

Abstract
The extracellular matrix (ECM) and its turnover play a crucial role in the pathogenesis of several inflammatory diseases, including age-related macular degeneration (AMD). Elastin, a critical protein component of the ECM, not only provides structural and mechanical support to tissues, but also mediates several intracellular and extracellular molecular signaling pathways. Abnormal turnover of elastin has pathological implications. In the eye elastin is a major structural component of Bruch’s membrane (BrM), a critical ECM structure separating the retinal pigment epithelium (RPE) from the choriocapillaris. Reduced integrity of macular BrM elastin, increased serum levels of elastin-derived peptides (EDPs), and elevated elastin antibodies have been reported in AMD. Existing reports suggest that elastases, the elastin-degrading enzymes secreted by RPE, infiltrating macrophages or neutrophils could be involved in BrM elastin degradation, thus contributing to AMD pathogenesis. EDPs derived from elastin degradation can increase inflammatory and angiogenic responses in tissues, and the elastin antibodies are shown to play roles in immune cell activity and complement activation. This review summarizes our current understanding on the elastases/elastin fragments-mediated mechanisms of AMD pathogenesis.
Keywords: Age-related macular degeneration, Extracellular matrix, Elastin, Elastase, Elastolytic enzymes, Elastin-derived peptides, Inflammation, Immune cell activity, Complement activation
1. Introduction
Age-related macular degeneration (AMD) is a major cause of irreversible vision loss among the elderly, currently affecting 170 million people worldwide (Pennington and DeAngelis, 2016). The United Nations 2019 report documented 703 million persons aged 65 years or older in the world, a number that is projected to double by 2050 (https://www.un.org/en/development/desa/population/publications/pdf/ageing/WorldPopulationAgeing2019-Highlights.pdf). Based on the current percentages of 25% early or intermediate AMD and 2.4% late AMD prevalence in the European Population 60 years or older (Li et al., 2020), or 0.37% late stage AMD prevalence in the 45-85-years-old global population (Schultz et al., 2021), the number of AMD patients is expected to soar. AMD is characterized by the central vision loss caused by the degeneration of rod and cone photoreceptors in the macula (Curcio et al., 1996), the area of the retina with the highest cone density, enabling central vision. Main risk factors of AMD include aging (Hussain et al., 2010), cigarette smoking (Seddon et al., 1996; Thornton et al., 2005; Velilla et al., 2013), oxidative stress, cardiovascular diseases, and genetic variants (Lim et al., 2012), which include mutations of genes regulating the inflammatory (Klein et al., 2005; Rohrer et al., 2019) and the extracellular matrix (ECM) turnover pathways (Kondo et al., 2008; Yamashiro et al., 2011).
Macular photoreceptor degeneration in AMD is mainly driven by the reduced or failing support from the outer retinal components, such as the retinal pigment epithelium (RPE)/Bruch’s membrane (BrM)/choriocapillaris (CC) complex. Based on the progression of the disease AMD is classified into two forms: dry and wet. In dry AMD or geographic atrophy (GA), RPE dysfunction appears first, followed debilitation of photoreceptors and choriocapillaris. In wet AMD, also called neovascular (n)AMD or exudative AMD, loss or dysfunction of the choroidal vasculature occurs first (Bhutto and Lutty, 2012), resulting in subsequent neovascularization, RPE dysfunction, and photoreceptor cell loss. The most prevalent form of AMD is dry AMD with severe cases of GA in the central retina, (Fleckenstein et al., 2018). Although wet AMD represents only 10–15% of total AMD cases, it accounts for ~90% of the blindness or severe vision loss associated with AMD (Ambati and Fowler, 2012; Ferris et al., 1984).
The underlying molecular mechanisms of AMD are not yet fully understood; thus, limited treatment strategies are available. Intravitreal injections of anti-VEGF are the only current FDA-approved treatment available for nAMD (Bloch et al., 2012). While anti-VEGF treatments show significant short-term benefits on best-corrected visual acuity and retinal structure in nAMD, continuous anti-VEGF exposure can cause drug intolerance, increased fibrosis, occasional development of GA, and hence fails to have long-term benefits in patients (Binder, 2012; Kaynak et al., 2018). Additional treatment paradigms include, but are not limited to, complement inhibitors that have been studied in intermediate AMD and late-stage GA (Wu and Sun, 2019) or an autophagy inducer such as rapamycin (sirolimus) tested to improve late-stage, persistent nAMD (Ambati and Fowler, 2012; Dejneka et al., 2004; Ishikawa et al., 2015) or GA (Gensler et al., 2018). However, other than anti-VEGF therapeutics, no other pathways have been proven as an effective strategy to reduce the progression of AMD. Therefore, a better understanding of AMD-associated pathways is crucial to developing an effective treatment strategy.
Aging and its associated accumulation of damage at the ECM are major risk factors for AMD. During aging many changes occur at the ECM including the accumulation of protein aggregates, ECM component crosslinking, glycation or fragmentation, with every cell type and tissue type producing its own unique ECM (Ewald, 2020). Studies have shown that damage of ECM or its abnormal turnover, especially at the RPE/BrM/CC complex has significant role in the pathogenesis of AMD (Nita et al., 2014). BrM is a complex five-layered elastin and collagen rich ECM structure located between RPE and CC, and is composed of the RPE basal lamina (RPE-BL), the inner collagenous later (ICL), the elastin layer (EL), the outer collagenous later (OCL) and the choriocapillaris basal lamina (CC-BL) (Curcio and Johnson, 2013). Degradation of EL at the macular BrM has been reported in AMD patients, and this EL loss has been postulated to enable choroidal blood vessels to break through BrM and cause CNV (Chong et al., 2005). Overall, EL thickness is less in the macula than in the periphery; and even less thick in eyes with early AMD and active CNV. Thus, this reduction in BrM EL integrity, which is important for BrM’s biomechanical properties and for maintaining a physical barrier to prevent the invasion of blood vessels, might provide some rationale why CNV occurs in the macula (Chong et al., 2005). In connection with this observation, serum elastin-derived peptides (EDPs), the byproducts of elastin degradation are significantly higher in moderate-severe AMD patients, especially AMD patients with retinal vascular abnormalities (Sivaprasad et al., 2005). Correspondingly, elevated levels of α-elastin antibodies (Morohoshi et al., 2012) and elastin gene polymorphisms are also reported in nAMD patients (Kondo et al., 2008; Yamashiro et al., 2011). Together these lines of evidence suggest that elastin metabolism plays a critical role in the pathogenesis of AMD and retinal angiogenesis. This review summarizes our current understanding of the role of elastin turnover in dry and wet forms of AMD.
2. Elastic fiber biogenesis, turnover and elastase inhibitors
Elastic fibers are made up of two distinct ECM components: elastin and microfibrils. The fibrous protein elastin makes up 90% of elastic fiber, forming its internal core, and the microfibrils provide a scaffold and organize the core elastin to form the typical elastic fiber. Elastin is encoded by a single mRNA transcript of 3–5 kb size (Davis and Mecham, 1998). The translated tropoelastin monomers are chaperoned by elastin binding protein (EBP; 67 kDa), which prevents intracellular self-segregation and premature degradation of elastin. The intracellularly formed tropoelastin monomers are then transferred to the extracellular space, where they will be polymerized to insoluble mature elastic fibers with the help of cross-linking enzymes lysyl oxidases (LOX) and LOX-like proteins (LOXL) 1–4(Mithieux and Weiss, 2005).
Microfibrils are mainly made up of fibrillin 1, fibrillin 2, and microfibril-associated glycoprotein-1 (MAGP-1) (Brown-Augsburger et al., 1996; Mithieux and Weiss, 2005; Sakai et al., 1986; Zhang et al., 1994). Another major group of ECM glycoproteins involved in elastin fiber assembly are fibulins-4 and −5. Fibulin-4 contributes to elastin cross-linking by activating and tethering LOX to tropoelastin (Horiguchi et al., 2009), and fibulin-4 deficient mice have disrupted elastin and collagen fibers and die shortly after birth (Noda et al., 2020). Interactions between fibulin-5 and tropoelastin as well as LOX, LOXL 1, 2 and 4 have been also reported. Fibulins 4, 5 can also bind to fibrillin 1 (Godwin et al., 2019). Together these non-redundant and essential interactions between fibulins, LOX, tropoelastin and fibrillins facilitate the formation of highly crosslinked insoluble elastic fibers (Fig. 1). Extensive cross-linking makes elastin one of the extremely durable proteins in the body with a half-life of ~70 years (Petersen et al., 2002) and minimal turnover rate in the body, with two exceptions, pathological conditions, or aging. Factors that affect elastin turnover during disease and aging are still emerging.
Fig. 1. Elastic fiber formation and degradation.
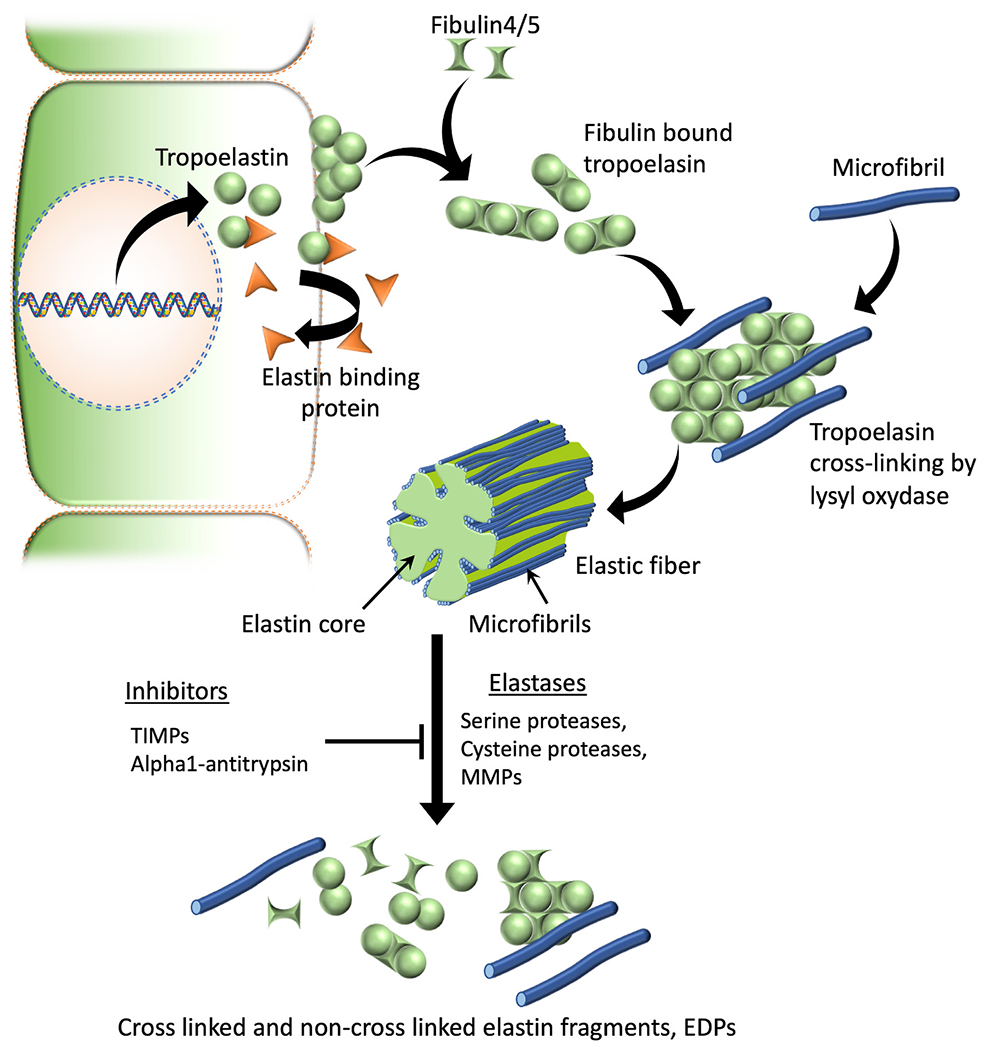
Tropoelastin monomers are formed intracellularly, which then will be chaperoned and transported to the ECM by elastin binding proteins. At the cell surface elastin binding proteins will be released leading to tropoelastin self-aggregation or coacervation. Coacervation leads to next stages of elastic fiber assembly such as binding of fibulins, microfibrillar deposition, recruitment of lysyl oxidase, elastin cross-linking and mature fiber formation. Serine proteases such as neutrophil elastase & HTRA1, cysteine proteases like cathepsins K, V, L, S & B, and several metalloproteases (MMPs 2, 3, 7, 9, 12 & 14) are known to have elastolytic capability and can contribute to the degradation of insoluble elastic fibers to generate elastin fragments or EDPs. TIMPs and alpha-1 proteases such as alpha-1 antitrypsin are known elastase inhibitors that can prevent the degradation of elastic fibers.
Abbreviations: TIMPs, Tissue inhibitors of metalloproteinases; MMPs, Matrix metalloproteinases; EDPs, Elastin-derived peptides.
Major factors that affect elastin turnover at the post-translational level are the elastases and their inhibitors. Elastases are defined as the elastin solubilizing enzymes as they can degrade insoluble mature elastic fibers and highly crosslinked elastin polymers into soluble elastin fragments (Bieth, 1986) (Fig. 1). Based on the differences in catalytic sites and enzyme activity elastases can be of serine, cysteine, or metalloproteinase category (Table 2). Major elastases, such as pancreatic and neutrophil elastases fall under the category of serine proteinases, whose catalytic activity depends on the serine group present on their catalytic sites and can be inhibited by alpha 1 proteinase inhibitors. Elastases produced by macrophages, which are the phagocytes derived from circulating monocytes, fall under the category of matrix metalloproteinase 12 (MMP12). Unlike serine protease, MMP12 is resistant to the inhibition by alpha-1 proteinase inhibitors (Banda and Werb, 1981; Werb et al., 1982), but their activity can be blocked by tissue inhibitors of metalloproteinases (TIMPs 1–4) and also by chelating agents such as EDTA, as they require metallic presence for their catalytic activity (Brew and Nagase, 2010).
Table 2.
List of enzymes with elastase activity. Abbreviations: VSMC, vascular smooth muscle cells; RPE, retinal pigment epithelium; MMP, matrix metalloproteinase; HTRA1, High-Temperature Requirement A Serine Peptidase 1.
Studies indicate that inflammation is a major trigger for increased elastase activity. TNF-α increases elastase activity in smooth muscle cells (Kothapalli and Ramamurthi, 2010), and IL-1β exposure increases elastase activity in RPE (Elner, 2002) and fibroblast cells (Croute et al., 1991). In addition, increased elastase activity can further exacerbate inflammation. It has been shown that neutrophil elastase increases insulin resistance and inflammation in mouse hepatocytes and adipocytes, and in support of this data, under conditions of high-fat diet and obesity, neutrophil elastase deficient mice exhibited increased insulin sensitivity and reduced inflammatory response (Talukdar et al., 2012). Thus, inflammation and elastase activities are interconnected, and can function as major triggers of abnormal elastin turnover in aging and diseases.
2.1. Neutrophil elastase inhibitors
Human neutrophil elastase is a basic, 218 amino acid single polypeptide glycoprotein with a molecular weight of 29.5 kDa. Its primary structure shows considerable homology with proteinase 3 (54%) and cathepsin G (37%). There are some endogenous protein inhibitors of elastase that include the two serpins, the α-1-proteinase inhibitor (α-1-PI) and monocyte/neutrophil elastase inhibitor (Serpin B1), and the chelonianin family of canonical inhibitors, secretory leukocyte proteinase inhibitor (SLPI) and elafin (Groutas et al., 2011). Elastase inhibitors have been the focus for drug development due to the involvement of elastases in pathologies of several prevalent diseases including chronic obstructive pulmonary disease (COPD) and fibrosis. However, it has been a challenge to specifically target the activity of elastase, due to its similarity and its overlapping functions with other serine proteases. A review article provides “an overview of major developments in COPD research with emphasis on low molecular weight neutrophil elastase inhibitors”, and the readers are referred to this excellent work (Groutas et al., 2011). In short, the design has focused on the development of so-called transition-state inhibitors, that bind to the enzyme’s active site serine and thereby prevent activation, or on agents that acylate the active site serine and have shown efficacy in the rat acute lung injury model (Groutas et al., 2011). Recently several anti-elastase polypeptides of bacterial, fungal, plant, and animal origin have been reported. These polypeptides are cyclic depsi-peptides containing modified and unusual amino acid residues, which developed as inhibitors of digestive enzymes and chemical defense against predators. These polypeptides have only been tested in vitro and are awaiting in vivo functional assays (Ahmad et al., 2020). Currently, the three FDA-approved neutrophil elastase inhibitors include Prolastin (Bayer), Aralast (Baxter), and Zemira (Aventis Behring), which are human plasma-derived alpha 1-antitrypsin (A1AT) therapies. A1AT is a 52 kDa serpin and was first described as a trypsin inhibitor; however, it acts as a more general potent protease inhibitor that can also limit elastase activity (Stockley, 2015).
3. Elastin in Bruch’s membrane (BrM) and other ocular tissues
In the eye, elastin is widely distributed and has been detected in the cornea, conjunctiva, muscle tendons, sclera, choroid, BrM, meninges (Gelman et al., 2010), lamina cribrosa (Hernandez et al., 2000) and retina (Chen and Weiland, 2014). In the anterior segment of the eye, elastin is critical for maintaining the shape of the cornea (Feneck et al., 2018; White et al., 2017a) and for controlling the outflow resistance in the trabecular meshwork (Umihira et al., 1994). Posteriorly, elastic fibers of the sclera protect the globe from mechanical stresses like elevated intraocular pressure (Gelman et al., 2010) and those present at the BrM appear to work as a structural barrier preventing choroidal vessels from growing into the outer retina (Chong et al., 2005). In human BrM, elastic fibers are stacked together with an interfibrillar space of ~1 μm to form a perforated elastin sheet or elastin layer (EL). This elastin sheet spans from the optic nerve to the pars plana of the ciliary body. In addition to elastin, the BrM EL also contains other ECM proteins such as collagen type VI and fibronectin. Human BrM thickness is reported as 2–4 μm, the EL making up the central 0.8 μm; similarly, in murine models, a total BrM thickness of 0.22–0.64 μm has been reported (Annamalai et al., 2020; Volland et al., 2015), with apparently a larger component of the overall thickness being contributed by the EL (0.13–0.17 μm) (Volland et al., 2015).
Human BrM forms by 6–7 weeks of gestation, with the BrM EL forming and becoming histologically visible by 11–12 weeks. Embryologically, RPE-BL and ICL are derived from ectoderm, OCL and CC-BL from mesoderm, with the EL being deposited by invading fibroblasts and endothelial cells of the CC (Curcio and Johnson, 2013). Consistently, tropoelastin mRNA levels are significantly higher in the embryonic and early postnatal days in murine eyes involving two phases of EL development, BrM elastin layer formation followed by maturation (Mori et al., 2019).
RPE has a crucial role in synthesizing several ECM proteins that make up BrM, including collagen I, III, IV, V, fibronectin, and laminin (Newsome et al., 1988). Although the RPE’s role in BrM elastin synthesis and degradation is not clearly stated, some studies performed in cultured RPE cells give us some clues. Cultured human RPE cells (ARPE 19 cells) express fibrillin 1 and LOX, suggesting that they can facilitate elastic fiber assembly (Wachi et al., 2005; Yeo et al., 2012), although the elastin gene expression is minimal (Sharma et al., 2005). Also, tropoelastin mRNA and protein expression has been reported in heat exposed ARPE19 cells, suggest their contribution in elastogenesis under stress (Sekiyama et al., 2012), and single cell RNA-seq profiling data demonstrated elastin expressing in freshly isolated primary human RPE cells (Dwight Stambolian, personal communication, 3-11-2022). Additionally, a single-cell RNA sequencing analysis performed on infant and aged RPE/choroid samples has reported a ~10-fold higher elastin (ELN, chromosome 7) gene expression in infant pericytes compared to the aged samples, supportive of the pericyte’s contribution to elastin production and reemphasizing the early occurrence of elastogenesis in the eye during development (Voigt et al., 2020).
Elastin makes up ~4% of the dry retinal tissue weight and has been detected at the basement membrane of retinal arterial walls (Chen and Weiland, 2014; Chong et al., 2005; Sagaties and Raviola, 1989). It has been suggested that smooth muscle cells and endothelial cells of retinal vessel walls might be involved in retinal elastin synthesis as they stain positive for elastin (Chen and Weiland, 2014). Another major retinal cell type that expresses elastin are the astrocytes present at the lamina cribrosa, which express elastin during development as well as in glaucoma (Hernandez et al., 2000; Pena et al., 2001).
4. Elastin turnover in ocular diseases
Abnormal elastin turnover/elastic fiber degradation/elastase activity has been associated with several ocular diseases (Table 1). Elastic fibers distributed throughout the sclera and the cornea play critical role in maintaining the structural integrity of the eye globe. Scleral elastin is most dense in the peripapillary region or near the optic nerve head and is predicted to have role in maintaining the intraocular pressure (Gelman et al., 2010). In primary open-angle glaucoma (POAG) (Hernandez, 1992), elastin loss and fragmentation are reported at the lamina cribrosa of the optic nerve head, a tissue required to maintain the ocular pressure gradient. Additionally, reduced LOX1, elastin protein expression and structural alterations of the elastic fiber network are reported in the lamina cribrosa of pseudoexfoliation glaucomatous eyes, a type of POAG which is caused by the abnormal deposition of proteins in the drainage system or other parts of the eye (Schlötzer-Schrehardt et al., 2012).
Table 1.
Role of elastin turnover in ocular diseases. Abbreviations: MMP, Matrix Metalloproteinase; LOX, Lysyl oxidase; BrM EL, Bruch’s membrane elastin layer; CNV, choroidal neovascularization; TIMP, tissue inhibitors of metalloproteinases; EDP, elastin derived peptides; HTRA1, High-Temperature Requirement A Serine Peptidase 1; LoxL1, Lysyl oxidase like 1; POAG, primary open angle glaucoma. The arrows indicate the direction of change.
| Disease | Pathological implications | References |
|---|---|---|
| Floppy eyelid syndrome | ↓ tarsal elastin level, ↑ elastin degradation, ↑ MMP 7 and 9 levels | (Netland et al., 1994; Schlötzer-Schrehardt et al., 2005) |
| Keratoconus | ↓ LOX distribution/activity in the cornea, ↓ elastic fiber system in the cornea | (Bykhovskaya et al., 2012; Dudakova and Jirsova, 2013; Dudakova et al., 2012; White et al., 2017b) |
| Ocular components of Marfan syndrome | ↑ectopia lentis, ↑glaucoma, ↓ retinal/choroidal vascular health | (Di Marino et al., 2020; Nahum and Spierer, 2008; Zadeh et al., 2011) |
| Ocular components of Pseudoxanthoma elasticum | ↑ Calcification and fragmentation of BrM EL, ↑ risk of CNV and macular atrophy | (Charbel Issa et al., 2009; Risseeuw et al., 2020b) |
| Sorsby’s fundus dystrophy | ↑ Subretinal deposits of mutated TIMP-3, ↑ BrM thickening and fragmentation of BrM EL, ↑CNV | (Anand-Apte et al., 2019; Chong et al., 2000) |
| Rhegmatogenous retinal detachment | ↓ LOX activity and ↑ MMP 2 and 9 levels in the vitreous | (Coral et al., 2008) |
| Diabetic retinopathy | ↑Neutrophil elastase levels and ↑vascular permeability, ↓ LOX activity and ↑ MMP 2 and 9 in the vitreous, ↑ serum levels of anti-elastin IgA antibodies | (Coral et al., 2008; Liu et al., 2019; Nicoloff et al., 2000) |
| AMD | ↓ integrity of macular BrM EL, ↑ thinning of macular BrM EL, ↑ serum levels of EDPs, ↑ serum levels of elastin antibodies, ↑Elastin and HTRA1 gene polymorphisms in polypoidal choroidal vasculopathy (PCV) and AMD | (Chong et al., 2005; Dewan et al., 2006; He et al., 2021; Kondo et al., 2008; Morohoshi et al., 2012; Sivaprasad et al., 2005; Tong et al., 2010; Yamashiro et al., 2011; Yanagisawa et al., 2015) |
| Glaucoma (POAG) | ↓LoxL1 and ↑elastin fragmentation/aggregate depositions at the lamina cribrosa, ↑Thickening of the elastic fiber sheath in trabecular meshwork, ↑elastin deposition in the Schlemm’s canal endothelial cells | (Pena et al., 1998; Quigley et al., 1994; Schlötzer-Schrehardt et al., 2012; Tektas and Lütjen-Drecoll, 2009; Umihira, 1993; Umihira et al., 1994) |
In POAG abnormal elastin turnover has also been reported at the trabecular meshwork (TM) and Schlemm’s canal endothelial cells, tissues which are critical for maintaining the aqueous outflow resistance. In particular, the elastin fiber sheath in the TM has been proposed to play a significant role in managing outflow resistance (Segawa, 1995), as thickening of the sheath has been observed in the aged TM. Age-related TM elastic fiber thickening may increase the risk of POAG in aging population (Tektas and Lütjen-Drecoll, 2009). Moreover, increased elastin expression has been reported in Schlemm’s canal endothelial cells of POAG patients compared to the age matched healthy eyes (Umihira, 1993; Umihira et al., 1994). Collectively, these data suggest that abnormal elastin turnover and its deposition at the TM/Schlemm’s canal complex could contribute in POAG pathogenesis by increasing the aqueous outflow resistance.
Upregulated elastolytic enzymes (MMP7 and MMP9), elastic fiber damage and reduced elastin levels are evident in floppy eyelid syndrome, an eye disease characterized by the diminished mechanical properties of the upper eyelid (Netland et al., 1994; Schlötzer-Schrehardt et al., 2005).
Corneal thinning (White et al., 2017a), ectopia lentis/dislocation of the lens (Zadeh et al., 2011), retinal detachment (Dietz, 1993), and impairment of retinal and choroidal vasculature (Di Marino et al., 2020) are reported in patients with Marfan syndrome, a hereditary connective tissue disorder caused by the mutation of fibrillin-1, a key microfibril protein essential for elastic fiber assembly.
Studies indicate that abnormal elastin turnover is a key player in keratoconus pathogenesis, a corneal disorder that is associated with progressive corneal thinning, ultimately leading to astigmatism and vision loss. Abnormal microfibril bundle distribution and the absence of the elastic fibril system reported in the posterior stroma of keratoconus corneas (White et al., 2017b) might be associated with the decreased distribution of LOX protein, and LOX gene polymorphisms (Dudakova and Jirsova, 2013; Dudakova et al., 2012; Hasanian-Langroudi et al., 2015). Reduced corneal transcript levels of LOX, together with reduced LOX activity in keratoconus patient tears have been associated with disease severity, as were reduced collagen and increased MMP9 transcript levels (Shetty et al., 2015).
Age-related visual impairment, increased BrM EL calcification, foveal CNV, macular atrophy and outer retinal atrophy and RPE atrophy (Gliem et al., 2016) are reported in pseudoxanthoma elasticum (PXE) pathogenesis (Jensen, 1977; Risseeuw et al., 2019, 2020a), a rare multi-system genetic disorder that primarily accompanies with increased calcification and fragmentation of elastic fibers (Roach and Islam, 2015). Cardiovascular, dermatological, and ophthalmological symptoms are common in these patients. The BrM breaks or angioid streaks due to elastin calcification (Charbel Issa et al., 2009) were co-localized with the CNV lesions and macular atrophy in PXE (Risseeuw et al., 2020b), indicating that BrM EL integrity is important for RPE survival and for preventing pathological CNV.
Sorsby’s Fundus Dystrophy (SFD), an ocular disease caused by mutations in the TIMP3 gene, shares many clinical features with AMD. Accumulation of mutated TIMP3 at BrM, and a thickening of BrM and BrM elastin layer abnormalities/abnormal arrangement of elastic fiber components, are reported in SFD pathogenesis along with CNV (Anand-Apte et al., 2019; Chong et al., 2000; Gourier and Chong, 2015). The elastin layer of BrM is irregular, thickened and broken in SFD compared to healthy subjects (Chong et al., 2000), indicating that mutated TIMP3 in SFD is failing to protect the EL or it has abnormal functions. BrM EL damage caused by the TIMP3 mutation may contribute to CNV invasion in SFD (Gourier and Chong, 2015).
Elastase activity and elastin turnover are also critical in diabetic retinopathy. Decreased LOX activity and increased MMP levels (MMP2, MMP9) are reported in the vitreous of proliferative diabetic retinopathy and rhegmatogenous retinal detachment, diseases for which inadequate collagen cross-linking and ECM alternations have been reported (Coral et al., 2008). Diabetes increases neutrophil elastase levels in both the retina and serum. Increased anti-elastin IgA antibodies were found in pediatric diabetic retinopathy patients (Nicoloff et al., 2000). Based on these observations, elastase inhibitors such as A1AT have been suggested as a possible therapeutic strategy for diabetic retinopathy. Interestingly, intraperitoneal administration of A1AT improved retinal thickness and retinal ganglion cell (RGC) loss in an animal model of diabetic retinopathy, likely via attenuating the level of pro-inflammatory cytokines such as TNF-α produced by macrophages (Ortiz et al., 2014, 2018; Potilinski et al., 2020). On the other hand, A1AT deficiency is associated with optic neuropathy (Younger et al., 2006) as well as ocular inflammatory conditions such as conjunctivitis and uveitis (Leonardi et al., 2010). Interestingly, deletion of IL-17 significantly reduced diabetes-induced increase of neutrophil elastase in retina and serum (Liu et al., 2019), indicative of cytokine modulation of elastin turnover in the diabetic retina.
A plethora of studies mentioned above emphasize a strong correlation of abnormal elastin turnover with multiple ocular diseases. One critical question arising here is that whether the elastin loss/disruption/abnormalities are part of the disease pathogenesis or a primary cause. Current evidence suggests a significant role of elastin turnover in the etiology of ocular diseases, as well as that altered biomechanics of elastic fibers and the inflammatory or the proangiogenic role of EDPs (Figs. 1 and 3) generated by the perturbed elastin turnover/increased elastase activity, could contribute to the pathology of ocular diseases.
Fig. 3. Proposed mechanisms of elastin turnover in the pathogenesis of dry and wet AMD.
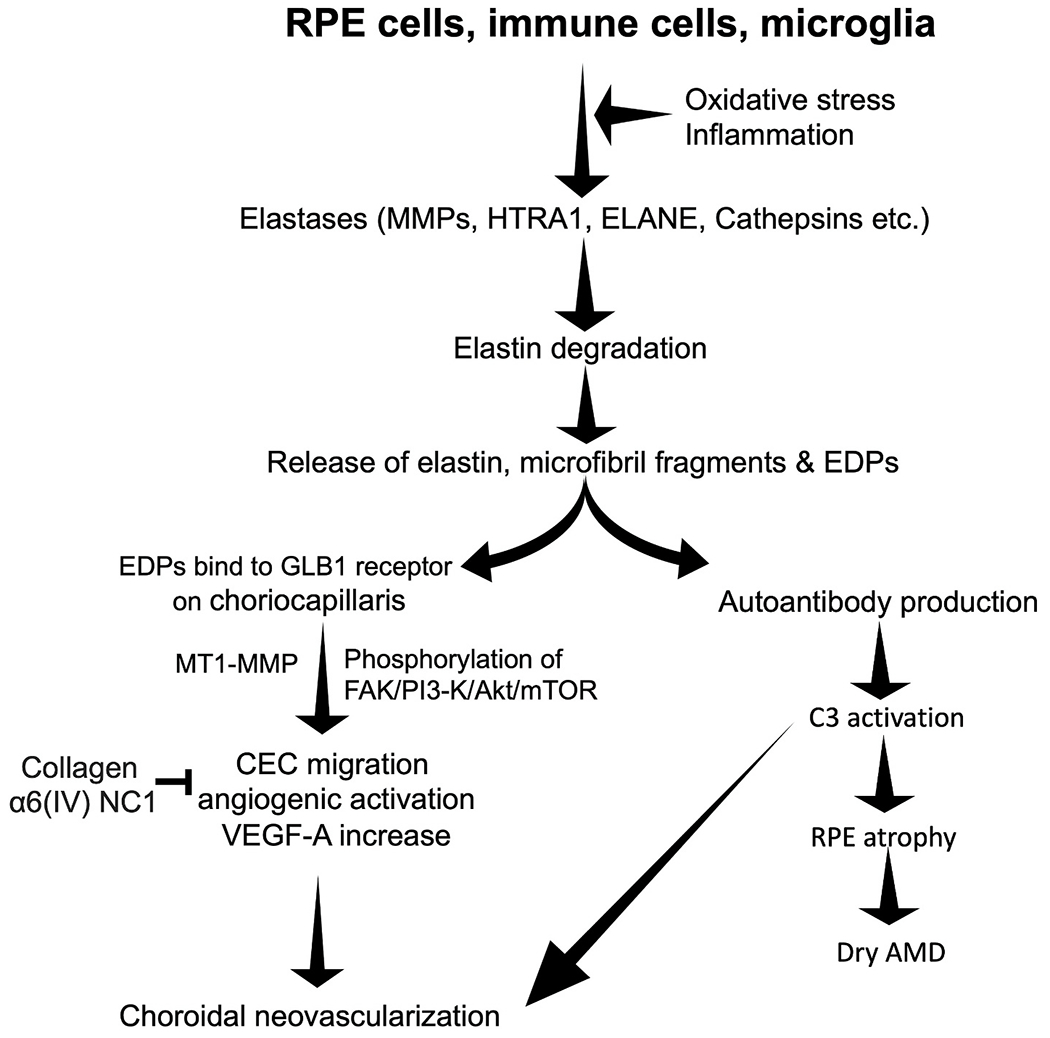
During AMD pathogenesis local inflammation or oxidative stress can upsurge the elastase secretion from RPE cells, infiltrating immune cells such as macrophages/neutrophils and microglia leading to elastin degradation and EDP generation. EDPs can bind to the GLB1 receptors present on the choroidal endothelial cells (Skeie et al., 2012). EDP binding to the endothelial cells can increase the MT1-MMP level (Robinet et al., 2005) and the phosphorylation of FAK/-PI3–K/Akt/mTOR (Gunda et al., 2013) pathway, leading to VEGF upregulation (Sounni et al., 2004), endothelial cell migration and pathological neovascularization. Collagen fragment α6(IV) NC1 can inhibit this elastin peptide mediated endothelial cell migration and angiogenesis (Gunda et al., 2013). Products of elastin degradation can also increase the autoantibody production and complement C3 overactivation (Annamalai et al., 2020), suggesting its possible role in RPE death and dry AMD pathogenesis. Abbreviations: RPE, Retinal Pigment Epithelium; MMP, matrix metalloproteinase; HTRA1, High-Temperature Requirement A Serine Peptidase 1; ELANE, Neutrophil Elastase/leukocyte elastase; EDP, Elastin Derived Peptides; GLB1, Galactosidase beta 1 receptor; MT1 MMP, Membrane Type 1 MMP; Collagen α6(IV) NC1, type IV collagen α-6 chain-derived non-collagenous domain; VEGF-A, Vascular Endothelial Growth Factor A; FAK/-PI3–K/Akt/mTOR, Focal adhesion kinase(FAK)/-phosphoinositide 3-kinase/protein kinase B (PI3K/AKT)/mammalian target of rapamycin (mTOR).
5. Elastin turnover and elastase activity in AMD
Thickening of BrM, and the relative decrease of elastin and increased calcification of EL are characteristics of aging BrM along with lipid buildup (Curcio, 2018; Curcio et al., 2011), concomitant with the development of basal laminar deposits (BLamD; localized between the RPE basement membrane and its plasma membrane) (Sarks, 1976) and basal linear deposits (BLinD; localized between the RPE basement membrane and the inner collagenous zone) (Sarks et al., 1980). Reduced elasticity (Booij et al., 2010) and macromolecular diffusion characteristics of the aging BrM caused by these structural alterations (Hussain et al., 2010) can contribute to RPE dysfunction and make the retina susceptible to CNV (Chong et al., 2005) (Fig. 2). A pathological increase of MMPs with elastase activity can exacerbate the age-related elastic fiber degradation and calcification (Basalyga et al., 2004) leading to increased brittleness of BrM and AMD pathology. Evidence indicates that abnormal elastin turnover has a significant role in the pathogenesis of wet AMD and pathological angiogenesis, but its role in dry AMD cannot be excluded. In disease, RPE cells (Elner et al., 2003), infiltrating immune cells such as neutrophils and macrophages (Beguier et al., 2020; Ghosh et al., 2019; Liu et al., 2019; Nakajima et al., 1992), as well as microglia (Ma et al., 2012; Nakajima et al., 1992) can upregulate their elastase secretion in the outer retina. This can exacerbate the age-related elastin degradation, and may contribute to both wet and dry AMD pathology (Fig. 3).
Fig. 2. Macular Bruch’s membrane elastin layer degradation in AMD.
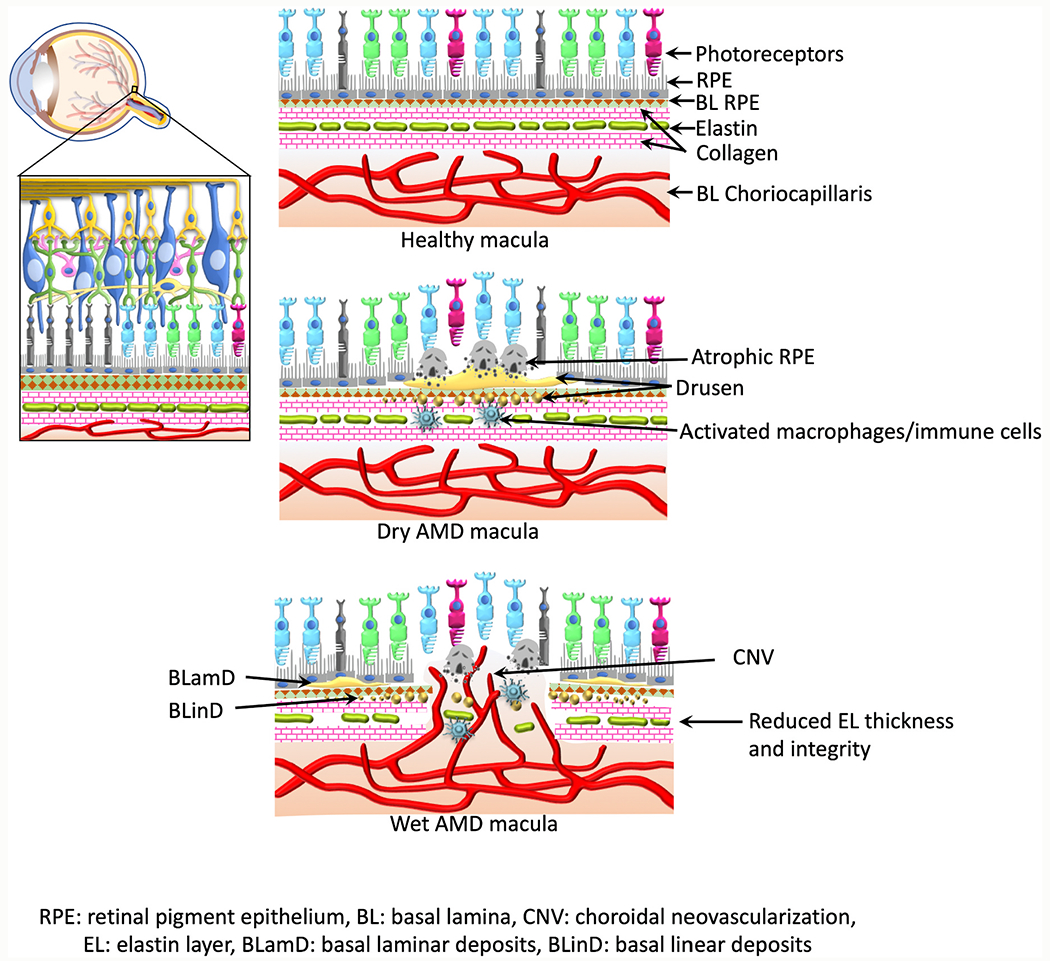
Figure indicates schematic representation of eyeball, retina, and macula during health and AMD. Reduced macular elastin layer integrity and thickness have been reported in AMD patients compared to the age matched healthy subjects; BrM elastin layer thinning, calcification, and porosity were more evident and were corresponding to the distribution of CNV lesions in wet AMD macula.
5.1. Elastin turnover in dry AMD
In the normal healthy retina, the mean macular EL thickness at the BrM is approximately 1/3 of the thickness of the mean extramacular EL thickness (Chong et al., 2005). The integrity of the EL, which is defined as the total length of visually detectable elastin divided by the overall length of visible BrM, is almost 100% in the extramacular region but drops to ~40% in the macula itself. In early AMD, the integrity of BrM EL is significantly reduced compared to age-matched healthy controls, whereas in late-stage AMD patients, including donors with choroidal neovascular membranes, both macular BrM EL integrity and thickness are significantly reduced compared to the controls, with a strong relationship between these two parameters in all regions measured. Surprisingly, this pathology-related decrease in macular elastin at the BrM was not evident in AMD patients with GA, a phenomenon that is currently not understood (Chong et al., 2005). Nevertheless, significantly reduced elastin loss and consequent vessel hardening/inelasticity are reported in the retinal vessels of dry AMD (GA) patients compared to the healthy control subjects (Chen and Weiland, 2014). Authors interpret that the retinal vessel damage caused by abnormal elastin turnover could contribute to fluid accumulation and impaired waste removal in the retina, leading to drusen deposition and GA progression.
Together, these findings suggest that loss of mechanical integrity of the EL, as well as the inflammation triggered by production of elastin peptides, might play a role in dry AMD progression. A thinner and more porous EL could lead to impaired RPE attachment to its basement membrane, promoting detachment of RPE cells, accumulation of basal laminar deposits as well as drusen formation (Chong et al., 2005). This hypothesis is supported by the use of elastin-based recombinant polymers that provide a good substrate for RPE cells (Shadforth et al., 2015; Sreekumar et al., 2018; Srivastava et al., 2011), indicating that degradation of the EL in BrM could lead to RPE dysfunction/detachment and could contribute to dry AMD pathogenesis. Evidence from animal studies also support the contribution of abnormal elastin turnover in dry AMD progression. A cigarette smoke induced dry AMD mouse model exhibited thickened BrM, especially outer collagenous layer thickening along with BrM elastin loss (Annamalai et al., 2020). EDPs are reported to increase collagen depositions in the retina (Skeie et al., 2012), suggesting that EL degradation and consequent production of EDPs could be a cause of increased collagen deposition leading to BrM thickening reported in AMD retinas. Also, mutations of fibulin-3 and −5, proteins involved in elastic fiber formation (Nakamura et al., 2002), have been associated with dry AMD characteristics, such as RPE detachment and sub-RPE drusen formation (Stone et al., 2004). Honeycomb retinal dystrophy, a retinal disease exhibiting many of the characteristics of dry AMD such as BrM thickening, sub-RPE depositions, and choroidal/choriocapillaris atrophy, along with photoreceptor cell disruption in the subfoveal space, is caused by a single missense mutation in fibulin-3 (Fu et al., 2007; Gerth et al., 2009). Fibulin-3 interacts with several ECM proteins including tropoelastin and collagen, and it stimulates the expression of TIMP 1 and 3, and inhibits the activities of MMP 2, 3 and 9 as well as angiogenesis (Zhang and Marmorstein, 2010). Whether fibulin-3 mutations are merely associated with drusen formation or whether its elastase inhibitory effect also contributes to dry AMD pathogenesis is not yet clear.
5.2. Role of elastin turnover and elastases in RPE cells
At the posterior eye, the RPE could play a critical role in elastin remodeling. The RPE cells secrete many of the ECM proteins needed for the generation of RPE basal lamina and regulate its remodeling with protease enzymes and their inhibitors (TIMPs 1, 2) (Alexander et al., 1990). RPE cells secrete low levels of several enzymes that have collagenase (MMP1), gelatinase (MMP2 and MMP9), and elastase activities (Guo et al., 1999; Hollborn et al., 2007). Cultured RPE cells secrete metalloproteinases and tissue inhibitors of metalloproteinases (TIMPs) into the media (Alexander et al., 1990), and in polarized monolayers, specifically towards the apical side (Greene et al., 2017; Kay et al., 2013).
Studies have shown that oxidative stress and cytokine exposure can modulate the elastase secretion from the RPE. Oxidative stress upregulates MMPs 1 and 3 levels in RPE cells (Alge-Priglinger et al., 2009) without altering the expression of TIMP-1. Similarly, TNF-α, IL-1β, and TGF-β2 exposures also increase the expression of MMP1, MMP2, and MMP3 and increase elastase activity (Elner et al., 2003) in cultured human RPE cells (Eichler et al., 2002). Elastases secreted by the RPE are not only involved in the elastic fiber breakdown, but can also contribute to retinal angiogenesis. Notably, increased MMP9 activity in cultured RPE cells induced by complement activation (H2O2 plus normal human serum) is associated with increased mobilization of VEGF and decreased mobilization of PEDF from the RPE ECM (Bandyopadhyay and Rohrer, 2012).
5.3. Role of elastin turnover in angiogenesis and wet AMD
Interestingly, Blumenkranz and colleagues have observed a correlation between wet AMD and an increased susceptibility of elastic fibers in general to photic or other degenerative stimuli (Blumenkranz et al., 1986). Several studies later supported this observation by reporting significant associations between elastin gene (ELN) polymorphisms and AMD pathogenesis, particularly in wet AMD (Kondo et al., 2008; Tanaka et al., 2011; Yamashiro et al., 2011; Yanagisawa et al., 2015). Additionally, the association of a polymorphism in HTRA1, an enzyme with elastase-like activity with all forms of AMD (Beguier et al., 2020; Cameron et al., 2007; Grob et al., 2012; Martínez-Velasco et al., 2020; Mohamad et al., 2019; Sundaresan et al., 2012; Tang et al., 2009; Tuo et al., 2008), and an HTRA1 promoter polymorphism association with wet AMD (Dewan et al., 2006), further supports the importance of elastin metabolism in wet AMD. It is interesting to investigate why elastin metabolism abnormalities are more prominent in wet AMD cases compared to dry AMD.
RPE-specific overexpression of HTRA1 in mouse RPE provides some mechanistic clues. Overexpression of human HTRA1 was associated with degradation of BrM elastin (Jones et al., 2011; Vierkotten et al., 2011), in combination with smoke exposure resulted in CNV (Nakayama et al., 2014), triggered PCV, a variant of wet AMD (Jones et al., 2011), and promoted inflammation and macrophage infiltration essential for ocular VEGF expression (Lu et al., 2019). In this mouse model, the authors observed the development of PCV in aged mice, and documented that elastase activity and proteolytic degradation triggered the initiation of CNV, whereas PCV progression was driven by the elevated immune response (Kumar et al., 2017). This data suggests multiple concepts about why abnormal elastin metabolism might be more important in wet AMD. First, weakening of the BrM EL by increased elastase activity together with increased elastin degradation evident in normal aging, can weaken the macular BrM, enabling CNV. Or, second, elastases and EDPs exhibit a varying range of pro-inflammatory and pro-angiogenic roles (Antonicelli et al., 2007) that could trigger CNV or PCV.
HTRA1 is a multifunctional serine protease produced by the RPE and secreted basally towards the choroid (Jones et al., 2011). In addition to its ability to degrade the elastic lamina of BrM and choroidal blood vessels (Jones et al., 2011), it can also cleave EFEMP1, an extracellular matrix protein associated with Doyne honeycomb retinal dystrophy, and thrombospondin 1 (TSP1), an inhibitor of angiogenesis (Lin et al., 2018). Surprisingly, HTRA1 protein levels have been found to increase with age in the RPE-BrM interface (Williams et al., 2021), but only in eyes from donors without the SNP in Chr10 (nonrisk). In donors with homozygous risk at the 10q26 locus, this age-related increase fails to occur, suggesting that loss of function is associated with AMD risk. These results are contrary to those obtained in mouse models, in which expression of human HTRA1 in RPE is associated with pathology (Kumar et al., 2014; Nakayama et al., 2014; Vierkotten et al., 2011). Likewise, the stated goals of the current phase II clinical trial by Genentech, which aims to inhibit HTRA1 in GA using blocking antibodies (NCT03972709), imply that ain of function is associated with AMD risk.
The study performed by Chong and colleagues (Chong et al., 2005) shows that areas that lack structural integrity of BrM EL corresponded to the sites of macular lesions in AMD patients. Hence a disruption at the level of BrM, may cause a breach of the barrier, triggering or enabling choroidal endothelial cells to migrate and form CNV. Experimental laser photocoagulation models confirm this hypothesis that disruption of BrM can induce CNV. The data by Schwesinger and colleagues confirm the notion that angiogenesis alone is not sufficient to induce CNV; their study showed that VEGF-induced angiogenesis leads to CNV only in the presence of a disrupted BrM (Schwesinger et al., 2001). Additionally, mice lacking LOXL-1 an enzyme essential for elastin polymerization have fragmented BrM EL and are more susceptible to CNV lesions following laser induction compared to the wild type with an intact EL (Yu et al., 2008).
Consistent with significant BrM elastin loss in the CNV eye, significantly higher serum levels of EDPs (Sivaprasad et al., 2005) and elastin antibodies (Morohoshi et al., 2012) are reported in wet AMD patients compared to the non-disease controls and dry AMD subjects. It has been shown that EDPs generated from vascular elastin degradation by proteases, attract T cells and macrophages that can contribute to vascular smooth muscle cell differentiation, proliferation and migration, overall contributing to vascular occlusive diseases (Brooke et al., 2003; Karnik et al., 2003). Similarly, EDPs generated from the degradation of BrM EL or retinal vessels could increase inflammation and angiogenesis in the AMD eye. Direct effects of EDPs have also been shown in endothelial cells, as EDPs increase cell migration, tubulogenesis, and proangiogenic MT1-MMP expression in choroidal endothelial cells (Gunda et al., 2013; Skeie and Mullins, 2008). These EDP-mediated angiogenic responses in choroidal endothelial cells could be prevented by the exposure of collagen fragments (Gunda et al., 2013), suggesting the opposite roles of elastin and collagen fragments in retinal angiogenesis. Moreover, it has been shown that circulating EDPs can bind to the beta-galactosidase (GLB-1) receptor present on choroidal endothelial cells and contribute to increased deposition of collagen IV in the RPE/choroid (Skeie et al., 2012), which ultimately can lead to BrM thickening.
Additionally, mutations of genes essential for elastic fiber formation are also associated with CNV (Lamande and Bateman, 2020). Mutations in EGF-containing fibrillin-like ECM protein 1 (EFEMP1) lead to Doyne Honeycomb Retinal Dystrophy, a disease in which drusen formation can lead to CNV (Terai et al., 2011); mutations in FBLN5, which leads to Cutis laxa, a disease characterized by loose skin, is associated with cuticular drusen, retinal detachment and CNV (Lotery et al., 2006). The deficiency of TIMP-3, an inhibitor of several MMPs with elastase-like activity has been reported in wet AMD patients (Krogh Nielsen et al., 2019) and RPE-specific overexpression of TIMP3 interestingly has been shown to ameliorate CNV in experimental models of AMD (Takahashi et al., 2000). Finally, anti HTRA1 antibody has been shown to reduce the lesion size in the photocoagulation murine models of CNV (Lu et al., 2019). Taken together, modulating elastin turnover via the elastase inhibitors appears to be a potential target in wet AMD.
A critical question already mentioned above is whether abnormal elastin turnover associated with AMD is an age-related phenomenon or a causative factor. Although aging can contribute to elastin turnover abnormalities, BrM elastin layer degradation or elastin turnover abnormalities reported in AMD (summarized in this review) are not just age-related occurrences. Data indicate more specific and prominent BrM changes such as reduced EL integrity and thinning in CNV or disciform scar formation in AMD patients compared to age matched controls (Chong et al., 2005). Correspondingly, serum EDPs (Sivaprasad et al., 2005) and elastin antibody levels (Morohoshi et al., 2012) were also found to be significantly higher in wet AMD patients compared to the age matched controls. Moreover, increased lung elastin degradation in smokers has been linked to AMD progression; also the administration of smoke induced oxidized elastin peptides has exacerbated the AMD phenotype in older mice (Annamalai et al., 2020), further reaffirming the association of elastin turnover abnormalities to AMD. Aging-associated elastin turnover could be a contributing factor in AMD; however, other factors such as increased elastase secretion caused by pathological oxidative stress/inflammation might be involved in worsening the phenotypes that lead to AMD progression.
Another interesting question is why people suffering from other ocular diseases with reduced elastin fibers do not also develop AMD. One possible reason could be the tissue-specific degradation of elastic fibers in different ocular disease. For example, in the case of AMD, elastic fiber degradation at BrM appears to play a critical role in CNV progression, whereas in keratoconus elastin loss occurs at the cornea (Table 1). Local inflammation or oxidative stress in the outer retina during AMD can increase elastase secretion from RPE cells and infiltrating immune cells, contributing to BrM EL degradation/abnormal elastin turnover. CNV development corresponding to the BrM/EL damage reported in Sorsby’s fundus dystrophy and Pseudoxanthoma elasticum PXE (Table 1), supports this observation in AMD. Reduced biomechanical properties of the BrM elastin layer and the inflammatory role of generated EDPs may contribute to AMD pathology, causing RPE atrophy and CNV, as postulated in Sorsby’s and PXE.
5.4. Targeting the EDP-anti-elastin antibody-complement axis in a mouse model
Elevated serum levels of EDPs (Sivaprasad et al., 2005) and elastin IgG and IgM antibodies (Morohoshi et al., 2012), reported in early and neovascular AMD, led us to suggest that abnormalities in elastin homeostasis and the corresponding antibody production may play a role in AMD. Specifically, IgGs and/or IgMs that bind to ligands on cell surfaces, basement membranes, or extracellular matrices can participate in tissue damage involving either the engagement of the complement system (complement-dependent cytotoxicity; CDC) or Fcγ-receptor (FcγR) dependent cell-mediated cytotoxicity (antibody-dependent cell-mediated cytotoxicity; ADCC) (Saeed et al., 2017). Both of these mechanisms have been shown to participate in chronic, slowly progressing diseases (Natoli et al., 2018; Wang and Ravetch, 2015). We have previously shown that long-term smoke exposure in C57BL/6J mice leads to features of human dry AMD, including a loss in retinal function, thickening of BrM, loss of the elastin layer integrity as well as lipid deposition in the area of BrM, and these changes are dependent on the activity of the alternative pathway of complement (Kunchithapautham et al., 2014; Woodell et al., 2013, 2016). Hence, we speculated that immunizing C57BL/6J mice with elastin peptide oxidatively modified by cigarette smoke (ox-elastin) would augment pathology in smoke-exposed mice (Annamalai et al., 2020). As expected, immunization of mice with ox-elastin resulted in a large increase in anti-ox-elastin IgG and IgM antibodies, whereas, elastin immunization had a smaller effect. Importantly, and in agreement with our hypothesis, ox-elastin immunization led to exacerbated smoke-induced vision loss and thicker BrM and increased levels of IgM, IgG2b, IgG3, and complement activation products in the RPE/choroid when compared to controls. Based on the specificity of the antibodies produced, pathology could be driven by a mixed effect, with IgM and IgG3 antibodies activating complement leading to CDC, whereas IgG2b could engage FcγR on effector cells eliciting ADCC. CDC and ADCC involvement have been confirmed in mice in which the alternative pathway of complement (factor B, fB−/−) or the Fcγ receptor (FcγR−/−) were eliminated (Rohrer et al., 2021; Woodell et al., 2013). Despite not being conclusive as to the relative contributions of the two pathways being engaged by the anti-elastin antibodies, these results support the growing body of evidence linking oxidative stress, smoking, complement activation, and autoimmunity to AMD pathogenesis.
Peptide immunotherapy has been studied extensively for the treatment of various autoimmune diseases, allergies as well as cancers (Larche, 2014; Romano et al., 2019; Shakya and Nandakumar, 2018; Smith and Peakman, 2018). While the exact mechanisms of immune modulation and suppression of disease by peptide immunotherapy has not yet been fully defined (Sabatos-Peyton et al., 2010), peripheral tolerance towards certain antigens in laboratory animals is generated by repeated exposure to the antigen, and is thought to involve deletion of reactive T cells, the induction T cell activation and/or generation of a regulatory T (Treg) cell response, or an altered response of macrophages (Butcher et al., 2018). If the generation of EDPs and their corresponding antibodies were to be causally related to pathology in this model, one could argue that attenuation of disease with peptide immunotherapy should be possible. We tested this hypothesis in smoke-exposed mice injected with oxidized mouse elastin peptide over the course of smoke exposure (Annamalai et al., 2020). Importantly, animals treated with the low dose of oxidized elastin peptide exhibited a reduced humoral immune response, producing lower levels of ox-elastin-specific IgG and IgM antibodies when compared to control smoke-exposed mice. This reduced level of serum anti-ox-elastin antibodies was associated with reduced levels of IgG/IgM deposition in RPE/choroid, and blunted complement activation. Structurally and functionally, low dose peptide immunotherapy resulted in preservation of BrM and improved contrast sensitivity compared to the non-tolerized mice. Finally, cytokine levels in the RPE/choroid fraction in response to treatment is suggestive of an induction of tolerance. Activation of the immune system by non-self cells or proteins (i.e., peptides leading to an immune response) involves a Th1 response driven by IFNγ; where as exposure to self cells or proteins, triggers tolerance, invloving a Th2 response activated by IL-4. Interestingly, peptide immunotherapy decreased IFN and increased IL-4. Our observations support the hypothesis by Nussenblatt and colleagues, that AMD might be suitable for tolerance therapy, which would re-align the adaptive immune response by suppressing T cell responses (Nussenblatt et al., 2014). Unfortunately, the smoke model in C57BL/6J mice does not lead to breakthrough choroidal neovascularization, such that the concept of tolerance for the prevention of CNV could not be tested.
6. Concluding remarks
Elastin and enzymes regulating its turnover play crucial roles in the pathogenesis of many ocular diseases including AMD, and especially neovascular AMD. BrM elastin turnover is critical in the pathogenesis of AMD. Degradation of, in particular, macular BrM elastin and thus reducing macular BrM integrity and its increasing porosity can aid the migration of choroidal endothelial cells into the subretinal space causing neovascularization. While the origin of the elastases breaking down the elastin layer is unclear, RPE and infiltrating immune cells are known to secrete proteases such as elastase, metalloproteinases, gelatinases, and their inhibitors (TIMPs 1, 2), and elastase activity was found to be increased with aging, contributing to the age-related degradation of BrM. Additionally, elastin fragments or EDPs generated by the elastin degeneration can have several cell-signaling roles; this includes EDP-mediated cell migration, differentiation, proliferation, chemotaxis, and angiogenesis. In endothelial cells in particular, EDPs induces angiogenesis via upregulating MMPs (MT1-MMP). Finally, we have proposed a mechanism whereby which EDPs act as ligands in the aged BrM, leading to the binding of anti-elastin antibodies, triggering complement activation. Elastin degradation, serum EDP levels, and anti-elastin antibody levels were increased in AMD patients compared to age matched healthy subjects, suggesting that abnormal elastin turnover reported in AMD is not just an age-related affect, but rather it can directly contribute to the pathology. It would be of great interest to further investigate the key roles played by proteases, oxidative stress, and inflammation in AMD pathogenesis. Insights gained from other conditions such as COPD suggest that elastase inhibitors should be explored as a new therapeutic approach. Altogether, the current evidence suggests that inhibition of elastin degradation could be a promising therapeutic strategy in neovascular AMD by preventing pathological angiogenesis and complement activation.
Acknowledgements
Funding for this project was provided in part by the National Institutes of Health (NIH) R01EY030072, the Department of Veterans Affairs IK6 BX004858, RX000444 and BX003050 and the South Carolina SmartState Endowment. We would like to acknowledge the help from Navneet Ammal Kaidery PhD, Department of Pediatrics for creating the figures for this manuscript and Kyrie Wilson, Department of Ophthalmology for editorial comments.
Data availability
No data was used for the research described in the article.
References
- Ahir A, Guo L, Hussain AA, Marshall J, 2002. Expression of metalloproteinases from human retinal pigment epithelial cells and their effects on the hydraulic conductivity of Bruch’s membrane. Invest. Ophthalmol. Vis. Sci 43, 458–465. [PubMed] [Google Scholar]
- Ahmad S, Saleem M, Riaz N, Lee YS, Diri R, Noor A, Almasri D, Bagalagel A, Elsebai MF, 2020. The natural polypeptides as significant elastase inhibitors. Front. Pharmacol 11, 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JP, Bradley JM, Gabourel JD, Acott TS, 1990. Expression of matrix metalloproteinases and inhibitor by human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci 31, 2520–2528. [PubMed] [Google Scholar]
- Alge-Priglinger CS, Kreutzer T, Obholzer K, Wolf A, Mempel M, Kernt M, Kampik A, Priglinger SG, 2009. Oxidative stress-mediated induction of MMP-1 and MMP-3 in human RPE cells. Invest. Ophthalmol. Vis. Sci 50, 5495–5503. [DOI] [PubMed] [Google Scholar]
- Ambati J, Fowler BJ, 2012. Mechanisms of age-related macular degeneration. Neuron 75, 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand-Apte B, Chao JR, Singh R, Stöhr H, 2019. Sorsby fundus dystrophy: insights from the past and looking to the future. J. Neurosci. Res 97, 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annamalai B, Nicholson C, Parsons N, Stephenson S, Atkinson C, Jones B, Rohrer B, 2020. Immunization against oxidized elastin exacerbates structural and functional damage in mouse model of smoke-induced ocular injury. Invest. Ophthalmol. Vis. Sci 61, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonicelli F, Bellon G, Debelle L, Hornebeck W, 2007. Elastin-elastases and inflamm-aging. Curr. Top. Dev. Biol 79, 99–155. [DOI] [PubMed] [Google Scholar]
- Banda MJ, Werb Z, 1981. Mouse macrophage elastase. Purification and characterization as a metalloproteinase. Biochem. J 193, 589–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay M, Rohrer B, 2012. Matrix metalloproteinase activity creates proangiogenic environment in primary human retinal pigment epithelial cells exposed to complement. Invest. Ophthalmol. Vis. Sci 53, 1953–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basalyga DM, Simionescu DT, Xiong W, Baxter BT, Starcher BC, Vyavahare NR, 2004. Elastin degradation and calcification in an abdominal aorta injury model: role of matrix metalloproteinases. Circulation 110, 3480–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguier F, Housset M, Roubeix C, Augustin S, Zagar Y, Nous C, Mathis T, Eandi C, Benchaboune M, Drame-Maigne A, Carpentier W, Chardonnet S, Touhami S, Blot G, Conart JB, Charles-Messance H, Potey A, Girmens JF, Paques M, Blond F, Leveillard T, Koertvely E, Roger JE, Sahel JA, Sapieha P, Delarasse C, Guillonneau X, Sennlaub F, 2020. The 10q26 risk haplotype of age-related macular degeneration aggravates subretinal inflammation by impairing monocyte elimination. Immunity 53, 429–441 e428. [DOI] [PubMed] [Google Scholar]
- Bhutto I, Lutty G, 2012. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Aspect. Med 33, 295–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieth JG, 1986. Elastases: catalytic and biological properties. In: Mecham, R.P. (Ed.), Regulation of matrix accumulation. Academic Press, Inc, Orlando, FL, pp. 217–320. [Google Scholar]
- Binder S, 2012. Loss of reactivity in intravitreal anti-VEGF therapy: tachyphylaxis or tolerance? Br. J. Ophthalmol 96, 1–2. [DOI] [PubMed] [Google Scholar]
- Bloch SB, Larsen M, Munch IC, 2012. Incidence of legal blindness from age-related macular degeneration in Denmark: year 2000 to 2010. Am. J. Ophthalmol 153, 209–213 e202. [DOI] [PubMed] [Google Scholar]
- Blumenkranz MS, Russell SR, Robey MG, Kott-Blumenkranz R, Penneys N, 1986. Risk factors in age-related maculopathy complicated by choroidal neovascularization. Ophthalmology 93, 552–558. [DOI] [PubMed] [Google Scholar]
- Booij JC, Baas DC, Beisekeeva J, Gorgels TG, Bergen AA, 2010. The dynamic nature of Bruch’s membrane. Prog. Retin. Eye Res 29, 1–18. [DOI] [PubMed] [Google Scholar]
- Boudier C, Godeau G, Hornebeck W, Robert L, Bieth JG, 1991. The elastolytic activity of cathepsin G: an ex vivo study with dermal elastin. Am. J. Respir. Cell Mol. Biol 4, 497–503. [DOI] [PubMed] [Google Scholar]
- Brew K, Nagase H, 2010. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim. Biophys. Acta 1803, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke BS, Bayes-Genis A, Li DY, 2003. New insights into elastin and vascular disease. Trends Cardiovasc. Med 13, 176–181. [DOI] [PubMed] [Google Scholar]
- Brown-Augsburger P, Broekelmann T, Rosenbloom J, Mecham RP, 1996. Functional domains on elastin and microfibril-associated glycoprotein involved in elastic fibre assembly. Biochem. J 318 (Pt 1), 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher SK, O’Carroll CE, Wells CA, Carmody RJ, 2018. Toll-like receptors drive specific patterns of tolerance and training on restimulation of macrophages. Front. Immunol 9, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykhovskaya Y, Li X, Epifantseva I, Haritunians T, Siscovick D, Aldave A, Szczotka-Flynn L, Iyengar SK, Taylor KD, Rotter JI, Rabinowitz YS, 2012. Variation in the lysyl oxidase (LOX) gene is associated with keratoconus in family-based and case-control studies. Invest. Ophthalmol. Vis. Sci 53, 4152–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DJ, Yang Z, Gibbs D, Chen H, Kaminoh Y, Jorgensen A, Zeng J, Luo L, Brinton E, Brinton G, Brand JM, Bernstein PS, Zabriskie NA, Tang S, Constantine R, Tong Z, Zhang K, 2007. HTRA1 variant confers similar risks to geographic atrophy and neovascular age-related macular degeneration. Cell Cycle 6, 1122–1125. [DOI] [PubMed] [Google Scholar]
- Charbel Issa P, Finger RP, Holz FG, Scholl HP, 2009. Multimodal imaging including spectral domain OCT and confocal near infrared reflectance for characterization of outer retinal pathology in pseudoxanthoma elasticum. Invest. Ophthalmol. Vis. Sci 50, 5913–5918. [DOI] [PubMed] [Google Scholar]
- Chen K, Weiland JD, 2014. Discovery of retinal elastin and its possible role in age-related macular degeneration. Ann. Biomed. Eng 42, 678–684. [DOI] [PubMed] [Google Scholar]
- Chong NH, Alexander RA, Gin T, Bird AC, Luthert PJ, 2000. TIMP-3, collagen, and elastin immunohistochemistry and histopathology of Sorsby’s fundus dystrophy. Invest. Ophthalmol. Vis. Sci 41, 898–902. [PubMed] [Google Scholar]
- Chong NH, Keonin J, Luthert PJ, Frennesson CI, Weingeist DM, Wolf RL, Mullins RF, Hageman GS, 2005. Decreased thickness and integrity of the macular elastic layer of Bruch’s membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am. J. Pathol 166, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocciolone AJ, Hawes JZ, Staiculescu MC, Johnson EO, Murshed M, Wagenseil JE, 2018. Elastin, arterial mechanics, and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol 315, H189–h205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coral K, Angayarkanni N, Madhavan J, Bharathselvi M, Ramakrishnan S, Nandi K, Rishi P, Kasinathan N, Krishnakumar S, 2008. Lysyl oxidase activity in the ocular tissues and the role of LOX in proliferative diabetic retinopathy and rhegmatogenous retinal detachment. Invest. Ophthalmol. Vis. Sci 49, 4746–4752. [DOI] [PubMed] [Google Scholar]
- Croute F, Delaporte E, Bonnefoy JY, Fertin C, Thivolet J, Nicolas JF, 1991. Interleukin-1 beta stimulates fibroblast elastase activity. Br. J. Dermatol 124, 538–541. [DOI] [PubMed] [Google Scholar]
- Curcio CA, 2018. Soft drusen in age-related macular degeneration: biology and targeting via the oil spill strategies. Invest. Ophthalmol. Vis. Sci 59, AMD160–AMD181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio CA, Johnson M, 2013. Chapter 20 - structure, function, and pathology of Bruch’s membrane. In: Ryan SJ, Sadda SR, Hinton DR, Schachat AP, Sadda SR, Wilkinson CP, Wiedemann P, Schachat AP (Eds.), Retina, fifth ed. W.B. Saunders, London, pp. 465–481. [Google Scholar]
- Curcio CA, Medeiros NE, Millican CL, 1996. Photoreceptor loss in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 37, 1236–1249. [PubMed] [Google Scholar]
- Curcio CA, Johnson M, Rudolf M, Huang JD, 2011. The oil spill in ageing Bruch membrane. Br. J. Ophthalmol 95, 1638–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis EC, Mecham RP, 1998. Intracellular trafficking of tropoelastin. Matrix Biol. 17, 245–254. [DOI] [PubMed] [Google Scholar]
- Dejneka NS, Kuroki AM, Fosnot J, Tang W, Tolentino MJ, Bennett J, 2004. Systemic rapamycin inhibits retinal and choroidal neovascularization in mice. Mol. Vis 10, 964–972. [PubMed] [Google Scholar]
- DeVera C, Tosini G, 2020. Circadian analysis of the mouse retinal pigment epithelium transcriptome. Exp. Eye Res 193, 107988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewan A, Liu M, Hartman S, Zhang SS, Liu DT, Zhao C, Tam PO, Chan WM, Lam DS, Snyder M, Barnstable C, Pang CP, Hoh J, 2006. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science 314, 989–992. [DOI] [PubMed] [Google Scholar]
- Di Marino M, Cesareo M, Aloe G, Nucci C, Giannini C, Martucci A, Aiello F, Pisano C, Ruvolo G, Mancino R, 2020. Retinal and choroidal vasculature in patients with Marfan syndrome. Transl. Vis. Sci. Technol 9, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz H, 1993. Marfan syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A (Eds.), GeneReviews(®). University of Washington, Seattle. [PubMed] [Google Scholar]
- Dudakova L, Jirsova K, 2013. The impairment of lysyl oxidase in keratoconus and in keratoconus-associated disorders. J. Neural. Transm 120, 977–982. [DOI] [PubMed] [Google Scholar]
- Dudakova L, Liskova P, Trojek T, Palos M, Kalasova S, Jirsova K, 2012. Changes in lysyl oxidase (LOX) distribution and its decreased activity in keratoconus corneas. Exp. Eye Res 104, 74–81. [DOI] [PubMed] [Google Scholar]
- Eichler W, Friedrichs U, Thies A, Tratz C, Wiedemann P, 2002. Modulation of matrix metalloproteinase and TIMP-1 expression by cytokines in human RPE cells. Invest. Ophthalmol. Vis. Sci 43, 2767–2773. [PubMed] [Google Scholar]
- Elner SG, 2002. Human retinal pigment epithelial lysis of extracellular matrix: functional urokinase plasminogen activator receptor, collagenase, and elastase. Trans. Am. Ophthalmol. Soc 100, 273–299. [PMC free article] [PubMed] [Google Scholar]
- Elner SG, Elner VM, Kindzelskii AL, Horino K, Davis HR, Todd RF 3rd, Glagov S, Petty HR, 2003. Human RPE cell lysis of extracellular matrix: functional urokinase plasminogen activator receptor (uPAR), collagenase and elastase. Exp. Eye Res 76, 585–595. [DOI] [PubMed] [Google Scholar]
- Ewald CY, 2020. The matrisome during aging and longevity: a systems-level approach toward defining Matreotypes promoting healthy aging. Gerontology 66, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feneck EM, Lewis PN, Ralphs J, Meek KM, 2018. A comparative study of the elastic fibre system within the mouse and human cornea. Exp. Eye Res 177, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris FL 3rd, Fine SL, Hyman L, 1984. Age-related macular degeneration and blindness due to neovascular maculopathy. Arch. Ophthalmol 102, 1640–1642. [DOI] [PubMed] [Google Scholar]
- Fleckenstein M, Mitchell P, Freund KB, Sadda S, Holz FG, Brittain C, Henry EC, Ferrara D, 2018. The progression of geographic atrophy secondary to age-related macular degeneration. Ophthalmology 125, 369–390. [DOI] [PubMed] [Google Scholar]
- Fu L, Garland D, Yang Z, Shukla D, Rajendran A, Pearson E, Stone EM, Zhang K, Pierce EA, 2007. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum. Mol. Genet 16, 2411–2422. [DOI] [PubMed] [Google Scholar]
- Gelman S, Cone FE, Pease ME, Nguyen TD, Myers K, Quigley HA, 2010. The presence and distribution of elastin in the posterior and retrobulbar regions of the mouse eye. Exp. Eye Res 90, 210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensler G, Clemons TE, Domalpally A, Danis RP, Blodi B, Wells J 3rd, Rauser M, Hoskins J, Hubbard GB, Elman MJ, Fish GE, Brucker A, Margherio A, Chew EY, 2018. Treatment of geographic atrophy with intravitreal sirolimus: the age-related eye disease study 2 ancillary study. Ophthalmology 2, 441–450. Retina. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth C, Zawadzki RJ, Werner JS, Héon E, 2009. Retinal microstructure in patients with EFEMP1 retinal dystrophy evaluated by Fourier domain OCT. Eye 23, 480–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Padmanabhan A, Vaidya T, Watson AM, Bhutto IA, Hose S, Shang P, Stepicheva N, Yazdankhah M, Weiss J, Das M, Gopikrishna S, Aishwarya, Yadav N, Berger T, Mak TW, Xia S, Qian J, Lutty GA, Jayagopal A, Zigler JS Jr., Sethu S, Handa JT, Watkins SC, Ghosh A, Sinha D, 2019. Neutrophils homing into the retina trigger pathology in early age-related macular degeneration. Commun. Biol 2, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliem M, Müller PL, Birtel J, Hendig D, Holz FG, Charbel Issa P, 2016. Frequency, phenotypic characteristics and progression of atrophy associated with a diseased Bruch’s membrane in pseudoxanthoma elasticum. Invest. Ophthalmol. Vis. Sci 57, 3323–3330. [DOI] [PubMed] [Google Scholar]
- Godwin ARF, Singh M, Lockhart-Cairns MP, Alanazi YF, Cain SA, Baldock C, 2019. The role of fibrillin and microfibril binding proteins in elastin and elastic fibre assembly. Matrix Biol. J. Int. Soc. Matrix Biol 84, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourier HC, Chong NV, 2015. Can novel treatment of age-related macular degeneration Be developed by better understanding of sorsby’s fundus dystrophy. J. Clin. Med 4, 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene WA, Burke TA, Kaini RR, Por ED, Wang HC, 2017. Polarized secretion of matrix metalloproteinases and their inhibitors by retinal pigment epithelium derived from induced pluripotent stem cells during wound healing. J. Ocul. Pharmacol. Therapeut. Off. J. Assoc. Ocular Pharmacol. Therapeut 33, 132–140. [DOI] [PubMed] [Google Scholar]
- Grob S, Luo J, Hughes G, Lee C, Zhou X, Lee J, Du H, Ferreyra H, Freeman WR, Kozak I, Zhang K, 2012. Genetic analysis of simultaneous geographic atrophy and choroidal neovascularization. Eye 26, 1106–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groutas WC, Dou D, Alliston KR, 2011. Neutrophil elastase inhibitors. Expert Opin. Ther. Pat 21, 339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunda V, Verma RK, Sudhakar YA, 2013. Inhibition of elastin peptide-mediated angiogenic signaling mechanism(s) in choroidal endothelial cells by the α6(IV)NC1 collagen fragment. Invest. Ophthalmol. Vis. Sci 54, 7828–7835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Hussain AA, Limb GA, Marshall J, 1999. Age-dependent variation in metalloproteinase activity of isolated human Bruch’s membrane and choroid. Invest. Ophthalmol. Vis. Sci 40, 2676–2682. [PubMed] [Google Scholar]
- Hasanian-Langroudi F, Saravani R, Validad MH, Bahari G, Yari D, 2015. Association of lysyl oxidase (LOX) polymorphisms with the risk of keratoconus in an Iranian population. Ophthalmic Genet. 36, 309–314. [DOI] [PubMed] [Google Scholar]
- Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD, 1997. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science (New York, N.Y.) 277, 2002–2004. [DOI] [PubMed] [Google Scholar]
- He F, Li X, Cai S, Lu L, Zhang T, Yang M, Fan N, Wang X, Liu X, 2021. Polymorphism Rs11200638 Enhanced HtrA1 Responsiveness and Expression Are Associated with Age-Related Macular Degeneration. Eye, London, England. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz A, Taddese S, Sippl W, Neubert RH, Schmelzer CE, 2011. Insights into the degradation of human elastin by matrilysin-1. Biochimie 93, 187–194. [DOI] [PubMed] [Google Scholar]
- Hernandez MR, 1992. Ultrastructural immunocytochemical analysis of elastin in the human lamina cribrosa. Changes in elastic fibers in primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci 33, 2891–2903. [PubMed] [Google Scholar]
- Hernandez MR, Pena JD, Selvidge JA, Salvador-Silva M, Yang P, 2000. Hydrostatic pressure stimulates synthesis of elastin in cultured optic nerve head astrocytes. Glia 32, 122–136. [DOI] [PubMed] [Google Scholar]
- Hollborn M, Stathopoulos C, Steffen A, Wiedemann P, Kohen L, Bringmann A, 2007. Positive feedback regulation between MMP-9 and VEGF in human RPE cells. Invest. Ophthalmol. Vis. Sci 48, 4360–4367. [DOI] [PubMed] [Google Scholar]
- Horiguchi M, Inoue T, Ohbayashi T, Hirai M, Noda K, Marmorstein LY, Yabe D, Takagi K, Akama TO, Kita T, Kimura T, Nakamura T, 2009. Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc. Natl. Acad. Sci. U. S. A 106, 19029–19034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain AA, Starita C, Hodgetts A, Marshall J, 2010. Macromolecular diffusion characteristics of ageing human Bruch’s membrane: implications for age-related macular degeneration (AMD). Exp. Eye Res 90, 703–710. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Jin D, Sawada Y, Abe S, Yoshitomi T, 2015. Future therapies of wet age-related macular degeneration. J. Ophthalmol, 138070, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen OA, 1977. Bruch’s membrane in pseudoxanthoma elasticum. Histochemical, ultrastructural, and x-ray microanalytical study of the membrane and angioid streak areas. Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. Albrecht Von Graefes Arch. Clin. Exp. Ophthalmol 203, 311–320. [DOI] [PubMed] [Google Scholar]
- Jones A, Kumar S, Zhang N, Tong Z, Yang JH, Watt C, Anderson J, Amrita, Fillerup H, McCloskey M, Luo L, Yang Z, Ambati B, Marc R, Oka C, Zhang K, Fu Y, 2011. Increased expression of multifunctional serine protease, HTRA1, in retinal pigment epithelium induces polypoidal choroidal vasculopathy in mice. Proc. Natl. Acad. Sci. U. S. A 108, 14578–14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY, 2003. A critical role for elastin signaling in vascular morphogenesis and disease. Development (Cambridge, U. K.) 130, 411–423. [DOI] [PubMed] [Google Scholar]
- Kay P, Yang YC, Paraoan L, 2013. Directional protein secretion by the retinal pigment epithelium: roles in retinal health and the development of age-related macular degeneration. J. Cell Mol. Med 17, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaynak S, Kaya M, Kaya D, 2018. Is there a relationship between use of anti-vascular endothelial growth factor Agents and atrophic changes in age-related macular degeneration patients? Turkish J. Orthod 48, 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Haghighat L, Spiekerkoetter E, Sawada H, Alvira CM, Wang L, Acharya S, Rodriguez-Colon G, Orton A, Zhao M, Rabinovitch M, 2011. Neutrophil elastase is produced by pulmonary artery smooth muscle cells and is linked to neointimal lesions. Am. J. Pathol 179, 1560–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J, 2005. Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo N, Honda S, Ishibashi K, Tsukahara Y, Negi A, 2008. Elastin gene polymorphisms in neovascular age-related macular degeneration and polypoidal choroidal vasculopathy. Invest. Ophthalmol. Vis. Sci 49, 1101–1105. [DOI] [PubMed] [Google Scholar]
- Kothapalli CR, Ramamurthi A, 2010. Induced elastin regeneration by chronically activated smooth muscle cells for targeted aneurysm repair. Acta Biomater. 6, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh Nielsen M, Subhi Y, Rue Molbech C, Nilsson LL, Nissen MH, Sørensen TL, 2019. Imbalances in tissue inhibitors of metalloproteinases differentiate choroidal neovascularization from geographic atrophy. Acta Ophthalmol. 97, 84–90. [DOI] [PubMed] [Google Scholar]
- Kumar S, Berriochoa Z, Ambati BK, Fu Y, 2014. Angiographic features of transgenic mice with increased expression of human serine protease HTRA1 in retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci 55, 3842–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Nakashizuka H, Jones A, Lambert A, Zhao X, Shen M, Parker M, Wang S, Berriochoa Z, Fnu A, VanBeuge S, Chévez-Barrios P, Tso M, Rainier J, Fu Y, 2017. Proteolytic degradation and inflammation play critical roles in polypoidal choroidal vasculopathy. Am. J. Pathol 187, 2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunchithapautham K, Atkinson C, Rohrer B, 2014. Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation. J. Biol. Chem 289, 14534–14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamande SR, Bateman JF, 2020. Genetic disorders of the extracellular matrix. Anat. Rec 303, 1527–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larche M, 2014. Mechanisms of peptide immunotherapy in allergic airways disease. Ann. Am. Thorac. Soc 11 (Suppl. 5), S292–S296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi A, Urban F, Bortolotti M, 2010. Role of alpha-1 antitrypsin (AAT) in ocular allergy and uveitis. Anti-Inflammatory Anti-Allergy Agents Med. Chem 9, 304–313. [Google Scholar]
- Li JQ, Welchowski T, Schmid M, Mauschitz MM, Holz FG, Finger RP, 2020. Prevalence and incidence of age-related macular degeneration in Europe: a systematic review and meta-analysis. Br. J. Ophthalmol 104, 1077–1084. [DOI] [PubMed] [Google Scholar]
- Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY, 2012. Age-related macular degeneration. Lancet (London, England) 379, 1728–1738. [DOI] [PubMed] [Google Scholar]
- Lin MK, Yang J, Hsu CW, Gore A, Bassuk AG, Brown LM, Colligan R, Sengillo JD, Mahajan VB, Tsang SH, 2018. HTRA1, an age-related macular degeneration protease, processes extracellular matrix proteins EFEMP1 and TSP1. Aging Cell 17, e12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Lessieur EM, Saadane A, Lindstrom SI, Taylor PR, Kern TS, 2019. Neutrophil elastase contributes to the pathological vascular permeability characteristic of diabetic retinopathy. Diabetologia 62, 2365–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotery AJ, Baas D, Ridley C, Jones RP, Klaver CC, Stone E, Nakamura T, Luff A, Griffiths H, Wang T, Bergen AA, Trump D, 2006. Reduced secretion of fibulin 5 in age-related macular degeneration and cutis laxa. Hum. Mutat 27, 568–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Lin V, May A, Che B, Xiao X, Shaw DH, Su F, Wang Z, Du H, Shaw PX, 2019. HTRA1 synergizes with oxidized phospholipids in promoting inflammation and macrophage infiltration essential for ocular VEGF expression. PLoS One 14, e0216808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Zhao L, Wong WT, 2012. Microglia in the outer retina and their relevance to pathogenesis of age-related macular degeneration. Adv. Exp. Med. Biol 723, 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Velasco A, Perez-Ortiz AC, Antonio-Aguirre B, Martínez-Villaseñor L, Lira-Romero E, Palacio-Pastrana C, Zenteno JC, Ramirez I, Zepeda-Palacio C, Mendoza-Velásquez C, Camacho-Ordóñez A, Ortiz Bibriesca DM, Estrada-Mena FJ, 2020. Assessment of CFH and HTRA1 polymorphisms in age-related macular degeneration using classic and machine-learning approaches. Ophthalmic Genet. 41, 539–547. [DOI] [PubMed] [Google Scholar]
- Menon M, Mohammadi S, Davila-Velderrain J, Goods BA, Cadwell TD, Xing Y, Stemmer-Rachamimov A, Shalek AK, Love JC, Kellis M, Hafler BP, 2019. Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat. Commun 10, 4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miekus N, Luise C, Sippl W, Baczek T, Schmelzer CEH, Heinz A, 2019. MMP-14 degrades tropoelastin and elastin. Biochimie 165, 32–39. [DOI] [PubMed] [Google Scholar]
- Mithieux SM, Weiss AS, 2005. Elastin. Adv. Protein Chem 70, 437–461. [DOI] [PubMed] [Google Scholar]
- Mohamad NA, Ramachandran V, Mohd Isa H, Chan YM, Ngah NF, Ching SM, Hoo FK, Wan Sulaiman WA, Inche Mat LN, Mohamed MH, 2019. Association of HTRA1 and ARMS2 gene polymorphisms with response to intravitreal ranibizumab among neovascular age-related macular degenerative subjects. Hum. Genom 13, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Yamada H, Toyama K, Takahashi K, Akama T, Inoue T, Nakamura T, 2019. Developmental and age-related changes to the elastic lamina of Bruch’s membrane in mice. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 257, 289–301. [DOI] [PubMed] [Google Scholar]
- Morohoshi K, Patel N, Ohbayashi M, Chong V, Grossniklaus HE, Bird AC, Ono SJ, 2012. Serum autoantibody biomarkers for age-related macular degeneration and possible regulators of neovascularization. Exp. Mol. Pathol 92, 64–73. [DOI] [PubMed] [Google Scholar]
- Murphy G, Cockett MI, Ward RV, Docherty AJ, 1991. Matrix metalloproteinase degradation of elastin, type IV collagen and proteoglycan. A quantitative comparison of the activities of 95 kDa and 72 kDa gelatinases, stromelysins-1 and −2 and punctuated metalloproteinase (PUMP). Biochem. J 277 (Pt 1), 277–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafi D, Kevany BM, Genoud C, Bai X, Palczewski K, 2013. Photoreceptor phagocytosis is mediated by phosphoinositide signaling. FASEB J. 27, 4585–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahum Y, Spierer A, 2008. Ocular features of Marfan syndrome: diagnosis and management. Isr. Med. Assoc. J 10, 179–181. [PubMed] [Google Scholar]
- Nakajima K, Shimojo M, Hamanoue M, Ishiura S, Sugita H, Kohsaka S, 1992. Identification of elastase as a secretory protease from cultured rat microglia. J. Neurochem 58, 1401–1408. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lozano PR, Ikeda Y, Iwanaga Y, Hinek A, Minamisawa S, Cheng CF, Kobuke K, Dalton N, Takada Y, Tashiro K, Ross J Jr., Honjo T, Chien KR, 2002. Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature 415, 171–175. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Iejima D, Akahori M, Kamei J, Goto A, Iwata T, 2014. Overexpression of HtrA1 and exposure to mainstream cigarette smoke leads to choroidal neovascularization and subretinal deposits in aged mice. Invest. Ophthalmol. Vis. Sci 55, 6514–6523. [DOI] [PubMed] [Google Scholar]
- Natoli R, Mason E, Jiao H, Chuah A, Patel H, Fernando N, Valter K, Wells CA, Provis J, Rutar M, 2018. Dynamic interplay of innate and adaptive immunity during sterile retinal inflammation: insights from the transcriptome. Front. Immunol 9, 1666. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Netland PA, Sugrue SP, Albert DM, Shore JW, 1994. Histopathologic features of the floppy eyelid syndrome. Involvement of tarsal elastin. Ophthalmology 101, 174–181. [DOI] [PubMed] [Google Scholar]
- Newsome DA, Pfeffer BA, Hewitt AT, Robey PG, Hassell JR, 1988. Detection of extracellular matrix molecules synthesized in vitro by monkey and human retinal pigment epithelium: influence of donor age and multiple passages. Exp. Eye Res 46, 305–321. [DOI] [PubMed] [Google Scholar]
- Nicoloff G, Baydanoff S, Stanimirova N, Petrova C, Christova P, 2000. An association of anti-elastin IgA antibodies with development of retinopathy in diabetic children. Gen. Pharmacol 35, 83–87. [DOI] [PubMed] [Google Scholar]
- Nita M, Strzałka-Mrozik B, Grzybowski A, Mazurek U, Romaniuk W, 2014. Age-related macular degeneration and changes in the extracellular matrix. Med. Sci. Mon. Int. Med. J. Exp. Clin. Res. Int. Med. J. Exp. Clin. Res 20, 1003–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda K, Kitagawa K, Miki T, Horiguchi M, Akama TO, Taniguchi T, Taniguchi H, Takahashi K, Ogra Y, Mecham RP, Terajima M, Yamauchi M, Nakamura T, 2020. A matricellular protein fibulin-4 is essential for the activation of lysyl oxidase. Sci. Adv 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenblatt RB, Lee RW, Chew E, Wei L, Liu B, Sen HN, Dick AD, Ferris FL, 2014. Immune responses in age-related macular degeneration and a possible long-term therapeutic strategy for prevention. Am. J. Ophthalmol 158, 5–11 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz G, Salica JP, Chuluyan EH, Gallo JE, 2014. Diabetic retinopathy: could the alpha-1 antitrypsin be a therapeutic option? Biol. Res 47, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz G, Lopez ES, Salica JP, Potilinski C, Fernández Acquier M, Chuluyan E, Gallo JE, 2018. Alpha-1-antitrypsin ameliorates inflammation and neurodegeneration in the diabetic mouse retina. Exp. Eye Res 174, 29–39. [DOI] [PubMed] [Google Scholar]
- Pena JD, Netland PA, Vidal I, Dorr DA, Rasky A, Hernandez MR, 1998. Elastosis of the lamina cribrosa in glaucomatous optic neuropathy. Exp. Eye Res 67, 517–524. [DOI] [PubMed] [Google Scholar]
- Pena JD, Agapova O, Gabelt BT, Levin LA, Lucarelli MJ, Kaufman PL, Hernandez MR, 2001. Increased elastin expression in astrocytes of the lamina cribrosa in response to elevated intraocular pressure. Invest. Ophthalmol. Vis. Sci 42, 2303–2314. [PubMed] [Google Scholar]
- Pennington KL, DeAngelis MM, 2016. Epidemiology of age-related macular degeneration (AMD): associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. (Lond) 3, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen E, Wågberg F, Angquist KA, 2002. Serum concentrations of elastin-derived peptides in patients with specific manifestations of atherosclerotic disease. Eur. J. Vasc. Endovasc. Surg. Off. J. Eur. Soc. Vasc. Surg 24, 440–444. [DOI] [PubMed] [Google Scholar]
- Potilinski MC, Lorenc V, Perisset S, Gallo JE, 2020. Mechanisms behind retinal ganglion cell loss in diabetes and therapeutic approach. Int. J. Mol. Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley H, Pease ME, Thibault D, 1994. Change in the appearance of elastin in the lamina cribrosa of glaucomatous optic nerve heads. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 232, 257–261. [DOI] [PubMed] [Google Scholar]
- Rabkin SW, 2017. The role matrix metalloproteinases in the production of aortic aneurysm. Prog. Mol. Biol. Trans. Sci 147, 239–265. [DOI] [PubMed] [Google Scholar]
- Risseeuw S, Ossewaarde-van Norel J, Klaver CCW, Colijn JM, Imhof SM, van Leeuwen R, 2019. Visual Acuity in Pseudoxanthoma Elasticum, vol. 39. Retina, Philadelphia, Pa, pp. 1580–1587. [DOI] [PubMed] [Google Scholar]
- Risseeuw S, Bennink E, Poirot MG, de Jong PA, Spiering W, Imhof SM, van Leeuwen R, Ossewaarde-van Norel J, 2020a. A reflectivity measure to quantify Bruch’s membrane calcification in patients with pseudoxanthoma elasticum using optical coherence tomography. Transl. Vis. Sci. Technol 9, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risseeuw S, Ossewaarde-van Norel J, van Buchem C, Spiering W, Imhof SM, van Leeuwen R, 2020b. The extent of angioid streaks correlates with macular degeneration in pseudoxanthoma elasticum. Am. J. Ophthalmol 220, 82–90. [DOI] [PubMed] [Google Scholar]
- Roach ES, Islam MP, 2015. Pseudoxanthoma elasticum. Handb. Clin. Neurol 132, 215–221. [DOI] [PubMed] [Google Scholar]
- Robinet A, Fahem A, Cauchard JH, Huet E, Vincent L, Lorimier S, Antonicelli F, Soria C, Crepin M, Hornebeck W, Bellon G, 2005. Elastin-derived peptides enhance angiogenesis by promoting endothelial cell migration and tubulogenesis through upregulation of MT1-MMP. J. Cell Sci 118, 343–356. [DOI] [PubMed] [Google Scholar]
- Rohrer B, Frazer-Abel A, Leonard A, Ratnapriya R, Ward T, Pietraszkiewicz A, O’Quinn E, Adams K, Swaroop A, Wolf BJ, 2019. Association of age-related macular degeneration with complement activation products, smoking, and single nucleotide polymorphisms in South Carolinians of European and African descent. Mol. Vis 25, 79–92. [PMC free article] [PubMed] [Google Scholar]
- Rohrer B, Parsons N, Annamalai B, Nicholson C, Obert E, Jones B, Dick AD, 2021. Peptide-based immunotherapy against oxidized elastin ameliorates pathology in mouse model of smoke-induced ocular injury. Exp. Eye Res, 108755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G, 2019. Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front. Immunol 10, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatos-Peyton CA, Verhagen J, Wraith DC, 2010. Antigen-specific immunotherapy of autoimmune and allergic diseases. Curr. Opin. Immunol 22, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AF, Wang R, Ling S, Wang S, 2017. Antibody engineering for pursuing a healthier future. Front. Microbiol 8, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagaties MJ, Raviola G, 1989. Immunogold staining of elastin in the wall of the retinal arteries in Macaca mulatta. Curr. Eye Res 8, 55–59. [DOI] [PubMed] [Google Scholar]
- Sakai LY, Keene DR, Engvall E, 1986. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J. Cell Biol 103, 2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarks SH, 1976. Ageing and degeneration in the macular region: a clinico-pathological study. Br. J. Ophthalmol 60, 324–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarks SH, Van Driel D, Maxwell L, Killingsworth M, 1980. Softening of drusen and subretinal neovascularization. Trans. Ophthalmol. Soc. U. K 100, 414–422. [PubMed] [Google Scholar]
- Schlötzer-Schrehardt U, Stojkovic M, Hofmann-Rummelt C, Cursiefen C, Kruse FE, Holbach LM, 2005. The Pathogenesis of floppy eyelid syndrome: involvement of matrix metalloproteinases in elastic fiber degradation. Ophthalmology 112, 694–704. [DOI] [PubMed] [Google Scholar]
- Schlötzer-Schrehardt U, Hammer CM, Krysta AW, Hofmann-Rummelt C, Pasutto F, Sasaki T, Kruse FE, Zenkel M, 2012. LOXL1 deficiency in the lamina cribrosa as candidate susceptibility factor for a pseudoexfoliation-specific risk of glaucoma. Ophthalmology 119, 1832–1843. [DOI] [PubMed] [Google Scholar]
- Schultz NM, Bhardwaj S, Barclay C, Gaspar L, Schwartz J, 2021. Global burden of dry age-related macular degeneration: a targeted literature review. Clin. Therapeut 43, 1792–1818. [DOI] [PubMed] [Google Scholar]
- Schwesinger C, Yee C, Rohan RM, Joussen AM, Fernandez A, Meyer TN, Poulaki V, Ma JJ, Redmond TM, Liu S, Adamis AP, D’Amato RJ, 2001. Intrachoroidal neovascularization in transgenic mice overexpressing vascular endothelial growth factor in the retinal pigment epithelium. Am. J. Pathol 158, 1161–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon JM, Willett WC, Speizer FE, Hankinson SE, 1996. A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA 276, 1141–1146. [PubMed] [Google Scholar]
- Segawa K, 1995. [Trabecular meshwork and elastin]. Nippon. Ganka Gakkai Zasshi 99, 1291–1302. [PubMed] [Google Scholar]
- Sekiyama E, Saint-Geniez M, Yoneda K, Hisatomi T, Nakao S, Walshe TE, Maruyama K, Hafezi-Moghadam A, Miller JW, Kinoshita S, D’Amore PA, 2012. Heat treatment of retinal pigment epithelium induces production of elastic lamina components and antiangiogenic activity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 26, 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadforth AM, Suzuki S, Alzonne R, Edwards GA, Richardson NA, Chirila TV, Harkin DG, 2015. Incorporation of human recombinant tropoelastin into silk fibroin membranes with the view to repairing Bruch’s membrane. J. Funct. Biomater 6, 946–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya AK, Nandakumar KS, 2018. Antigen-specific tolerization and targeted delivery as therapeutic strategies for autoimmune diseases. Trends Biotechnol. 36, 686–699. [DOI] [PubMed] [Google Scholar]
- Sharma RK, Orr WE, Schmitt AD, Johnson DA, 2005. A functional profile of gene expression in ARPE-19 cells. BMC Ophthalmol. 5, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty R, Sathyanarayanamoorthy A, Ramachandra RA, Arora V, Ghosh A, Srivatsa PR, Pahuja N, Nuijts RM, Sinha-Roy A, Mohan RR, Ghosh A, 2015. Attenuation of lysyl oxidase and collagen gene expression in keratoconus patient corneal epithelium corresponds to disease severity. Mol. Vis 21, 12–25. [PMC free article] [PubMed] [Google Scholar]
- Sivaprasad S, Chong NV, Bailey TA, 2005. Serum elastin-derived peptides in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 46, 3046–3051. [DOI] [PubMed] [Google Scholar]
- Skeie JM, Mullins RF, 2008. Elastin-mediated choroidal endothelial cell migration: possible role in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 49, 5574–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeie JM, Hernandez J, Hinek A, Mullins RF, 2012. Molecular responses of choroidal endothelial cells to elastin derived peptides through the elastin-binding protein (GLB1). Matrix Biol. 31, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EL, Peakman M, 2018. Peptide immunotherapy for type 1 diabetes-clinical advances. Front. Immunol 9, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounni NE, Roghi C, Chabottaux V, Janssen M, Munaut C, Maquoi E, Galvez BG, Gilles C, Frankenne F, Murphy G, Foidart JM, Noel A, 2004. Up-regulation of vascular endothelial growth factor-A by active membrane-type 1 matrix metalloproteinase through activation of Src-tyrosine kinases. J. Biol. Chem 279, 13564–13574. [DOI] [PubMed] [Google Scholar]
- Sreekumar PG, Li Z, Wang W, Spee C, Hinton DR, Kannan R, MacKay JA, 2018. Intra-vitreal αB crystallin fused to elastin-like polypeptide provides neuroprotection in a mouse model of age-related macular degeneration. J. Contr. Release Off. J. Contr. Release Soc 283, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava GK, Martín L, Singh AK, Fernandez-Bueno I, Gayoso MJ, Garcia-Gutierrez MT, Girotti A, Alonso M, Rodríguez-Cabello JC, Pastor JC, 2011. Elastin-like recombinamers as substrates for retinal pigment epithelial cell growth. J. Biomed. Mater. Res 97, 243–250. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z, 1999. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 98, 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockley RA, 2015. The multiple facets of alpha-1-antitrypsin. Ann. Transl. Med 3,130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EM, Braun TA, Russell SR, Kuehn MH, Lotery AJ, Moore PA, Eastman CG, Casavant TL, Sheffield VC, 2004. Missense variations in the fibulin 5 gene and age-related macular degeneration. N. Engl. J. Med 351, 346–353. [DOI] [PubMed] [Google Scholar]
- Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P, 1998. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J. Clin. Investig 102, 576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan P, Vashist P, Ravindran RD, Shanker A, Nitsch D, Nonyane BA, Smeeth L, Chakravarthy U, Fletcher AE, 2012. Polymorphisms in ARMS2/HTRA1 and complement genes and age-related macular degeneration in India: findings from the INDEYE study. Invest. Ophthalmol. Vis. Sci 53, 7492–7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart CC, Greene CM, Carroll TP, O’Neill SJ, McElvaney NG, 2005. Elastolytic proteases: inflammation resolution and dysregulation in chronic infective lung disease. Am. J. Respir. Crit. Care Med 171, 1070–1076. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nakamura T, Hayashi A, Kamei M, Nakabayashi M, Okada AA, Tomita N, Kaneda Y, Tano Y, 2000. Inhibition of experimental choroidal neovascularization by overexpression of tissue inhibitor of metalloproteinases-3 in retinal pigment epithelium cells. Am. J. Ophthalmol 130, 774–781. [DOI] [PubMed] [Google Scholar]
- Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, Ofrecio J, Lin M, Brenner MB, Olefsky JM, 2012. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med 18, 1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Nakayama T, Yuzawa M, Wang Z, Kawamura A, Mori R, Nakashizuka H, Sato N, Mizutani Y, 2011. Analysis of candidate genes for age-related macular degeneration subtypes in the Japanese population. Mol. Vis 17, 2751–2758. [PMC free article] [PubMed] [Google Scholar]
- Tang NP, Zhou B, Wang B, Yu RB, 2009. HTRA1 promoter polymorphism and risk of age-related macular degeneration: a meta-analysis. Ann. Epidemiol 19, 740–745. [DOI] [PubMed] [Google Scholar]
- Tektas OY, Lütjen-Drecoll E, 2009. Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp. Eye Res 88, 769–775. [DOI] [PubMed] [Google Scholar]
- Terai N, Sandner D, Hadjiraftis S, Pillunat LE, 2011. [Maculopathy with subretinal yellow deposits]. Ophthalmologe 108, 467–472. [DOI] [PubMed] [Google Scholar]
- Thornton J, Edwards R, Mitchell P, Harrison RA, Buchan I, Kelly SP, 2005. Smoking and age-related macular degeneration: a review of association. Eye 19, 935–944. [DOI] [PubMed] [Google Scholar]
- Tong Y, Liao J, Zhang Y, Zhou J, Zhang H, Mao M, 2010. LOC387715/HTRA1 gene polymorphisms and susceptibility to age-related macular degeneration: a HuGE review and meta-analysis. Mol. Vis 16, 1958–1981. [PMC free article] [PubMed] [Google Scholar]
- Tong B, Wan B, Wei Z, Wang T, Zhao P, Dou Y, Lv Z, Xia Y, Dai Y, 2014. Role of cathepsin B in regulating migration and invasion of fibroblast-like synoviocytes into inflamed tissue from patients with rheumatoid arthritis. Clin. Exp. Immunol 177, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo J, Ross RJ, Reed GF, Yan Q, Wang JJ, Bojanowski CM, Chew EY, Feng X, Olsen TW, Ferris FL 3rd, Mitchell P, Chan CC, 2008. The HtrA1 promoter polymorphism, smoking, and age-related macular degeneration in multiple case-control samples. Ophthalmology 115, 1891–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umihira J, 1993. [Ultrastructural immunohistochemical localization of elastin in the human trabecular meshwork]. Nippon. Ganka Gakkai Zasshi 97, 1143–1150. [PubMed] [Google Scholar]
- Umihira J, Nagata S, Nohara M, Hanai T, Usuda N, Segawa K, 1994. Localization of elastin in the normal and glaucomatous human trabecular meshwork. Invest. Ophthalmol. Vis. Sci 35, 486–494. [PubMed] [Google Scholar]
- Velilla S, Garcia-Medina JJ, Garcia-Layana A, Dolz-Marco R, Pons-Vazquez S, Pinazo-Duran MD, Gomez-Ulla F, Arevalo JF, Diaz-Llopis M, Gallego-Pinazo R, 2013. Smoking and age-related macular degeneration: review and update. J. Ophthalmol, 895147, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierkotten S, Muether PS, Fauser S, 2011. Overexpression of HTRA1 leads to ultrastructural changes in the elastic layer of Bruch’s membrane via cleavage of extracellular matrix components. PLoS One 6, e22959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt AP, Whitmore SS, Mulfaul K, Chirco KR, Giacalone JC, Flamme-Wiese MJ, Stockman A, Stone EM, Tucker BA, Scheetz TE, Mullins RF, 2020. Bulk and single-cell gene expression analyses reveal aging human choriocapillaris has pro-inflammatory phenotype. Microvasc. Res 131, 104031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volland S, Esteve-Rudd J, Hoo J, Yee C, Williams DS, 2015. A comparison of some organizational characteristics of the mouse central retina and the human macula. PLoS One 10, e0125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachi H, Sato F, Murata H, Nakazawa J, Starcher BC, Seyama Y, 2005. Development of a new in vitro model of elastic fiber assembly in human pigmented epithelial cells. Clin. Biochem 38, 643–653. [DOI] [PubMed] [Google Scholar]
- Wang TT, Ravetch JV, 2015. Immune complexes: not just an innocent bystander in chronic viral infection. Immunity 42, 213–215. [DOI] [PubMed] [Google Scholar]
- Werb Z, Banda MJ, McKerrow JH, Sandhaus RA, 1982. Elastases and elastin degradation. J. Invest. Dermatol 79 (Suppl. 1), 154s–159s. [DOI] [PubMed] [Google Scholar]
- White TL, Lewis P, Hayes S, Fergusson J, Bell J, Farinha L, White NS, Pereira LV, Meek KM, 2017a. The structural role of elastic fibers in the cornea investigated using a mouse model for Marfan syndrome. Invest. Ophthalmol. Vis. Sci 58, 2106–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TL, Lewis PN, Young RD, Kitazawa K, Inatomi T, Kinoshita S, Meek KM, 2017b. Elastic microfibril distribution in the cornea: differences between normal and keratoconic stroma. Exp. Eye Res 159, 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BL, Seager NA, Gardiner JD, Pappas CM, Cronin MC, Amat di San Filippo C, Anstadt RA, Liu J, Toso MA, Nichols L, Parnell TJ, Eve JR, Bartel PL, Zouache MA, Richards BT, Hageman GS, 2021. Chromosome 10q26-driven age-related macular degeneration is associated with reduced levels of HTRA1 in human retinal pigment epithelium. Proc. Natl. Acad. Sci. U. S. A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodell A, Coughlin B, Kunchithapautham K, Casey S, Williamson T, Ferrell WD, Atkinson C, Jones BW, Rohrer B, 2013. Alternative complement pathway deficiency ameliorates chronic smoke-induced functional and morphological ocular injury. PLoS One 8, e67894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodell A, Jones BW, Williamson T, Schnabolk G, Tomlinson S, Atkinson C, Rohrer B, 2016. A targeted inhibitor of the alternative complement pathway accelerates recovery from smoke-induced ocular injury. Invest. Ophthalmol. Vis. Sci 57, 1728–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun X, 2019. Complement system and age-related macular degeneration: drugs and challenges. Drug Des. Dev. Ther 13, 2413–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro K, Mori K, Nakata I, Tsuchihashi T, Horie-Inoue K, Nakanishi H, Tsujikawa A, Saito M, Iida T, Yarnada R, Matsuda F, Inoue S, Awata T, Yoneya S, Yoshimura N, 2011. Association of elastin gene polymorphism to age-related macular degeneration and polypoidal choroidal vasculopathy. Invest. Ophthalmol. Vis. Sci 52, 8780–8784. [DOI] [PubMed] [Google Scholar]
- Yanagisawa S, Sakurada Y, Miki A, Matsumiya W, Imoto I, Honda S, 2015. The association of elastin gene variants with two angiographic subtypes of polypoidal choroidal vasculopathy. PLoS One 10, e0120643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y, Li Z, Greenbaum D, Bogyo M, Weber E, Brömme D, 2004. Cathepsin V, a novel and potent elastolytic activity expressed in activated macrophages. J. Biol. Chem 279, 36761–36770. [DOI] [PubMed] [Google Scholar]
- Yeo GC, Baldock C, Tuukkanen A, Roessle M, Dyksterhuis LB, Wise SG, Matthews J, Mithieux SM, Weiss AS, 2012. Tropoelastin bridge region positions the cell-interactive C terminus and contributes to elastic fiber assembly. Proc. Natl. Acad. Sci. U. S. A 109, 2878–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger JR, McHenry JG, Hogan RN, Jones FR, 2006. Optic neuropathy and alpha–1 antitrypsin deficiency. Invest. Ophthalmol. Vis. Sci 47, 732–732. [Google Scholar]
- Yu Q, Stamenkovic I, 2000. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 14, 163–176. [PMC free article] [PubMed] [Google Scholar]
- Yu HG, Liu X, Kiss S, Connolly E, Gragoudas ES, Michaud NA, Bulgakov OV, Adamian M, DeAngelis MM, Miller JW, Li T, Kim IK, 2008. Increased choroidal neovascularization following laser induction in mice lacking lysyl oxidaselike 1. Invest. Ophthalmol. Vis. Sci 49, 2599–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadeh N, Bernstein JA, Niemi AK, Dugan S, Kwan A, Liang D, Hyland JC, Hoyme HE, Hudgins L, Manning MA, 2011. Ectopia lentis as the presenting and primary feature in Marfan syndrome. Am. J. Med. Genet 155a, 2661–2668. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Marmorstein LY, 2010. Focus on molecules: fibulin-3 (EFEMP1). Exp. Eye Res 90, 374–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Apfelroth SD, Hu W, Davis EC, Sanguined C, Bonadio J, Mecham RP, Ramirez F, 1994. Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices. J. Cell Biol 124, 855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang Y, Xiao H, Liang X, Sun D, Peng S, 2012. A gene expression profile of the developing human retinal pigment epithelium. Mol. Vis 18, 2961–2975. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.