

Abstract

Synthetic analogues of the DNA-alkylating cytotoxins of the duocarmycin class have been extensively investigated in the past 40 years, driven by their high potency, their unusual mechanism of bioactivity, and the beautiful modularity of their structure–activity relationship (SAR). This Perspective analyzes how the molecular designs of synthetic duocarmycins have evolved: from (1) early SAR studies, through to modern applications for directed cancer therapy as (2) prodrugs and (3) antibody–drug conjugates in late-stage clinical development. Analyzing 583 primary research articles and patents from 1978 to 2022, we distill out a searchable A0-format “Minard map” poster of ca. 200 key structure/function-tuning steps tracing chemical developments across these three key areas. This structure-based overview showcases the ingenious approaches to tune and target bioactivity, that continue to drive development of the elegant and powerful duocarmycin platform.
Keywords: duocarmycin, cancer prodrug, CC-1065, antibody-drug-conjugates (ADC), CBI therapeutics, structural evolution
1. Introduction
The natural products CC-1065 and duocarmycin SA are irreversible DNA alkylators that react after docking in the minor groove. Since their isolation from Streptomyces from 1978 onward,1,2 their picomolar cytotoxic potency has attracted continuous attention. Several total syntheses have been reported,3−5 and biochemical research has shown how their site-selectivity of DNA alkylation depends on structural features and stereochemistry.6,7 Clinical drug8 and prodrug9 candidates for cancer treatment quickly advanced to phase I and II clinical trials.10−13 Even after initial trials were discontinued due to narrow therapeutic index or strong side effects, an entire “duocarmycin family” of synthetic analogues with a broad range of aims and applications have been pursued. This minireview aims to distill this diversity of duocarmycin development into a rapidly grasped, yet comprehensive, format.
Medicinal chemistry around duocarmycins has focused on three key areas (Figure 1). (1) SAR studies have explored the relationship of pharmacophore structure to DNA alkylation, and simplified synthetic analogues such as the cyclopropabenz[e]indoles (CBIs)14 have been developed, to retain the parent functionality but with greater chemical tractability.15(2) Prodrugs aiming to direct activity better toward target cancer cells have explored activatable alkylation motifs and bifunctional conjugates.16(3) Antibody-drug conjugates (ADCs) have also been developed for improved targeting, and in this incarnation the first duocarmycin derivative was recently FDA-approved.17
Figure 1.

(a) 583 duocarmycin research reports were classified by a nine-point scheme, then structurally analyzed. (b) The A0-sized Poster S1 “Minard map” summarizes the structural evolution of >200 duocarmycin-derived agents from SAR to prodrugs and ADC.
In this Perspective we present a focused digest of the chemistry in these areas, collating most of the prolific research on duocarmycins using a systematic literature review workflow (SLR;18Figure 1a, details in Supporting Information), then graphically summarizing it for rapid analysis. We classify the SLR database according to research focus, use it for meta-analysis, and provide it for future researchers with an interest in the field to orient their molecular designs. We then provide a searchable, dynamic datafile in A0 poster format (Figure 1b; Poster S1) which groups and analyzes structural design features, with particular focus on duocarmycin family (1) SAR, (2) prodrugs and bifunctional conjugates, and (3) ADCs.
2. Systematic Literature Review (SLR)
SLR18 was conducted to collate and group the vast majority of experimental literature concerning duocarmycins. Two groups with low structural diversity were split off: (a) reports of the isolation, characterization, and mechanism of action of natural products structurally related to CC-1065; and (b) reports of preclinical and clinical trials of early cancer drug candidates. Three groups with high structural diversity are analyzed here: (1) SAR: synthesis and cellular evaluation of derivatives in structure–activity-relationship (SAR) studies; (2) Prodrugs: synthesis, evaluation, and/or therapeutic use of prodrugs, mainly based on bioactivation of the seco-duocarmycin latent alkylator functional unit, or of bifunctional small molecule conjugates bearing at least one (seco-)duocarmycin; (3) ADCs: synthesis, conjugation, and therapeutic efficacy of (multi)functional ADCs incorporating a synthetic duocarmycin or its seco-precursor.
Literature screening was first done by Boolean keyword search initiated with, e.g., [“CC-1065” or “duocarmycin”] AND [“analog” or “prodrug” or “derivative”] then refined with more specific keywords (see Supporting Information). From this, the major academic groups or pharmaceutical companies in each area of research were identified. For each group, all references reporting duocarmycin family agents were manually collected and categorized. Lastly, selected recent reviews on specific topics within the field of duocarmycins16,17,19−24 were harvested for additional references. Thus, a comprehensive duocarmycin structural library was assembled, from 583 reports—mainly of primary research (Figure 2).
Figure 2.
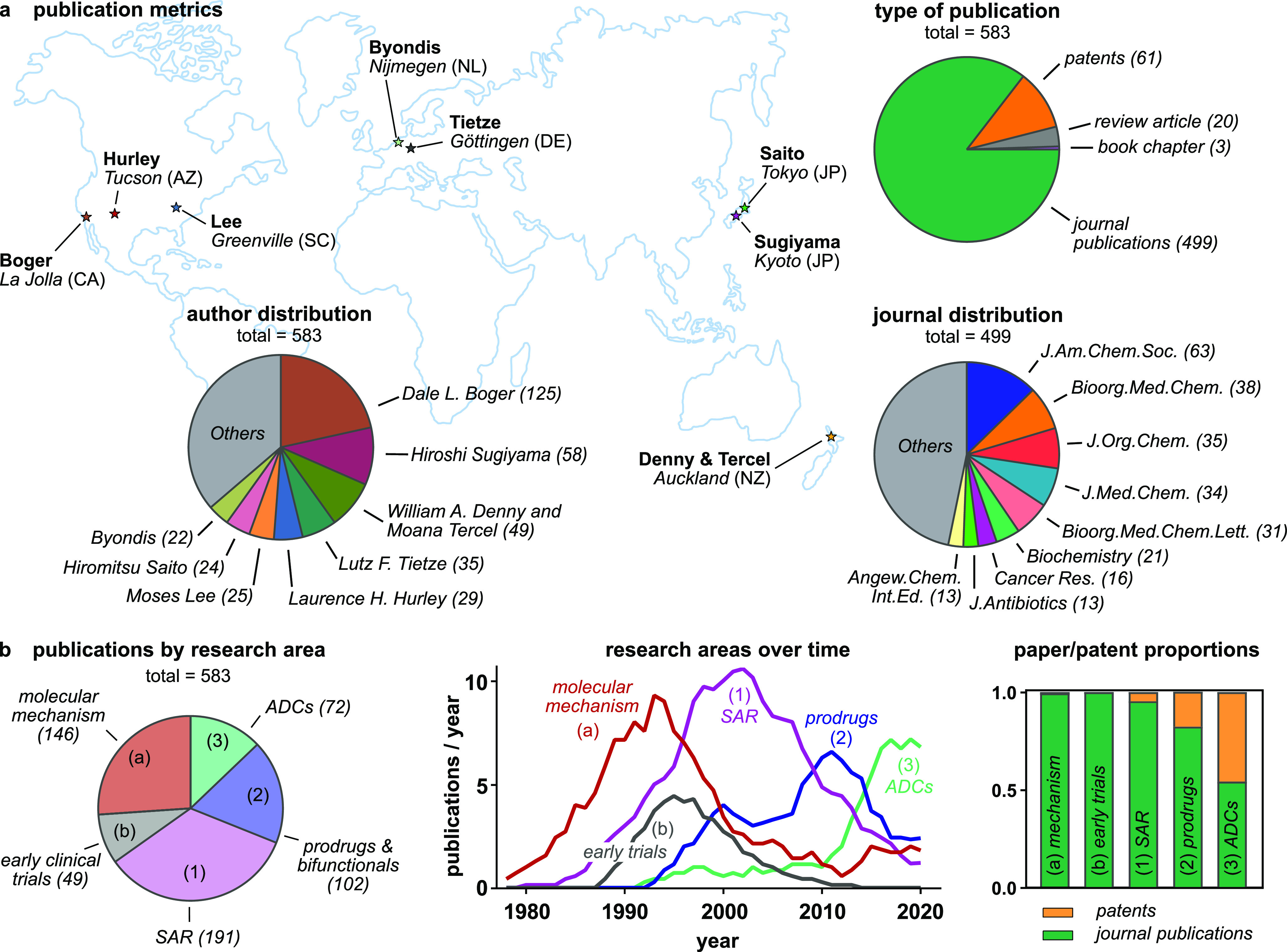
Literature metrics. (a) 583 primary research items form the duocarmycin literature database reviewed here. Charts show the major research groups (>20 publications) and journals (>10 publications). (b) The literature was grouped as: (a) natural products, biochemistry, and molecular mechanism of CC-1065 and close analogues; (b) initial clinical trial compounds; then the focus groups of this Perspective: (1) synthetic analogues and SAR; (2) prodrugs and bifunctional conjugates; (3) ADCs. Group histograms reveal the chronological progress of duocarmycin research. Paper/patent ratios may indicate perceived commercialisation chances.
2.1. Literature Metrics
A bibliographic overview of this library is given in Figure 2. Of the 583 total research items, the vast majority were published in scientific journals (123 journals, 499 publications, 86%) covering all areas from basic biology, biochemistry, medicinal chemistry, and molecular sciences, to physical chemistry, theoretical chemistry, and preclinical or clinical oncology. Major progress in chemical design and SAR has been published in chemistry (JACS 63, JOC 35, JMC 33, ANIE 13, Chem. Eur. J. 11) and bioorganic chemistry journals (BMC 38, BMCL 31); isolation and mechanism reports cluster in Biochemistry (21) and J. Antibiotics (13); and clinical results in oncology journals (Cancer Res. 16, Cancer Chemother. Pharmacol. 11, Mol. Cancer Ther. 9). 61 patents or patent applications filed by academic groups and pharmaceutical companies also entered this library (Figure 2a).
2.2. Evolution of the Focus of Duocarmycin Research
The sequence of duocarmycin development is easily visible when analyzing the five report groups by date (Figure 2b). Isolation and early molecular mechanism studies (group a; 146 items) dominate the 1980s and 1990s and have been key for further molecular designs. Rapidly following initial cytotoxicity studies, small molecule drugs (adozelesin and bizelesin) and hydrolytic prodrugs (carzelesin and pibrozelesin) were taken into initial clinical anticancer trials, that were discontinued during the 2000s (group b; 49 items). Hurley (29), Krueger (17), and others were the major academic groups driving both these developments.
Exhaustive and creative structural variations during the 1990s and 2000s largely mapped the SAR in this molecular class (Group 1, SAR: 192 items) with major contributions by Boger (125), Sugiyama (58), and Lee (25). Innovation increasingly focused on targeting, with activatable prodrugs and bifunctional small molecule conjugates taking off during the 2000s and 2010s (Group 2, Prodrugs: 102 items) led by Denny and Tercel (49), Tietze (35), Saito (23), and others. Finally, since the 2010s, conjugates of duocarmycins with monoclonal antibodies (Group 3, ADCs: 71 items) have opened up a new future for this class of bioactives. Combining the tunable potency and molecular flexibility of the duocarmycins, with the potential for enriched delivery to cancers, has led to a new wave of duocarmycin antibody–drug conjugates in clinical trials, driven by Byondis B.V. (22), Medarex Inc. (7), and others. With their increasing therapeutic relevance, the share of patents in the last two areas of research is also significantly higher (Figure 2b).
3. Structural Evolution of Duocarmycin Analogues
The structural evolution of duocarmycins across these groups can also be best understood along a time axis, that resolves both the stepwise and disruptive innovations that have driven this field from 1978 to 2022. Figure 3 is a cartoon representation showing a data point for each research item in the three focus groups (circle: journal; star: patent); the A0-size Poster S1 in the Supporting Information maps these data points one-to-one onto representative chemical structures from each research item, color-coded for functionality, and DOI-hyperlinked for access to the original papers. We also encourage interested chemists to print a copy for easy reference.
Figure 3.
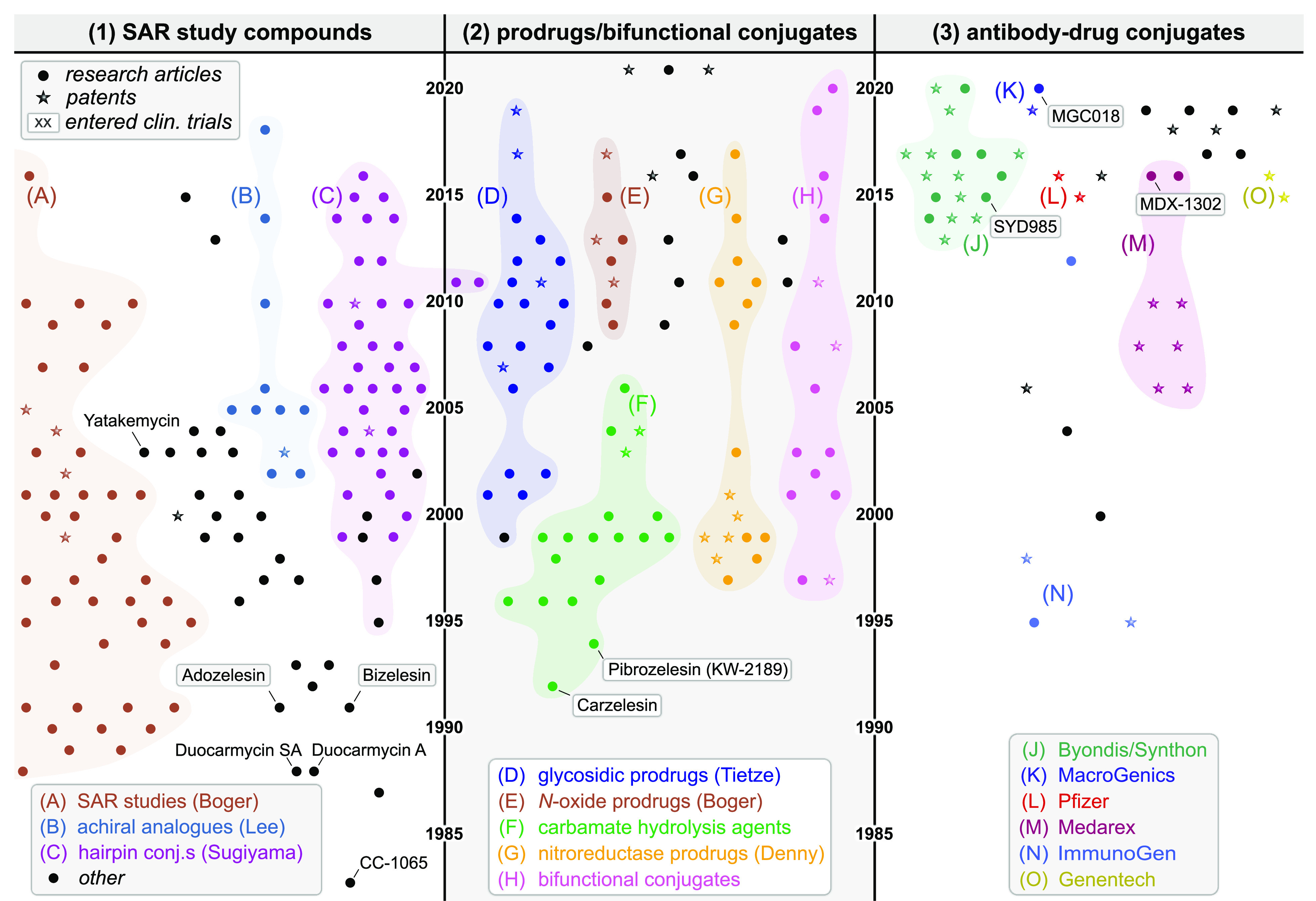
Structural developments of the duocarmycins (cartoon; all chemical structures in Poster S1). In Group 1 (SAR compounds), studies resolved the molecular motifs crucial for rational tuning of bioactivity. In Group 2 (Prodrugs), non-natural prodrugs (glycosides, nitroaryls, carbamates, N-oxides) and bifunctional conjugates expanded the scope of duocarmycins. In Group 3 (ADCs), industry has been a main driver of research.
3.1. Group 1: SAR Compounds
The lead natural product CC-1065 was isolated in 1978,1 and its first total synthesis was reported in 1988, laying the grounds for much synthetic development.3 During the 1990s and 2000s, systematic variations of both the core alkylator motif (“segment A”, typically an activated cyclopropane) and the DNA-docking motif (“segment B”) led to our current understanding of the structural features that need to be arranged for DNA association and sequence-selective alkylation (succinctly described by Hurley7).
Many heterocyclic systems beyond the native cyclopropa[e]pyrroloindole (CPI) of duocarmycin SA25 can serve as segment A. Much research has focused on the chemically tractable CBI, with optional substitutions;14 cyclopropaindole26 (CI) and others27 also alkylate DNA with the reactivity trend (CBI ∼ CPI > CI) (Figure 4a).
Figure 4.
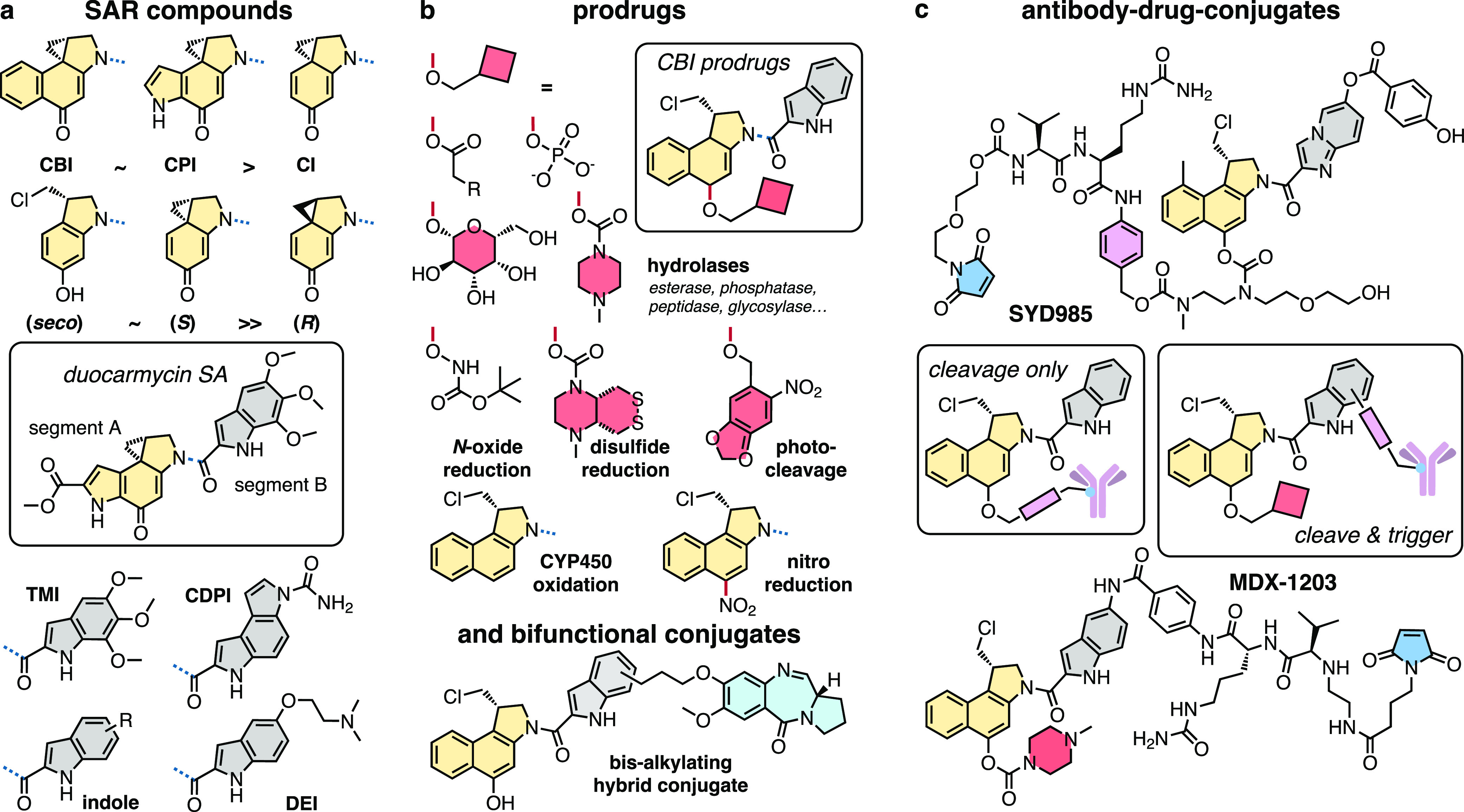
Structural elements of duocarmycin therapeutics. (a) SAR analysis: variations of segments A and B. (b) Activatable prodrugs: strategies to trigger bioactivity. (c) Antibody–drug conjugates: CBI-ADCs including clinical candidates SYD985 and MDX-1302. See also Poster S1.
The activated cyclopropane electrophile must be in its native (S)-configuration for DNA alkylation,28,29 but high potency can be maintained with “proagent” seco-variants, that use in situ intramolecular Winstein spirocyclization to unfurl their activated cyclopropane, relying on the para-phenol.30 Good leaving groups (-Cl, -Br, -OMs)9,31 and several alternatives to the dihydroindole (5-, 6-, 7-membered rings)32 are all tolerated. Alternatively, masking this phenol suppresses spirocyclization:33 a disruptive step that opened the door for rational tuning of prodrug candidates in later years (see below). The group of Lee also introduced achiral seco-variants that are similarly reactive, but structurally simpler and more accessible.34,35
Segment B heterocycles have mainly clustered around indole-based rings that strengthen DNA binding. Stepwise simplification of the native dimeric segment B (in CC-1065) gave variously the deoxygenated CDPI dimer,36 3,4,5-trimethoxyindole (TMI),29 and monoalkoxylated (DEI),37 or even simple mono/oligoindoles; and other heterocycles38 can also be used. These do impact DNA binding, alkylation site-selectivity, and potency; but overall, the tolerance for segment B variance is high (Figure 4a).
Assembling the A and B segments has also received attention. A remarkable class of hairpin duocarmycin conjugates was driven by Lown and Sugiyama in the 2000s.39 Using synthetic oligo-pyrroles/imidazoles from the minor-groove binder distamycin A as segment B binding domains gave potent duocarmycin analogues allowing sequence-selective alkylation in specific areas of DNA.40,41 “Standard” duocarmycins consist of segments A and B linked by an amide bond: but the natural product Yatakemycin42,43 has revealed that multiple B segments may be used, and randomly shuffled around without losing bioactivity.44 Dimeric bisalkylators with two A segments, allowing interstrand DNA cross-linking, also give extremely high potency.45,46
3.2. Group 2: Activatable Prodrugs and Bifunctionals
Early trials already exploited duocarmycin prodrugs where seco-duocarmycin bioactivity was to be triggered in situ by unmasking a para-phenol, to avoid parasitic loss of a preformed cyclopropane en route to target tissues. These carbamate hydrolysis designs (Carzelesin,47,48 Pibrozelesin/KW-21899,49) were discontinued in clinical trials due to side effects and low therapeutic index.13,50 Follow-up work mined esters, solubilized carbamates, phosphates, and others as other hydrolytic activation methods (Figure 4b),51−56 although none of these promise any greater mechanistic selectivity for cancer, since the activating hydrolases are ubiquitously expressed.
Key steps toward cancer-selective prodrugs were initiated by the lab of Denny in the late 1990s. They introduced nitro-seco-CBIs that can be irreversibly reduced to the amino-seco-CBI in the low-oxygen conditions found in solid tumors. These amines then undergo Winstein cyclization becoming DNA-alkylators (Figure 4b).57−59 In the early 2000s, Tietze developed glycosidic prodrugs that can be built modularly, aiming at antitumor uses relying on glycosidases.37,60,61 Adopting novel chemistries in the 2010s, Boger introduced O-amino-N-acyl-seco-CBIs that are also subject to bioreductive activation.62−64 The field of masked seco-CBIs has by now exploited the full arsenal of chemical biology, passing through reducible Co-complexes,65 Fe(II)-reactive peroxides,66 photoactivated designs,67,68 and oxidizable naphthalenes.69 Recently, cyclic dichalcogenides (that resist monothiol exchange, but can be reductively activated by specific oxidorectases such as thioredoxin) have joined this panoply of prodrugs.70,71 Finally, bifunctional conjugates of duocarmycins with other pharmaceuticals (glucuronide,72 biotin,73 antibiotics,74 pyrrolobenzodiazepine (Figure 4b),75,76 albumin,77 peptides78) show the wide applicability and adaptability of this unique class of bioactives.
3.3. Group 3: Antibody–Drug Conjugates (ADCs)
Monoclonal antibodies against cancer-selective biomarkers have the potential to deliver high-potency cytotoxic cancer drugs to tumors in a targeted and therapeutically effective manner. The duocarmycins’ outstanding potency has motivated much ADC research, with two general designs emerging. Type A designs mask the seco-duocarmycin phenol with a linker conjugated to the antibody: allowing spirocyclization-based activation after linker cleavage. Intracellular cleavage of these linkers (dipeptides like ValCit that are prone to lysosomal proteolysis; hydrolyzable phosphates; reducible disulfides) can directly liberate the key Winstein cyclization phenol, but additional self-immolative spacers, that undergo cyclization or elimination cascades to liberate this phenol, are common.79−83 Type B designs attach a phenolic prodrug of the duocarmycin, via a peripheral site, to the antibody: permitting an extra layer of prodrug-based selectivity if prerelease activation can be avoided (Figure 4c). ADCs of Type B are less clearly reported, and many are IP-protected by pharmaceutical companies.84−87
The late-2000s rebirth of preclinical/clinical development in the duocarmycin class has essentially been driven by these ADCs, with a variety of designs achieving in vivo efficacy in mouse cancer models.56,88−92 Beyond the choice of biomarker and payload, ADC development must balance factors from conjugation site, degree of labeling, and linker nature,93,94 through to chemical conjugation method, making refinement of ADCs more complex than that of prodrugs.95 Nevertheless, the ADCs SYD985, MGC018, and MDX-1203 all reached clinical trials with promising results and high efficacy.96−98 While MDX-1203 was halted due to insufficient improvement of therapeutic benefit compared to alternative therapeutics, SYD985 was recently given fast-track approval as a follow-up or cotreatment for patients with HER2-positive metastatic breast cancer.99 This is the first duocarmycin approved for clinical use; its success will spur the developments of the future (Figure 5).
Figure 5.
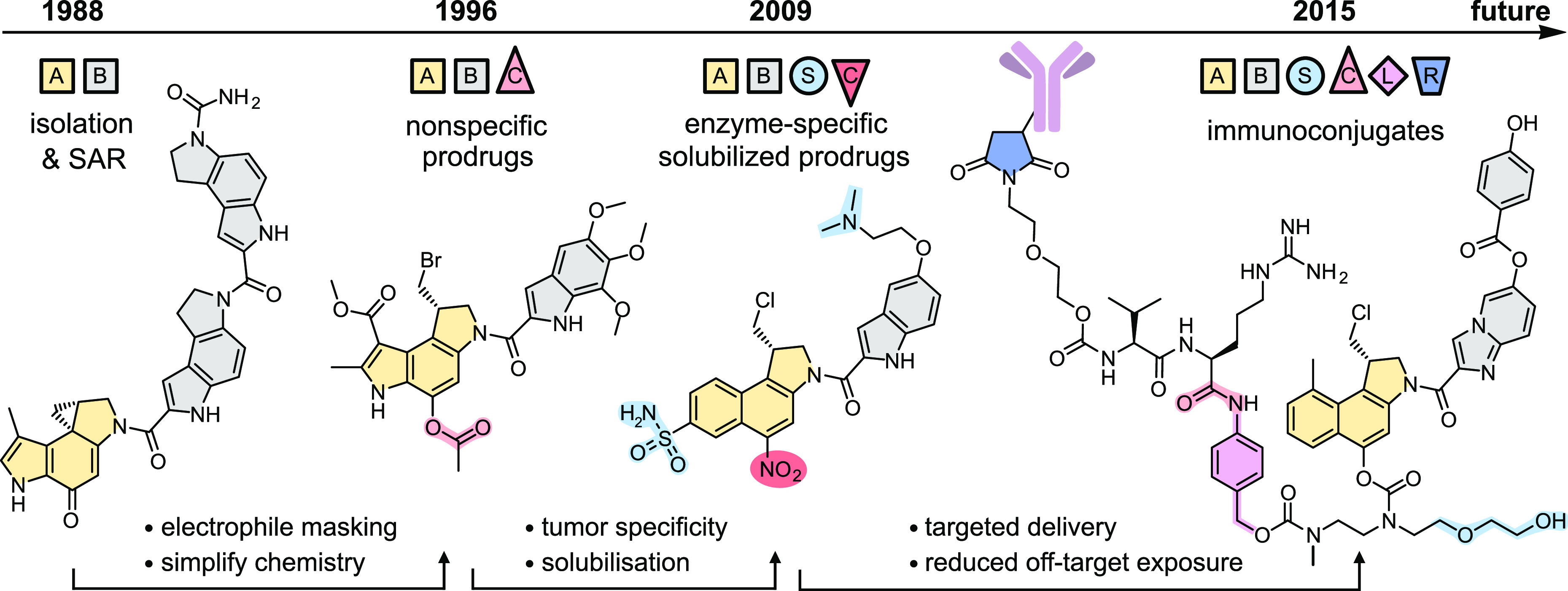
Color-coded highlights of the disruptive chemical steps that have led the duocarmycins from isolation to the clinic (see also Poster S1). (Key: A,B = A-,B-segments. C = intracellular cleavage site. S = solubilizer. L = self-immolative spacer. R = reactive group for antibody conjugation.)
3.4. Guiding Principles for Future Developments
Predicting the future of drug development is a challenge, but this structure/function-based Perspective highlights trends that can drive the duocarmycins’ next decades. First, duocarmycins will remain high-value targets in cancer therapy. If their bioactivity can be directed, then their outstanding potency and their binding-triggered covalent-reactive mechanism, promise high efficacy with limited resistance in a broad scope of indications. Second, progress will continue to rely on disruptive innovations in duocarmycin chemistry. Key strategies so far include (i) SAR simplification for synthetic access, (ii) protecting the cyclopropane warhead by forming it in situ, (iii) chemical mechanisms for tumor-selective prodrug activation, and (iv) antibody-based mechanisms for tumor-selective prodrug delivery. Solubilization and self-immolative spacers have also proven critical. It is perhaps no accident that the first duocarmycin to be clinically successful had built in nearly all these strategies (Figure 5). We see much potential for new therapeutics that also harness these strategies but tackle other indications with different target expression profiles and biodistribution needs. We also believe that finding sufficiently selective yet sufficiently high-turnover chemical mechanisms for tumor-specific activation would revolutionize both ADC and small molecule prodrug applications, and we await developments in this still-underexplored chemical space.
4. Conclusions
Duocarmycins have undergone great efforts toward developing targeted cancer therapeutics. A careful understanding of their unusual mechanism of bioactivity, leveraging spirocyclization and docking for high-potency site-selective DNA alkylation, has enabled many creative approaches using the duocarmycins as a modular bioactive platform. Here we have provided a structured literature review tracking the chemical developments of the last 40 years, that have led from isolation to basic understanding, early trials and setbacks, re-engineering, and ultimately a first clinical anticancer agent.
We hope this concise overview will promote a structure/function-based understanding, allowing rational design and use of duocarmycin-based bioactives. It also follows the didactic tradition of Njardarson’s Posters100 with the A0-size Poster S1, that can be printed and hung up in hallways for graphical overview and discussions, or used digitally for easy followup of its 200 embedded key structures (DOI hyperlinks).
The modularity of duocarmycin bioactivity should encourage researchers to design in structural features à la carte. A structure-based overview to guide the choice and understanding of these features, with easy direction to the corresponding references, may be very helpful for gaining a coup d’oeil when entering new scientific territory: particularly where the frontiers of research are increasingly interdisciplinary. We can still expect much from the duocarmycins; and we hope this Perspective and its Poster bring a graphic understanding of how to design, incorporate, and exploit this powerful molecular class.
Acknowledgments
We thank Julia Schulz (Institute for Machine Tools and Industrial Management, TU Munich) for conceptual input concerning structured literature review, Dale Boger (Scripps) for references and kind discussion, Marc-André Kasper (Tubulis GmbH, Martinsried) for collegial discussion about the therapeutic impact of antibody-drug-conjugates.
Glossary
Abbreviations
- CBI
cyclopropabenz[e]indole
- CDPI
3-carbamoyl-1,2-dihydro-3H-pyrrolo[3,2-e]indole-7-carboxylate
- CPI
cyclopropa[e]pyrroloindole
- CI
cyclopropaindole
- DEI
N,N-dimethylaminoethoxyindole
- TMI
3,4,5-trimethoxyindole
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.2c00448.
Details of how the SLR has been performed, including grouped lists of the major contributors to SAR/prodrug/ADC research, and containing the hyperlinked full-format bibliography of the 583 references in the SLR, organized by theme (PDF)
Poster S1 (>200 structures with hyperlinked references) (PDF)
ChemDraw file corresponding to the Poster, which contains all the duocarmycin structures without hyperlinks in CDX format for use/reuse of structures by copy-paste, or for mining the structures by SMILES (CDX)
∼600-work literature citation database provided as an RIS that can be imported into any reference manager software with one double-click, to make all the duocarmycin citations available (ZIP)
Author Contributions
CRediT: Jan Gabriel Felber conceptualization, data curation, investigation, methodology, software, validation, visualization, writing-original draft, writing-review & editing; Oliver Thorn-Seshold conceptualization, supervision, visualization, writing-original draft, writing-review & editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Hanka L. J.; Dietz A.; Gerpheide S. A.; Kuentzel S. L.; Martin D. G. CC-1065 (NSC-298223), A New Antitumor Antibiotic. Production, in Vitro Biological Activity, Microbiological Assays and Taxonomy of the Producing Microorganism. J. Antibiot. 1978, 31 (12), 1211–1217. 10.7164/antibiotics.31.1211. [DOI] [PubMed] [Google Scholar]
- Takahashi I.; Takahashi K.-I.; Ichimura M.; Morimoto M.; Asano K.; Kawamoto I.; Tomita F.; Nakano H. Duocarmycin A, a New Antitumor Antibiotic from Streptomyces. J. Antibiot. 1988, 41 (12), 1915–1917. 10.7164/antibiotics.41.1915. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Coleman R. S. Total Synthesis of (+)-CC-1065 and Ent-(−)-CC-1065. J. Am. Chem. Soc. 1988, 110 (4), 1321–1323. 10.1021/ja00212a067. [DOI] [Google Scholar]
- Boger D. L.; Machiya K.; Hertzog D. L.; Kitos P. A.; Holmes D. Total Synthesis and Preliminary Evaluation of (+)- and Ent-(−)-Duocarmycin SA. J. Am. Chem. Soc. 1993, 115 (20), 9025–9036. 10.1021/ja00073a019. [DOI] [Google Scholar]
- Okano K.; Tokuyama H.; Fukuyama T. Total Synthesis of (+)-Yatakemycin. J. Am. Chem. Soc. 2006, 128 (22), 7136–7137. 10.1021/ja0619455. [DOI] [PubMed] [Google Scholar]
- Hurley L. H.; Reynolds V. L.; Swenson D. H.; Petzold G. L.; Scahill T. A. Reaction of the Antitumor Antibiotic CC-1065 with DNA: Structure of a DNA Adduct with DNA Sequence Specificity. Science 1984, 226 (4676), 843–844. 10.1126/science.6494915. [DOI] [PubMed] [Google Scholar]
- Hurley L. H.; Lee C. S.; McGovren J. P.; Warpehoski M. A.; Mitchell M. A.; Kelly R. C.; Aristoff P. A. Molecular Basis for Sequence-Specific DNA Alkylation by CC-1065. Biochemistry 1988, 27 (10), 3886–3892. 10.1021/bi00410a054. [DOI] [PubMed] [Google Scholar]
- Bhuyan B. K.; Smith K. S.; Adams E. G.; Wallace T. L.; Von Hoff D. D.; Li L. H. Adozelesin, a Potent New Alkylating Agent: Cell-Killing Kinetics and Cell-Cycle Effects. Cancer Chemother. Pharmacol. 1992, 30 (5), 348–354. 10.1007/BF00689961. [DOI] [PubMed] [Google Scholar]
- Kobayashi E.; Okamoto A.; Asada M.; Okabe M.; Nagamura S.; Asai A.; Saito H.; Gomi K.; Hirata T. Characteristics of Antitumor Activity of KW-2189, a Novel Water-Soluble Derivative of Duocarmycin, against Murine and Human Tumors. Cancer Res. 1994, 54, 2404–2410. [PubMed] [Google Scholar]
- Shamdas G. J.; Alberts D. S.; Modiano M.; Wiggins C.; Power J.; Kasunic D. A.; Elfring G. L.; Earhart R. H. Phase I Study of Adozelesin (U-73,975) in Patients with Solid Tumors. Anti-Cancer Drugs 1994, 5, 10–14. 10.1097/00001813-199402000-00002. [DOI] [PubMed] [Google Scholar]
- Alberts S. R.; Erlichman C.; Reid J. M.; Sloan J. A.; Ames M. M.; Richardson R. L.; Goldberg R. M. Phase I Study of the Duocarmycin Semisynthetic Derivative KW-2189 given Daily for Five Days Every Six Weeks. Clin. Cancer Res. 1998, 4, 2111–2117. [PubMed] [Google Scholar]
- Cristofanilli M.; Bryan W. J.; Miller L. L.; Chang A. Y.-C.; Gradishar W. J.; Kufe D. W.; Hortobagyi G. N. Phase II Study of Adozelesin in Untreated Metastatic Breast Cancer. Anti-Cancer Drugs 1998, 9, 779–782. 10.1097/00001813-199810000-00006. [DOI] [PubMed] [Google Scholar]
- Markovic S. N.; Suman V. J.; Vukov A. M.; Fitch T. R.; Hillman D. W.; Adjei A. A.; Alberts S. R.; Kaur J. S.; Braich T. A.; Leitch J. M.; Creagan E. T. Phase II Trial of KW2189 in Patients With Advanced Malignant Melanoma. Am. J. Clin. Oncol. 2002, 25 (3), 308–312. 10.1097/00000421-200206000-00022. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Ishizaki T.; Zarrinmayeh H.; Kitos P. A.; Suntornwat O. A Potent, Simple Derivative of an Analog of the CC-1065 Alkylation Subunit. Bioorg. Med. Chem. Lett. 1991, 1 (1), 55–58. 10.1016/S0960-894X(01)81090-8. [DOI] [Google Scholar]
- MacMillan K. S.; Boger D. L. Fundamental Relationships between Structure, Reactivity, and Biological Activity for the Duocarmycins and CC-1065. J. Med. Chem. 2009, 52 (19), 5771–5780. 10.1021/jm9006214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes Z.; Morais G. R.; Loadman P. M.; Pors K. How Can the Potential of the Duocarmycins Be Unlocked for Cancer Therapy?. Drug Discovery Today 2021, 26 (2), 577–584. 10.1016/j.drudis.2020.11.020. [DOI] [PubMed] [Google Scholar]
- Yao H.-P.; Zhao H.; Hudson R.; Tong X.-M.; Wang M.-H. Duocarmycin-Based Antibody–Drug Conjugates as an Emerging Biotherapeutic Entity for Targeted Cancer Therapy: Pharmaceutical Strategy and Clinical Progress. Drug Discovery Today 2021, 26 (8), 1857–1874. 10.1016/j.drudis.2021.06.012. [DOI] [PubMed] [Google Scholar]
- Boell S. K. Cecez-Kecmanovic Dubravka. On Being “Systematic” in Literature Reviews in IS. Journal of Information Technology 2015, 30 (2), 161–173. 10.1057/jit.2014.26. [DOI] [Google Scholar]
- Ghosh N.; Sheldrake H.; Searcey M.; Pors K. Chemical and Biological Explorations of the Family of CC-1065 and the Duocarmycin Natural Products. Curr. Top. Med. Chem. 2009, 9 (16), 1494–1524. 10.2174/156802609789909812. [DOI] [PubMed] [Google Scholar]
- Patil P.; Satam V.; Lee M. A Short Review on the Synthetic Strategies of Duocarmycin Analogs That Are Powerful DNA Alkylating Agents. Anti-Cancer Agents in Med. Chem. 2015, 15 (5), 616–630. 10.2174/1871520615666141216144116. [DOI] [PubMed] [Google Scholar]
- Searcey M. Duocarmycins - Natures Prodrugs?. Curr. Pharm. Des 2002, 8 (15), 1375–1389. 10.2174/1381612023394539. [DOI] [PubMed] [Google Scholar]
- Cacciari B.; Romagnoli R.; Baraldi P. G.; Ros T. D.; Spalluto G. CC-1065 and the Duocarmycins: Recent Developments. Exp. Op. Ther. Patents 2000, 10 (12), 1853–1871. 10.1517/13543776.10.12.1853. [DOI] [Google Scholar]
- Tietze L. F.; Krewer B. Antibody-Directed Enzyme Prodrug Therapy: A Promising Approach for a Selective Treatment of Cancer Based on Prodrugs and Monoclonal Antibodies. Curr. Biol. Drug Des. 2009, 74 (3), 205–211. 10.1111/j.1747-0285.2009.00856.x. [DOI] [PubMed] [Google Scholar]
- F. Tietze L.; Schmuck K. Prodrugs for Targeted Tumor Therapies: Recent Developments in ADEPT, GDEPT and PMT. Curr. Pharm. Des. 2011, 17 (32), 3527–3547. 10.2174/138161211798194459. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Coleman R. S. Total Synthesis of (.+-.)-N2-(Phenylsulfonyl)-CPI, (.+-.)-CC-1065, (+)-CC-1065, Ent-(−)-CC-1065, and the Precise, Functional Agents (.+-.)-CPI-CDPI2, (+)-CPI-CDPI2, and (−)-CPI-CDPI2 [(.+-.)-(3bR*,4aS*)-, (+)-(3bR,4aS)-, and (−)-(3bS,4aR)-Deoxy-CC-1065]. J. Am. Chem. Soc. 1988, 110 (14), 4796–4807. 10.1021/ja00222a043. [DOI] [Google Scholar]
- Boger D. L.; Zarrinmayeh H.; Munk S. A.; Kitos P. A.; Suntornwat O. Demonstration of a Pronounced Effect of Noncovalent Binding Selectivity on the (+)-CC-1065 DNA Alkylation and Identification of the Pharmacophore of the Alkylation Subunit. Proc. Natl. Acad. Sci. U.S.A. 1991, 88 (4), 1431–1435. 10.1073/pnas.88.4.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger D. L.; Garbaccio R. M.; Jin Q. Synthesis and Evaluation of CC-1065 and Duocarmycin Analogues Incorporating the Iso-CI and Iso-CBI Alkylation Subunits: Impact of Relocation of the C-4 Carbonyl. J. Org. Chem. 1997, 62 (25), 8875–8891. 10.1021/jo971686p. [DOI] [Google Scholar]
- Boger D. L.; Yun W.; Terashima S.; Fukuda Y.; Nakatani K.; Kitos P. A.; Jin Q. DNA Alkylation Properties of the Duocarmycins: (+)-Duocarmycin A, Epi-(+)-Duocarmycin A, Ent-(−)-Duocarmycin A and E&Ent-(−)-Duocarmycin A. Bioorg. Med. Chem. Lett. 1992, 2 (7), 759–765. 10.1016/S0960-894X(00)80407-2. [DOI] [Google Scholar]
- Boger D. L.; Yun W. CBI-TMI: Synthesis and Evaluation of a Key Analog of the Duocarmycins. Validation of a Direct Relationship between Chemical Solvolytic Stability and Cytotoxic Potency and Confirmation of the Structural Features Responsible for the Distinguishing Behavior of Enantiomeric Pairs of Agents. J. Am. Chem. Soc. 1994, 116 (18), 7996–8006. 10.1021/ja00097a006. [DOI] [Google Scholar]
- Baird R.; Winstein S. Neighboring Carbon and Hydrogen. Li. Dienones from Ar1[UNK]-3 Participation. Isolation and Behavior of Spiro(2,5)Octa-1,4-Diene-3-One. J. Am. Chem. Soc. 1963, 85, 567. 10.1021/ja00888a020. [DOI] [Google Scholar]
- Boger D. L.; Munk S. A.; Zarrinmayeh H. (+)-CC-1065 DNA Alkylation: Key Studies Demonstrating a Noncovalent Binding Selectivity Contribution to the Alkylation Selectivity. J. Am. Chem. Soc. 1991, 113 (10), 3980–3983. 10.1021/ja00010a046. [DOI] [Google Scholar]
- Boger D. L.; Hertzog D. L.; Bollinger B.; Johnson D. S.; Cai H.; Goldberg J.; Turnbull P. Duocarmycin SA Shortened, Simplified, and Extended Agents: A Systematic Examination of the Role of the DNA Binding Subunit. J. Am. Chem. Soc. 1997, 119 (21), 4977–4986. 10.1021/ja9637208. [DOI] [Google Scholar]
- Tietze L. F.; Lieb M.; Herzig T.; Haunert F.; Schuberth I. A Strategy for Tumor-Selective Chemotherapy by Enzymatic Liberation of Seco-Duocarmycin SA-Derivatives from Nontoxic Prodrugs. Bioorg. Med. Chem. 2001, 9 (7), 1929–1939. 10.1016/S0968-0896(01)00098-0. [DOI] [PubMed] [Google Scholar]
- Daniell K.; Stewart M.; Madsen E.; Le M.; Handl H.; Brooks N.; Kiakos K.; Hartley J. A.; Lee M. Design, Synthesis, and Biological Evaluation of Achiral Analogs of Duocarmycin SA. Bioorg. Med. Chem. Lett. 2005, 15 (1), 177–180. 10.1016/j.bmcl.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Sato A.; McNulty L.; Cox K.; Kim S.; Scott A.; Daniell K.; Summerville K.; Price C.; Hudson S.; Kiakos K.; Hartley J. A.; Asao T.; Lee M. A Novel Class of in Vivo Active Anticancer Agents: Achiral Seco -Amino- and Seco -Hydroxycyclopropylbenz[ e ]Indolone (Seco -CBI) Analogues of the Duocarmycins and CC-1065. J. Med. Chem. 2005, 48 (11), 3903–3918. 10.1021/jm050179u. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Ishizaki T.; Wysocki R. J.; Munk S. A.; Kitos P. A.; Suntornwat O. Total Synthesis and Evaluation of (.+-.)-N-(Tert-Butoxycarbonyl)-CBI, (.+-.)-CBI-CDPI1, and (.+-.)-CBI-CDPI2: CC-1065 Functional Agents Incorporating the Equivalent 1,2,9,9a-Tetrahydrocyclopropa[1,2-c]Benz[1,2-e]Indol-4-One (CBI) Left-Hand Subunit. J. Am. Chem. Soc. 1989, 111 (16), 6461–6463. 10.1021/ja00198a089. [DOI] [Google Scholar]
- Tietze L. F.; Major F.; Schuberth I. Antitumor Agents: Development of Highly Potent Glycosidic Duocarmycin Analogues for Selective Cancer Therapy. Angew. Chem., Int. Ed. 2006, 45 (39), 6574–6577. 10.1002/anie.200600936. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Yun W.; Han N. 1,2,9,9a-Tetrahydrocyclopropa[c]Benz[e]Indol-4-One (CBI) Analogs of CC-1065 and the Duocarmycins: Synthesis and Evaluation. Bioorg. Med. Chem. 1995, 3 (11), 1429–1453. 10.1016/0968-0896(95)00130-9. [DOI] [PubMed] [Google Scholar]
- Fregeau N. L.; Wang Y.; Pon R. T.; Wylie W. A.; Lown J. W. Characterization of a CPI-Lexitropsin Conjugate-Oligonucleotide Covalent Complex by 1H NMR and Restrained Molecular Dynamics Simulation. J. Am. Chem. Soc. 1995, 117 (35), 8917–8925. 10.1021/ja00140a004. [DOI] [Google Scholar]
- Bando T.; Narita A.; Asada K.; Ayame H.; Sugiyama H. Enantioselective DNA Alkylation by a Pyrrole–Imidazole S -CBI Conjugate. J. Am. Chem. Soc. 2004, 126 (29), 8948–8955. 10.1021/ja049398f. [DOI] [PubMed] [Google Scholar]
- Tao Z.-F.; Fujiwara T.; Saito I.; Sugiyama H. Rational Design of Sequence-Specific DNA Alkylating Agents Based on Duocarmycin A and Pyrrole–Imidazole Hairpin Polyamides. J. Am. Chem. Soc. 1999, 121 (21), 4961–4967. 10.1021/ja983398w. [DOI] [Google Scholar]
- Igarashi Y.; Futamata K.; Fujita T.; Sekine A.; Senda H.; Naoki H.; Furumai T. Yatakemycin, a Novel Antifungal Antibiotic Produced by Streptomyces Sp. TP-A0356. J. Antibiot. 2003, 56 (2), 107–113. 10.7164/antibiotics.56.107. [DOI] [PubMed] [Google Scholar]
- Tichenor M. S.; Kastrinsky D. B.; Boger D. L. Total Synthesis, Structure Revision, and Absolute Configuration of (+)-Yatakemycin. J. Am. Chem. Soc. 2004, 126 (27), 8396–8398. 10.1021/ja0472735. [DOI] [PubMed] [Google Scholar]
- Tichenor M. S.; MacMillan K. S.; Trzupek J. D.; Rayl T. J.; Hwang I.; Boger D. L. Systematic Exploration of the Structural Features of Yatakemycin Impacting DNA Alkylation and Biological Activity. J. Am. Chem. Soc. 2007, 129 (35), 10858–10869. 10.1021/ja072777z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y.; Seto S.; Furuta H.; Ebisu H.; Oomori Y.; Terashima S. The Novel Cyclopropapyrroloindole(CPI) Bisalkylators Bearing 3,3′-(1,4-Phenylene)Diacryloyl Group as a Linker. Bioorg. Med. Chem. Lett. 1998, 8 (15), 2003–2004. 10.1016/S0960-894X(98)00346-1. [DOI] [PubMed] [Google Scholar]
- Bando T.; Iida H.; Saito I.; Sugiyama H. Sequence-Specific DNA Interstrand Cross-Linking by Imidazole–Pyrrole CPI Conjugate. J. Am. Chem. Soc. 2001, 123 (21), 5158–5159. 10.1021/ja003660c. [DOI] [PubMed] [Google Scholar]
- Li L. H.; DeKoning T. F.; Kelly R. C.; Krueger W. C.; McGovren J. P.; Padbury G. E.; Petzold G. L.; Wallace T. L.; Ouding R. J.; Prairie M. D.; Gebhard I. Cytotoxicity and Antitumor Activity of Carzelesin, a Prodrug Cyclopropylpyrroloindole Analogue. Cancer Res. 1992, 52, 4904–4913. [PubMed] [Google Scholar]
- van Tellingen O.; Nooijen W. J.; Schaaf L. J.; Henrar R. E. C.; Beijnen J. H. Comparative Pharmacology of the Novel Cyclopropylpyrroloindole-Prodrug Carzelesin in Mice, Rats, and Humans. Cancer Res. 1998, 58, 2410–2416. [PubMed] [Google Scholar]
- Ogasawara H.; Nishio K.; Takeda Y.; Ohmori T.; Kubota N.; Funayama Y.; Ohira T.; Kuraishi Y.; Isogai Y.; Saijo N. A Novel Antitumor Antibiotic, KW-2189 Is Activated by Carboxyl Esterase and Induces DNA Strand Breaks in Human Small Cell Lung Cancer Cells. Jpn. J. Cancer Res. 1994, 85, 418–425. 10.1111/j.1349-7006.1994.tb02375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis N.; Aamdal S.; Awada A.; Calvert H.; Fumoleau P.; Sorio R.; Punt C.; Verweij J.; van Oosterom A.; Morant R.; Wanders J.; Hanauske A.-R. Carzelesin Phase II Study in Advanced Breast, Ovarian, Colorectal, Gastric, Head and Neck Cancer, Non-Hodgkin’s Lymphoma and Malignant Melanoma: A Study of the EORTC Early Clinical Studies Group (ECSG). Cancer Chemother. Pharmacol. 2000, 46 (2), 167–171. 10.1007/s002800000134. [DOI] [PubMed] [Google Scholar]
- Boger D. L. CBI Prodrug Analogs of CC-1065 and the Duocarmycins. Synthesis 1999, 1999 (S1), 1505–1509. 10.1055/s-1999-3658. [DOI] [Google Scholar]
- Nagamura S.; Kanda Y.; Kobayashi E.; Gomi K.; Saito H. Synthesis and Antitumor Activity of Duocarmycin Derivatives. Chem. Pharm. Bull. 1995, 43 (9), 1530–1535. 10.1248/cpb.43.1530. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Li L.; Tian Z.; Jiang W.; Larrick J. W. Synthesis and Antitumor Activity of CBI-Bearing Ester and Carbamate Prodrugs of CC-1065 Analogue. Bioorg. Med. Chem. 2006, 14 (23), 7854–7861. 10.1016/j.bmc.2006.07.062. [DOI] [PubMed] [Google Scholar]
- Junutula J. R.; Smith S. W.; Borkin D.; Degrado S.. Isoquinolidinobenzodiazepine (ICQ) - 1-(Chloromethyl)-2,3-Dihydro-1H-Benzo8e9indole (CBI) Dimers. WO2018/071455A1, April 19, 2018.
- Zhao R. Y.; Chari R. V. J.. Prodrugs of CC-1065 Analogs. US007655660B2, June 29, 2004.
- Zhao R. Y.; Erickson H. K.; Leece B. A.; Reid E. E.; Goldmacher V. S.; Lambert J. M.; Chari R. V. J. Synthesis and Biological Evaluation of Antibody Conjugates of Phosphate Prodrugs of Cytotoxic DNA Alkylators for the Targeted Treatment of Cancer. J. Med. Chem. 2012, 55 (2), 766–782. 10.1021/jm201284m. [DOI] [PubMed] [Google Scholar]
- Atwell G. J.; Wilson W. R.; Denny W. A. Synthesis and Cytotoxicity of Amino Analogues of the Potent DNA Alkylating Agent Seco-CBI-TMI. Bioorg. Med. Chem. Lett. 1997, 7 (12), 1493–1496. 10.1016/S0960-894X(97)00259-X. [DOI] [Google Scholar]
- Hay M. P.; Sykes B. M.; Denny W. A.; Wilson W. R. A 2-Nitroimiazole Carbamate Prodrug of 5-Amino-1-(Chloromethyl)-3-[(5,6,7-Trimethoxyindol-2-Yl)Carbonyl]-1,2-Dihydro-3H-Benz[e]Indole (Amino-Seco-CBI-TMI) for Use with ADEPT and GDEPT. Bioorg. Med. Chem. Lett. 1999, 9, 2237–2242. 10.1016/S0960-894X(99)00381-9. [DOI] [PubMed] [Google Scholar]
- Tercel M.; Atwell G. J.; Yang S.; Ashoorzadeh A.; Stevenson R. J.; Botting K. J.; Gu Y.; Mehta S. Y.; Denny W. A.; Wilson W. R.; Pruijn F. B. Selective Treatment of Hypoxic Tumor Cells In Vivo: Phosphate Pre-Prodrugs of Nitro Analogues of the Duocarmycins. Angew. Chem., Int. Ed. 2011, 50 (11), 2606–2609. 10.1002/anie.201004456. [DOI] [PubMed] [Google Scholar]
- Tietze L. F.; von Hof J. M.; Müller M.; Krewer B.; Schuberth I. Glycosidic Prodrugs of Highly Potent Bifunctional Duocarmycin Derivatives for Selective Treatment of Cancer. Angew. Chem., Int. Ed. 2010, 49 (40), 7336–7339. 10.1002/anie.201002502. [DOI] [PubMed] [Google Scholar]
- Tietze L. F.; Schuster H. J.; Krewer B.; Schuberth I. Synthesis and Biological Studies of Different Duocarmycin Based Glycosidic Prodrugs for Their Use in the Antibody-Directed Enzyme Prodrug Therapy. J. Med. Chem. 2009, 52 (2), 537–543. 10.1021/jm8009102. [DOI] [PubMed] [Google Scholar]
- Lajiness J. P.; Robertson W. M.; Dunwiddie I.; Broward M. A.; Vielhauer G. A.; Weir S. J.; Boger D. L. Design, Synthesis, and Evaluation of Duocarmycin O -Amino Phenol Prodrugs Subject to Tunable Reductive Activation. J. Med. Chem. 2010, 53 (21), 7731–7738. 10.1021/jm1010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielhauer G. A.; Swink M.; Parelkar N. K.; Lajiness J. P.; Wolfe A. L.; Boger D. Evaluation of a Reductively Activated Duocarmycin Prodrug against Murine and Human Solid Cancers. Cancer Biol. & Ther. 2013, 14 (6), 527–536. 10.4161/cbt.24348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe A. L.; Duncan K. K.; Parelkar N. K.; Brown D.; Vielhauer G. A.; Boger D. L. Efficacious Cyclic N -Acyl O -Amino Phenol Duocarmycin Prodrugs. J. Med. Chem. 2013, 56 (10), 4104–4115. 10.1021/jm400413r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G.-L.; Stevenson R. J.; Chang J. Y.-C.; Brothers P. J.; Ware D. C.; Wilson W. R.; Denny W. A.; Tercel M. N-Alkylated Cyclen Cobalt(III) Complexes of 1-(Chloromethyl)-3-(5,6,7-Trimethoxyindol-2-Ylcarbonyl)-2,3-Dihydro-1H-Pyrrolo[3,2-f]Quinolin-5-Ol DNA Alkylating Agent as Hypoxia-Activated Prodrugs. Bioorg. Med. Chem. 2011, 19 (16), 4861–4867. 10.1016/j.bmc.2011.06.076. [DOI] [PubMed] [Google Scholar]
- Spangler B.; Fontaine S. D.; Shi Y.; Sambucetti L.; Mattis A. N.; Hann B.; Wells J. A.; Renslo A. R. A Novel Tumor-Activated Prodrug Strategy Targeting Ferrous Iron Is Effective in Multiple Preclinical Cancer Models. J. Med. Chem. 2016, 59 (24), 11161–11170. 10.1021/acs.jmedchem.6b01470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietze L. F.; Müller M.; Duefert S.-C.; Schmuck K.; Schuberth I. Photoactivatable Prodrugs of Highly Potent Duocarmycin Analogues for a Selective Cancer Therapy. Chem.—Eur. J. 2013, 19 (5), 1726–1731. 10.1002/chem.201202773. [DOI] [PubMed] [Google Scholar]
- Nani R. R.; Gorka A. P.; Nagaya T.; Yamamoto T.; Ivanic J.; Kobayashi H.; Schnermann M. J. In Vivo Activation of Duocarmycin–Antibody Conjugates by Near-Infrared Light. ACS Cent. Sci. 2017, 3 (4), 329–337. 10.1021/acscentsci.7b00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pors K.; Loadman P. M.; Shnyder S. D.; Sutherland M.; Sheldrake H. M.; Guino M.; Kiakos K.; Hartley J. A.; Searcey M.; Patterson L. H. Modification of the Duocarmycin Pharmacophore Enables CYP1A1 Targeting for Biological Activity. Chem. Commun. 2011, 47 (44), 12062. 10.1039/c1cc15638a. [DOI] [PubMed] [Google Scholar]
- Thorn-Seshold O.; Felber J.; Thorn-Seshold J.; Zeisel L.. Disulfide-Based Prodrug Compounds. PCT/EP2022/057483, 2021.
- Thorn-Seshold O.; Zeisel L.; Felber J. G.. Dichalcogenide Prodrugs. PCT/EP2022/059280, 2021.
- Wang Y.; Yuan H.; Wright S. C.; Wang H.; Larrick J. W. Synthesis and Preliminary Cytotoxicity Study of Glucuronide Derivatives of CC-1065 Analogues. Bioorg. Med. Chem. 2003, 11 (7), 1569–1575. 10.1016/S0968-0896(02)00603-X. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Yuan H.; Wright S. C.; Wang H.; Larrick J. W. Synthesis and Cytotoxicity of a Biotinylated CC-1065 Analogue. BMC Chem. Bio. 2002, 2, 1–4. 10.1186/1472-6769-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Yuan H.; Wright S. C.; Wang H.; Larrick J. W. Synthesis and Preliminary Cytotoxicity Study of a Cephalosporin-CC-1065 Analogue Prodrug. BMC Chem. Bio. 2001, 1, 4–8. 10.1186/1472-6769-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q.; Duan W.; Simmons D.; Shayo Y.; Raymond M. A.; Dorr R. T.; Hurley L. H. Design and Synthesis of a Novel DNA–DNA Interstrand Adenine–Guanine Cross-Linking Agent. J. Am. Chem. Soc. 2001, 123 (20), 4865–4866. 10.1021/ja005658r. [DOI] [PubMed] [Google Scholar]
- Purnell B.; Sato A.; O’Kelley A.; Price C.; Summerville K.; Hudson S.; O’Hare C.; Kiakos K.; Asao T.; Lee M.; Hartley J. A. DNA Interstrand Crosslinking Agents: Synthesis, DNA Interactions, and Cytotoxicity of Dimeric Achiral Seco-Amino-CBI and Conjugates of Achiral Seco-Amino-CBI with Pyrrolobenzodiazepine (PBD). Bioorg. Med. Chem. Lett. 2006, 16 (21), 5677–5681. 10.1016/j.bmcl.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Jiang J.; Jiang X.; Cai S.; Han H.; Li L.; Tian Z.; Jiang W.; Zhang Z.; Xiao Y.; Wright S. C.; Larrick J. W. Synthesis and Antitumor Activity Evaluations of Albumin-Binding Prodrugs of CC-1065 Analog. Bioorg. Med. Chem. 2008, 16 (13), 6552–6559. 10.1016/j.bmc.2008.05.025. [DOI] [PubMed] [Google Scholar]
- Krall N.; Pretto F.; Decurtins W.; Bernardes G. J. L.; Supuran C. T.; Neri D. A Small-Molecule Drug Conjugate for the Treatment of Carbonic Anhydrase IX Expressing Tumors. Angew. Chem., Int. Ed. 2014, 53 (16), 4231–4235. 10.1002/anie.201310709. [DOI] [PubMed] [Google Scholar]
- Gangwar S.; Sufi B.. Cytotoxic Compounds and Conjugates with Cleavable Substrates. US 20060247295A1, November 2, 2006.
- Boyd S. E.; Chen L.; Gangwar S.; Guerlavais V.; Horgan K.; Sufi B.; Huang H.; King D. J.; Pan C.; Cardarelli J. M.. Antibody-Drug Conjugates and Methods of Use. US20080279868A1, November 13, 2008.
- Maderna A.; Doroski M. D.; Chen Z.; Risley H. L.; Casavant J. M.; O’Donnell C. J.; Porte A. M.; Subramanyam C.. Bifunctional Cytotoxic Agents. US20150209445A1, July 30, 2015.
- Maderna A.; Subramanyam C.; Tumey L. N.; Chen Z.; Casavant J. M.. Bifunctional Cytotoxic Agents Containing the CTI Pharmacophore. WO 2016/151432A1, September 29, 2016.
- Beusker P. H.; Coumans R. G. E.; Elgersma R. C.; Menge W. M. P. B.; Joosten A. F.; Spijker H. J.; de Groot F. M. H.. Novel Conjugates of CC-1065 Analogs and Bifunctional Linkers. US20130224227A1, August 29, 2013.
- Janda K. D.; Wirsching P.; Boger D. L.. Compositions and Methods for Delivery of Antitumor Agents. WO2006/002895A2, January 12, 2006.
- Chen L.; Gangwar S.; Guerlavais V.; Lonberg N.; Zhang Q.. Chemical Linkers with Single Amino Acids and Conjugates Thereof. US20100113476A1, May 6, 2010.
- Jackson P. M. J.; Thurston D. E.; Rahman K. M.. Asymmetric Conjugate Compounds. US20190144443A1, May 16, 2019.
- Helin J.; Saarinen J.; Satomaa T.; Ekholm F. S.. Saccaride Derivative of a Toxic Payload and Antibody Conjugates Thereof. US20180228906A1, August 16, 2018.
- Dokter W.; Ubink R.; van der Lee M.; van der Vleuten M.; van Achterberg T.; Jacobs D.; Loosveld E.; van den Dobbelsteen D.; Egging D.; Mattaar E.; Groothuis P.; Beusker P.; Coumans R.; Elgersma R.; Menge W.; Joosten J.; Spijker H.; Huijbregts T.; de Groot V.; Eppink M.; de Roo G.; Verheijden G.; Timmers M. Preclinical Profile of the HER2-Targeting ADC SYD983/SYD985: Introduction of a New Duocarmycin-Based Linker-Drug Platform. Mol. Cancer Ther. 2014, 13 (11), 2618–2629. 10.1158/1535-7163.MCT-14-0040-T. [DOI] [PubMed] [Google Scholar]
- Yu L.; Lu Y.; Yao Y.; Liu Y.; Wang Y.; Lai Q.; Zhang R.; Li W.; Wang R.; Fu Y.; Tao Y.; Yi S.; Gou L.; Chen L.; Yang J. Promiximab-Duocarmycin, a New CD56 Antibody-Drug Conjugates, Is Highly Efficacious in Small Cell Lung Cancer Xenograft Models. Oncotarget 2018, 9 (4), 5197–5207. 10.18632/oncotarget.23708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Urban D. J.; Nani R. R.; Zhang Y.-F.; Li N.; Fu H.; Shah H.; Gorka A. P.; Guha R.; Chen L.; Hall M. D.; Schnermann M. J.; Ho M. Glypican-3-Specific Antibody Drug Conjugates Targeting Hepatocellular Carcinoma. Hepatology 2019, 70 (2), 563–576. 10.1002/hep.30326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scribner J. A.; Brown J. G.; Son T.; Chiechi M.; Li P.; Sharma S.; Li H.; De Costa A.; Li Y.; Chen Y.; Easton A.; Yee-Toy N. C.; Chen F. Z.; Gorlatov S.; Barat B.; Huang L.; Wolff C. R.; Hooley J.; Hotaling T. E.; Gaynutdinov T.; Ciccarone V.; Tamura J.; Koenig S.; Moore P. A.; Bonvini E.; Loo D. Preclinical Development of MGC018, a Duocarmycin-Based Antibody–Drug Conjugate Targeting B7-H3 for Solid Cancer. Mol. Cancer Ther. 2020, 19 (11), 2235–2244. 10.1158/1535-7163.MCT-20-0116. [DOI] [PubMed] [Google Scholar]
- Tong X.-M.; Feng L.; Suthe S. R.; Weng T.-H.; Hu C.-Y.; Liu Y.-Z.; Wu Z.-G.; Wang M.-H.; Yao H.-P. Therapeutic Efficacy of a Novel Humanized Antibody-Drug Conjugate Recognizing Plexin-Semaphorin-Integrin Domain in the RON Receptor for Targeted Cancer Therapy. J. Immunotherapy Cancer 2019, 7 (1), 250. 10.1186/s40425-019-0732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCombs J. R.; Owen S. C. Antibody Drug Conjugates: Design and Selection of Linker, Payload and Conjugation Chemistry. AAPS 2015, 17 (2), 339–351. 10.1208/s12248-014-9710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters C.; Brown S. Antibody–Drug Conjugates as Novel Anti-Cancer Chemotherapeutics. Bioscience Reports 2015, 35 (4), e00225. 10.1042/BSR20150089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper M.; Stengl A.; Ochtrop P.; Gerlach M.; Stoschek T.; Schumacher D.; Helma J.; Penkert M.; Krause E.; Leonhardt H.; Hackenberger C. P. R. Ethynylphosphonamidates for the Rapid and Cysteine-Selective Generation of Efficacious Antibody–Drug Conjugates. Angew. Chem., Int. Ed. 2019, 58 (34), 11631–11636. 10.1002/anie.201904193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji U.; van Herpen C. M. L.; Saura C.; Thistlethwaite F.; Lord S.; Moreno V.; Macpherson I. R.; Boni V.; Rolfo C.; de Vries E. G. E.; Rottey S.; Geenen J.; Eskens F.; Gil-Martin M.; Mommers E. C.; Koper N. P.; Aftimos P. Trastuzumab Duocarmazine in Locally Advanced and Metastatic Solid Tumours and HER2-Expressing Breast Cancer: A Phase 1 Dose-Escalation and Dose-Expansion Study. Lancet Oncology 2019, 20 (8), 1124–1135. 10.1016/S1470-2045(19)30328-6. [DOI] [PubMed] [Google Scholar]
- Owonikoko T. K.; Hussain A.; Stadler W. M.; Smith D. C.; Kluger H.; Molina A. M.; Gulati P.; Shah A.; Ahlers C. M.; Cardarelli P. M.; Cohen L. J. First-in-Human Multicenter Phase I Study of BMS-936561 (MDX-1203), an Antibody-Drug Conjugate Targeting CD70. Cancer Chemother. Pharmacol. 2016, 77 (1), 155–162. 10.1007/s00280-015-2909-2. [DOI] [PubMed] [Google Scholar]
- Jang S.; Powderly J. D.; Spira A. I.; Bakkacha O.; Loo D.; Bohac G. C.; Sharma M. Phase 1 Dose Escalation Study of MGC018, an Anti-B7-H3 Antibody-Drug Conjugate (ADC), in Patients with Advanced Solid Tumors. J. Clin. Onc. 2021, 39 (15_suppl), 2631–2631. 10.1200/JCO.2021.39.15_suppl.2631. [DOI] [Google Scholar]
- Fast Track Designation Granted for Breast Cancer Treatment. Oncology Times 2018, 40 (5), 16. 10.1097/01.COT.0000531194.80509.6d. [DOI] [Google Scholar]
- McGrath N. A.; Brichacek M.; Njardarson J. T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87 (12), 1348–1349. 10.1021/ed1003806. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.