such as the B‐factors, will stay the same. However, since different metals have different preferred geometry and atomic contacts, changes to the other parameters should be inspected.
Crystallographic refinement always incorporates constraints on bond lengths and angles, and a simple substitution model is always suboptimal. To circumvent this limitation, CMM uses REFMAC5 (Brown et al., 2015) to refine the model with an alternative metal. For each site, the “Radius of refinement” is the radius of the sphere of residues allowed to move during refinement. For instance, if the radius is set to 1 Å, then the only atom permitted to move during refinement will be the metal ion itself. “Radius of refinement” selection roughly corresponds to the metal itself, the first coordination sphere, the second coordination sphere, and larger. Limiting the movable region can prevent refinement from unintentionally changing distant parts of the macromolecule. Files downloaded from CMM should be treated by a few cycles of refinement using an appropriate refinement protocol (Shabalin et al., 2018).
The refinement module allows for interactive modeling of metal ions and fast evaluation of how the selected changes alter each parameter and affect the electron density maps, especially the difference map. This type of analysis is especially important for substitutions that have similar binding site geometry yet differ in the number of electrons, that is, Na+ and Ca2+.
Within the PDB, some metals have been modeled with an occupancy of less than one, indicating that only a fraction of the metal‐binding sites within the crystal lattice is occupied. However, it seems that sometimes the occupancy of such metals has been refined to account for peaks in the difference map resulting from metal misidentification. Thus, CMM provides an option to set the occupancy to one when checking alternate metals.
After refinement, CMM returns to the validation module. The maps resulting from refinement are shown in the molecular display window. The graphs of the distribution of atomic distances represent the new metal, and the validation color‐coding represents the refined structural model.
2.3. Testing of CMM functionalities
To thoroughly test the new CMM methodology and functionalities, we performed various checks of metal‐containing structures downloaded from the PDB. Out of ~62,000 PDB deposits downloaded from the PDB in April 2022, only 22,270 were selected for the final test. We eliminated deposits that (1) did not have structure factors, (2) contained metal ions that were in contact with fewer than two residues that were not water, (3) had resolution worse than 2.0 Å, and (4) belonged to the so‐called group depositions, like PanDDA structures (Jaskolski et al., 2022). In cases when the same macromolecule had more than one deposit, all deposits were checked independently. Only the last version was considered when the deposit had several versions (wwPDB consortium, 2019). Figure 4 shows examples of cases that satisfy the above criteria (Figure 4a–c) and those excluded from further analyses (Figure 4d–f). For example, Figure 4d shows a metal that is not bound to any macromolecule residue, Figure 4e shows no electron density around the Zn metal and is a PanDDA structure, and Figure 4f has a resolution too poor to use as a test case.
FIGURE 4.
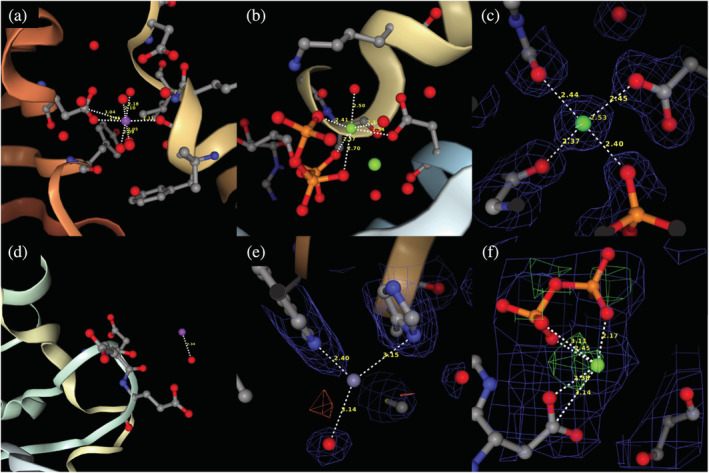
Examples of metal binding sites included (a‐c) and excluded (d‐f) from the test set of structures. (a) 1PJ5, (b) 7ESB, (c) 1HN4, (d) 1G8H (metal not bound to the protein), (e) 5R66 (no density for Zn, PanDDA structure (Jaskolski et al., 2022)), (f) 1AON (poor resolution, 3.0 Å)
In this set of 22,270 structures, we scrutinized structures that had obvious issues related to metal ion binding sites. We first checked if the use of alternative metals could fit and produce better validation parameters, starting with the metals at the top of the ranked metal list (Figure 3b). We simultaneously examined the difference electron density map at the binding site. Clearly, it is impossible to examine 22,270 cases visually, so we used the program MAPMAN (Kleywegt & Jones, 1996) to obtain the electron density value of the difference map within 1.4 Å from the metal center. Whenever there was a significant difference map around such metal, we examined the possibility of alternate metals occupying the site. Last, we look carefully into the metals listed in the crystallization conditions. For many of these metals, we performed refinement and validation once again.
3. ACCOUNTS, SECURITY, AND DATA PRIVACY
CMM has no accounting system; all uploaded data become inaccessible when the application window is closed. The only information collected for statistical purposes is the internet address and the country where the IP address is registered. Only the Virtual Private Network (VPN) provider's IP address is collected when data come through a VPN. As with any other method, the lack of an accounting system has its negatives. The most inconvenient aspect is that users cannot save sessions and return to previous results. The advantage of this system is the security of data submitted by scientists using CMM. According to majority of users, the advantages of the lack of an accounting system outweigh the disadvantages. Bug reports and suggestions for new functionalities are handled by a sophisticated system that tracks user submitted comments, requests, program inconsistencies, and errors. The list of reported errors is permanent, visible to anybody, and shows the delay between report and correction of the application. Such an implementation allows inspection of previous error submissions and makes new submission in the context of entire effort necessary to keep CMM and any other web application alive. Bug reports, but not requests for new functionality, can be filed anonymously.
3.1. Applications and examples
In this section, we discuss the analysis of PDB 1PVF in CMM to show the new functionalities. More examples can be found in “Help and Examples” on the CMM web page.
3.2. 1PVF
The 1PVF deposit is an isopentenyl diphosphate: dimethylallyl diphosphate isomerase in complex with pyrophosphate PPi (residue name DPO). Frequently, magnesium ions are modeled near PPi, ATP, or ADP to compensate for the significant negative charge around the protein. The 1PVF structure has four metal ion binding sites. Two of these sites have magnesium ions in contact with PPi. The previous version of CMM suggested that magnesium is a good choice for these metal sites; however, the new version of CMM reveals these metal sites have significant positive density in the difference maps around the magnesium, which suggests that these sites contain a metal ion with more electrons. In the ranked list of possible alternative metals, manganese has the same score as magnesium, and MnCl2 is reported in the crystallization conditions. Examination of the manganese‐containing model (alternate) shows that it has equally good geometrical validation scores. After modeling and refinement, the geometrical parameters were still good (Table 2) and the positive density in the difference map had vanished as presented in Figure 5.
TABLE 2.
Table 1PVF
| Residue | Metal | Occupancy | B‐factor (env.) | Atom contacts | Valence | nVECSUM | Geometry | gRMSD | Vacancy | |
|---|---|---|---|---|---|---|---|---|---|---|
| A401 | Original | Mg | 1 | 14.6 (16.6) | O6 | 2.1 | 0.071 | Octahedral | 6° | 0 |
| Refined | Mn | 1 | 14.6 (16.6) | O6 | 2.7 | 0.045 | Octahedral | 5.4° | 0 | |
| B401 | Original | Mg | 1 | 16.4 (16.1) | O6 | 1.7 | 0.054 | Octahedral | 6.3° | 0 |
| Refined | Mn | 1 | 16.4 (16.1) | O6 | 2.3 | 0.06 | Octahedral | 6.2° | 0 |
FIGURE 5.
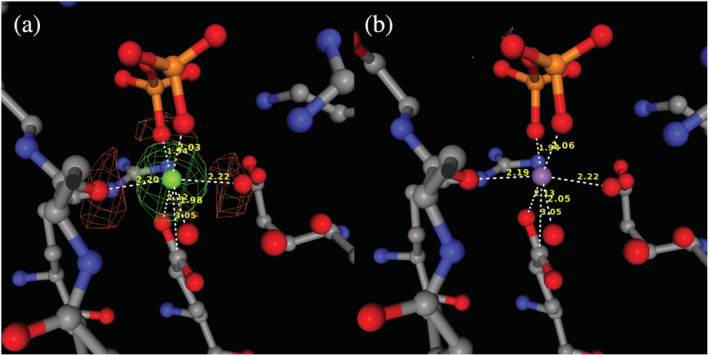
Metal ion binding site (residue: 401, chain: a) of 1PVF before and after refinement of the manganese substitution. The difference map is shown. (a) Site with originally modeled magnesium ion with significant density in difference map. (b) After modeling with manganese and refinement, there is no density in the difference map.
4. CONCLUSIONS
CMM is particularly useful when the experimenter works on structure determination and refinement, that is, when experimental data are already collected. One must realize that identification of metal ions bound to protein/present in a crystal structure should be initiated during the experiment planning and continue during sample preparation, diffraction experiment, structure determination, validation, and analysis. Thus, the expression and purification of metal‐containing proteins, handling of metals with more than one oxidation state, and crystallization are essential for studying the metal active site. For example, the pH of the crystallization solution influences the binding properties of many amino acids in the metal binding environment. It was shown that histidine's protonation state and conformation induced by pH changes the geometry of the main zinc‐binding site in albumin, which is responsible for transporting 98% of Zn2+ in mammalian blood plasma (Handing et al., 2016).
The selection of a tunable synchrotron beamline for the diffraction experiment allows for almost unambiguous identification of metal ions. Collecting two diffraction data sets, one above and one below a metal absorption edge, can confirm the metal's identity and location. The presence of an anomalous signal above the edge and the absence of a signal below the edge confirm the identity of the metal. Two wavelength experiments for zinc or cobalt are straightforward for most tunable synchrotron stations, which usually operate in the range of 0.8–2.3 Å. The absorption edges for zinc and cobalt are 9.6586 keV (1.2837 Å) and 7.7089 (1.6083 Å), respectively. However, experiments with long wavelengths are tricky because of the high x‐ray absorption by the air. Many experiments are performed on fixed wavelength beamlines or home sources (in 2021, approximately 10% of deposits came from the data collected at home sources). For single wavelength experiments, the new version of CMM is an extremely valuable tool that allows checking the correctness of the metal identification and provides the ability to model alternate metals and perform simple refinement within the same web application.
Researchers modeling metal sites in macromolecules can only benefit from checking the quality of metal binding using CMM. However, the models coming from CMM will almost certainly need additional rounds of refinement before the structure is deposited in the PDB, especially if refinement in CMM has been performed with only a portion of the structure. The refinement performed in CMM uses many default parameters, and researchers are encouraged to use their preferred refinement protocols (Shabalin et al., 2018). Optimal refinement protocols can have a significant impact on the quality of the structure. Modeling metal binding sites can be difficult because they lie at the interface of organic and inorganic chemistry. The motivation behind the further development of this already very popular web application is the recognition that “good enough” is not good enough to improve the reproducibility of biomedical research.
5. MATERIALS AND METHODS
The server has been implemented using the CakePHP 4 framework. Electron density maps are generated from structure factors using the FFT program from CCP4 (Winn et al., 2011) using a grid sampled at 1/2.5 of the maximum resolution. The refinement is performed using REFMAC5 (Murshudov et al., 2011; Nicholls, 2017). The interactive parts of the website (project editing and viewing) are built using the JavaScript framework. CMM uses the NGL viewer for displaying models and electron density maps. CMM is currently optimized for desktop presentation and has been tested on current versions of Google Chrome (on Linux, Windows, and macOS), Mozilla Firefox (on Linux, Windows, and MacOS), Microsoft Edge (on Windows), and Apple Safari (on MacOS). The CMM application is currently being transferred to the Django framework, allowing for new, better functionalities.
AUTHOR CONTRIBUTIONS
Michal Gucwa: Formal analysis (equal); investigation (equal); software (equal); validation (equal); writing – original draft (equal). Joanna Lenkiewicz: Data curation (equal); formal analysis (equal); investigation (equal); software (equal). Heping Zheng: Formal analysis (equal); methodology (equal); software (equal); writing – review and editing (equal). Marcin Cymborowski: Formal analysis (equal); investigation (equal); software (equal). David R. Cooper: Investigation (equal); resources (equal); software (equal); validation (equal); writing – original draft (equal); writing – review and editing (equal). Krzysztof Murzyn: Conceptualization (equal); investigation (equal); methodology (equal); supervision (equal); writing – original draft (equal); writing – review and editing (equal). Wladek Minor: Conceptualization (equal); data curation (equal); formal analysis (equal); funding acquisition (equal); investigation (equal); methodology (equal); project administration (equal); resources (equal); supervision (equal); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal).
CONFLICT OF INTEREST
Wladek Minor notes that he has also been involved in the development of software and data management and data‐mining tools; some of these have been commercialized by HKL Research. Wladek Minor is the cofounder of HKL Research and a member of the board. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
ACKNOWLEDGMENTS
The authors thank Zbyszek Dauter, Alex Wlodawer, Ivan Shabalin, Vanessa Bijak, and Shrisha Poonuganti for valuable discussions and software testing. Funding for this research was provided by NIH grants GM 117325 and GM132595.
Gucwa M, Lenkiewicz J, Zheng H, Cymborowski M, Cooper DR, Murzyn K, et al. CMM—An enhanced platform for interactive validation of metal binding sites. Protein Science. 2023;32(1):e4525. 10.1002/pro.4525
Review Editor: Nir Ben‐Tal
Funding information National Institutes of Health, Grant/Award Numbers: GM117325, GM132595
Contributor Information
Krzysztof Murzyn, Email: krzysztof.murzyn@uj.edu.pl.
Wladek Minor, Email: wladek@iwonka.med.virginia.edu.
DATA AVAILABILITY STATEMENT
All data are available at: https://cmm.minorlab.org
REFERENCES
- Brown A, Long F, Nicholls RA, Toots J, Emsley P, Murshudov G. Tools for macromolecular model building and refinement into electron cryo‐microscopy reconstructions. Acta Crystallogr D Biol Crystallogr. 2015;71:136–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley SK, Berman HM, Bhikadiya C, Bi C, Chen L, Di Costanzo L, et al. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019;47:D464–74. 10.1093/nar/gky1004/5144139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chruszcz M, Domagalski M, Osinski T, Wlodawer A, Minor W. Unmet challenges of structural genomics. Curr Opin Struct Biol. 2010;20:587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DR, Porebski PJ, Chruszcz M, Minor W. X‐ray crystallography: assessment and validation of protein‐small molecule complexes for drug discovery. Expert Opin Drug Discov. 2011;6:771–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domagalski MJ, Zheng H, Zimmerman MD, Dauter Z, Wlodawer A, Minor W. The quality and validation of structures from structural genomics. Methods Mol Biol. 2014;1091:297–314. 10.1007/978-1-62703-691-7_21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington TM, Mathur M, Soderberg CK, Denis A, Perfito N, Iorns E, et al. Investigating the replicability of preclinical cancer biology. Elife. 2021;10:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington TM, Denis A, Perfito N, Iorns E, Nosek BA. Challenges for assessing replicability in preclinical cancer biology. Elife. 2021;10:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handing KB, Shabalin IG, Kassaar O, Khazaipoul S, Blindauer CA, Stewart AJ, et al. Circulatory zinc transport is controlled by distinct interdomain sites on mammalian albumins. Chem Sci. 2016;7:6635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handing KB, Niedzialkowska E, Shabalin IG, Kuhn ML, Zheng H, Minor W. Characterizing metal‐binding sites in proteins with X‐ray crystallography. Nat Protoc. 2018;13:1062–90. 10.1038/nprot.2018.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding MM, Nowicki MW, Walkinshaw MD. Metals in protein structures: a review of their principal features. Crystallogr Rev. 2010;16:247–302. [Google Scholar]
- Jaskolski M, Wlodawer A, Dauter Z, Minor W, Rupp B. Group depositions to the Protein Data Bank need adequate presentation and different archiving protocol. Protein Sci. 2022;31:784–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA. xdlMAPMAN and xdlDATAMAN ‐ programs for reformatting, analysis and manipulation of biomacromolecular electron‐density maps and reflection data sets. Acta Crystallogr D Biol Crystallogr. 1996;52:826–8. [DOI] [PubMed] [Google Scholar]
- Kovalevskiy O, Nicholls RA, Murshudov GN. Automated refinement of macromolecular structures at low resolution using prior information. Acta Crystallogr Sect D Struct Biol. 2016;72:1149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppuraj G, Dudev M, Lim C. Factors governing metal‐ligand distances and coordination geometries of metal complexes. J Phys Chem B. 2009;113:2952–60. [DOI] [PubMed] [Google Scholar]
- Majorek K, Kuhn ML, Chruszcz M, Anderson WF, Minor W. Double trouble‐buffer selection and his‐tag presence may be responsible for nonreproducibility of biomedical experiments. Protein Sci. 2014;23:1359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls RA. Ligand fitting with CCP 4. Acta Crystallogr D Struct Biol. 2017;73:158–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedzialkowska E, Gasiorowska O, Handing KB, Majorek KA, Porebski PJ, Shabalin IG, et al. Protein purification and crystallization artifacts: the tale usually not told. Protein Sci. 2016;25:720–33. 10.1002/pro.2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosek BA, Errington TM. The best time to argue about what a replication means? Before you do it. Nature. 2020;583:518–20. [DOI] [PubMed] [Google Scholar]
- Pozharski E, Weichenberger CX, Rupp B. Techniques, tools and best practices for ligand electron‐density analysis and results from their application to deposited crystal structures. Acta Crystallogr Sect D Biol Crystallogr. 2013;69:150–67. [DOI] [PubMed] [Google Scholar]
- Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov. 2011;10:712. 10.1038/nrd3439-c1 [DOI] [PubMed] [Google Scholar]
- Rose AS, Hildebrand PW. NGL viewer: a web application for molecular visualization. Nucleic Acids Res. 2015;43:W576–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AS, Bradley AR, Valasatava Y, Duarte JM, Prlic A, Rose PW. NGL viewer: web‐based molecular graphics for large complexes. Bioinformatics. 2018;34:3755–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalin IG, Porebski PJ, Minor W. Refining the macromolecular model – achieving the best agreement with the data from X‐ray diffraction experiment. Crystallogr Rev. 2018;24:236–62. 10.1080/0889311X.2018.1521805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart OS, Horský V, Gore S, Svobodová VR, Bendová V, Kleywegt GJ, et al. Validation of ligands in macromolecular structures determined by X‐ray crystallography. Acta Crystallogr Sect F Struct Biol. 2018;74:228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure‐factor data and their agreement with the atomic model. Acta Crystallogr Sect D Biol Crystallogr. 1999;55:191–205. [DOI] [PubMed] [Google Scholar]
- Weichenberger CX, Pozharski E, Rupp B. Visualizing ligand molecules in twilight electron density. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013;69:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodawer A, Minor W, Dauter Z, Jaskolski M. Protein crystallography for non‐crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J. 2008;275:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodawer A, Minor W, Dauter Z, Jaskolski M. Protein crystallography for aspiring crystallographers or how to avoid pitfalls and traps in macromolecular structure determination. FEBS J. 2013;280:5705–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- wwPDB consortium . Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019;47:D520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Chruszcz M, Lasota P, Lebioda L, Minor W. Data mining of metal ion environments present in protein structures. J Inorg Biochem. 2008;102:1765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Hou J, Zimmerman MD, Wlodawer A, Minor W. The future of crystallography in drug discovery. Expert Opin Drug Discov. 2014;9:125–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Chordia MD, Cooper DR, Chruszcz M, Müller P, Sheldrick GM, et al. Validation of metal‐binding sites in macromolecular structures with the CheckMyMetal web server. Nat Protoc. 2014;9:156–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available at: https://cmm.minorlab.org