

Abstract
The transient receptor potential melastatin 8 (TRPM8) channel is the primary molecular transducer responsible for the cool sensation elicited by menthol and cold in mammals. TRPM8 activation is controlled by cooling compounds together with the membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2). Our knowledge of cold sensation and the therapeutic potential of TRPM8 for neuroinflammatory diseases and pain will be enhanced by understanding the structural basis of cooling agonists and PIP2-dependent TRPM8 activation. We present cryo–electron microscopy structures of mouse TRPM8 in closed, intermediate, and open states along the ligand- and PIP2-dependent gating pathway. Our results uncover two discrete agonist sites, state-dependent rearrangements in the gate positions, and a disordered-to-ordered transition of the gate-forming S6–elucidating the molecular basis of chemically induced cool sensation in mammals.
One Sentence Summary
Structures in the closed, intermediate, and open states elucidate PIP2 and cooling agonist dependent TRPM8 activation.
The transient receptor potential melastatin member 8 (TRPM8) is a menthol and cold receptor expressed in dorsal root ganglia and trigeminal ganglia neurons (1, 2). It is required for sensing cold temperatures as TRPM8-knockout mice show severe behavioral deficiency in response to cold stimuli (3–5). Recent studies show that TRPM8 is also critical for warm sensing, as TRPM8-deficient mice are unable to perceive warmth (6). TRPM8 is the principal mediator in menthol-induced analgesia of acute and inflammatory pain (7). In sensory fibers innervating the ocular surface, this channel regulates basal tear secretion by sensing evaporation-induced cooling and changes in osmolality (8, 9). In addition to its critical role in human temperature sensing, increasing evidence suggests that TRPM8 is a promising therapeutic target of dry eye disease, migraine, cold-related pain, chronic pain, and chronic cough (10–16).
TRPM8 is a polymodal calcium-permeable cation channel whose gating is controlled by various physical and chemical stimuli (17, 18). It is activated by innocuous cold temperatures and by “cooling” compounds, such as menthol found in peppermint and the synthetic supercooling agonist icilin. TRPM8 activation requires the signaling phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2); the depletion of PIP2 causes desensitization of channel activity (19–21). Channel function is also activated by voltage (22). However, the molecular bases underlying channel opening by the interplay of these stimuli remain elusive.
Previous structural studies on avian TRPM8 channels from flycatcher Ficedula albicollis and great tit Parus major (TRPM8FA and TRPM8PM, respectively) revealed the architecture of TRPM8 and the binding sites for PIP2, the cooling agonists icilin and menthol derivative WS–12 (TRPM8FA) (18, 23, 24), and the antagonist AMTB (TRPM8PM) (25). All reported ligand-bound TRPM8 structures adopt non-conducting states; thus, it is unclear how ligand binding is transduced to opening of the ion conduction pathway of TRPM8. Moreover, despite the high sequence identity (~80%) between avian and mammalian TRPM8 orthologs, it is well known that they exhibit substantial differences in chemical and thermal sensitivity (26, 27).
In this study, we used PIP2 and type I and type II agonists to open the mouse (Mus musculus) TRPM8 channel (TRPM8MM), capturing distinct closed, intermediate, and open states by single-particle cryo–electron microscopy (cryo-EM). We observed notable mobility and structural rearrangements in the pore domain along with unexpected changes in the state-dependent intracellular gate. Our study combines mutagenesis, electrophysiology, and molecular dynamics (MD) simulations to elucidate the molecular basis of ligand-dependent gating of a mammalian TRPM8 channel.
Synergistic TRPM8 activation by C3 and AITC
A dialkylphosphorylalkane cooling agent, 1-diisopropylphosphorylnonane [also called cryosim-3 (C3)] (Fig. 1A) was reported to relieve dry eye disease by inducing cooling to promote tear secretion through TRPM8 activation (15). We characterized the agonist effects of C3 using two-electrode voltage clamp (TEVC) recordings on Xenopus laevis (African clawed frog) oocytes expressing TRPM8 channels (Fig. 1B). Independent of Ca2+ (fig. S1), C3 elicited robust outward-rectifying currents with a half-maximal effective concentration (EC50) of 10.3 ± 0.4 μM (Fig. 1B). In contrast to menthol or icilin (27, 28), no appreciable Ca2+-dependent desensitization of TRPM8MM by C3 was observed either in the TEVC recordings (fig. S1A) or in the inside-out patch recordings at the concentrations tested (500 and 200 μM, respectively) (fig. S1B). C3 is therefore a good chemical tool for structural studies of ligand gating in TRPM8 as a result of its apparent independence of Ca2+ in activation and desensitization.
Figure 1. Functional characterization and structure determination of the mouse TRPM8MM channel.
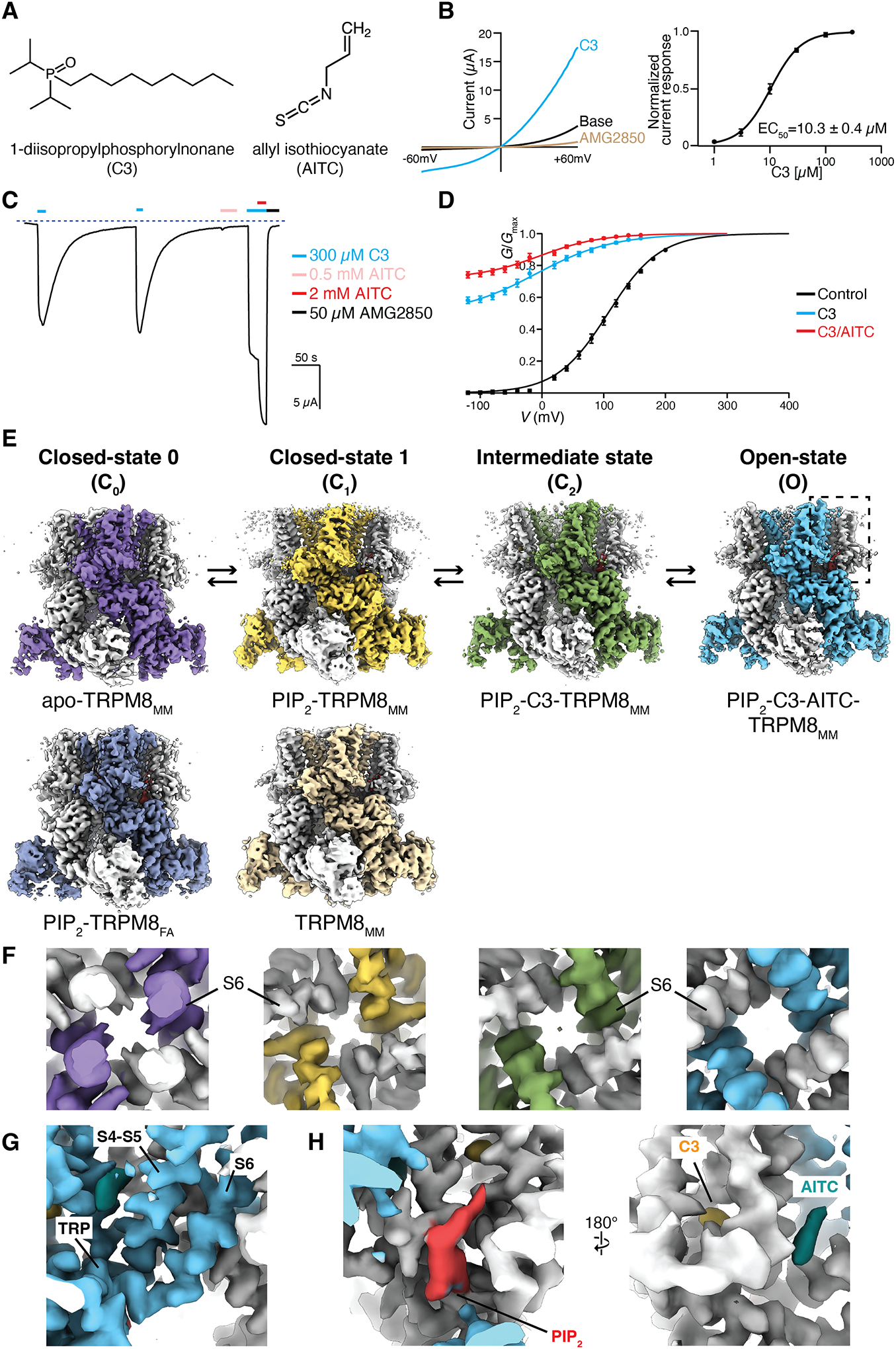
(A) Chemical structures of 1-diisopropylphosphorylnonane (C3) and allyl isothiocyanate (AITC).
(B) (Left) TRPM8MM current-voltage (I-V) plots obtained from 600-ms voltage ramps, following the sequential application of 300 μM C3 then 50 μM TRPM8 antagonist AMG2850. (Right) Mean normalized concentration-response relations for C3. Data are shown as mean ± SEM (n = 6). The curves are fit to the Hill equation with EC50 = 10.3 ± 0.4 μM, and s (slope) = 1.59 ± 0.1.
(C) Representative time course recording of TRPM8MM currents elicited by 300 μM C3, 0.5 mM AITC, 300 μM C3/2 mM AITC, and 50 μM AMG2850, as indicated by the colored horizontal lines. The dotted line indicates the zero-current level. The voltage was initially held at −60 mV and ramped to +60 mV over 300 ms every 2 seconds; plotted here are the currents at −60 mV.
(D) Conductance-voltage (G-V) relationships of TRPM8MM in the absence of agonist (control, black; n = 5) and in the presence of 300 μM C3 only (blue; n = 7) and 300 μM C3/2 mM AITC (red; n = 7), respectively. Error bars indicate SEM.
(E) Cryo-EM reconstructions of TRPM8MM and TRPM8FA channels in the C0, C1, C2, and O states, as indicated. Thresholding, 0.25 (purple), 0.36 (silver-gray), 0.22 (orange), 0.2 (light orange), 0.24 (green), and 0.22 (blue).
(F) Close-up view at the S6 gate of 3D reconstructions from (E), viewed from the intracellular side, at thresholding 0.24, 0.25, 0.18, and 0.22, respectively.
(G) Close-up view of the EM densities for the S4-S5 junction, S6, and TRP domain in the O state from (E), at thresholding 0.24.
(H) Close-up view of the EM densities for PIP2 (red), C3 (yellow), AITC (teal) from the dashed box in the O state from (E), at thresholding 0.25. All thresholding values were obtained at the isosurface level.
Janssens et al. (29) defined two distinct agonist types for TRPM8 based on kinetic analysis: type I (menthol-like) agonists stabilize the open state, whereas type II [allyl isothiocyanate (AITC)-like] agonists destabilize the closed state. C3-activated TRPM8 slows voltage-dependent deactivation substantially, a feature of the type I agonist (fig. S1, C and D). We hypothesized that combining a type I and a type II agonist should synergistically increase the TRPM8 channel open probability. In TEVC recordings, coapplication of 300 μM C3 and 2 mM AITC increased the inward current by ~50 % compared with the current elicited by 300 μM C3 alone and increased the relative conductance at 0 mV (Fig. 1, C and D). AITC produced very weak agonistic effects at low millimolar concentrations (0.5 mM in Fig. 1C). We formed a strategy to coapply C3 and AITC to enhance the open probability of TRPM8MM to visualize the ligand-dependent conformational changes leading to the open state.
Structure determination of mouse TRPM8
TRPM8MM shares 82 and 94% sequence identity with TRPM8FA and human TRPM8, respectively (fig. S2). We determined six structures (five TRPM8MM and one TRPM8FA) capturing four distinct conformational states along the gating pathway (Fig. 1, E and F): ligand-free TRPM8MM (apo-TRPM8MM) and PIP2-bound TRPM8FA (PIP2-TRPM8FA) in the closed (C) C0 state, PIP2-bound TRPM8MM and TRPM8MM in the C1 state, PIP2- and C3-bound TRPM8MM (PIP2-C3-TRPM8MM) in the intermediate C2 state, and the open (O) state TRPM8MM in complex with PIP2, C3, and AITC (PIP2-C3-AITC-TRPM8MM). The three-dimensional (3D) reconstructions for the six structures were determined to overall resolutions of 3.07 to 3.59 Å (figs. S3 and S4). The high-quality electron microscopy (EM) densities for the pore lining S6 helices and the TRP domains enabled us to unambiguously model the register and assign the gating residues (Fig. 1G and fig. S5). The outer pore is typically less well resolved as a result of its intrinsic flexibility. Particle subtraction followed by focused 3D classification on the transmembrane region (see fig. S6 and methods) facilitated model building for the outer pore region. The TRPM8MM channel adopts a three-layered homotetrameric architecture (fig. S7A). The transmembrane channel region at the top layer constitutes the pre-S1 domain, the transmembrane domain (TMD), and the TRP domain (fig. S7B). The TMD consists of the voltage-sensor–like domain (VSLD), formed by transmembrane helices S1 to S4, and the pore domain, constituting the S4-S5 junction, S5, pore helix (PH), and the pore-lining helix S6.
For the PIP2-C3-AITC-TRPM8MM dataset, we resolved two 3D classes (fig. S6A): Class 1 contains strong EM densities for PIP2, C3, and AITC (Fig. 1H) whereas class 2 shows EM densities for PIP2 and C3 but not for AITC. Importantly, this class 2 adopts a nearly identical conformation to the EM map for the PIP2-C3-TRPM8MM structure (map correlation 0.98; fig. S6B). We define both the class 2 and the PIP2-C3-TRPM8MM dataset as the intermediate C2 state because its conformation is between the C1 and the O states (see discussion below). PIP2-C3-TRPM8MM was used for structural analysis of the C2 state owing to its better map quality around S6.
PIP2 binding sensitizes mouse TRPM8
PIP2 is essential for TRPM8 function (19–21). Our previous TRPM8FA study showed that PIP2 binds to the interfacial cavity at the S4b, TRP domain, and pre-S1. TRPM8FA exhibits two PIP2-associated conformations: (i) loose attachment to a wider binding site formed by α-helical S4b and straight S5, also observed in apo-TRPM8FA and (ii) full engagement with the cavity consisting of 310-helical S4b and bent S5 (fig. S8, A and B) (23). In the current study, we determined two TRPM8MM structures with PIP2 bound: PIP2-TRPM8MM and TRPM8MM. Strong EM density for PIP2 was present at the interfacial cavity of PIP2-TRPM8MM (Fig. 2A). In the TRPM8MM structure, an EM density consistent with the shape of PIP2 was resolved, although no PIP2 was included, suggesting that endogenous PIP2 is copurified with the channel (Fig. 2A). The two structures are very similar (fig. S8C; TMD Cα RMSD = 0.6 Å), exhibiting the fully PIP2 engaged conformation with 310-helical S4b and bent S5, and revealing drastic conformational differences with those of the apo or the loose attachment mode in avian TRPM8 (fig. S8D; TMD Cα RMSD = 6.0 Å).
Figure 2. PIP2 binding primes the TRPM8MM channel.
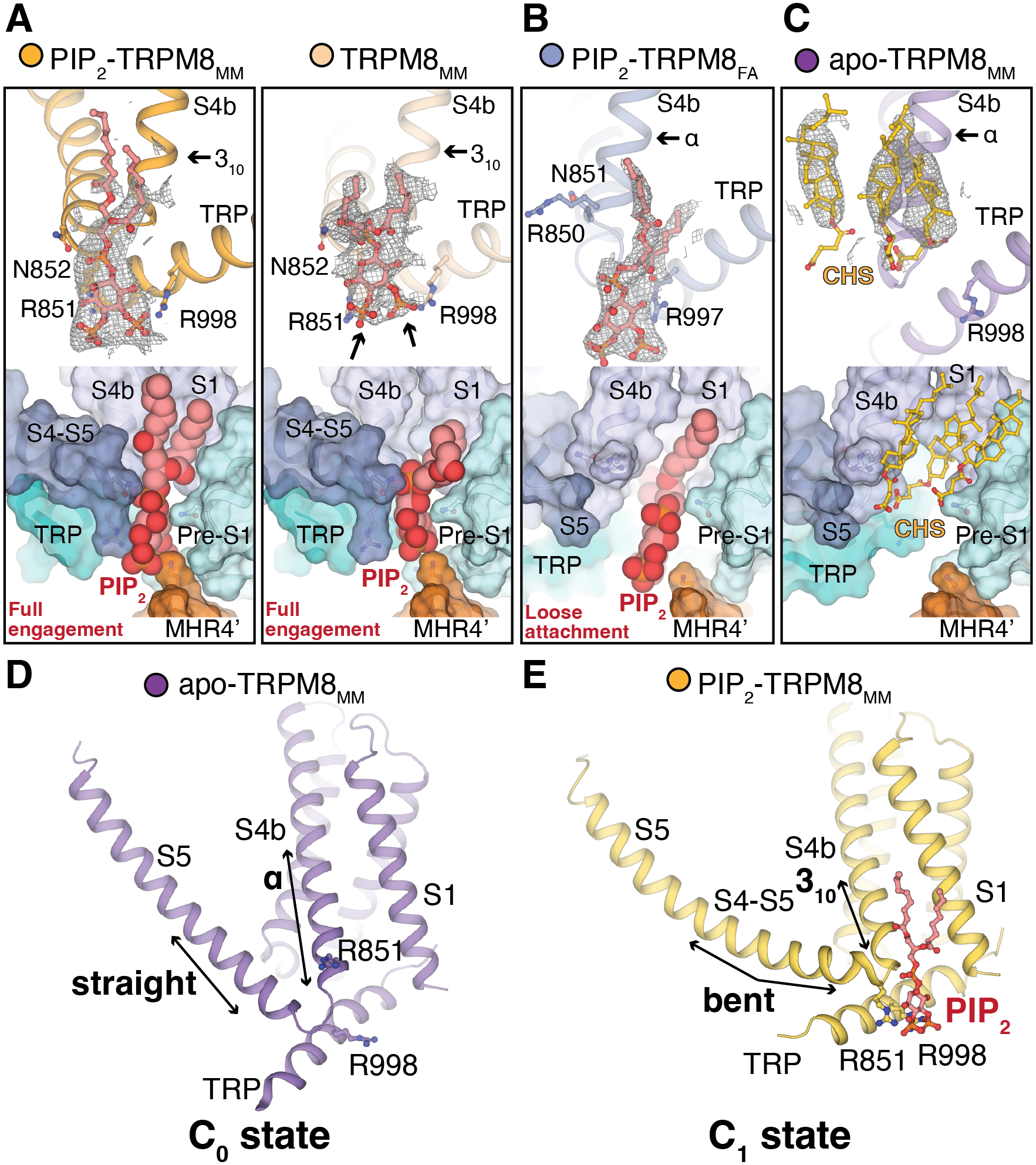
(A to C) (Top) EM densities (gray mesh) for PIP2 (red sticks) in PIP2-TRPM8MM, TRPM8MM (A), and PIP2-TRPM8FA (B). CHS (yellow sticks) binds to apo-TRPM8MM (C) at the interfacial cavity. Densities are contoured at 0.16 and 0.15 (A), 0.31 (B), and 0.22 to 0.24 (C) thresholding, respectively. Residue sidechains are shown in sticks. (Bottom) Surface representations showing the two PIP2 binding modes [(A) and (B)] and CHS (C) in the interfacial cavity. PIP2 is shown as red spheres.
(D to E) Conformations of S4b and S5 in apo-TRPM8MM (D) and PIP2-TRPM8MM (E). Arrows indicate key structural differences. PIP2 (red) and residues are shown in sticks.
Because we do not observe the loose attachment mode in mouse TRPM8 in the presence of PIP2, we hypothesized that distinct from avian orthologs, mammalian TRPM8 channels are readily primed by PIP2 binding and undergo TMD rearrangement. To test this proposal, we first determined the TRPM8FA structure in complex with PIP2 alone (PIP2-TRPM8FA), which closely resembles the conformation of avian apo TRPM8 (Fig. 2B and fig. S8E; TMD Cα RMSD = 1.0 Å). PIP2 binds in the loosely attached mode in TRPM8FA, in contrast to its binding mode in PIP2-TRPM8MM (fig. S8F; TMD Cα RMSD = 4.9 Å). Second, we posited that the removal of endogenous PIP2 copurified with mouse TRPM8 would convert the channel conformation to that of the apo-TRPM8FA structure. For channel purification, we included cholesteryl hemisuccinate (CHS), a detergent that was shown to occupy the interfacial cavity in the TRPM8 structures determined in the presence of CHS (25, 30). From this cryo-EM dataset, we resolved three subclasses with distinct conformations (fig. S9A). Class I (defined as apo-TRPM8MM), the interfacial cavity of which is occupied by CHS molecules in place of PIP2, resembles the apo-TRPM8FA conformation (Fig. 2C). Class II shows residual PIP2 density at the binding site and resembles PIP2-TRPM8MM (fig. S9, B and C). Class III resembles the Ca2+-bound and PIP2-depleted desensitized TRPM8 structures (25, 30), which is likely a result of the additional interaction of the CHS molecule to the outer pore region (fig. S9, B and D). Our results suggest that, in the absence of PIP2, apo-TRPM8MM adopts the same conformation as apo-TRPM8FA which we define as the C0 state. PIP2 binding triggers conformational transition to a fully engaged mode in PIP2-TRPM8MM, which we define as the C1 state (Fig. 2, D and E). Our data show that both avian and mammalian TRPM8s share a common PIP2- and agonist-dependent gating landscape with distinct differences in the energetic barriers for PIP2 binding, resulting in different preferred ground states between avian (C0 state) and mammalian (C1 state) TRPM8.
Synergistic agonist and lipid binding centered at helix S4b
Inside the VSLD cavity, we resolved a pronounced EM density consistent with the shape of the diisopropylphosphoryl head group of C3 positioned below Tyr745 on S1 and near Arg842, His845 on S4b (Fig. 3A and fig. S10, A and B). Tyr745 is critical for menthol binding (31), and the diisopropylphosphoryl head group position with respect to Tyr745 is similar to that of the menthol analog WS–12 in TRPM8FA (23). Based on MD simulations, we modeled a headgroup binding pose that enables C3 to interact with polar residues and water molecules at the base of the VSLD (fig. S10, C and D). Mutations Tyr745His, Arg842Ala, and His845Ala all suppress TRPM8 activation by C3 relative to AITC (Fig. 3B). Based on the binding pose, the mutational effects, and the kinetics of C3 deactivation (fig. S1, C and D), we conclude that C3 is a type I agonist.
Figure 3. Ligand binding to TRPM8MM.
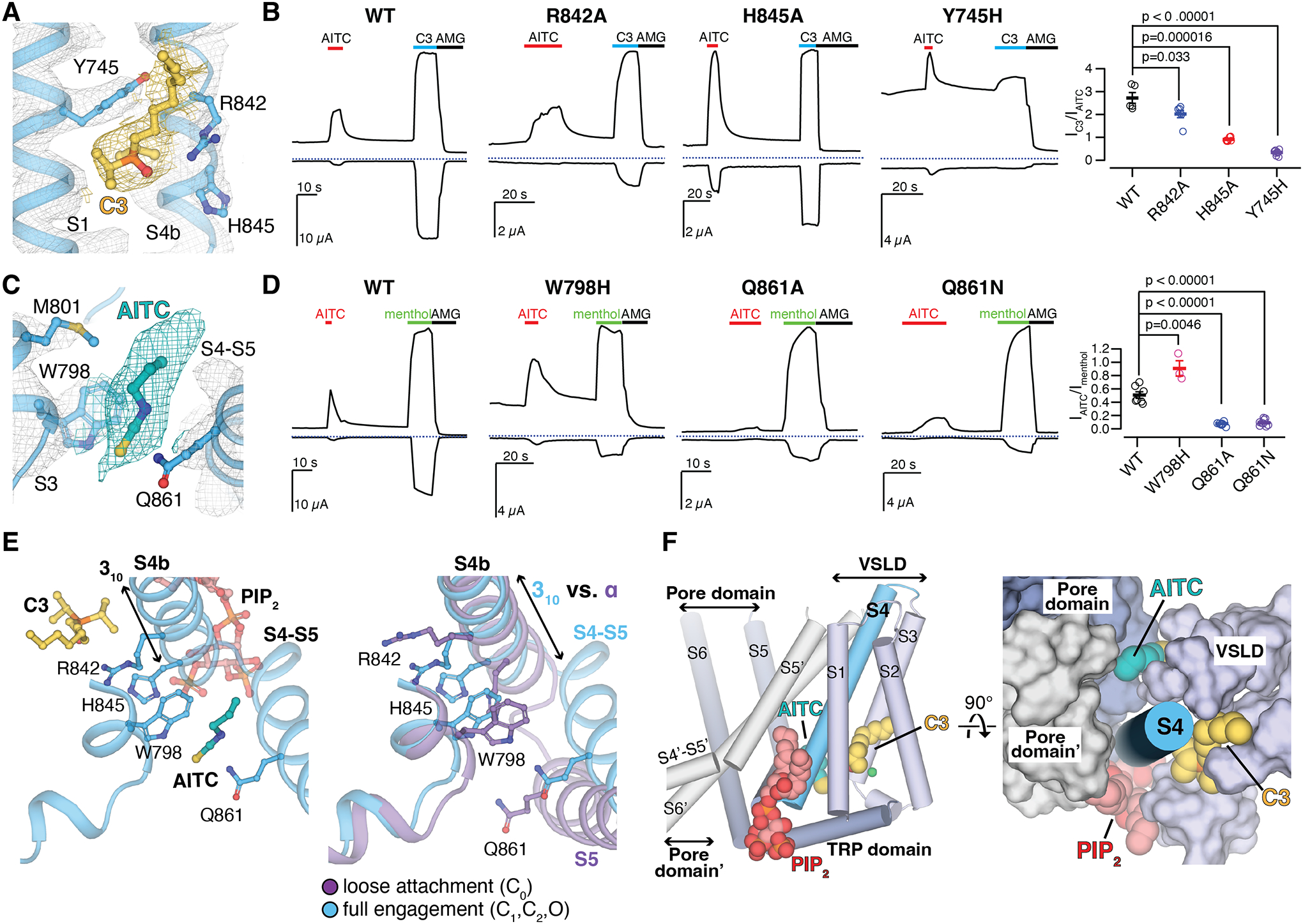
(A and C) EM densities for C3 [(A) yellow] and AITC [(C) teal] in the O state PIP2-C3-AITC-TRPM8MM structure. Ligands and residue sidechains are shown as sticks. Densities are contoured at thresholding 0.11 for C3 [(A) yellow mesh], 0.200 for AITC [(C) teal mesh], and 0.27 for TRPM8MM channel (gray mesh), respectively.
(B and D) Representative TEVC recordings on wildtype and mutant TRPM8MM channels at −60 mV (lower traces) and +60 mV (upper traces). Horizontal colored lines denote the application of 2 mM AITC (red), 300 μM C3 (blue), and 50 μM AMG2850 (AMG) (black). The dotted lines denote the zero current. Summary of the current magnitudes in response to 300 μM C3 relative to 2 mM AITC at +60 mV from experiments in the left panels [(B), rightmost panel] and the current magnitudes in response to 2 mM AITC relative to 1 mM menthol at +60 mV [(D) rightmost panel], respectively. Values for individual oocytes are shown as open circles along with mean ± S.E.M. (n = 3 to 10 oocytes). P-values are calculated by two-tailed Student’s t-test.
(E) Comparison of conformational changes at S4b and S5 upon PIP2, C3, and AITC binding reveals that AITC cannot bind to TRPM8 in the C0 state (right). Ligands and residue sidechains are shown in sticks.
(F) PIP2, C3, and AITC bind surrounding the S4b helix (blue cylinder). TMD helices are shown in either cylinder (left) or surface (right). The neighboring pore domain (indicated by an apostrophe) is colored gray.
In the 3D reconstruction of the O state structure, we observed a strong EM density consistent with the size and shape of AITC in front of the S4-S5 junction (Fig. 3C). The isothiocyanate group of AITC is sandwiched between Trp798 on S3 and Gln861 at the S4-S5 junction and mutation of Gln861 abolished AITC activation. The Trp798Ala mutant could not be activated by C3 or AITC, but substitution of Trp798 to His increased the efficacy of AITC activation (Fig. 3D). MD simulations are consistent with weak AITC binding at this site (fig. S10, E and F), which we term the type II agonist site. Type II agonist binding is possible only in the full engagement mode (the 310-helix of S4b), which is induced by PIP2 and C3 binding (Fig. 3E).
Taken together, the binding sites for PIP2 and cooling agonists (C3 and AITC) required for TRPM8 activation are positioned at three different sides surrounding the S4b helix, which is the nexus of structural elements important for gating: the VSLD, the pore domain (S5), and the TRP domain (Fig. 3F). The effects of the individual compounds can thus allosterically enhance each other and provide synergistic transmission of the effects towards the gate to increase the open probability of the channel.
Pore conformation changes during gating
The C0 conformation of TRPM8FA and the current apo-TRPM8MM structures have a wide-set pore and selectivity filter, unlike other TRP channels (23–25, 32–34). It was not previously clear how ions would be coordinated in such a large radius pore. Our structures now offer a clue to this question. In the mouse structures, we find that extensive conformational rearrangements take place in the pore during gating (Fig. 4A). Analysis of pore cavity volumes (Fig. 4, B and C) reveals a noticeable decrease as the channel transitions from the C0 to the C1/C2 state, and then to the O state. When the channel is liganded by PIP2 and agonists, the pore-lining S6 helices rotate and the pore helices move towards the center of the pore. The pore cavity progressively decreases in size while the selectivity filter gradually forms, as illustrated in the HOLE plot (Fig. 4B), changing shape to become a canonical ion conduction pathway akin to that of other homo-tetrameric cation channels. The surface charge distribution in the pore changes as a result of conformational changes in S6 in response to ligand gating. Namely, the hydrophobic or charge neutral S6 residues, which face the center of the pore in C0, are replaced by negatively charged or polar residues in the C1/C2 and O states (Fig. 4A and fig. S11). Concomitant reduction in pore size and a progressive increase in electronegative surface potential of the pore cavity likely facilitate the conduction of cations through the pore.
Figure 4. Structural changes in the pore domain and S6 gate position during gating.
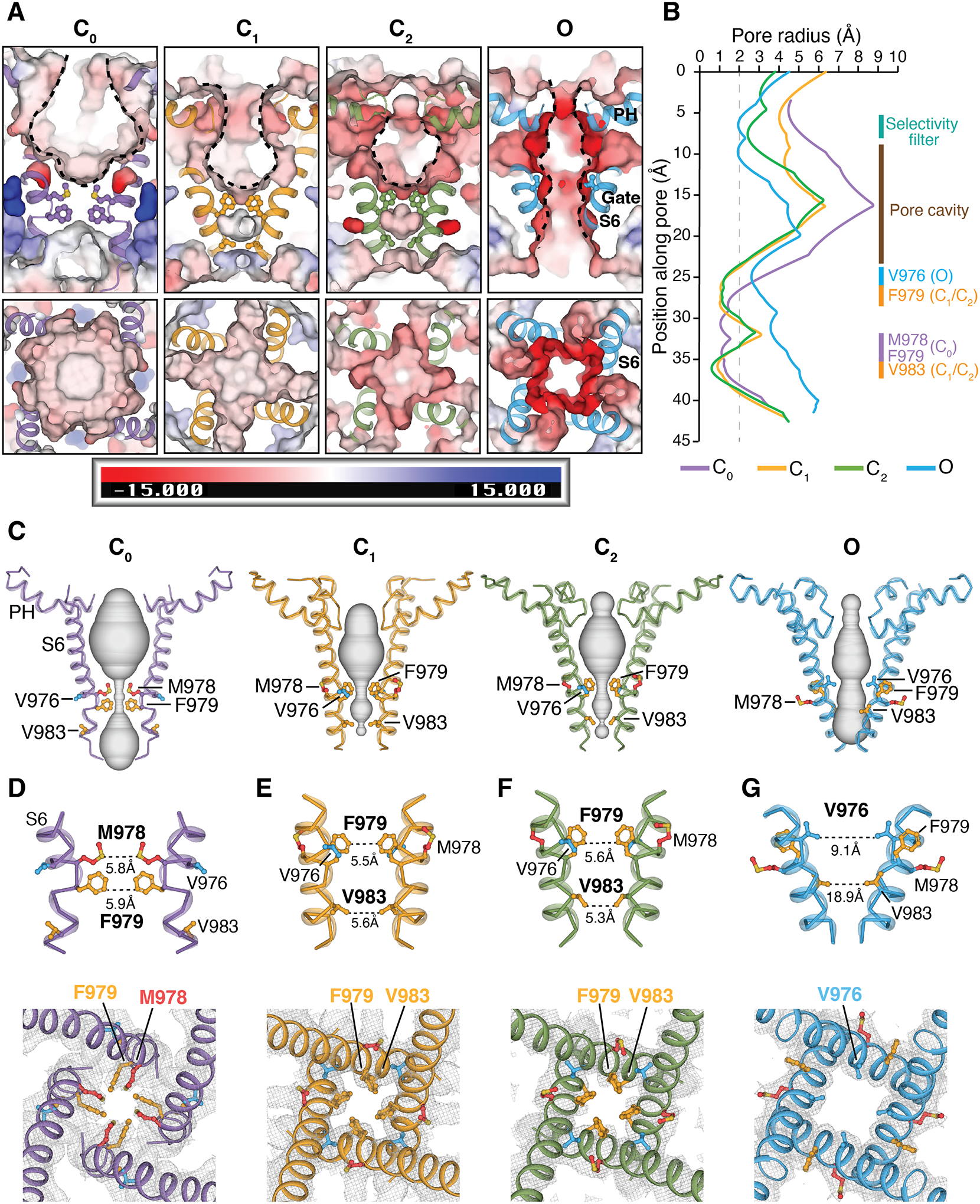
(A) Viewed from the membrane plane (top) and from the extracellular side (bottom), electrostatic potential of the pore-lining surface in the C0, C1, C2, and O states, calculated by APBS (74) implemented in PyMOL. S6 helices are shown in cartoon and gating residues as sticks. The scale bar indicates electrostatics from −15 to +15 kT/e.
(B) Pore radii calculated using the HOLE program (75) as colored in (A) (see methods). Regions spanning the selectivity filter, pore cavity, and S6 gate in distinct conformation states are denoted. The dotted line denotes a 2.0 Å-radius.
(C) Ion permeation pathway in the C0, C1, C2, and O states shown as gray surfaces.
(D to G) Close-up views of the S6 gate from the membrane plane (top panels) and from the extracellular side (bottom panels) for the C0 state [(D) purple], C1 state [(E) orange], C2 state [(F) green], and O state [(G) blue]. Diagonal distances in Å between opposing gating residues are labeled in the top panels. Gray meshes represent EM densities at the S6 gate region, contoured at 0.19 (D), 0.16 (E), 0.16 (F), and 0.16 (G) thresholding, respectively. Gating residues are shown in sticks and colored red for Met978, orange for Phe979 and Val983, and blue for Val976. The apo-TRPM8MM (purple), PIP2-TRPM8MM (orange), PIP2-C3-TRPM8MM (green), and PIP2-C3-AITC-TRPM8MM (blue) structures are used for illustration and analysis.
S6 gate changes upon opening
Comparing structures of the different conformations along the gating pathway reveals spatial alterations of the narrowest constriction point at the intracellular S6 gate (Figs. 4, D to G and 5A). In the C0 state structure, Met978 and Phe979 protrude towards the channel lumen and constitute a double-layered hydrophobic seal of 5.8 Å and 5.9 Å in diameter (Fig. 4D). Upon the binding of PIP2 and/or C3 in the C1 and C2 states, Met978 rotates about 90° counterclockwise away from the pore, whereas Phe979 and Val983 subsequently rotate in and form a second double-layered hydrophobic gate distinct from that in the C0 state (Fig. 4, E and F). This structure reveals the role of Phe979 and Val983 as gating residues. In the O state, Met978 and Phe979 rotate another 90° counterclockwise away from the pore, whereas Val976 swings into the ion pathway forming a wider conduction point of ~9.1 Å in diameter, permitting passage of hydrated cations (Fig. 4G) (35).
Figure 5. State-dependent interface in the O state of TRPM8MM.
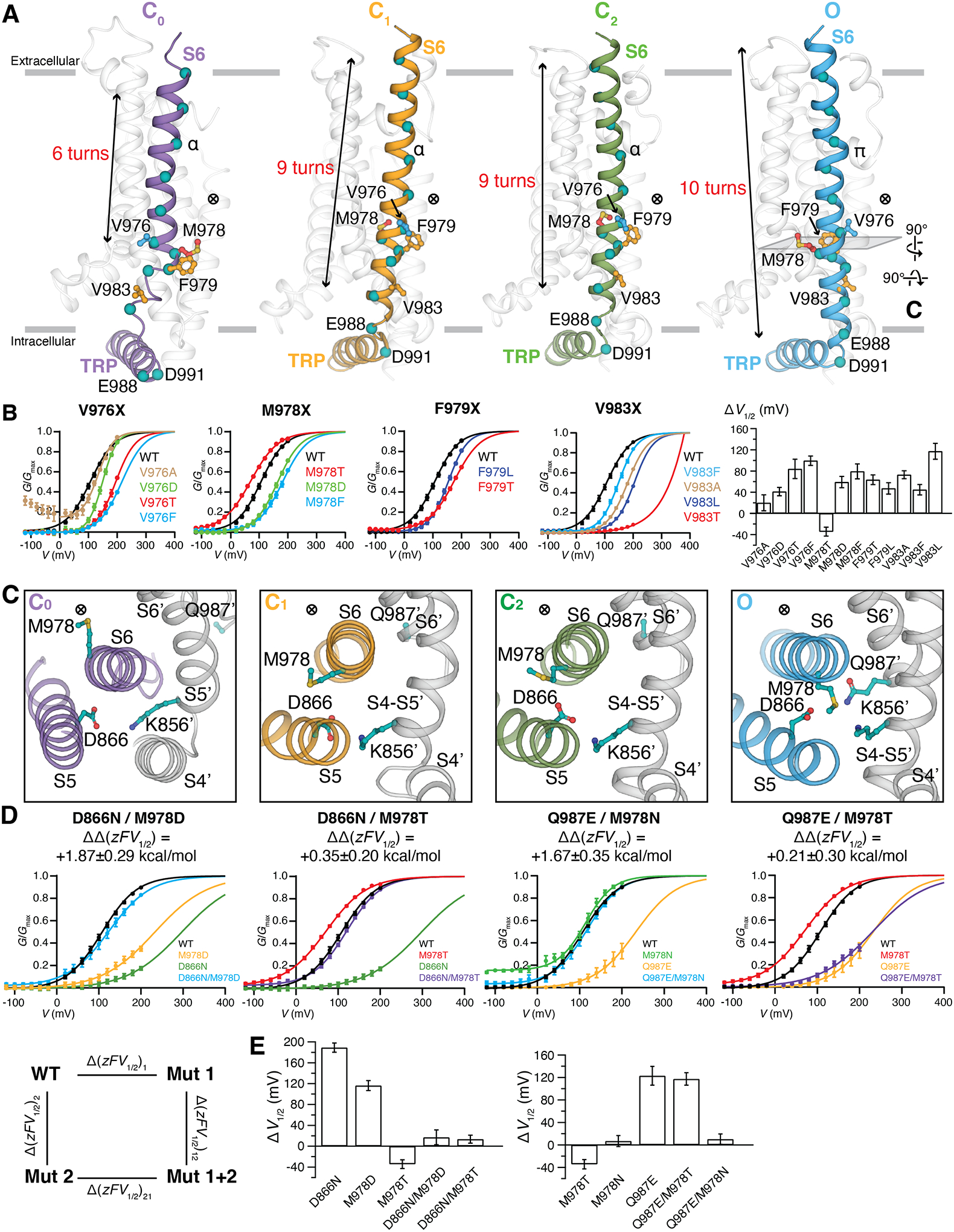
(A) Side-by-side comparison of the S6 rearrangement in the C0, C1, C2, and O state structures viewed from the membrane plane. Gating residues are shown as sticks and colored as in Fig. 4. The ⨂ symbol next to the gate residue indicates the location of the pore. Teal colored spheres represent residues from every helical turn along S6. S1 to S5 and PH are colored transparent gray for clarity. Gray bars indicate the membrane bilayer position. The number of helical turns in S6 is denoted.
(B) G-V relationships of gating residue mutants. The data were fit with Boltzmann functions, with extrapolation to +400 mV. Bar chart (rightmost panel) quantifying the effect of mutations on the V1/2 of activation with respect to wildtype (ΔV1/2) as mean ± SEM (n = 4 to 11 oocytes). V1/2 of V983T is far right-shifted so its ΔV1/2 is omitted in the bar chart.
(C) Close-up extracellular view at the MDQK interface sliced from (A). Gray cartoon and (´) represent structural domains from the neighboring protomer. ⨂ indicates the location of the ion conduction pore.
(D) G-V relationship for the double mutant cycle analysis. G-V curves for D866N, M978D, Q987E, Q987E/M978T were obtained with Gmax values measured in the presence of menthol (fig. S14). Coupling energy ΔΔzFV1/2 (mean ± S.E.M.) was calculated using Equations #2 to #7 in methods.
(E) Bar chart quantifying the difference of V1/2 between mutant and wildtype (ΔV1/2) as mean ± S.E.M. (n = 4 to 11 oocytes).
The S6 gate transition is accompanied by rotation, vertical and lateral translation, and secondary structural changes in the pore lining S6 helix (Fig. 5A). From the C0 to C1/C2 states the entire S6 rotates along the vertical axis by ~90° counterclockwise, followed by an additional 90° rotation from the C1/C2 states to the O state, which is associated with an α- to π-helical transition in S6 (36). As a result Met978, which gates the C0 state, flips nearly 180° in the O state to interact with residues on the intrasubunit S5 and the neighboring S4-S5 junction and S6. The sidechain of Val976 must also rotate 180° from the C0 state to gate the channel in the O state. These 90° to 180° rotations result in solvent-accessibility changes of the pore-lining S6 residues between the solvent-accessible lumen and the solvent-inaccessible membrane bilayer.
Besides rotation, S6 is coupled to S5 movement away from the channel lumen during channel activation (fig. S12, A and B). In going from C1/C2 to O, a ~2 to 5 Å shift in residue position indicates the downward movement of S6 (Fig. 5A and fig. S12, B and C). Notably, coils at the N- and C-termini of S6 in the C0 state become partially helical in the C1/C2 state and adopt fully α-helical structures in the O state (Fig. 5A and fig. S12C). These cumulative effects thereby extend S6 by four helical turns (Fig. 5A) accompanied by a subdomain structure shift at the linkage between S6 and the TRP domain, where the N-terminal Glu988 on the TRP domain in the C0 state relocates to the C-terminus of the S6 helix in the O state (Fig. 5A and fig. S12C).
To further probe the O state structure, we performed MD simulations. Based on the wetting or dewetting status at the gate inside the pore domain, the O state was stable in five out of the total eight simulation replicates (200 nanoseconds each), whereas transient opening and closure were observed in two, and gate closure was observed in one (fig. S13). Transient opening and closing are typically observed for TRPM8 at the single channel level (2). To further interrogate the observed pore conformation, we systematically mutated the four gate residues (Val976, Met978, Phe979, and Val983) to Ala, Thr/Ser (polar), Asp (charged), and Phe/Leu (nonpolar), respectively, then measured the conductance-voltage (G-V) relationships in response to voltage activation (Fig. 5B). The channel function is highly sensitive to mutagenesis at these positions: (i) mutation of Val976 to Asp, Thr, and Phe, Met978 to Asp and Phe, Phe979 to Thr and Leu, and Val983 to Ala, Thr, Phe, and Leu profoundly shifted G-V curves to more positive membrane potentials compared with the wildtype (>~42 mV). Val976Ala showed increased basal currents at negative potential. (ii) We could not detect appreciable currents for Phe979Ala, Phe979Asp, and Val983Asp mutants. There appears to be no correlation in sidechain properties (size and polarity) of mutated residues, likely because these gate residues are involved in the interaction networks of multiple states.
An O state-dependent interface at the S6 gate region
The observed state-dependent changes of the S6 gate rearrange Met978 on S6 to form an interface with Asp866 on S5, Gln987 on S6′, and Lys856 on the S4′-S5′ junction from the neighboring pore domain in the O state. This is an unusual interface arrangement as hydrophobic Met978 faces three hydrophilic residues (Fig. 5C). This interface indicates that mutation of Met978 to Thr would introduce hydrogen bonding with Asp866 and/or Gln987, thus stabilizing the O state conformation. Consistent with this idea, we observed an appreciable left-shift of the G-V curve (ΔV1/2 = −34.41 ± 8.37 mV) in the Met978Thr mutant (Fig. 5B).
We used double-mutant cycle analysis to further examine the coupling between paired residues among Met978, Asp866, and Gln987 (Fig. 5, D and E). We first probed Asp866 and its coupling with Met978. The V1/2 values of Asp866Asn and Met978Asp right-shifted about 189 mV and 116 mV, respectively, compared with that of the wildtype, indicating that these point mutations destabilize the O state. However, the V1/2 of the double mutant Asp866Asn/Met978Asp is similar to that of the wildtype (ΔV1/2 = +13.63 ± 14.14 mV) with a large coupling energy (ΔΔzFV1/2) of >1.87 ± 0.29 kcal/mol, corroborating that these two sites are energetically coupled in the O state (see methods). For mutants with large V1/2 shifts, additional G-V curves were measured in the presence of menthol to obtain more accurate Gmax (fig. S14). We also found that Gln987Glu and Met978Asn are strongly energetically coupled (~1.67 ± 0.35 kcal/mol). However, the double mutant pairs Gln987Glu and Met978Thr, Asp866Asn and Met978Thr did not show a substantial coupling energy, indicating the requirement for a specific set of interactions at the S6 interface.
Structural basis of ligand-induced TRPM8 activation
Comparisons of the C1, C2, and O state structures allow us to grasp the molecular basis of TRPM8 activation by agonists (Fig. 6 and figs. S15 and S16). A small local change at the ligand binding site in the VSLD propagates throughout the TMD and triggers conformational rearrangements in the pore domain for gate opening. Upon C3 binding, residues in the 310-helical S4b rotate slightly and stretch downward by ~1.4 Å (Fig. 6A). This minimal change at S4b is transduced through the connecting S4-S5 junction helix which rotates slightly closer to S3 and S4 (Fig. 6B). The bending point in S5 changes from Ile865 in the C1 state to Phe868 in the O state, which amplifies the translation and rotation in S5. More importantly, S5 maintains extensive but differing interactions with S6; the movement in S5 being tightly coupled with the ~10.7 Å translocation of S6 away from the central axis, leading to the dilation of the ion conduction pore (Fig. 6B). This coupled movement is not a simple rigid body movement, as the interfacial residue contacts also change to accommodate the changes in the S6 gate (fig. S16A). The pore helix, located between S5 and the neighboring S6, is tilted and displaced toward the intracellular side by ~5.5 Å and moves closer to the neighboring protomer in the O state relative to the C1 state (Fig. 6, B and C).
Figure 6. Structural basis of ligand-dependent gating of TRPM8MM.
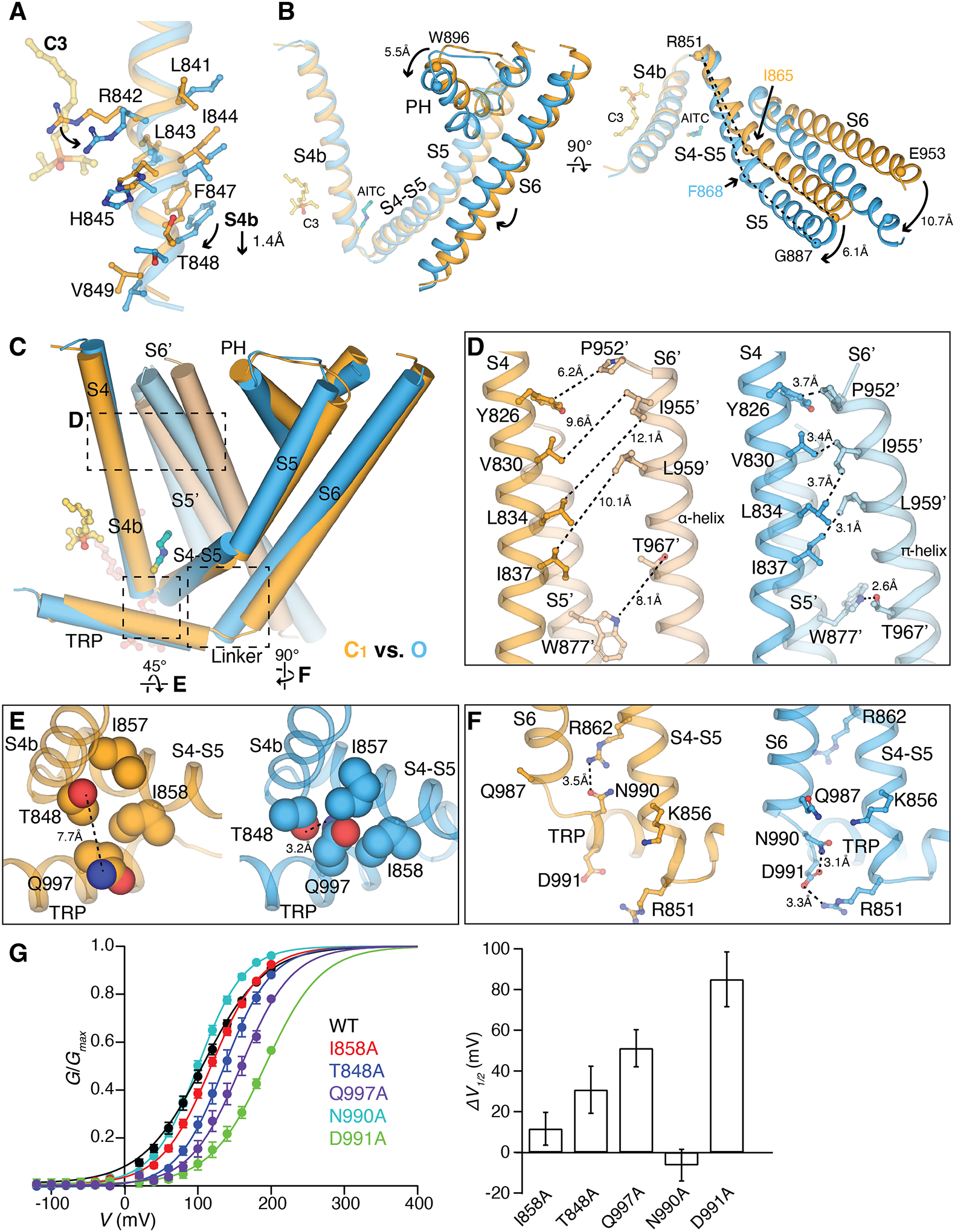
(A) Structural overlay of the C1 (orange) and O (blue) states at S4b. C3 (yellow) and residue sidechains are shown in sticks. Arrows indicate downward movement and sidechain rotations.
(B) Comparison of the C1 (orange) and O (blue) state structures at the TMD, aligned at the VSLD. Arrows indicate movement in S5, PH, and S6 by the marked distances. Key residues for comparison are shown as spheres. Dashed lines (right panel) compare the change in the bending points (Ile865 and Phe868 as spheres) on S5. C3 and AITC are shown as sticks for orientation reference. S1 to S3 are removed for clarity.
(C) Comparison of two neighboring protomers in the C1 (orange and light orange) and O state (blue and light blue) at the TMDs. The channel is shown as cylinders and ligands as sticks. S1 to S3 are removed in chain A and only S5 and S6 are shown in chain B for clarity. Dashed regions are zoomed in for comparison in (D to F). Alignment was done using the entire tetrameric channel.
(D to F) Close-up views showing side-by-side comparison of the C1 (orange) and O state (blue) structures at inter- and intra-subunit interfaces marked in (C). Sidechains are shown in sticks [(D) and (F)] or spheres (E). Dashed lines indicate the minimum distances between the corresponding residues.
(G) (Left) G-V relationship of mutants at interfaces as indicated. (Right) Bar chart quantifying the difference of V1/2 between mutant and wildtype (ΔV1/2) as mean ± S.E.M. (n = 4 to 6 oocytes).
Contact analysis suggests there are three interfaces that undergo the most notable changes during TRPM8 activation (fig. S15). First, the lateral displacement and helical rotation in S6 from C1 to O requires stabilization by intersubunit hydrophobic contacts with the neighboring S4 (S4′) plus an intrasubunit hydrogen bonding between Thr967 on S6 and Trp877 on S5 (Fig. 6, C and D, and fig. S16B). This network is absent in the intermediate C2 state, where although PIP2 and C3 binding is propagated to coupled movements of S5 and S6 (partially), the gate remains closed, suggesting that this interfacial contact is necessary to maintain the O state pore domain. Second, the interface amongst the TRP domain, S4b, and the S4-S5 junction rearranges during TRPM8 gating (Fig. 6E and fig. S16C). Compared with the C1 state, the TRP domain packs tighter with the VSLD (S4b) and the S4-S5 junction in the O state. The hydrophilic sidechain of Gln997 on the TRP domain flips inward and inserts into a hydrophobic groove formed by Ile857 and Ile858 on the S4-S5 junction and hydrogen bonds with Thr848 on S4b (Fig. 6E and fig. S16C), suggesting a change in solvent accessibility. Third, the S6-TRP connection region undergoes a coil-to-helix transition during channel gating, rearranging its interface with the S4-S5 junction and S5. In the C1 state, Asn990 on the N-terminal end of the TRP domain hydrogen bonds with Arg862 on the S4-S5 junction. During activation this interaction breaks and Asn990 and Asp991 on the TRP domain and Arg851 on S5 (which also binds to PIP2) form an interaction triad involving hydrogen bonding and a salt bridge, thus stabilizing the helical formation at the S6-TRP linkage in the O state (Fig. 6F and fig. S16D). Mutations of many residues involved in these interfaces lead to substantial changes in the G-V curves (Fig. 6G). Our findings are consistent with a previous study that reported residues Val986 to Asn990 on the C-terminal end of S6 mediate interactions with the TRP domain and the S4-S5 junction and are critical for TRPM8 activation (37). Notably, the C2 state conformation is between those of the C1 and O states along the conformational pathway towards gating (fig. S16, E and F). In contrast to the TMD, the cytoplasmic domain (CD) does not undergo substantial conformational changes during channel activation (fig. S16, H to K).
Discussion
Our open, closed, and intermediate structures show that mammalian and avian TRPM8 share the allosteric networks necessary for ligand activation but differ in their PIP2 binding affinity and/or propensity for PIP2-induced conformational changes. Under physiological conditions, the more populated ground state is likely the C1 state for mammalian TRPM8 and the C0 state for avian TRPM8. Mammalian and avian TRPM8 have distinct temperature and chemical sensitivities with mammalian orthologs substantially more sensitive to cold and exhibiting stronger cold-induced responses (26). Higher cold sensitivity in mammals may thus be caused by a higher sensitivity to PIP2 or enhanced allosteric coupling between PIP2 and temperature sensing.
Our structural data reveal noncanonical changes in the pore domain during TRPM8 channel gating. The pore helices move concomitantly with a decrease in pore cavity size and increased electronegativity. This transition at the pore in TRPM8 gating (Fig. 4A) is distinct from TRPV1, in which the selectivity filter and intracellular gate dilate for pore opening with minimal change in the pore cavity (38, 39), or potassium channels, in which no change in the selectivity filter and minimal change in the pore cavity occur during gating (40, 41). It is still unclear why TRPM8 undergoes such noncanonical changes in the pore domain. Quallo et al. showed that TRPM8 is an osmosensor in corneal afferent neurons, where it activates in response to hyperosmolality (9). When extracellular osmolality increases, water molecules in the pore cavity are driven out down the gradient and the cavity size decreases. Although it will require experimental testing to evaluate, we speculate that the pore cavity of TRPM8 could potentially act as a sensor to detect osmolality change.
We observe large, state-dependent conformational changes in the key interfaces as well as coil-to-helix transition in the pore-forming S6 helix of TRPM8 for ligand and PIP2 gating, which are associated with electrostatics and solvent accessibility changes in the pore. Because TRPM8 is a polymodal sensor for both thermally and chemically induced cooling, the conformational changes observed in ligand activation may also play a role in TRPM8 cold sensing. For example, lowering temperature would favor the coil-to-helix transition in the S6 helix for TRPM8 activation (42). Our study thus provides structural and mechanistic insights into the interplay of diverse chemical and physical stimuli on TRPM8 functions.
Materials and Methods
Protein expression and purification
The cDNA sequence for the full-length wildtype mouse TRPM8 (TRPM8MM) was cloned into a modified pEG BacMam vector (43) in frame with a preScission Protease cleavage site, a FLAG tag, and a 10x His tag at the C-terminus. TRPM8MM channel was expressed by baculovirus-mediated transduction of the human embryonic kidney (HEK) 293F suspension cells. Cells were cultured in Freestyle 293 medium (Life Technologies) and maintained at 37 °C in the presence of 8% CO2. The baculovirus was generated and amplified in accordance with the standard protocol for the Bac-to-Bac® Baculovirus Expression System (Life Technologies). 3–4% P2 virus for TRPM8MM was added to HEK293F cells at a cell density of ~2.5 M mL−1. After 18 h, 10 mM sodium butyrate was added to the cell culture and the growth temperature was lowered to 30 °C. After ~ 45–48 h of expression, cells were harvested, resuspended in buffer A (50 mM Tris-HCl pH 8, 150 mM NaCl, 12 μg mL−1 leupeptin, 12 μg mL−1 pepstatin, 12 μg mL−1 aprotinin, 1.2 mM phenylmethylsulfonyl fluoride, and DNase I), and lysed by a Dounce tissue grinder. 1% glycol-diosgenin (GDN; Anatrace) and 0.2 mg ml−1 soybean polar lipid extract (Avanti Polar Lipids) was added to the cell lysate and protein was solubilized at 4 °C by gentle agitation for 2 h. Insoluble materials were removed by centrifugation at 8,000g for 30 min at 4 °C. The supernatant was incubated with anti-FLAG M2 resin (Sigma-Aldrich) for 40 min at 4 °C with gentle agitation. The resin was harvested, packed into a gravity-flow column (Bio-Rad), and washed with 10 column volumes (CV) of buffer B (20 mM Tris-HCl pH 8, 150 mM NaCl, 0.02% GDN, 5 mM ATP, 10 mM MgCl2) followed by 10 CV of buffer C (20 mM Tris-HCl pH 8, 150 mM NaCl, 0.02% GDN). Protein was eluted by 5 CV of buffer C supplemented with 0.128 mg mL−1 FLAG peptide. Protein elution was concentrated and further purified on a Superose 6 Increase column (Cytiva Life Science) equilibrated with buffer C at 4 °C.
To determine the TRPM8MM and PIP2-TRPM8MM structures in the C1 state, protein was solubilized in buffer A supplemented with 1% GDN and 5 mM EDTA (pH 8) at 4 °C for 2 h. After anti-FLAG M2 resin binding, the column was washed with 10 CV buffer D (20 mM Tris-HCl pH 8, 300 mM NaCl, 0.02% GDN, 5 mM ATP, 10 mM MgCl2, 5 mM EDTA) and 10 CV buffer E (20 mM Tris-HCl pH 8, 150 mM NaCl, 0.02% GDN, 5 mM EDTA), followed by 5 CV elution in buffer E with FLAG peptide. Protein elution was further purified on a Superose 6 Increase column equilibrated with buffer E at 4 °C.
To determine the apo-TRPM8MM structure in the C0 state, protein was solubilized in buffer A supplemented with 1% GDN and 5 mM EDTA (pH 8) at 4 °C for 2 h. After anti-FLAG M2 resin binding, the column was washed with 10 CV of buffer F [20 mM Tris-HCl pH 8, 300 mM NaCl, 0.02% GDN, 0.004% cholesteryl hemisuccinate (CHS; Anatrace), 5 mM ATP, 10 mM MgCl2, 5 mM EDTA], 10 CV of buffer G (20 mM Tris-HCl pH 8, 300 mM NaCl, 0.02% GDN, 0.004% CHS, 5 mM EDTA), 10 CV of buffer H (20 mM Tris-HCl pH 8, 150 mM NaCl, 0.02% GDN, 0.004% CHS, 5 mM EDTA), and eluted with 5 CV of buffer H plus FLAG peptide. Protein elution was further purified on a Superose 6 Increase column equilibrated with buffer H at 4 °C.
For the collared flycatcher TRPM8 (TRPM8FA), a codon-optimized gene for the full-length channel with mutations Phe535Ala, Tyr538Asp, and Tyr539Asp was synthesized and cloned into the same pEG expression vector (Bio Basic Inc.). The functional integrity of this mutant TRPM8FA was confirmed previously (24). The protein expression and purification methods were identical to the previous report (23). In brief, the TRPM8FA channel was expressed in HEK293S GnTi− cells and solubilized and purified in digitonin detergent (Sigma-Aldrich).
All mammalian cell lines were authenticated and tested negative for mycoplasma by the Duke Cell Culture Facility. All mutations in the current study were introduced using the QuikChange mutagenesis kit (Agilent) and verified by Sanger sequencing (Genewiz Inc. and Azenta Life Science).
Cryo-EM specimen preparation
The peak fractions of TRPM8MM eluted from size exclusion chromatography in distinct buffer conditions as described above were pooled and concentrated to 0.6–0.8 mg mL−1 for cryo-EM analysis. Protein was equilibrated at ambient temperature (20 °C) for ~5 min followed by incubation with different ligand conditions at 20 °C for 2–5 min. For the apo-TRPM8MM and TRPM8MM samples, no ligand was added to the purified protein. For the PIP2-TRPM8MM sample, protein was incubated with 1 mM water-soluble diC8-PIP2 (Echelon Biosciences). For the PIP2-C3-TRPM8MM sample, protein was incubated with 1 mM diC8-PIP2 and 1 mM C3. For the PIP2-C3-AITC-TRPM8MM sample, protein was incubated with 1 mM diC8-PIP2, 1 mM C3, and 500 μM AITC (Sigma-Aldrich). For the PIP2-TRPM8FA sample, protein was incubated with 1 mM diC8-PIP2 at 4 °C.
3 μL of protein was applied to freshly glow-discharged Quantifoil R 1.2/1.3 300-mesh Cu holey carbon grids with a 2 nm continuous carbon layer (Quantifoil). For the apo-TRPM8MM, TRPM8MM, PIP2-TRPM8MM, and PIP2-C3-TRPM8MM samples, 100 μM fluorinated octyl maltoside (FOM; Anatrace) was quickly mixed with protein before applying to grids. Grids were prepared with a Mark IV Vitrobot (FEI) at 20 °C and 100% humidity. Grids were blotted for 2.5–6 s at blot force 0 followed by immediate plunge freezing in liquid ethane. The blotting time varied depending on the specific ligand conditions to achieve optimal image quality for data collection. Cryo-EM grids were stored in liquid nitrogen before data acquisition.
Cryo-EM data acquisition
Grids were screened on a Talos Arctica (FEI) operated at 200 keV equipped with a Ceta, K2, or K3 detector.
Cryo-EM datasets for the apo-TRPM8MM, TRPM8MM, and PIP2-C3-AITC-TRPM8MM structures were collected on a Titan Krios (FEI) operating at 300 keV equipped with a K3 detector (Gatan) with GIF BioQuantum energy filter (20 eV slit width; Gatan) in counting mode, using the Latitude-S automated data acquisition software (Gatan). Movies were collected at a nominal magnification of 81,000× with a physical pixel size of 1.08 Å pixel−1 using a nominal defocus range of −0.7 to −2.2 μm. Each movie stack (60 frames) was acquired with a total dose of ~60 e− Å−2. The exposure time and dose rate were 3.7 s and ~20 e− pixel−1 s−1, 3.7 s and ~20 e− pixel−1 s−1, 4.6 s and ~15 e− pixel−1 s−1, respectively.
Cryo-EM datasets for the PIP2-TRPM8MM and PIP2-C3-TRPM8MM structures were collected on a Titan Krios (FEI) operating at 300 keV equipped with a K3 detector (Gatan) with GIF BioQuantum energy filter (20 eV slit width; Gatan), using the Serial-EM automated data acquisition software (44). Movies were acquired at a nominal magnification of 81,000× in super-resolution mode with a pixel size of 0.528 Å pixel−1 and 0.535 Å pixel−1, respectively, using a nominal defocus range of −0.8 to −2.2 μm. For the PIP2-TRPM8MM data, each movie stack (50 frames) was acquired over 3.1 s exposure time, using a dose rate of 17.5 e− pixel−1 s−1 and a total dose of ~50 e− Å−2. For the PIP2-C3-TRPM8MM data, each movie stack (40 frames) was acquired over 3.5 s exposure time, using a dose rate of 17 e− pixel−1 s−1 and a total dose of ~50 e− Å−2.
The PIP2-TRPM8FA dataset was collected on a Titan Krios (FEI) operating at 300 keV equipped with a Falcon III detector in counting mode, using the EPU automated data-acquisition program. Movies were acquired at a nominal magnification of 75,000× with a physical pixel size of 1.08 Å pixel−1 using a nominal defocus range of −1.25 to −3 μm. Each movie (30 frames) was acquired with a dose rate of 0.8 e− pixel−1 s−1 with a total dose of ~42 e− Å−2.
Cryo-EM data processing
A total of 4283, 10147, 9536, 12071, 9135, and 13218 movies were collected for the PIP2-TRPM8FA, apo-TRPM8MM, TRPM8MM, PIP2-TRPM8MM, PIP2-C3-TRPM8MM, and PIP2-C3-AITC-TRPM8MM structures, respectively. All six datasets were processed in a similar format as illustrated in fig. S3B using RELION 3.1 and 4.0 (45, 46) and CryoSPARC (47). Beam-induced motion correction and dose-weighting were performed using MotionCor2 (48) in RELION. For PIP2-TRPM8MM and PIP2-C3-TRPM8MM data, the movies were Fourier-binned 2×2 to a physical pixel size of 1.056 Å/pixel and 1.07 Å/pixel, respectively. Gctf (49) was used for CTF estimation of non–dose-weighted summed images. Micrographs were selected based on the astigmatism, CTF fit quality, and defocus values. Particles were auto-picked for the entire dataset using template-free Laplacian-of-Gaussian (LoG) algorithm in RELION after optimizing the picking threshold with a small subset of micrographs. 2D templates from manual particle picking were used for template-based auto-picking the PIP2-TRPM8FA dataset in RELION. Typically, there were on average ~350–400 particles per micrographs. Particles were re-centered and re-extracted Fourier-binned 4×4 (64- or 80-pixel box size) and imported to CryoSPARC. After one or two round(s) of 2D classification (100 classes), false picks and chaperone or ice contaminant classes were excluded. The remaining particles were transferred back to RELION and input to 3D auto-refinement with C4 symmetry imposed. The published EM map of apo-TRPM8FA (EMD-7127) was rescaled and low-pass filtered to 30 Å as the initial reference without a mask. The refined particles, if the reconstruction resolution reached Nyquist, were re-centered, re-extracted Fourier binned 2×2 for 3D auto-refinement with a soft mask covering the entire channel. If not, 3D classification with alignment (K=3, T=8) was performed, from which the class showing clear shape of TRPM8 channel was selected and proceeded in the same manner. Refined particles at 2×2 Fourier binning were classified by 3D classification without image alignment (K=2–4, T=8 or 16). Particles from the class with the best resolved density for transmembrane helices were re-centered and re-extracted without binning and were input to 3D auto-refinement with a soft full mask. The refined particles were subject to CTF refinement (50) and Bayesian polishing (51) to yield a decent consensus 3D reconstruction. Furthermore, to sort out higher-resolution 3D classes with stronger density at the TMD and to dissect conformational heterogeneity, we performed particle subtraction followed by focused 3D classification. In brief, a tight mask (fig. S6A) was made to subtract out signals of detergent belt and cytoplasmic domains (CDs; including MHR1–4 and post-TRP domain CTD) from the consensus 3D. Next, the subtracted particles were subject to a masked 3D classification without image alignment (K=2–4, T=8 or 16). Particles comprising the best resolved class or distinct conformations at TMD were reverted to original full particles, which were input to 3D auto-refinement with a full mask. Additional CTF refinement and Bayesian polishing were done to improve the map quality. This particle subtraction, along with focused 3D classification, were reiterated as necessary. Finally, particles yielding the best 3D reconstruction from RELION were transferred to CyroSPARC and input to non-uniform (NU) refinement (52) and local refinement. To improve the EM density quality at the pore helix and outer pore region for the PIP2-C3-TRPM8MM and PIP2-C3-AITC-TRPM8MM structures, we performed focused refinement at the TMD after subtracting signals of detergent belt and the CDs. The subtracted particles were refined using local refinement in CryoSPARC. A detailed data processing flowchart for the O state PIP2-C3-AITC-TRPM8MM structure is depicted in fig. S6A. Local resolution estimation and the Fourier shell correlation (FSC) validation of the final 3D reconstruction were calculated using the gold-standard 0.143 FSC (53) in CryoSPARC (47).
Model building, refinement, and validation
The published PIP2-icilin-Ca2+-TRPM8FA structure (PDB 6NR3) was docked into the cryo-EM map for the O state PIP2-C3-AITC-TRPM8MM structure followed by manual model building in Coot (54). The residue sequence was replaced with the wildtype mouse TRPM8 sequence. Secondary structures were first rigid body fit into the EM densities. Side chains were adjusted to optimal rotamer conformations and loops were rebuilt to fit into the density. Residues with bulky side chains guided the correct registers of helices and β-strands. Residues and side chains missing in the previous structure but resolved in the current data were built in. The high-quality EM density at the TRP domain and its linkage with S6 facilitated register assignment at the S6 gate (Fig. 1G and fig. S5F). Ideal geometry restraints were imposed on secondary structures and rotamer conformation as much as possible during the initial manual model building in Coot.
The PIP2-C3-AITC-TRPM8MM structure served as the initial reference for model building of the TRPM8MM, PIP2-TRPM8MM, and PIP2-C3-TRPM8MM structures. The published PIP2-WS–12-TRPM8FA structure (PDB 6NR2) was used as the initial model for building the PIP2-TRPM8FA and apo-TRPM8MM structures. Subsequent manual model building in Coot was performed following the procedure described above. In addition, to facilitate model building of the pore helices (PHs) and the selectivity filters (SFs), the TMD focused EM maps were used for the PIP2-C3-AITC-TRPM8MM and PIP2-C3-TRPM8MM structures.
Ligand geometry restraint files were generated from isomeric or canonical SMILES strings using the eLBOW tool (55) in PHENIX using fixed bond lengths and angles. For the TRPM8MM, PIP2-TRPM8MM, and PIP2-TRPM8FA structures, a PIP2 molecule was modeled into the PIP2 density in each protomer of the structure, respectively (Fig. 2, A and B). For the apo-TRPM8MM structure, three CHS molecules were modeled into the elongated shape densities at the interfacial cavity per protomer (Fig. 2C and fig. S9B). For the PIP2-C3-TRPM8MM structure, a PIP2 molecule, a C3 molecule, and a Ca2+ ion were modeled into each protomer (fig. S10B). For the PIP2-C3-AITC-TRPM8MM structure, a PIP2 molecule, a C3 molecule, an AITC molecule, and a Ca2+ ion were modeled into each protomer (Fig. 3, A and C, fig S10A).
The manually built structure models were subjected to real-space refinement in PHENIX against cryo-EM maps along with ligand restraints, using global minimization, rigid body refinement, and B-factor refinement with secondary structure restraints (56). Geometry outliers in the real-space refined models were identified by the Molprobity server (http://molprobity.biochem.duke.edu/) (57) and were further manually fixed in Coot. The FSCs between the structure model against the full map and both half-maps were calculated in PHENIX (56), and showed good agreement to each other, indicating the models were not over-refined.
Structure analysis and illustration were performed in Coot and PyMOL (Schrödinger) (58). The Cα RMSD values in fig. S8 were calculated in PyMOL, using the “align” command and specifying Cα-only alignment at TMD residues 721 to 1027. Analysis and presentation of cryo-EM 3D reconstructions and EM densities were performed in UCSF Chimera (59), UCSF ChimeraX (60), and PyMOL. For the HOLE plot in Fig. 4B, to ensure a similar vertical position along the ion conduction pore, structures were aligned at the tetrameric TMD level. Gly913, Pro916, Val919, Asp918 were chosen as the starting point and Thr982, Glu988, Glu988, and Glu988 as the ending point for the C0, C1, C2, O state structures, respectively.
Structure-based analysis of protein interfacial contact
The analysis of intra- and inter-subunit residue contact was performed using the CONTACT/ACT program supported in the CCP4 suite (61) for the PIP2 -TRPM8MM, PIP2-C3-TRPM8MM, and PIP2-C3-AITC-TRPM8MM structures. Hydrogen atoms in the structure coordinates were first removed by the PDB Tools in PHENIX (56) before inputting to CCP4. Inter-chain contacts were probed between residues 502–1026 (covering MHR4 to CTDH1 domains in TRPM8 structures) from chain A and chain B. Contacts between any atom type within 0.0–4.1 Å distance were reported. The list of contacts was visually inspected. Source atom from residues in chain A and target atom from residues in chain B were selected when the distance between the two atoms allows for prominent interactions: maximum 3.3 Å for hydrogen bonding, maximum 4.0 Å for salt bridge interaction, and maximum 4.0 Å for hydrophobic interactions (van der Waals interactions). Interacting residue pairs which are located on S1–S6, PH, PL, and the TRP domain were highlighted and plotted in fig. S15. For intra-chain contacts, contact distances within 0.0–4.1 Å range for all residues were calculated. The list of contact for atoms from residue 502–1026 in chain A only was visually inspected using the same criteria described above. Interacting residue pairs from S4, the pore domain, and the TRP domain were illustrated in fig. S15.
Molecular dynamics (MD) simulation
Eight replicates of the O state PIP2-C3-AITC-TRPM8MM structure with AITC, C3, and PIP2 bound were embedded in a mixed membrane of POPC : POPE : Cholesterol = 2:1:1. The 8 replicates started with the exact same atom configuration with exception of two atoms in the C3 ligand. In 4 replicates, the P=O bond of C3 was oriented upward toward Tyr745; in the remaining 4 replicates, the P=O bond was oriented downward away from Tyr745, to study the proper ligand binding pose. Sodium and chloride ions were added to neutralize the charge of the entire system and maintain a salt concentration of ~0.15 M. PIP2 was modelled as SAPI24, phosphatidylinositol-4,5-bisphosphate with protonation on 4′-phosphate group and stearic (18:0) and arachidonic (20:4) acid as tails. The simulations were performed using the CHARMM36m force field (lipid and protein) (62–65), TIP3P water model (66), and CGenFF (AITC and C3) (67). The initial simulation system was assembled in CHARMM-GUI Membrane Builder (68–71) and equilibrated using the standard CHARMM-GUI six-step protocol. After that, additional restrained simulation of 30 ns was performed for protein backbone in each replicate to relax the whole protein, with the harmonic force constant gradually reducing from 50 to 0 kJ mol−1 nm−2. A 1-fs time step was used for production using OpenMM (72). The van der Waals interactions were cut off at 12 Å with a force-switching function between 10 and 12 Å. Each system was held at a constant particle number, 1 bar pressure, and 300.15 K temperature (NPT) with hydrogen mass repartitioning (73), and simulated for 200 ns.
For C3 ligand orientation analysis, the tilting angle between the P=O bond in C3 regarding the membrane bilayer plane was calculated along the time course. For characterizing the pore opening state, the region spanning from Asn973 to Ala977 was selected. The number of water molecules in this region was counted and averaged across the 10 frames for every ns.
Two-electrode voltage-clamp electrophysiology in Xenopus laevis oocytes
The full-length wildtype mouse TRPM8 gene was cloned into a pGEM-HE vector. The plasmid was linearized with XbaI restriction enzyme and complementary RNA (cRNA) was synthesized by in vitro transcription using T7 RNA polymerase (Thermo Fisher). All defolliculated oocytes were ordered from Xenocyte. cRNAs were injected into Xenopus laevis oocytes and were incubated at 17 °C for 2–4 days in ND96 solution [96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2 and 5 mM HEPES, pH 7.6 (with NaOH)]. Oocyte membrane voltage was controlled using an OC-725C oocyte clamp (Warner Instruments). Data were filtered at 1–3 kHz and digitized at 20 kHz using pClamp software (Molecular Devices) and a Digidata 1440A digitizer (Axon Instruments). Microelectrode resistances were 0.1–0.3 MΩ when filled with 3 M KCl. The external recording solution contained 100 mM KCl, 2 mM MgCl2, 5 mM HEPES, pH 7.6 (with KOH). All TRPM8 agonists (AITC, menthol, WS–12 from Sigma-Aldrich and C3 from Edward Wei at U.C. Berkeley) and antagonist AMG2850 (Alomone Labs) were applied using a gravity-fed perfusion system. For time course recording, the voltage was initially held at −60 mV and ramped to +60 mV for 300 ms every second.
For the conductance-voltage (G-V) recording, a voltage-step protocol covering the voltage range from −120 to +200 mV with +20 mV increment was used. After initial recording, 50 μM AMG2850 was applied to the chamber to block TRPM8 currents; then the same voltage-step protocol was repeated to measure background endogenous currents, which were subtracted from the initial recording to obtain TRPM8-speficic currents. For Val976Ala mutant, 200 μM AMG2850 was used. The steady-state currents at different test potentials were quantified and the apparent channel open probability (Po) was determined as G/Gmax. The maximum conductance Gmax values were obtained by extrapolating G-V curves to +400 mV. For mutants M978D, D866N, Q987E, Q987E/M978T, and V983T (fig. S14) which showed strongly right-shifted G-V relationships, additional G-V recordings in the presence of 200–500 μM menthol were done to measure their Gmax values. The steady state conductance G measured in the absence of menthol was normalized to Gmax.
The G-V curves were constructed from current amplitudes and voltages being measured then by fitting the conductance to a two-state Boltzmann equation:
| (1) |
where G/Gmax is the normalized channel conductance, z is the number of equivalent gating charge, kB is the Boltzmann constant, T is the absolute temperature, V is voltage, and V1/2 is the voltage for half-maximal activation.
Double-mutant cycle analysis and coupling energy
Structure-based design of double mutations and relevant single mutations were created by site-directed mutagenesis. A total of 10 constructs (Fig. 5D) were expressed in Xenopus laevis oocytes. The G-V relationships of mutant and wildtype channels were measured following the methods in the previous section and the V1/2 and z values were extracted. The free energy change for the channel to transition from the closed to the open state was calculated as
| (2) |
The perturbation in free energy change upon mutation was calculated as
| (3) |
| (4) |
Finally, the nonadditive coupling free energy was calculated as
| (5) |
The standard error in ΔG was calculated as
| (6) |
The propagated standard errors in ΔΔG were calculated as
| (7) |
The standard errors in coupling energy ΔΔΔG were calculated by linear error propagation as above in (7).
Cell culture and transfection
HEK293T cells up to passage 25 were grown in DMEM culture medium supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% Antibiotic-Antimycotic (Gibco), and were maintained at 37 °C in the presence of 5% CO2. Cells seeded in 6-well plates (Genesee Scientific) were transiently transfected at ~50% confluency using X-tremeGENE 9 DNA transfection reagent (Sigma-Aldrich) at a 1:3 DNA: transfection reagent ratio (3 μL reagent for every 1 μg DNA). 800 ng of TRPM8MM DNA and 200 ng eGFP DNA were transfected for each well. About 24 h following transfection, TRPM8MM-expressing cells were trypsinized and plated onto 12mm-dimater coverslips pre-coated with 0.1 mg mL−1 Poly-L-lysine (PLL; Sigma-Aldrich) and 50 μg mL−1 laminin (Sigma-Aldrich) inside 12-well culture plates. Cells were used for inside-out patch clamp ~24 h after split onto coverslips. The same TRPM8MM construct cloned in pEG vector for cryo-EM study was used for HEK293T cell transfection.
For whole-cell recording, cells seeded in 6-well plates were transiently transfected at ~20% confluency using FuGENE® 6 DNA transfection reagent (Promega) at a 1:3 of DNA: transfection reagent ratio (0.3 μL reagent for every 0.1 μg DNA). 100 ng of TRPM8MM-GFP fusion cloned in a pcDNA4 vector were transfected for each well and cells were used 16–24 h after transfection.
Patch clamp electrophysiology
All patch-clamp recordings were performed at ambient temperature (20–24 °C). Pipettes were pulled from borosilicate glass with resistances between 2 to 2.5 MΩ. Current responses were lowpass filtered at 2 kHz (Axopatch 200B), digitally sampled at 5–10 kHz (Digidata 1440A) and converted to digital files in Clampex10.4 (Clampfit10.4, Molecular Devices; Igor Pro 6.34A, Wavemetrics). Electrodes were filled with an intracellular solution containing 140 mM NaCl, 5 mM MgCl2, 10 mM HEPES, 5 mM EGTA and adjusted to pH 7.4 (NaOH). For inside-out patch-clamp recordings, extracellular solutions contained 140 mM NaCl, 5 mM HEPES pH 7.4 (NaOH), and 200 μM Ca2+. Following formation of giga-ohm seal, inside-out configuration was formed by excising the membrane patches from cells co-expressing TRPM8 and eGFP. TRPM8 currents were elicited by focal perfusion of test solutions containing 300 μM C3 in the presence of 200 μM Ca2+ to the cytoplasmic side of the excised patches for 2 min, followed by application of 50 μM of TRPM8 specific antagonist AMG2850. For time course recording of TRPM8 channels, a repeated ramp protocol from −120 mV to +120 mV at a 2-s interval was used to elicit channel activity, and the membrane was held at 0 mV. Peak currents at ±120 mV were used for data analysis presented in the right panel in fig. S1B.
For whole cell recording (fig. S1, C and D), the intracellular solution contained 140 mM NaCl, 5 mM MgCl2, 5 mM EGTA, and 10 mM HEPES pH 7.4 (NaOH), and the extracellular solution contained 140 mM NaCl and 10 mM HEPES pH 7.4 (NaOH). The published methods (29) was used to characterize TRPM8 activation by type I and type II agonists. TRPM8 current was recorded using voltage steps from −80 to +120 mV, in the absence of agonist, as well as in the presence of 3 mM AITC, 10 μM C3, 30 μM menthol, and 10 μM C3/ 3mM AITC. To analyze the gating kinetics during TRPM8 activation and deactivation in the presence of different agonist conditions, mono-exponential function was used to fit current traces, which yielded the time constants (τ) of current relaxation at +120 mV and −80 mV voltage, respectively.
Statistical analysis
Statistical analyses were performed in Igor 6.0, using two-tailed Student’s t test for single comparisons between two data groups. Comparisons with p-values <0.05 are considered statistically significant. Data in summary graphs are presented as mean ± S.E.M., where each data point presents an individual recording. p-values are denoted in the figures.
Supplementary Material
Acknowledgements:
We thank E.T.F. Wei from U.C. Berkeley for providing the C3 compound and encouraging us to test it for structural studies. We thank A. Patapoutian from TSRI and M. Bandell from GNF for sharing unpublished data and the plasmid containing mouse TRPM8-GFP. Cryo-EM data were screened and collected at the Duke University Shared Materials Instrumentation Facility (SMIF), the Pacific Northwest Center for Cryo-EM (PNCC) at OHSU, National Center for CryoEM Access and Training (NCCAT), the Simons Electron Microscopy Center at the New York Structural Biology Center, the National Cancer Institute’s National Cryo-EM Microscopy facility (NCEF) at the Frederick National Laboratory for Cancer Research, National Institute of Environmental Health Sciences (NIEHS), and the UNC Cryo-EM Core. We thank J. Myers at PNCC, A. Wier and T. Fox at NCEF, M. Aragon, H. Wei, H. Kuang at NCCAT, E. Viverette at NIEHS, J. Peck and J. Strauss at UNC, and N. Bhattacharya at SMIF for assistance with the microscope operation. We thank L. Zubcevic, J. Grandl, and J. Fedor for critical manuscript reading and Y. Suo for assistance with data collection at SMIF.
Funding:
This work was supported by the National Institutes of Health (R35NS097241 and R01EY031698 to S.-Y.L. and R01GM138472 to W.I.) and by the National Institute of Health Intramural Research Program; US National Institutes of Environmental Health Science (ZIC ES103326 to M.J.B.). Some of this research was supported by NIH grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research, by the NIH Common Fund Transformative High Resolution Cryo–Electron Microscopy program (U24 GM129539), by grants from the Simons Foundation (SF349247) and NY State Assembly, by the contract 75N91019D00024 (NCEF), and by NIH grant P30CA016086 (UNC).
Footnotes
Competing interests: The authors declare no competing interests.
Supplementary Materials
Data and materials availability:
For PIP2-TRPM8FA, apo-TRPM8MM, TRPM8MM, PIP2-TRPM8MM, PIP2-C3-TRPM8MM, and PIP2-C3-AITC-TRPM8MM structures, the coordinates are deposited in the Protein Data Bank with the accession codes 8E4Q, 8E4P, 8E4O, 8E4N, 8E4M, 8E4L; the cryo-EM density maps are deposited in the Electron Microscopy Data Bank (EMDB) with the accession codes EMD-27896, EMD-27895, EMD-27894, EMD-27893, EMD-27892, and EMD-27891. The cryo-EM maps for the Class II C1-state and the Class III putative desensitized state from TRPM8MM purified in ligand- and PIP2-free condition are deposited in EMDB with accession codes EMD-27889 and EMD-27890.
References and Notes
- 1.Peier AM et al. , A TRP channel that senses cold stimuli and menthol. Cell 108, 705–715 (2002). [DOI] [PubMed] [Google Scholar]
- 2.McKemy DD, Neuhausser WM, Julius D, Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416, 52–58 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Bautista DM et al. , The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 448, 204–208 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Dhaka A et al. , TRPM8 is required for cold sensation in mice. Neuron 54, 371–378 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Colburn RW et al. , Attenuated cold sensitivity in TRPM8 null mice. Neuron 54, 379–386 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Paricio-Montesinos R et al. , The Sensory Coding of Warm Perception. Neuron 106, 830–841 e833 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu B et al. , TRPM8 is the principal mediator of menthol-induced analgesia of acute and inflammatory pain. Pain 154, 2169–2177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parra A et al. , Ocular surface wetness is regulated by TRPM8-dependent cold thermoreceptors of the cornea. Nat Med 16, 1396–1399 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Quallo T et al. , TRPM8 is a neuronal osmosensor that regulates eye blinking in mice. Nat Commun 6, 7150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alcalde I et al. , Morphological and functional changes in TRPM8-expressing corneal cold thermoreceptor neurons during aging and their impact on tearing in mice. J Comp Neurol 526, 1859–1874 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Chasman DI et al. , Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet 43, 695–698 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horne DB et al. , Discovery of TRPM8 Antagonist (S)-6-(((3-Fluoro-4-(trifluoromethoxy)phenyl)(3-fluoropyridin-2-yl)methyl)carbamoy l)nicotinic Acid (AMG 333), a Clinical Candidate for the Treatment of Migraine. J Med Chem 61, 8186–8201 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Weyer AD, Lehto SG, Development of TRPM8 Antagonists to Treat Chronic Pain and Migraine. Pharmaceuticals 10, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knowlton WM et al. , A sensory-labeled line for cold: TRPM8-expressing sensory neurons define the cellular basis for cold, cold pain, and cooling-mediated analgesia. J Neurosci 33, 2837–2848 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang JM et al. , A novel TRPM8 agonist relieves dry eye discomfort. BMC Ophthalmol 17, 101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Millqvist E, TRPV1 and TRPM8 in Treatment of Chronic Cough. Pharmaceuticals 9, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKemy DD, in TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades, Liedtke WB, Heller S, Eds. (Boca Raton (FL), 2007). [PubMed] [Google Scholar]
- 18.Yin Y, Lee SY, Current View of Ligand and Lipid Recognition by the Menthol Receptor TRPM8. Trends Biochem Sci 45, 806–819 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohacs T, Lopes CM, Michailidis I, Logothetis DE, PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat Neurosci 8, 626–634 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Zakharian E, Cao C, Rohacs T, Gating of transient receptor potential melastatin 8 (TRPM8) channels activated by cold and chemical agonists in planar lipid bilayers. J Neurosci 30, 12526–12534 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu B, Qin F, Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J Neurosci 25, 1674–1681 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voets T et al. , The principle of temperature-dependent gating in cold- and heat-sensitive TRP channels. Nature 430, 748–754 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Yin Y et al. , Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 363, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin Y et al. , Structure of the cold- and menthol-sensing ion channel TRPM8. Science 359, 237–241 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diver MM, Cheng Y, Julius D, Structural insights into TRPM8 inhibition and desensitization. Science 365, 1434–1440 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pertusa M, Rivera B, Gonzalez A, Ugarte G, Madrid R, Critical role of the pore domain in the cold response of TRPM8 channels identified by ortholog functional comparison. J Biol Chem 293, 12454–12471 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chuang HH, Neuhausser WM, Julius D, The super-cooling agent icilin reveals a mechanism of coincidence detection by a temperature-sensitive TRP channel. Neuron 43, 859–869 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Kuhn FJ, Kuhn C, Luckhoff A, Inhibition of TRPM8 by icilin distinct from desensitization induced by menthol and menthol derivatives. J Biol Chem 284, 4102–4111 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Janssens A et al. , Definition of two agonist types at the mammalian cold-activated channel TRPM8. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao C et al. , Structures of a mammalian TRPM8 in closed state. Nat Commun 13, 3113 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bandell M et al. , High-throughput random mutagenesis screen reveals TRPM8 residues specifically required for activation by menthol. Nat Neurosci 9, 493–500 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Zubcevic L et al. , Conformational ensemble of the human TRPV3 ion channel. Nat Commun 9, 4773 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirschi M et al. , Cryo-electron microscopy structure of the lysosomal calcium-permeable channel TRPML3. Nature 550, 411–414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suo Y et al. , Structural Insights into Electrophile Irritant Sensing by the Human TRPA1 Channel. Neuron 105, 882–894 e885 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rao S, Klesse G, Stansfeld PJ, Tucker SJ, Sansom MSP, A heuristic derived from analysis of the ion channel structural proteome permits the rapid identification of hydrophobic gates. Proc Natl Acad Sci U S A 116, 13989–13995 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zubcevic L, Lee SY, The role of pi-helices in TRP channel gating. Curr Opin Struct Biol 58, 314–323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taberner FJ, Lopez-Cordoba A, Fernandez-Ballester G, Korchev Y, Ferrer-Montiel A, The region adjacent to the C-end of the inner gate in transient receptor potential melastatin 8 (TRPM8) channels plays a central role in allosteric channel activation. J Biol Chem 289, 28579–28594 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwon DH et al. , Heat-dependent opening of TRPV1 in the presence of capsaicin. Nat Struct Mol Biol 28, 554–563 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao E, Liao M, Cheng Y, Julius D, TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee CH, MacKinnon R, Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures. Science 360, 508–513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansen SB, Tao X, MacKinnon R, Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang CY, Klemke JW, Getahun Z, DeGrado WF, Gai F, Temperature-dependent helix-coil transition of an alanine based peptide. J Am Chem Soc 123, 9235–9238 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Goehring A et al. , Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat Protoc 9, 2574–2585 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mastronarde DN, Automated electron microscope tomography using robust prediction of specimen movements. Journal of Structural Biology 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Zivanov J et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kimanius D, Dong L, Sharov G, Nakane T, Scheres SHW, New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochemical Journal 478, 4169–4185 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Zheng SQ et al. , MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang K, Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zivanov J, Nakane T, Scheres SHW, Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zivanov J, Nakane T, Scheres S, A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCRJ, 17, (2019). doi: 10.1107/S205225251801463X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Punjani A, Zhang H, Fleet DJ, Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nature Methods 17, 1214–1221 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Scheres SH, Chen S, Prevention of overfitting in cryo-EM structure determination. Nat Methods 9, 853–854 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P, Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Moriarty NW, Grosse-Kunstleve RW, Adams PD, electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D 65, 1074–1080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adams PD et al. , PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen VB et al. , MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Delano WL, The PyMol Molecular Graphics System. (DeLano Scientific, 2002). [Google Scholar]
- 59.Goddard TD, Huang CC, Ferrin TE, Visualizing density maps with UCSF Chimera. J Struct Biol 157, 281–287 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Pettersen EF et al. , UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Winn MD et al. , Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Best RB et al. , Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J Chem Theory Comput 8, 3257–3273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang J, MacKerell AD Jr., CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 34, 2135–2145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klauda JB et al. , Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114, 7830–7843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang J et al. , CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Durell SR, Brooks BR, Ben-Naim A, Solvent-Induced Forces between Two Hydrophilic Groups. The Journal of Physical Chemistry 98, 2198–2202 (1994). [Google Scholar]
- 67.Vanommeslaeghe K et al. , CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31, 671–690 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu EL et al. , CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J Comput Chem 35, 1997–2004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee J et al. , CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J Chem Theory Comput 12, 405–413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jo S, Kim T, Iyer VG, Im W, CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 71.Jo S, Lim JB, Klauda JB, Im W, CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys J 97, 50–58 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eastman P et al. , OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol 13, e1005659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gao Y et al. , CHARMM-GUI Supports Hydrogen Mass Repartitioning and Different Protonation States of Phosphates in Lipopolysaccharides. J Chem Inf Model 61, 831–839 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jurrus E et al. , Improvements to the APBS biomolecular solvation software suite. Protein Sci 27, 112–128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MS, HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph 14, 354–360, 376 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For PIP2-TRPM8FA, apo-TRPM8MM, TRPM8MM, PIP2-TRPM8MM, PIP2-C3-TRPM8MM, and PIP2-C3-AITC-TRPM8MM structures, the coordinates are deposited in the Protein Data Bank with the accession codes 8E4Q, 8E4P, 8E4O, 8E4N, 8E4M, 8E4L; the cryo-EM density maps are deposited in the Electron Microscopy Data Bank (EMDB) with the accession codes EMD-27896, EMD-27895, EMD-27894, EMD-27893, EMD-27892, and EMD-27891. The cryo-EM maps for the Class II C1-state and the Class III putative desensitized state from TRPM8MM purified in ligand- and PIP2-free condition are deposited in EMDB with accession codes EMD-27889 and EMD-27890.