

Abstract
The care and treatment of patients with craniosynostosis and the new developments were described for health care professionals involved in this in the guideline “Treatment and Management of Craniosynostosis”, which was revised in 2020. A patient version was written based on the professional guideline to make the information accessible to patients and parents too. In this patient version, each chapter consists of several sections. Firstly, an introduction and background information is provided in each chapter. Various questions are then answered based on scientific literature. Finally, the recommendations indicate the importance of the literature for care in practice and how this care should be provided in practice. This patient version is an abbreviated and simplified representation of the professional guideline. The introduction, conclusions, and recommendations sections of each chapter were revised and, where necessary, rewritten. With some surgical techniques, links to animation videos (recognizable by underlined references) have been added in the text for clarification. An attempt was made to stay as close as possible to the original guideline in terms of content, questions, numbering, and classification. The patient version can therefore easily be read side by side with the professional guideline if more information is required about a specific subject. As this patient version is a summary and does not deal with all aspects in detail, no rights can be derived from its content and the professional guideline takes precedence at all times. Originally, this patient version has been written in response to the established Dutch guideline on craniosynostosis for health care professionals.2 This professional guideline has been specifically tailored to the Dutch health care setting and policy. There are however differences between health care systems and national health policies of other countries and the Netherlands. It is important to keep in mind that this may, at some points, result in the management of care in your country and/or hospital different from outlined here.
Key Words: craniosynostosis, guideline, patient version, treatment
CHAPTER 1. GENERAL INTRODUCTION
Reasoning
Craniosynostosis is estimated to occur in 4.4 to 7.2 children per 10,000 live births. Syndromic craniosynostosis is expected to occur in 0.9 to 1.6 children per 10,000 live births.1,2 These ranges are defined by recent scientific studies in Norway3 and the Netherlands.4 Although we do not know the exact number of people with craniosynostosis across Europe, large differences across European countries are not expected. Different European countries have different health care systems and therefore the number of hospitals treating patients with craniosynostosis is different per country.5
Objective
This guideline document contains recommendations to support daily practice where craniosynostosis is suspected and after confirmation of this diagnosis. The guideline provides recommendations for health care providers in recognizing craniosynostosis, the logistics involved in parent referrals to craniofacial centres, multidisciplinary care within a craniofacial centre, and requirements that a craniofacial centre and its members must meet. Thus, the guideline provides a focus on uniform care in craniosynostosis and the implementation of this care in the Netherlands. This section looked at craniosynostosis of 1 cranial suture (isolated), multiple cranial sutures (multisuture), and syndromic craniosynostosis. The first guideline was issued in 2010. In 2017, the Dutch Society for Plastic and Reconstructive Surgery decided to revise the guideline as a number of items required an update based on recent scientific literature, and because the topics on prenatal detection and speech/language development were not yet included.
Target Group
This version of the guideline is primarily intended for parents and patients.
About Craniosynostosis
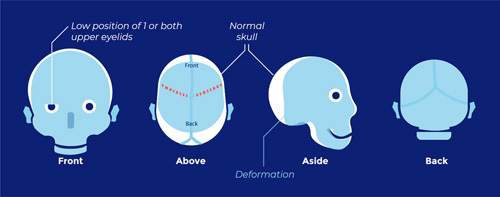
Craniosynostosis concerns a congenital skull defect, in which 1 or more cranial sutures are already fused before birth. The cranial sutures are located between the bone plates of the skull and allow for the rapid growth of the skull in the first 2 years of life. The growth of the skull is largely controlled by the growth of the brain.
Cranial sutures are essential for skull growth in the first 2 years (during the brain’s rapid growth). Premature fusion of cranial sutures prevents normal skull growth, resulting in characteristic shape deviations of the skull.
Craniosynostosis occurs in 1 in every 2100 to 2500 births and may occur as either nonsyndromic (also indicated as isolated) or syndromic. Syndromic craniosynostosis occurs when other birth defects are present in addition to craniosynostosis. In syndromic cases, several cranial sutures are often fused, usually involving both coronal sutures.
The distinction between nonsyndromic and syndromic is determined by a clinical geneticist doing a physical examination and through genetic testing.
The types of craniosynostosis are classified as follows:
Isolated (a single fused suture), nonsyndromic:
-
Sagittal suture synostosis (scaphocephaly; characterized by a long narrow head):
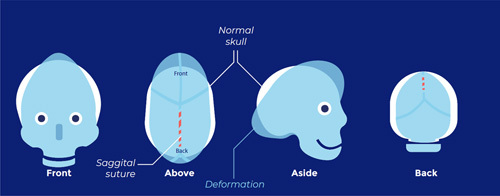
(All images: ERN CRANIO, accessed September 19, 2022).6
-
Metopic suture synostosis (trigonocephaly; characterized by a triangular forehead):
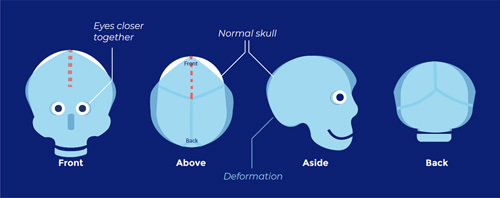
Coronal suture synostosis, 1-sided (frontal plagiocephaly; flattening of 1 side of the forehead):
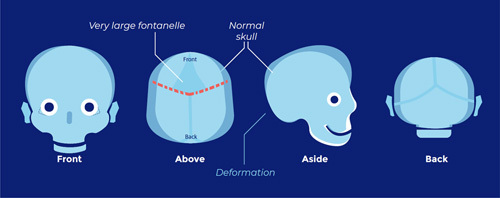
Lambdoid suture synostosis (pachycephaly; flattening of the back of the head):
Multisuture (multiple fused sutures) or syndromic:
-
Apert syndrome (FGFR2 mutation Ser252Trp and Pro253Arg, deletion exon IIIc, Alu insertion exon IIIc):
-
Crouzon or Pfeiffer syndrome (FGFR2 mutations except Apert mutations, rarely FGFR1 mutations or—if combined with the skin condition acanthosis nigricans—FGFR3 mutations):
-
Saethre-Chotzen syndrome (TWIST1 mutations or deletions):
Muenke syndrome (Pro250Arg FGFR3 mutation):
Craniofrontonasal dysplasia (EFNB1 mutations)
TCF12 associated craniosynostosis
ERF associated craniosynostosis
ILRA associated craniosynostoses
Multisuture craniosynostoses (also called complex craniosynostoses), often 2 or more fused cranial sutures with no known genetic cause.
CHAPTER 2. METHODOLOGY FOR GUIDELINE DEVELOPMENT
The following persons participated in updating and revising the guideline:
-
Nederlandse Vereniging voor Plastische Chirurgie (Dutch Association for Plastic Surgery)
Prof I.M.J. Mathijssen, Erasmus Universitair Medisch Centrum (Erasmus University Medical Center), Rotterdam
Dr S.L. Versnel, Erasmus University Medical Center, Rotterdam Patiënten- en oudervereniging LAPOSA (LAPOSA Patients and Parents Association)
Ms B. Lieuwen, Msc, Ma
-
Nederlands Instituut voor Psychologen (Dutch Association of Psychologists) + Landelijke Vereniging Medische Psychologie (National Association of Medical Psychology)
Dr J.M.E. Okkerse, Erasmus University Medical Center, Rotterdam
Mr J.J. Reuser, Radboud University Medical Center, Nijmegen Nederlands Oogheelkundig Gezelschap (Dutch Ophthalmic Society)
Dr S.E. Loudon, Erasmus University Medical Center, Rotterdam Nederlandse Vereniging voor Anesthesiologie (Dutch Society of Anesthesiology)
Mr A. Gonzalez Candel, Erasmus University Medical Center, Rotterdam Nederlandse Vereniging voor Keel-, Neus-en Oorheelkunde (Dutch Society for Ear Nose and Throat Surgery)
Dr M.P. van der Schroeff, Erasmus University Medical Center/Sophia, Rotterdam
Ms H.H.W. de Gier, Erasmus University Medical Center/Sophia, Rotterdam
-
Nederlandse Vereniging voor Kindergeneeskunde (Dutch Pediatric Association)
Dr K.F.M. Joosten, Erasmus University Medical Center/Sophia, Rotterdam
Dr N. Bannink, Franciscus Gasthuis and Vlietland, Rotterdam and Schiedam
Mr L.G.F.M. van ‘t Hek, Radboud University Medical Center, Nijmegen Nederlandse Vereniging voor Mondziekten, Kaak-en Aangezichtschirurgie (Dutch Association for Oral and Maxillofacial Surgery)
Prof E.B. Wolvius, Erasmus University Medical Center, Rotterdam
Dr W.A. Borstlap, Radboud University Medical Center, Nijmegen Nederlandse Vereniging voor Neurochirurgie (Dutch Society of Neurosurgery)
Dr M.L.C. van Veelen, Erasmus University Medical Center, Rotterdam
Dr H.H.K. Delye, Radboud University Medical Center, Nijmegen Vereniging Klinische Genetica Nederland (Dutch Society of Clinical Genetics)
Dr M.F. van Dooren, Erasmus University Medical Center, Rotterdam Vereniging Klinische Genetische Laboratoriumdiagnostiek (Association of Clinical Genetic Laboratory Diagnostics)
Dr R. Pfundt, Radboud University Medical Center, Nijmegen Nederlandse Vereniging voor Logopedie en Foniatrie (Dutch Society for Speech Therapy and Phoniatrics)
Dr M.C.J.P. Franken, Erasmus University Medical Center/Sophia, Rotterdam
Ms E. Kerkhofs, Radboud University Medical Center, Nijmegen Nederlandse Vereniging voor Obstetrie and Gynaecologie (Prenatale geneeskunde) (Dutch Society for Obstetrics and Gynecology (Prenatal Medicine))
Dr T.E. Cohen-Overbeek, Erasmus University Medical Center/Sophia, Rotterdam
Ms M. Woiski, Radboud University Medical Center, Nijmegen Nederlandse Vereniging voor Orthodontisten (Dutch Union of Orthodontists)
Dr S.T.H. Tjoa, Erasmus University Medical Center/Sophia, Rotterdam
-
Nederlandse Vereniging voor Radiologie (The Radiological Society of the Netherlands)
Dr M.H.G. Dremmen, Erasmus University Medical Center/Sophia, Rotterdam
-
Nederlandse Vereniging Relatie-en gezinstherapie (Dutch Association for Relationship and Family Therapy)
Ms F. Meertens, Erasmus University Medical Center/Sophia, Rotterdam Koninklijk Nederlands Genootschap voor Fysiotherapie/Nederlandse Vereniging voor Fysiotherapie in de Kinder- en Jeugdgezondheidszorg (Royal Dutch Society for Physiotherapy/Dutch Association for Physiotherapy in Child and Youth Health Care)
Dr L.A. van Vlimmeren, Radboud University Medical Center, Nijmegen Nederlandse Vereniging voor Psychiatrie (Dutch Society for Psychiatry)
Ms M.H.M. van Lier, Erasmus University Medical Center/Sophia, Rotterdam
-
Nederlandse Vereniging voor Neurologie (Dutch Society for Neurology)
Prof M.A.A.P. Willemsen, Radboud University Medical Center, Nijmegen Supported by:
Ms B.S. Niël-Weise, medical microbiologist (nonpracticing), independent guideline methodologist, Deventer
Dr J.J.A. de Beer, independent guideline methodologist, Utrecht
Mr H. Deurenberg, SIROSS, information specialist, Oss
CHAPTER 3. REFERRAL AND DIAGNOSTICS
Question 3.1: What are the implications for pregnancy care once craniosynostosis is diagnosed prenatally?
Craniosynostosis is very rarely diagnosed during pregnancy. Testing (prenatal diagnosis) can be done in an academic hospital if craniosynostosis is already suspected during pregnancy. Once craniosynostosis has been diagnosed, the counseling and possible treatment will be taken over by the specialist centre. This is necessary because there is a higher risk of obstructed labor during childbirth with all forms of craniosynostosis. Furthermore, syndromic craniosynostosis requires proper care due to possible breathing problems in the baby during birth.
Question 3.2: What is the policy on recognition, referral, and radiological diagnostics in primary or secondary care in children with suspected craniosynostosis?
Craniosynostosis should be recognized in time for optimal treatment. Craniosynostosis patients, however, often turn out not to be recognized or to be referred at a late stage. This is often because it is thought that the child’s preferred posture is the cause of an abnormal skull shape, which is far more common than craniosynostosis. Because a special flowchart is used more and more in primary or secondary care (general practitioners, clinic doctors, and pediatricians), diagnosis and referral have improved considerably. The presence of an abnormal skull shape immediately after birth, whether or not there is a preferred posture and whether improvement has occurred in the skull shape, are points that are included in the flowchart.
Excessive diagnostics are often carried out before referral to an academic hospital (tertiary care), which leads to a further delay in referral, an additional burden and uncertainty for the patient and parents, and unnecessary costs. Testing can take place in the specialist centre to determine whether and which cranial sutures are fused. This is preferably done with an ultrasound scan of the head as this does not generate radiation. However, it does require more experience from the radiologist. An x-ray of the head can also be taken, but the assessment of this also requires a lot of experience and the examination does generate some radiation. A 3-dimensional computed tomography (3D-CT) scan of the skull is the most reliable examination and can usually be done quickly, but does generate radiation. In the case of syndromic craniosynostosis, magnetic resonance imaging (MRI) of the brain may be necessary.
Question 3.3 What is the policy regarding genetic diagnostics in a child with confirmed or suspected craniosynostosis?
In principle, genetic diagnosis is done in a craniosynostosis specialist centre as soon as the diagnosis is confirmed and once parents have agreed to it. The role of the clinical geneticist within a multidisciplinary craniofacial team is aimed at being able to answer the questions of both the parents and the treating physicians.
The most important questions of the parents are whether their child is otherwise healthy, what the cause of the condition is, how big the chance of recurrence is for any further children in their family and/or future grandchildren, and what the possibilities are of prenatal diagnosis. As to conducting genetic diagnostics, this depends on the parent’s values and preferences.
Recommendations
Question 3.1
If the general practitioner or obstetrician suspects craniosynostosis when doing an ultrasound, the pregnant woman should be referred to an academic hospital for prenatal diagnosis. If the diagnosis of craniosynostosis is made there, the pregnant woman will be referred to the craniosynostosis specialist centre for counseling and guidance.
Question 3.2
Use the flowchart to ensure craniosynostosis is better recognized in primary and secondary care. A child suspected to be suffering from craniosynostosis is referred to a craniosynostosis specialist centre as soon as possible, without additional diagnostics. This increases the likelihood that children may still qualify for a minimally invasive operation before the age of 6 months.
Skull x-rays or an ultrasound of the skull are always done if there is a moderate suspicion of craniosynostosis. If there is a strong suspicion of craniosynostosis based on external features, a 3D-CT is immediately done for diagnostic purposes. Children with syndromic craniosynostosis are sometimes given additional MRI scans to assess other brain disorders and symptoms of increased intracranial pressure (ICP) before surgery.
Question 3.3
Genetic diagnosis is done in a specialist centre.
Targeted genetic testing is done in children with proven craniosynostosis and obvious external features.
More extensive and broader genetic testing is done in children with established craniosynostosis combined with other congenital disorders and/or developmental delays.
CHAPTER 4. PERIOPERATIVE CARE
Question 4.1 What is the perioperative surgical management of craniosynostosis?
The correction of craniosynostosis during childhood can be associated with relatively high blood loss. This risk increases with extensive and open skull surgery. Administering certain medications and collecting blood and returning it to the patient during surgery can reduce blood loss and blood transfusions. In addition to the surgical and anesthetic challenge, other conditions (comorbidities) that may be associated with the syndromic conditions must be taken into account. For this reason, strict organizational conditions must be imposed on the surgical process, before, during, and after the procedure.
Which medicines, blood products, or measures, like inducing low blood pressure or use of the cell saver are effective in reducing blood loss or the need for a blood transfusion?
The use of tranexamic acid (a medicine that prevents the breaking down of blood clots) probably ensures that far fewer blood products have to be administered as a result of blood loss.
The use of a cell saver (a device that collects lost blood and returns it to the patient) and erythropoietin (medicine that promotes the production of red blood cells) may result in fewer blood products having to be transfused due to the occurrence of less blood loss.
The effect of other strategies is still unproven.
By administering fibrinogen (a coagulation factor) based on an extensive coagulation measurement, in which a value of <13 mm is pursued on FIBTEM (part of the coagulation test), there may be less blood loss than when pursuing a value of <8 mm. Whether it is safe to administer fibrinogen at a limit value of 13 mm has not yet been demonstrated.
Administering fresh frozen plasma (blood plasma) before blood loss occurs, does not lead to less blood loss compared with when it is only administered when blood loss has already occurred and there is an immediate need.
No difference in actual blood loss is seen with a mean blood pressure between 55 mm Hg and 65 mm Hg. Therefore, striving for low-normal blood pressure is not of any added benefit.
It is unclear whether the administration of vitamin K1 (a substance that causes the production of clotting factors) leads to less blood loss and fewer blood transfusions.
Recommendations
Question 4.1
Children with craniosynostosis are only treated in a specialized pediatric centre.
Use tranexamic acid during surgery to limit blood loss.
Consider collecting the patient’s blood during surgery (using a cell saver) and then returning it to limit the number of blood transfusions.
Use fresh frozen plasma and/or fibrinogen as soon as signs of abnormal coagulation develop during surgery.
CHAPTER 5. SURGICAL TREATMENT OF ISOLATED, NONSYNDROMIC CRANIOSYNOSTOSIS
Question 5.1 What is the surgical management of nonsyndromic craniosynostosis?
The 4 most common forms of isolated, nonsyndromic craniosynostosis are in order of occurrence: (1) sagittal suture synostosis, (2) metopic suture synostosis, (3) unilateral coronal suture synostosis, and (4) unilateral lambdoid suture synostosis. Unilateral coronal suture synostosis may be associated with a syndrome, such as Muenke or Saethre-Chotzen syndrome, and a possible genetic cause should be considered.
(1) What is the indication for surgical treatment?
Nonsyndromic craniosynostosis can present with varying severity of skull abnormality. Surgical treatment seems to be assessed based on:
The associated risk of ICP
Preventing or limiting associated brain abnormalities
The external abnormality (with both esthetic and psychological consequences) Surgical treatment of sagittal suture, unilateral coronal suture, and unilateral lambdoid suture synostosis is indicated as no spontaneous improvement of the abnormal skull shape is expected.
In children with a mild or moderate triangle-shaped skull (metopic synostosis), there is doubt as to the usefulness and necessity of surgery. Only with a pronounced “severe” triangle-shaped skull does surgery actually improve the appearance.
(2) What are the patient-relevant effects of different surgical techniques, in particular minimally invasive surgery versus open skull surgery for the 4 types of nonsyndromic synostosis?
Many different surgical techniques have been described for the treatment of unilateral, nonsyndromic craniosynostosis. Minimally invasive surgery (the removal of the fused cranial suture and helmet therapy or spring-assisted distraction) and open skull correction are the current techniques being used. Minimally invasive surgery is associated with less blood loss, fewer blood transfusions, shorter surgery duration and admission time, and a similar esthetic result in both sagittal sutures,7 metopic suture,8 unicoronal suture,9 and unilambdoid suture synostosis compared with “classic” open skull surgery of the metopic suture,10 coronal suture,11 sagittal suture,12 and lambdoid suture.
Spring-assisted distraction in sagittal suture synostosis13 is probably less likely to lead to ICP in the years after surgery than an open skull correction. Ophthalmic results after a minimally invasive procedure with coronal suture synostosis may also be better than with an open correction. It is as yet unclear whether this is due to the type of operation, the severity of the abnormality, or the time, at which surgery is done.
However, there is still little information on the occurrence of ICP in follow-up, the development of long-term appearance and neurocognitive outcomes in children who have undergone minimally invasive surgery.
There is no scientific evidence to make a choice between the 2 methods of minimally invasive surgery (fused cranial suture removal and helmet therapy versus spring-assisted distraction).
(3) What are patient-relevant effects of the different timing of surgery, that is, “early” (under 6 mo of age) versus “late” (over 6 mo of age)?
Minimally invasive surgery is almost always performed before the age of 6 months. Open cranial corrections are mainly performed after this age. In children with sagittal suture synostosis, the likelihood of developing ICP increases over the course of the first year of life (from 2.5% at 6 mo to 10% at 11 mo).
Surgery is therefore advisable before the age of 6 months. With metopic suture synostosis, the probability of ICP within the first year of life remains low. There is no need to perform the operation before the age of 6 months.
An early endoscopic operation may lead to better ophthalmic results in coronal suture synostosis. This may be because of the early timing of the operation or because it is usually only children with a “mild” form who undergo this type of surgery.
The early and late treatment for unilateral lambdoid suture synostosis may lead to a similar esthetic result.
Results of the comparisons between open and minimally invasive corrections come from studies that collectively present weak evidence due to limitations in the studies themselves or because studies do not show quite the same results.
Recommendations
Question 5.1
Do not operate on children with a bony ridge over the metopic suture or with a mild form of trigonocephaly. No recommendation is given as to whether or not to operate on children with a moderate form of trigonocephaly.
At the age of 5 years, assess the appearance of children with a mild and moderate form of trigonocephaly who have not had surgery yet.
Surgical correction of the abnormality is indicated in all other forms of isolated, nonsyndromic craniosynostosis.
Perform minimally invasive surgery on a child with sagittal suture synostosis if younger than 5.5 to 6 months. If the child is older, an open correction is preferably done. No recommendation is given with regard to the type of surgery with metopic suture, unicoronal suture, and unilateral lambdoid suture synostosis.
Surgery for isolated nonsyndromic craniosynostosis is done within the first year of life.
With sagittal suture synostosis, the operation is preferably done before the age of 6 months.
For metopic suture, unicoronal suture, and unilateral lambdoid suture synostosis, there is no recommendation with regard to the timing of the operation.
Early referral to the specialist centre (well before the age of 6 mo) ensures the minimally invasive surgical option is possible.
CHAPTER 6. SURGICAL TREATMENT OF SYNDROMIC CRANIOSYNOSTOSIS—THE CRANIAL VAULT
Question 6.1 What is the policy on surgical treatment of the cranial vault in multisuture and syndromic craniosynostosis?
The distinction between multisuture craniosynostosis (multiple fused cranial sutures) and syndromic craniosynostosis is made based on external features. Multisuture craniosynostosis can occur in all variations of 2 or more affected cranial sutures. In this group, new genetic causes for craniosynostosis are still identified, such as the genes TCF12, ERF, and IL11RA. Multiple congenital defects are present in syndromic craniosynostosis. The 4 most common forms of syndromic craniosynostosis are: (1) Apert, (2) Crouzon (including Pfeiffer syndrome), (3) Saethre-Chotzen, and (4) Muenke syndrome.
(1) What are the patient-relevant effects of different indications for surgical treatment of multisuture and syndromic craniosynostosis, that is, routine treatment versus in response to signs of elevated intracranial pressure?
Internationally, there are different opinions regarding the type of operation that is carried out first and when this happens. In the various international centres, the first skull operation is sometimes an expansion of the occiput14 or the forward positioning of the forehead. The first skull expansion is often performed at a certain age, based on protocol, but in 1 specific centre, only when signs of ICP have been detected. The number of children with Apert or Crouzon syndrome who are operated on, when this is done according to protocol, is 10% to 20% higher than when surgery only takes place based on symptoms of ICP. The latter option can only be done safely if there is frequent testing for signs of ICP, such as through an ophthalmoscope or other ophthalmic examinations. However, these tests are not 100% reliable, so ICP can be missed.
In patients with Saethre-Chotzen syndrome or multisuture craniosynostosis, surgery is required for both abnormal skull shape and the risk of ICP. For Muenke syndrome, an abnormal skull shape is the main indication for surgery, given the low risk of ICP. There is no relevant difference in the occurrence of ICP in Apert and Crouzon syndrome after 5 years of follow-up among patients who have had protocol-based surgery than with surgery after signs of ICP. With Saethre-Chotzen syndrome, Muenke syndrome, and multisuture craniosynostosis, there is as yet no clarity about this.
(2) What are the long-term surgical specific outcomes of different surgical techniques, in particular minimally invasive surgery (endoscopic strip craniectomy with helmet therapy or spring-assisted distraction or conventional distraction (slowly twisting bone elements apart) of the occiput versus open cranial correction (of forehead or occiput)?
An occipital expansion14 (with distraction or springs) in patients with Apert and Crouzon syndrome probably results in an increased skull circumference, increased cranial volume, less deviation in the positioning of the cerebellum (tonsillar herniation), and a reduced occurrence of ICP compared with an expansion of the forehead or an expansion of the occiput without distraction. These better outcomes of occipital surgery are found up to 5 years after surgery.
Minimally invasive surgery through endoscopic removal of the fused coronal sutures with helmet therapy in syndromic craniosynostosis has a higher risk of repeat surgery due to delayed skull growth or signs of excessive intracranial pressure occurring within 1 year of surgery. An endoscopic operation does have less blood loss, shorter surgical time, and shorter hospitalization time than an open skull operation.
(3) What are the long-term results regarding cognition and esthetics (appearance) of the different timing of surgery, that is, “early”, defined as before the age of 12 months, versus “late”, that is, after the age of 12 months?
Patients with syndromic craniosynostosis or in whom both coronal sutures are fused, who undergo cranial surgery within the first 12 months, may have a higher Intelligence Quotient (IQ) than patients who are operated on after the first 12 months.
Cranial surgery from the age of 6 to 9 months gives a better esthetic result in patients with Muenke syndrome than an earlier operation. Excessive intracranial pressure is relatively rare in this syndrome, and therefore this “later” operation cannot harm. Cranial surgery between 6 and 9 months in patients with Apert, Crouzon, or Saethre-Chotzen syndrome leads to better esthetic results than surgery before or after that period.
Recommendations
Question 6.1
Operate on children with syndromic craniosynostosis or multisuture craniosynostosis.
Patients should be screened regularly for ICP if it was decided to wait to operate. If ICP then occurs, it is necessary to operate.
Evaluate the neurocognitive functioning and vision of children with multisuture or syndromic craniosynostosis at the age of 7.
In patients with Apert and Crouzon syndrome and in patients with multisuture craniosynostosis where at least both occipital sutures are fused, the first cranial surgery is done on the back of the head using cranial distraction.
Patients with Saethre-Chotzen and Muenke syndrome will have the first cranial surgery to enlarge the forehead with the upper half of the edge of the eye socket.
In other forms of syndromic craniosynostosis, the type of surgery depends on the cranial deformity.
Consider minimally invasive treatment in patients with nonsyndromic craniosynostosis in whom both coronal sutures are fused.
In other multisuture craniosynostosis, the type of surgery being done depends on the cranial deformity. There is no evidence available as to whether open or minimally invasive surgery is better.
In multisuture and syndromic craniosynostosis, the surgery takes place between 6 and 9 months. In Muenke syndrome, the surgery takes place between 9 and 12 months.
Minimally invasive surgery for multisuture craniosynostosis should be performed as early as possible, and at the latest before the age of 6 months.
CHAPTER 7. SURGICAL TREATMENT OF SYNDROMIC CRANIOSYNOSTOSIS—FACIAL
Question 7.1 What is the surgical management of the face in syndromic craniosynostosis with underdevelopment of the upper jaw and eye sockets?
Apert and Crouzon syndrome are associated with underdevelopment of the upper jaw, too shallow eye sockets, and too far apart eyes and to a lesser extent with underdevelopment of the lower jaw. The indication for surgical correction varies from an acute drop in vision, breathing problems, the lower jaw not fitting on the upper jaw or an esthetic problem, and the resulting psychological consequences. Various different techniques are possible to correct these deformities, the timing of which has a major influence on the final result.
(1) What are the surgical specific factors that influence the choice between the different surgical techniques (internal versus external distraction and Le Fort III osteotomy versus variations on Le Fort III osteotomy) for the treatment of an underdeveloped midface (midface hypoplasia)?
A Le Fort III operation15 (midface from upper jaw to lower eye socket edges) with distraction (slowly twisting bone elements apart) can move the middle face further forward than a Le Fort III without distraction. There is also less recurrence of the forward displacement after the use of distraction. It is preferable to place an external frame instead of internal distractors because the direction of displacement can be better influenced. Possible other advantages of an external frame are better facial concavity correction and fewer wound infections.
A monobloc operation (midface, forehead, and upper eye socket edges) with distraction16 (with external frame or internal distractors) corrects the too-shallow eye sockets and breathing problems. Complications such as the leakage of cerebrospinal fluid and problems with the equipment are hardly different from each other in both methods.
In Apert syndrome, a facial bipartition17 (monobloc and bringing together of the eye sockets) with distraction is preferably performed with an external frame. A Le Fort II18 operation (upper jaw to nose) with distraction combined with bringing both sides of the cheekbones (forward also has better facial contour results than a Le Fort III operation) with distraction.
(2) What are the long-term surgical specific results of the different timing of surgery in the absence of a hard indication, that is, “early”, defined as before the age of 6 to 8 years, versus “late”, that is, after the age of 6 to 8 years?
A Le Fort III without distraction, performed before the age of 6 years, probably leads to a high risk of recurrent midface hypoplasia in adulthood. A Le Fort III with external distraction performed before the age of 8 years without overcorrection (more correction than necessary at the age of surgery) increases the risk of recurrent midface hypoplasia in adulthood. A monobloc (bringing forward the entire face and forehead) with external distraction seems to provide a good forward movement of the face, regardless of the age at which surgery is performed. This procedure performed before the age of 8 years seems to lead to a higher risk of recurrence of respiratory problems.
Recommendations
Question 7.1
A midface advancement in children with Apert and Crouzon/Pfeiffer is actually always combined with distraction.
An external frame is preferably used for a Le Fort III distraction and a facial bipartition.
If an external frame is necessary, then the placement of internal distractors can also be considered. It is therefore possible to remove the frame earlier, as soon as the distraction is completed.
The type of surgery required is determined based on the facial deformity of the individual patient.
Perform a midface advancement with distraction in children with Apert syndrome and Crouzon between 8 and 12 years of age, or from 17 years of age.
The operation should be performed earlier if there are serious breathing problems during sleep or the eyes cannot be closed properly and damage to the cornea may occur.
A midface advancement should preferably not be carried out between the ages of 12 and 17 years, because there is a higher chance of psychosocial problems and unrealistic expectations of the treatment result.
CHAPTER 8. INCREASED INTRACRANIAL PRESSURE
Question 8.1 How is ICP in craniosynostosis treated?
The risk of ICP varies greatly depending on the type of craniosynostosis, with the multisuture form and syndromic form associated with a much higher risk than the isolated nonsyndromic form. However, the risk of these problems in the isolated nonsyndromic group is much less recognized and therefore possibly underdiagnosed if present. It is important to promptly detect and treat ICP. High intracranial pressure, for example, can lead to irreversible vision impairment. It is unclear, which method is most suitable for detecting ICP, which cutoff values should be used, and how often this examination should be carried out to detect problems in time.
Increased intracranial pressure is caused by craniocerebral imbalance, aberrant venous drainage, obstructive sleep apnea (OSA) syndrome, an abnormal location of the cerebellum, and hydrocephalus. The risk of ICP continues to increase as long as there is no surgery. Sometimes, ICP occurs in the years after cranial-expansion surgery.
(1) What is the occurrence of ICP in different types of craniosynostosis?
Sometimes, with unisutural craniosynostosis, ICP already exists before the operation. With sagittal suture synostosis, this is in 2.5% to 14% of children. With metopic suture synostosis, it occurs in 2% to 8% of children. With unicoronal suture synostosis, this is the case in 16% of children.
In some situations, ICP still occurs in the years after the skull operation. For sagittal suture synostosis, this occurs in 2% to 9% and with metopic suture, in 1.5% of cases. How often this occurs with unicoronal suture synostosis is unknown.
Before cranial surgery, ICP occurs in children with Apert syndrome in 9% to 83%, Crouzon syndrome in 53% to 64%, Saethre-Chotzen in 19% to 43%, and Muenke syndrome in 0% to 4% of cases.
After cranial surgery, ICP occurs in 35% to 45% of children with Apert, 20% to 47% with Crouzon, 17% to 42% with Saethre-Chotzen, and 0% to 5% of children with Muenke syndrome. When several cranial sutures are fused, ICP after cranial correction occurs in 58% to 67% of cases. In bicoronal suture synostosis after cranial correction, it is present in 31% of children.
(2) What is the diagnostic accuracy of the following diagnostic tools for detecting or excluding ICP: (1) (abnormal) head circumference growth curves, (2) presence of imprint of the brain/blood vessels on the inside of the skull on x-ray, (3) optic nerve ultrasound, (4) presence or absence of papilloedema (fluid around the optic nerve) detected by fundoscopy and (5) OCT (optical coherence tomography—measuring the thickness of the retina)?
A deviating growth curve of the cranial circumference can be used to demonstrate ICP in metopic suture synostosis. This method is less suitable for sagittal suture synostosis. This is because a deviating growth curve does not always indicate the presence of ICP. The usefulness of the cranial circumference growth curve has not yet been investigated and described for unicoronal suture synostosis. The growth curve of the cranial circumference can probably be used in syndromic craniosynostosis to determine ICP.
The presence or absence of visible imprints of the gyri of the brain on an x-ray, if a child is under 18 months of age may be unreliable for determining the presence of ICP. The presence of visible gyri of the brain on x-ray is a reliable sign of ICP, but the absence of these signs does not mean that intracranial pressure is normal. For children aged 18 months to 4 years using these signs as a screening method is more reliable.
If a coronal suture also closes after the operation for sagittal suture synostosis in the first 2 years, this may lead to a higher risk of ICP.
An ultrasound to check the thickness of the optic nerve does not seem to be a reliable screening method for determining ICP.
Papilledema in an ophthalmoscope (fundoscopy) may be a sign of ICP, but the absence of papilledema in children under 8 years of age does not exclude ICP.
Optical coherence tomography eye tests are probably a reliable method to screen for ICP, but can only be performed properly if the child cooperates properly.
(3) What are the craniosynostosis-specific factors at play in the choice between the different surgical techniques to treat ICP?
The reason for ICP in sagittal suture synostosis is often a skull that is too small. Therefore, treatment is aimed at enlarging the skull.
There are multiple causes for syndromic craniosynostosis, such as a too small skull volume, moderate to severe breathing problems, hydrocephalus, or too high pressure in the veins in the brain. The treatment is aimed at removing the main cause of the ICP.
Recommendations
Question 8.1
Screen annually for ICP in sagittal suture synostosis using an ophthalmoscope and/or OCT up to and including the age of 6 years.
Screen annually for ICP in the metopic suture, unicoronal suture, and unilateral lambdoid suture synostosis by measuring the cranial circumference. If there is a deviating growth curve, an ophthalmoscopic, or OCT eye test is also done.
Children with syndromic and multisuture craniosynostosis are screened for ICP up to and including the age of 6 years. In Crouzon syndrome, this is once every 4 months until the age of 2, then every 6 months until the age of 4, and then every year. Screen every 6 months with Apert syndrome, Saethre-Chotzen, and multisuture craniosynostosis, and every year for Muenke syndrome.
The treatment of ICP depends on the causative factors and treatment should be adapted accordingly.
CHAPTER 9. HYDROCEPHALUS
Question 9.1 What is the surgical management of hydrocephalus in craniosynostosis?
Hydrocephalus is an increase in the width of the cranial chambers that accompanies signs of ICP. This should be distinguished from enlarged chambers containing cerebrospinal fluid without ICP (ventriculomegaly). These disorders can cause problems in the functioning and development of children. Both disorders are almost nonexistent (0.88%) in nonsyndromic craniosynostosis and therefore are not discussed further in this chapter.
(1) How common is hydrocephalus in children with craniosynostosis and how can it be detected?
Ventriculomegaly occurs regularly (8% in Muenke; 6%–17% in Saethre-Chotzen; 24% in multisuture craniosynostosis) to common (13%–56% in Crouzon; 39%–71% in Apert) in syndromic craniosynostosis. Patients with Apert syndrome and a Chiari (partial sagging of the lower part of the cerebellum in the foramen magnum that impedes the flow of the cerebrospinal fluid) have a greater chance of ventriculomegaly.
Hydrocephalus occurs in 6% to 26% of children with Crouzon/Pfeiffer, 0% to 6% in Apert, and 5% to 12% in multisuture craniosynostosis, and does not occur or is very rare in Saethre-Chotzen and Muenke syndrome.
(2) What are the factors in the location and structure of the brain that influence the choice between different surgical techniques for the treatment of hydrocephalus?
The factors that predict a successful treatment are not known. Hydrocephalus in craniosynostosis can possibly be treated properly with an expansion of the skull or the placement of a drain from the ventricle to the abdomen to allow for the discharge of excessive cerebrospinal fluid (ventriculoperitoneal shunt). It is also possible to make a hole in the bottom of the third ventricle so that cerebrospinal fluid can flow off in another way (endoscopic third ventriculostomy) or to enlarge the foramen magnum (foramen magnum decompression). Both good and bad results have been described for all these treatments. If the treatment performed has not helped sufficiently, another follow-up treatment may be necessary.
Recommendations
Question 9.1
Screen all patients with Crouzon syndrome and multisuture craniosynostosis with MRI upon referral. Patients with ventriculomegaly should be given a second MRI to exclude hydrocephalus. When this is done, depends on the progress of the condition itself and what symptoms occur over time.
Treat hydrocephalus by cranial expansion with or without decompression of the foramen magnum by placing a ventriculoperitoneal shunt or making a hole in the bottom of the third ventricle. The treatment being used is adapted per patient, and depends on the MRI results, among other things.
The posttreatment effect is monitored well by MRI scans. If the treatment does not reflect the desired result, an additional treatment is used.
CHAPTER 10. CHIARI
Question 10.1 What is the management of Chiari in craniosynostosis?
The risk of Chiari I malformation (partial sagging of the lower part of the cerebellum in the foramen magnum) varies greatly per type of syndromic craniosynostosis and is hardly or not at all seen in nonsyndromic craniosynostosis. The occurrence, causes, consequences, and need for treatment are often unclear. Chiari I malformation is best imaged with an MRI scan, but as to how often it should be done for the different types of craniosynostosis, and when a specific treatment is indicated, is unclear.
(1) How common is Chiari in children with craniosynostosis, and what is needed to diagnose it?
Chiari occurs in children with isolated nonsyndromic craniosynostosis. This is in 3% to 8% with sagittal suture synostosis, 0% in metopic suture synostosis, 6% to 18% with coronal suture synostosis, and 25% to 60% with lambdoid suture synostosis. It is possible that the presence of Chiari rarely causes any symptoms in these children. It is preferably diagnosed by MRI.
Chiari may occur in Crouzon and Pfeiffer in 70% to 82% of cases, and in 2% to 29% in Apert. In multisuture craniosynostosis, where the lambdoid sutures are also fused, Chiari is found in 57% to 71% and without fused lambdoid sutures, in 7% to 11%. It is unknown how common this is with Saethre-Chotzen and Muenke syndrome. It is possible that Chiari may often be present without complaints and symptoms in multisuture and syndromic craniosynostosis. It can often only be determined with the help of an x-ray and preferably MRI.
(2) Which Chiari-specific factors play a role in the decision whether or not to treat?
It is possible that 17% to 50% of patients with Crouzon-Pfeiffer or multisuture craniosynostosis will still develop complaints and symptoms, therefore requiring surgery.
(3) What are the factors that determine whether an operation is necessary, which operation will be performed, and at what time this will be done?
The factors, which are important in determining whether an operation is necessary, which operation will be performed, and at what time this is to be done, are not known.
Several types of operations have been described for Chiari treatment (widening of the foramen magnum before or after an occipital expansion before the onset of or after the development of symptoms).
Recommendations
Question 10.1
-
(1)
Screen patients with nonsyndromic unilateral lambdoid suture synostosis, children with Crouzon/Pfeiffer syndrome, and multisuture craniosynostosis with fused lambdoid sutures immediately by MRI scan in the specialist centre.
Repeat the MRI at the age of 4 and 18 years, and when there are complaints that may indicate a Chiari.
Screen for the presence of a syrinx (a cavity formation in the spinal cord containing cerebrospinal fluid) by doing an MRI scan of the spinal cord at the neck, chest, and lower back level (cervical, thoracic, and lumbar myelum) if the diagnosed Chiari increases and/or becomes symptomatic.
-
(2 and 3)
Surgical treatment of Chiari is only recommended if the patient has complaints. Otherwise, an active follow-up policy is pursued by the pediatric neurosurgeon or pediatric neurologist by annual monitoring for neurological complaints or symptoms, doing an MRI when necessary, and instructions to the parents.
CHAPTER 11. VISUAL, REFRACTIVE, AND MOTILITY DISORDERS
Question 11.1 What screening is necessary to detect disorders related to vision or eye movement in the different types of nonsyndromic and syndromic craniosynostosis in a timely manner?
Vision loss in craniosynostosis is caused by damage to the optic nerve due to ICP, corneal deformation due to incomplete closure of the eyelids, or a lazy eye due to strabismus or refractive disorders.
A timely diagnosis and treatment are essential for maintaining vision.
(1) How common are vision or eye movement disorders in different types of nonsyndromic craniosynostosis?
It is possible that eye abnormalities (impaired vision, strabismus, etc.) occur regularly with a metopic suture and unilateral coronal suture synostosis. These abnormalities occur very regularly in syndromic craniosynostosis. Regular assessment can ensure the prevention of a lazy eye and that good vision is retained.
(2) Which screening tests are the most accurate?
There is little scientific evidence available as to what tests can best be used at specific times to determine eye problems in the presence of craniosynostosis.
Recommendations
Question 11.1
Because of the frequent occurrence of eye abnormalities, ophthalmic examinations are required in metopic suture synostosis, unicoronal suture synostosis, multisuture craniosynostosis involving 1 coronal suture, and all syndromic forms of craniosynostosis.
Referral is done at the first consultation in the tertiary centre. Depending on the results, follow-up tests are booked.
CHAPTER 12. RESPIRATORY DISORDERS
Question 12.1 What is the policy on respiratory disorders in syndromic craniosynostosis?
Obstructive sleep apnea syndrome is characterized by episodes of partial and/or complete upper obstruction of the airway while sleeping and thereby causing breathing pauses during sleep. The clinical symptoms are diverse and can be classified as symptoms at night: (1) restless sleep, (2) snoring, (3) apnea, (4) bedwetting, and (5) sweating, and during the day: (1) dry mouth when waking up, (2) fatigue, (3) impaired mental function, (4) drop in school performance, and (5) behavioral disorders. Growth disorders can occur in the long term.
Children with craniosynostosis syndromes are among the groups at risk of OSA. Respiratory disorders can also cause ICP. This is likely because the blood vessels in the brain dilate when breathing is interrupted due to more CO2 and more blood goes to the brain in response. Children with syndromic craniosynostosis often have ICP already. Breathing disorders can then cause this intracranial pressure to become even higher or too high.
Given the severity of OSA syndrome and the excellent treatment options, early diagnosis is of utmost importance.
(1) What respiratory disorders occur with craniosynostosis, how frequently do these occur, and how severe are they?
In children with multisuture and syndromic craniosynostosis, OSA occurs in 70% of patients. The clinical picture is the most severe and occurs most often in patients with Apert syndrome, Crouzon, and Pfeiffer syndrome.
Central apnea occurs in 4% of cases and decreases with age.
(2) What are the OSA-specific factors that weigh in the indication system for treatment, especially in the case of mild OSA?
Suffering from moderate or severe OSA may be associated with ICP and disturbed sleep and is therefore a reason for treatment. Mild OSA may not be associated with ICP and a disturbed ratio and duration of the various sleep depths and phases. This is only treated if the patient is being very negatively affected by it.
(3) What are the anatomical factors influencing the choice of surgical treatment to be used?
If enlarged tonsils and or adenoids are diagnosed in children with syndromic craniosynostosis along with OSA, removing the tonsils may reduce the severity of respiratory disorders. Unfortunately, this often turns out not to be enough to completely solve the problem.
A narrow upper respiratory tract can cause OSA at multiple levels. It is possible to search for the location of the cause or causes of OSA endoscopically (keyhole surgery in the airway).
If there is an underdevelopment of the face, a midface advancement may possibly reduce moderate to severe respiratory disorders to mild or no complaints. If the airway narrows at the base of the tongue, extending the lower jaw can reduce moderate to severe respiratory disorders to mild symptoms or eliminate them entirely.
Recommendations
Question 12.1
-
(2)
Refer children with syndromic craniosynostosis suspected of having OSA to a specialized centre for diagnostic sleep screening.
Screen children with syndromic craniosynostosis annually with a diagnostic sleep test (polysomnography type 1) at a specialist centre, until at least the age of 6 years.
Conduct a diagnostic sleep test if the discussion with the doctor points to complaints indicating respiratory disorders.
Perform an upper airway endoscopy (oral cavity, nose, and throat) if moderate or severe OSA has been identified to determine the levels of obstruction in the airway.
-
(3)
In case of mild OSA, treatment is started if there are other complaints too. Noninvasive surgical interventions, such as the removal of the tonsils and adenoids, are preferred.
Choose an OSA treatment based on OSA severity, the age of the patient, related factors, the feasibility of the treatment, and other physical complaints.
Consider Le Fort III surgery15 or monobloc advancement16 in children with syndromic craniosynostosis and severe OSA where respiratory support is required to treat the problems. If necessary, this procedure is combined with an advancement of the lower jaw.
Consider septum surgery from adulthood for additional improvement of nasal airflow and OSA-related complaints.
CHAPTER 13. HEARING IMPAIRMENTS AND SPEECH/LANGUAGE DEVELOPMENT
Question 13.1 What is the policy on hearing impairments and speech/language development in craniosynostosis?
There are several reasons why patients with craniosynostosis have hearing impairments and/or a delay in language/speech development. Hearing loss can be an additional cause of developmental delay in children who are already at increased risk.
What type of hearing loss occurs in patients with craniosynostosis and at what frequency?
Hearing loss should be considered in all children with syndromic craniosynostosis: (1) 61% to 71% in Muenke syndrome, (2) 44% to 80% in Apert syndrome, (3) 92% in Pfeiffer syndrome, (4) 29% to 74% in Crouzon syndrome, and (5) 29% in Saethre-Chotzen syndrome. ~7% of children with multisuture craniosynostosis have hearing loss.
Hearing loss in children with syndromic and multisuture craniosynostosis is caused by sounds not being properly conducted through the ear canal and/or middle ear to the inner ear (conductive hearing loss). In Muenke syndrome, hearing loss is mainly caused because the inner ear or auditory nerve is abnormal (sensorineural hearing loss).
Question 13.2 What is the indication for speech and language development screening?
Are children, adolescents, and adults with isolated or multisuture craniosynostosis, or with syndromic craniosynostosis, subject to an increased risk of speech and language problems compared with children without craniosynostosis?
Children with isolated craniosynostosis between the ages of 6 and 18 months have a slightly increased risk of speech and language problems compared with children without a craniofacial abnormality. Sagittal suture synostosis is the exception.
At the age of 36 months, children with isolated craniosynostosis have a moderately increased risk of speech and language problems. This is probably more true for unicoronal suture and lambdoid suture synostosis and to a lesser extent for metopic suture and sagittal suture synostosis.
At the age of 7 years, speech and language problems in children with metopic suture, unilateral lambdoid suture, and coronal suture synostosis are likely to be slightly more common than in children of the same age with sagittal suture synostosis. Children with sagittal suture synostosis are unlikely to have an increased risk of speech and language problems. No studies were found on children with multisuture craniosynostosis that can properly address the question about preventing speech and language problems.
Recommendations
Question 13.1
-
In children with craniosynostosis up to the age of 4:
Neonatal hearing screening is done as with all newborns. If further testing is needed after the results, this is done in an audiological centre.
The ENT doctor examines the ear canal and the eardrum every year and a hearing test is done.
-
In children with craniosynostosis from the age of 4:
Hearing is screened by doing an auditory examination if there is a reason to do so. This examination can be done in a local audiological centre or in the craniofacial specialist centre’s audiological centre. If the testing is done in a local audiological centre, the report is sent to the craniofacial specialist centre.
Question 13.2
-
In case of isolated craniosynostosis (sagittal suture or metopic suture synostosis):
If parents or health care professionals are concerned about speech and language development, the parents/health care providers are asked to complete a specific screening questionnaire (SNEL). If there are concerns based on the filled-in answers, additional speech-therapy-related tests are carried out, preferably in a craniofacial centre.
With isolated craniosynostosis (coronal suture and lambdoid suture synostosis) and multisuture craniosynostosis:
-
Younger than 36 months:
If parents or health care professionals are concerned about speech and language development, the parents/health care providers are asked to complete a specific screening questionnaire (SNEL). If there are concerns based on answers provided, additional speech-therapy-related tests are carried out, preferably in a craniofacial centre.
-
From 36 months:
Ask the parents/health care providers to fill in a specific screening questionnaire (SNEL). If there are concerns based on the answers provided, additional speech-therapy-related tests are carried out, preferably in a craniofacial centre.
-
5 to 6 years:
For children in grade 2, ask the parents to bring a printout of the school assessment system to the craniofacial centre visit.
When school performance indicates there are problems with reading and spelling, additional speech-therapy-related tests are carried out. If there is a suspicion that these problems are due to IQ or attention problems, further neuropsychological tests are carried out.
-
7 to 8 years:
For children ages 5 and 6, ask the parents to bring a printout of the school assessment system to the craniofacial centre visit because there is a greater risk of problems with reading and spelling.
When school performance indicates there are problems with reading and spelling, additional speech-therapy-related tests are carried out. If there is a suspicion that these problems are due to IQ or attention problems, further neuropsychological tests are carried out.
Syndromic Craniosynostosis
Conduct regular speech therapy examinations on speech and language development from the moment the child is first referred to the craniosynostosis specialist team.
If there is a suspicion that these problems are based on IQ or attention problems, further neuropsychological and psychological tests are carried out.
CHAPTER 14. DENTOFACIAL ABNORMALITIES
Question 14.1 What is the policy regarding orthodontic care with syndromic craniosynostosis?
Dentofacial abnormalities occur in almost all syndromic abnormalities and are reinforced by the necessary surgical procedures. There are often several phases in which the treatments take place. The final correction should always be aligned with the orthognathic surgical correction that is planned for the future.
Which dentofacial abnormalities occur in patients with syndromic craniosynostosis, and how often?
Orthodontic and dental problems in patients with Apert syndrome and Crouzon are likely caused by an abnormal and stunted growth of the upper jaw in length, width, and height, which causes the upper jaw to be too small in all directions.
It is likely that underdevelopment of the upper jaw and palate occurs frequently in patients with syndromic craniosynostosis.
Growth of the lower jaw is possibly affected by the premature fusing of the cranial sutures.
Nonsymmetrical growth of the lower jaw may be seen more often in patients with Apert syndrome and Crouzon than in control groups.
Cleft lip (open soft palate) and a cleft uvula occur in 75% of patients with Apert syndrome and in 5% of patients with Muenke syndrome.
The dental arch may fail to widen during growth in patients with Apert and Crouzon syndrome. Excessive swellings of the gums are also seen that may become larger with age.
Because of underdevelopment of the upper jaw in patients with Apert and Crouzon syndrome, the jaws may not fit together properly due to an underbite (68%). Often, there are additional problems such as an open bite or cross bite (upper and bottom teeth do not align correctly).
Dental development in patients with Apert and Crouzon syndrome may be delayed or seem to be rather late normal development, and lead to teeth erupting later and in a different manner and sequence.
Nonerupted teeth are more common in patients with Apert syndrome (46.4%) and Crouzon syndrome (35.9%) than in the control group.
More cavities, plaque, gum disease, and enamel defects may be seen in patients with craniosynostosis than in the normal population.
Recommendations
Question 14.1
A dentist or orthodontist who is not part of the craniofacial team should never treat a craniosynostosis patient without consulting the craniosynostosis specialist team.
Besides the recommendation to visit an oral care provider before the second year of life, the orthodontist advises the parents to regularly see the dentist, pediatric dentist, or dental hygienist if oral hygiene is inadequate.
Perform orthodontic checks within the craniosynostosis specialist team in patients with syndromic craniosynostosis around the age of: 4, 6, 9, 12, 15, and 17 years.
A long-term treatment plan is drawn up at the first contact, at the age of 4 years. This plan may be adjusted during orthodontic follow-up checks, depending on the results.
The treatment plan is drawn up by the orthodontist at the craniosynostosis specialist centre. This is always done in consultation with maxillofacial and plastic surgeons.
The orthodontic treatment plan does not necessarily have to be implemented in the craniofacial centre, but under the supervision of the orthodontist at the craniosynostosis specialist centre.
CHAPTER 15. (NEURO)COGNITIVE, SOCIO-EMOTIONAL, AND BEHAVIORAL FUNCTIONING
Question 15.1 What is the policy on (neuro)cognitive, socio-emotional, and behavioral problems with craniosynostosis?
A lot of research has been done on the mental function (cognition) and behavior of children with nonsyndromic craniosynostosis. However, the results of these studies vary widely: some researchers report hardly any cognitive and/or behavioral problems in children with nonsyndromic craniosynostosis, whereas other researchers mention very high percentages (up to 100%) of cognitive and/or behavioral problems. These differences in outcomes can often be explained by the limitations of the study design. In contrast, so far few studies have been done on the cognitive function and behavior of children with syndromic craniosynostosis.
What (neuro)cognitive, socio-emotional, and behavioral problems occur in children with single-suture nonsyndromic craniosynostosis, multisuture craniosynostosis, or syndromic craniosynostosis, and at what frequency?
Mental and motor developmental delays are more common in isolated craniosynostosis than in children without craniosynostosis. In a developmental study, these children under the age of 4 often score higher on the mental scale than on the motor scale. There are no noticeable differences in mental function between the different types of isolated craniosynostosis.
The IQ in children of primary school age with non-syndromic craniosynostosis is probably slightly lower than or comparable to the IQ of children without craniosynostosis. The verbal IQ, which relates to vocabulary, language sense, and reasoning ability is often lower than the performance IQ, which relates to practical insight, problem-solving, spatial, and visual insight. Intelligence Quotient scores below 80 to 85 are more common in children with metopic suture, lambdoid suture, and coronal suture synostosis than in children without craniosynostosis.
At the age of 3 years, parents of children with isolated nonsyndromic craniosynostosis may report behavioral problems more often on a behavioral questionnaire (Externalizing Child Behavior Check List scale) than parents of children without craniosynostosis (14.5% versus 7.6%).
At the age of 7 years, parents of children with isolated nonsyndromic craniosynostosis may report behavioral problems more often on a behavioral questionnaire (Total Problem Score Child Behavior Check List scale) than parents of children without craniosynostosis (33% versus 21%).
Children with metopic suture synostosis have the most behavioral problems (41%) and children with sagittal suture synostosis have the least (29%).
Children with Apert syndrome, Muenke syndrome, and multisuture craniosynostosis seem to have a (greatly) increased risk of intellectual disability.
Parents of children with syndromic or multisuture craniosynostosis report more social problems, attention problems and attention disorders, and internalizing problems in their child compared with the control group.
Children with Apert or Muenke syndrome have the most problems. Social, emotional, and behavioral problems are strongly linked to intelligence.
Parents of children with syndromic or multisuture craniosynostosis indicate a lower quality of life for their child than the control population. In children under the age of 4, this mainly concerns Apert syndrome and multisuture craniosynostosis, and in children older than 4 years, Apert and Muenke syndrome in particular.
Recommendations
Question 15.1
-
In children with isolated nonsyndromic craniosynostosis:
Screen these children between 18 months and 4 years for motor developmental delay, mental and intellectual, socio-emotional, and behavioral problems. If a screening is abnormal, further psychological and/or pediatric physiotherapeutic examination is called for.
-
In children with a metopic suture, coronal suture, or lambdoid suture synostosis:
If they are aged 7 or 8, screen these children on mental and intellectual, socio-emotional, and behavioral problems. Further psychological testing is conducted if the results are abnormal.
-
In children with sagittal suture synostosis:
If they are aged 8 or 9, screen these children for verbal comprehension (testing vocabulary), numeracy skills, inhibition (ability to control behavior and reconsider before an (impulsive) response), and the ability to perform multiple tasks simultaneously (divided attention). Further psychological testing is conducted if the results are abnormal.
-
In children with syndromic craniosynostosis or multisuture craniosynostosis:
Screen these children for mental and cognitive functioning, socio-emotional, and behavioral problems: if the child is 2 or 3 years old, around the time of primary school selection, and if the child is 8 or 9 years old.
Tests should always be conducted on these children if there are problems with regard to behavior, social, and cognitive functioning.
Measure the children’s quality of life by using specially designed questionnaires for parents. If a child is old enough (from the age of 12) he/she can fill it in themselves. The treatment policy is then aimed at the areas where the child scored poorly, where possible.
-
General information on screening:
Psychological screening and testing in children with craniosynostosis are preferably done by the psychologist in the craniosynostosis specialist team where the child is treated.
In case of a developmental delay, further testing and treatment can be done as described in the “Guideline for etiological diagnostics in children with a developmental delay/cognitive disability”.
CHAPTER 16. PSYCHOSOCIAL FUNCTIONING
Question 16.1 What is the policy on psychosocial functioning in children with craniosynostosis and their family?
The condition craniosynostosis itself affects psychosocial functioning, but its medical treatment also has an influence on the psychosocial side. It can affect the child, his or her parents, siblings, family, friends, school, the parent’s work, etc. A whole system is involved in the treatment of a child with a craniofacial disorder. Psychosocial concerns the psychological, relational, and social aspects of life.
In craniofacial care, there is a clear difference in the treatment of syndromic craniosynostoses and nonsyndromic craniosynostoses. In general, it seems that syndromic craniosynostosis has a much longer treatment process, requires more operations and has a longer-term impact on life, which could possibly lead to more psychosocial problems.
What psychosocial problems are involved in the patient and family? How often do these problems occur and what are the risk factors for the occurrence of these problems?
There is a greater risk of psychosocial problems in children with syndromic craniosynostosis.
The quality of life, when looking specifically at physical health, is lower in children with syndromic craniosynostosis than in children without craniosynostosis.
In particular, the scores on vision, hearing, and speech were lower. For Apert syndrome, scores on physical function, the emotional impact of the parent(s), family activities, and cognition were also lower than in families and children without craniosynostosis.
Parents of children with multisuture or syndromic craniosynostosis have a reduced quality of life compared with parents of children without craniosynostosis. They mainly score lower on the psychosocial level.
Posttraumatic stress disorder (PTSD) occurs in about 10% of children who have had an intensive care unit admission and their parents.
Stress responses from parents (mothers especially) are the main predictor of PTSD in children.
More than one-third of youths with a craniofacial condition experience problems related to appearance.
The most important predictors for psychosocial improvement after surgery include the age of the patient, the patient’s expectations in advance of the outcome of the surgery, and who made the decision to do surgery (especially in young adults).
Recommendations
Question 16.1
Support to parents and family from the craniosynostosis expertise team
-
Prevention of psychosocial problems
Inform patients and parents about the Patients and Parents Association LAPOSA.
Inform parents about the possibility of referral to a social worker/psychologist for support in raising the child.
Repeatedly offer parents with a child with syndromic craniosynostosis contact with a social worker/psychologist—mainly around the child’s transitional phases, such as when choosing a school.
Screen the family for the presence of psychosocial problems and symptoms of PTSD regularly throughout the course of treatment.
-
On indication
Refer the family to a social worker/psychologist in case of psychosocial problems.
Refer the parents and child with PTSD or suspected PTSD to the psychologist of the craniosynostosis specialist team or a psychologist in or near the place of residence.
-
Support to a patient with craniosynostosis from the craniosynostosis specialist team
Offer psychosocial care from the team throughout the treatment process.
Do psychosocial screening for long-term treatments that demand a lot of motivation from the patient. If necessary, offer support to improve the feasibility of treatment.
Offer counseling focused on psychosocial adaptation, self-understanding (how someone assesses themselves), social skills, and self-image for young people who experience problems in these areas.
Offer adolescents with a desire for surgical treatment at least 1 contact with a specialized psychosocial counselor to assess their expectations and motivation.
CHAPTER 17. CRITERIA FOR CRANIOSYNOSTOSIS EXPERTISE CENTRE AND TEAM MEMBERS
Question 17.1 What are the minimum requirements for a craniosynostosis expertise centre and its team members?
Care for patients with nonsyndromic or syndromic craniosynostosis requires a multidisciplinary approach, given the complex care these patients require. As it is a rare condition, centralization of this care is desirable to ensure maximum expertise is gained, the quality of care is high and to facilitate scientific research to improve care. Multidisciplinary care requires good coordination and communication within the team itself, with the practitioners involved outside the centre and with the patient and parents. Responsibility and division of tasks for the various care providers within the team should therefore be clearly defined.
Comparative studies of results from various craniofacial teams will be able to exert a positive effect on the quality of care. This can take place at both national and international levels. Consultation within the teams and collectively will also make an important contribution to the quality of care, but also to joint research and innovation.
Recommendations
Question 17.1
-
Composition of the craniosynostosis expertise team
Care for patients with craniosynostosis is provided from a multidisciplinary setting.
A craniosynostosis specialist centre has at least the following health care providers and facilities:
| Health care provider/facility | Unisutural nonsyndromic | Multisutural or syndromic |
|---|---|---|
| Pediatrician | x | x |
| Clinical geneticist | x | x |
| Pediatric anesthetist | x | x |
| Pediatric intensivist | x | x |
| Neurosurgeon | x | x |
| Pediatric neurologist | — | x |
| Ophthalmologist | x | x |
| Pediatric radiologist | x | x |
| Plastic surgeon | x | x |
| Maxillofacial surgeon | x | x |
| Orthodontist | — | x |
| Otorhinolaryngologist | — | x |
| Psychologist | x | x |
| Social worker | x | x |
| Speech therapist | x | x |
| Pedagogical employee | x | x |
| Team chairman (1 of the core specialists) | x | x |
| Care coordinator | x | x |
| Photogrammetry, x-ray, ultrasound, CT (3-dimensional) | x | x |
| Magnetic resonance imaging | — | x |
| Pediatric intensive care unit | x | x |
| Polysomnography (sleep study) | — | x |
-
Back up of the basic specialties (so at least 2 specialists for neurosurgery, plastic surgery, and oral surgery) is advised to ensure continuity of care.
Collaboration within the craniosynostosis specialist centre:
Care for patients with craniosynostosis should be provided from a multidisciplinary setting. A care path must be established.
The team roles should be clearly defined.
-
Joint consultations are held with the presence of the core specialists (plastic surgery, oral surgery, and neurosurgery) and the availability of the other specialists.
Collaboration outside the craniosynostosis expertise centre:
-
Patients with craniosynostosis are only treated in an accredited craniosynostosis expertise centre. Specific parts of the care program can be performed in your own region on request and under the coordination of the craniosynostosis expertise centre
Task division within the craniosynostosis expertise centre:
Care is provided on the basis of established protocols that are reviewed annually.
The multidisciplinary care per individual patient is coordinated between the care providers and communicated to the patient and parents and any care providers from outside the team.
A practitioner from a core specialty is the team leader. He or she is ultimately responsible for ensuring that the craniosynostosis expertise centre meets all criteria.
-
The care coordinator (usually a nursing specialist) is responsible for coordinating care and is the point of contact for patients and cotreatment providers from outside the team.
Centralization
Care for nonsyndromic, unisutural craniosynostosis in the Netherlands is centrally provided in 2 craniosynostosis expertise centres.
Care for syndromic craniosynostosis is centrally provided in 1 centre.
-
The minimum number of intracranial operations (skull surgery) for craniosynostosis is 20 per surgeon per year.
Reporting of results and activities
An internal audit takes place at least once a year. This involves looking at the performance quality and the working methods of the craniosynostosis expertise team and any necessary improvement actions take place.
Every craniosynostosis expert team issues an annual report:
| Item | Unisutural nonsyndromic | Multisutural or Syndromic |
|---|---|---|
| No. of operations per diagnosis | x | x |
| No. of procedures per type of operation | x | x |
| No. of patients treated according to protocol | x | x |
| Perioperative dural and brain injury | x | x |
| Excessive blood loss | x | x |
| Infections | x | x |
| Unplanned reoperations | x | x |
| Issues with equipment (springs, distractors, and helmet) | x | x |
| Quality of life/patient related outcome measure (PROM) | x | x |
| Appearance esthetic result | x | x |
| Behavior | — | x |
| Neurocognition and behavior | x | x |
| Obstructive sleep apnea | — | x |
| Increased intracranial pressure | x | x |
| Hydrocephalus | — | x |
| Hearing | — | x |
| Speech/language | x | x |
| Vision | x | x |
CHAPTER 18. FLOW CHART/PATIENT SUMMARY VISUAL
Authors: Karen Wilkinson-Bell, Olivia Spivack, MSc
Design: Jana Steerneman, MSc



Footnotes
European Reference Network Craniofacial Anomalies and Ear-Nose-Throat Disorders (ERN CRANIO), co-funded by the European Union.
The authors report no conflicts of interest.
Contributor Information
Mariët Faasse, Email: onderzoek@laposa.nl.
Irene M.J. Mathijssen, Email: onderzoek@laposa.nl.
REFERENCES
- 1.Mathijssen IMJ. Updated guideline on treatment and management of craniosynostosis. J Craniofac Surg 2021;32:371–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.NVPC. Guideline Treatment and care for craniosynostosis [Richtlijn behandeling en zorg voor craniosynostose] [Guideline database website]. February 17, 2020. Accessed March 24, 2022. https://richtlijnendatabase.nl/richtlijn/craniosynostose/verwijzing_en_diagnostiek_bij_craniosynostose.html [Google Scholar]
- 3.Tonne E, Due-Tonnessen BJ, Wiig U, et al. Epidemiology of craniosynostosis in Norway. J Neurosurg Pediatr 2020;26:68–75 [DOI] [PubMed] [Google Scholar]
- 4.Cornelissen M, Ottelander B, Rizopoulos D, et al. Increase of prevalence of craniosynostosis. J Craniomaxillofac Surg 2016;44:1273–1279 [DOI] [PubMed] [Google Scholar]
- 5.Spivack O, Gaillard L. ERN CRANIO hospital representatives. ERN CRANIO patient coverage of craniosynostosis in Europe. Orphanet J Rare Dis 2022;17:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.ERN CRANIO. Craniosynostosis [ERN CRANIO website]. Accessed September 19, 2022. https://ern-cranio.eu/diagnoses/craniosynostosis-and-other-craniofacial-anomalies/craniosynostosis/ [Google Scholar]
- 7.ERN CRANIO. Endoscopically assisted suturectomy for scaphocephaly. January 7, 2022. Accessed September 19, 2022. https://www.youtube.com/watch?v=gcpWY-9cCoM [Google Scholar]
- 8.ERN CRANIO. Endoscopically assisted suturectomy for trigonocephaly. January 7, 2022. Accessed September 19, 2022. https://www.youtube.com/watch?v=F2Ggi9YpdiY [Google Scholar]
- 9.ERN CRANIO. Endoscopically assisted suturectomy for anterior plagiocephaly. January 7, 2022. Accessed September 19, 2022. https://www.youtube.com/watch?v=Alb93ynobjI [Google Scholar]
- 10.ERN CRANIO. Correction of the forehead in trigonocephaly. March 17, 2022. Accessed September 19, 2022. https://www.youtube.com/watch?v=5SA3Bw7rBhE [Google Scholar]
- 11.ERN CRANIO. Correction of the forehead in plagiocephaly. March 17, 2022. Accessed September 19, 2022. https://www.youtube.com/watch?v=0LjSEPPrixA [Google Scholar]
- 12.ERN CRANIO. Classic correction in scaphocephaly. March 17, 2022. Accessed September 19, 2022. https://www.youtube.com/watch?v=f44u72MrsMc [Google Scholar]
- 13.ERN CRANIO. Spring distraction for scaphocephaly. September 25, 2020. Accessed September 19, 2022. https://www.youtube.com/watch?v=Rs53OtFKNRo [Google Scholar]
- 14.ERN CRANIO. Distraction of the back of the skull or occipital distraction. September 25, 2020. Accessed September 19, 2022. https://www.youtube.com/watch?v=wW-everrA8k [Google Scholar]
- 15.ERN CRANIO. Le Fort Face correction. March 17, 2022. Accessed September 19, 2022. https://www.youtube.com/watch?v=0KlCPkIWB_c [Google Scholar]
- 16.ERN CRANIO. Monobloc and faciotomy. September 25, 2020. Accessed September 19, 2022. https://www.youtube.com/watch?v=KLeh6ZnC3iY [Google Scholar]
- 17.ERN CRANIO. Monobloc and faciotomy. September 25, 2020. Accessed September 19, 2022. https://www.youtube.com/watch?v=KLeh6ZnC3iY [Google Scholar]
- 18.ERN CRANIO. Le Fort Face correction. March 17, 2022. Accessed September 19, 2022. https://www.youtube.com/watch?v=0KlCPkIWB_c [Google Scholar]