

Abstract
In this study, the synthesis of two new guanidine hydroquinoline ligands served as basis for six new zinc guanidine complexes. Two of these complexes showed very high activity in the lactide polymerization under industrial conditions. The lactide polymerization was demonstrated in solution and melt conditions observing high activity and molar masses up to 90 000 g mol−1. Density functional theory studies elucidated the high activity of the complexes associated with the influence of the ligand backbone and the use of triflate counterions. On the way towards a circular economy, polymerization and depolymerization go hand in hand. So far, guanidine complexes have only shown their good activity in the ring opening polymerization of esters, and guanidine complexes with pure N donors have not been tested in recycling processes. Herein, the excellent ability of zinc guanidine complexes to catalyze both polymerization and depolymerization was demonstrated. The two most promising zinc complexes efficiently mediated the methanolysis of polylactide into methyl lactate under mild reaction conditions.
Keywords: bioplastic, circular economy, polylactide, ring-opening polymerization, zinc complexes
Specific design: Using a hydroquinoline backbone for zinc guanidine complexes in the ring‐opening polymerization of lactide results in an enormous activity increase compared to their aromatic counterparts. Not only is the complex very active in the polymerization of lactide, but it also catalyzes the chemical recycling process of solvolysis. With this knowledge, complexes could be designed in a more targeted manner in the future.

Introduction
Plastics enable our modern lifestyle and are therefore part of our daily consumption habits. Most of the global plastic products are petroleum‐based plastics, which are designed for a linear economy. They turn to waste after single usage and often end up in landfills or the natural environment. [1] The long‐term accumulation of these plastics in the nature makes them a major environmental problem. [2] Additionally, the consumption of plastic is expected to continuously increase globally, rendering sustainable alternatives more and more important. [3]
A new strategy for a sustainable handling is therefore to create a circular economy for plastics including elimination, innovation, and circularity.[ 3a , 4 ] Innovation in this context includes renewable polymers as alternatives to petroleum‐based ones. Since some plastics are designed to become waste (e. g. packaging materials), biodegradable properties of bioplastics are particularly advantageous. [5] Ideally, bioplastics are additionally recyclable to return them to the feedstock as new raw material.
Polylactide (PLA) is a bio‐based, biodegradable, and biocompatible polymer and therefore one of the most promising bioplastics as renewable alternative fitting into the concept of a circular economy. [6] Several life cycle assessments have already shown the positive impact of using PLA instead of conventional polymers based on petrochemicals, like polyethylene terephthalate. [7]
The versatile usability of this polymer ranges from simple packaging materials to biomedical applications. [8] For a holistic circularity for plastics, the material itself also has to be free of hazardous chemicals, including the principles of green chemistry. [9] Currently, a toxic catalyst, tin(II) bis(2‐ethylhexanoate) [Sn(Oct)2], is used in the industrial production process of PLA and remains in the polymer. [10] The scientific desire to replace this catalyst with an environmentally benign one led to a large number of studies that address zinc, [11] magnesium, [12] or iron [13] complexes but also organocatalysts. [14] The challenge is not only to replace the catalyst but also to receive at least similar or better catalytic activity under industrially relevant conditions, meaning that the complex has to be stable towards impurities in the monomer or in the process. Among the complexes that can fulfill these requirements, many zinc complexes are represented.[ 11d , 11h , 15 ] Especially zinc guanidine complexes are well known as such suitable catalysts in the lactide polymerization. [15f] Their environmental compatibility has been further confirmed in eco‐toxicological studies. [16] In 2020, Herres‐Pawlis and co‐workers introduced the currently fastest, robust, and biocompatible zinc complex. With this zinc bisguanidine triflate complex a much higher polymerization activity than for the industrially used Sn(Oct)2 catalyst was observed. [11g] All these catalyst innovations enable an improved sustainability for PLA, but the reusing and recycling of this material still need to be tackled.
Indeed, the biodegradability of PLA allows industrial composting leading to enhanced waste strategies. However, this end‐of‐life scenario for PLA still belongs to a linear economic model. Instead, mechanical or chemical recycling offers an approach for plastic recycling towards a circular economy.[ 3a , 17 ] Mechanical recycling as one option is limited by the number of cycles through a loss of mechanical properties during the thermomechanical degradation process and a possible downgrading of the material. Conversely, chemical recycling can be either used to recover the polymer into monomeric units or to transform the polymer into reusable resources. [18] With a chemical recycling of PLA either lactic acid can be recovered as monomeric unit or commodity chemicals are produced directly. [19] Here, the alcoholysis of PLA producing lactate esters should be emphasized as such recycling method. The received lactate esters can be used as green solvents or to produce platform chemicals out of them, strengthening the circular economy approach. [20] However, examples of PLA degradation to lactate esters catalyzed by robust metal complexes with earth‐abundant metals are scarce in literature, mostly utilizing zinc(II) complexes for the methanolysis of PLA.[ 11e , 12d , 15d , 21 ]
As already mentioned, guanidine complexes are well investigated in the ring‐opening polymerization (ROP) of lactide, but only very few examples have been explored towards their use in degradation processes. [22]
In this study we present six novel, robust zinc guanidine complexes based on a guanidine hydroquinoline ligand system. In particular, two triflate complexes demonstrate very high activity in the ROP of lactide under industrially conditions. The application in solution is investigated as well as mild degradation studies of PLA into methyl lactate contributing to a better sustainability of bioplastics.
Results and Discussion
Recently, we have shown that an aliphatic ligand backbone of zinc guanidine complexes leads to a higher activity compared to its aromatic counterpart.[ 11g , 23 ] This raised our interest to design a new ligand system that is comparable to the TMGqu and DMEGqu ligand system published by us [24] with the difference of a hydroquinoline instead of a quinoline backbone. The TMGqu system stood out due to its high activity at that point in time and the elucidation of the full mechanism by experimental and theoretical methods. [25] The synthesis route for the new ligand was inspired by the publications of Uenishi and Hamada [26] and Bruns et al. [27] Azidation of the alcohol 1 yields the azide 2, which can be hydrogenated to the hydroquinoline 3. The known chiral hydroquinoline 3 was functionalized with two different Vilsmeier salts (DMEG: dimethylethyleneguanidino and TMG: tetramethylguanidino), yielding the new racemic ligands DMEGhydroqu (L1) and TMGhydroqu (L2) (Scheme 1).
Scheme 1.

Ligand synthesis of DMEGhydroqu (L1) and TMGhydroqu (L2).
Both ligands were combined afterwards with different zinc salts to achieve six new zinc guanidine complexes (C1–C6) crystallizing as racemates (Figure 1). Accordingly, two zinc chloride complexes [ZnCl2(DMEGhydroqu)] (C1) and [ZnCl2(TMGhydroqu)] (C4) were received, as well as two zinc bromide complexes [ZnBr2(DMEGhydroqu)] (C2) and [ZnBr2(TMGhydroqu)] (C5). The use of weakly‐coordinating anions facilitates the synthesis of the complexes [Zn(DMEGhydroqu)2](OTf)2 (C3) and [Zn(TMGhydroqu)2](OTf)2 (C6). In all complexes the Zn atom is four‐coordinated, but in the case of complexes C1–C2 and C4–C5 the zinc ion is coordinated by two N‐donors and two chlorides or bromides, whereby the zinc ion in C3 and C6 is coordinated twice by the N,N‐hybrid guanidine donor ligand paired with weakly coordinating triflate anions yielding a bis(chelate) complex.
Figure 1.
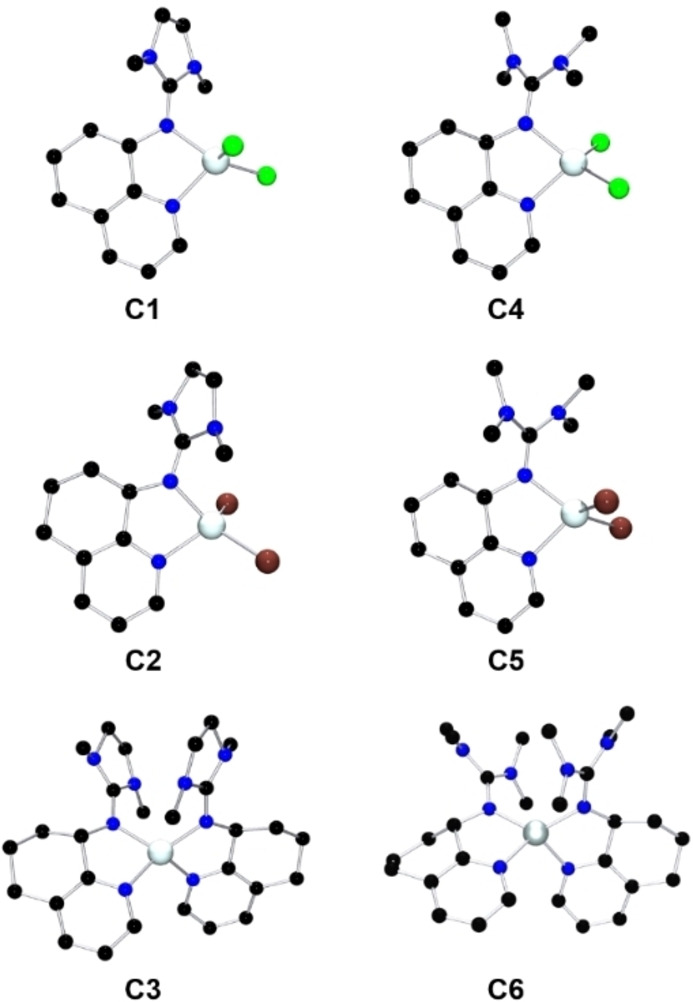
Molecular structures of the complexes C1–C2, C4–C5, and the cationic complex units in C3 and C6 in the solid state. H‐atoms and non‐coordinating anions are omitted for clarity.
The complexes’ molecular structure was elucidated by single‐crystal X‐ray diffraction (XRD) analysis (Table 1). Here, all complexes have a delocalized guanidine unit (described by the structure factor ρ [28] ) and a distorted tetrahedral coordination (see structure parameter τ 4 [29] ), whereby complex C3 and C6 are even more distorted than the other ones (Table 1). The molecular structures in the solid state of the six complexes show the general trend of a shorter Zn−Ngua bond length compared to the Zn−Npy bond length. This trend has already been described for complex analogues with a quinoline backbone (see Table S6). [24] However, these literature‐known quinoline complexes have coordinated one of the two triflate ions to the zinc center. Additionally, complexes C3 and C6 have a significant shorter Zn−Ngua bond length than the other four complexes (Table 1). Interestingly, the Zn−Ngua bond length of the hydroquinoline triflate complexes C3 and C6 are also considerably shorter than those of the aromatic counterparts reported previously. [24] In addition, these two complexes have a bite angle at least two degrees larger than the aromatic complexes (Table S6).
Table 1.
Key geometric data of complexes C1–C6.
|
Complex |
Zn−Ngua [Å] |
Zn−Npy [Å] |
Ngua−Zn−Npy [°] |
ρ [a] |
τ 4 [b] |
|---|---|---|---|---|---|
|
C1 |
2.033(2) |
2.055(1) |
81.1(1) |
0.98 |
0.74 |
|
C2 |
2.034(4) |
2.053(4) |
81.6(1) |
0.96 |
0.74 |
|
C3 |
1.963(2) 1.981(2) |
2.022(2) 2.031(2) |
85.0(1)/ 83.9(1)[c] |
0.98 0.98 |
0.65 |
|
C4 |
2.018(1) |
2.049(1) |
82.5(1) |
0.97 |
0.77 |
|
C5 |
2.012(2) |
2.049(2) |
82.6(1) |
0.98 |
0.73 |
|
C6 [d] |
1.959(3) 1.950(3) |
2.034(3) 2.042(3) |
84.5(1)/ 84.6(1)[c] |
0.99 0.99 |
0.71 |
[a] ρ=2a/(b+c). [28] [b] τ 4 =[360°−(α+β)]/141°, with τ 4=0 indicating square‐planar coordination and τ 4=1 for tetrahedral coordination. [29] [c] Two chelate angles of both ligands to the Zn atom are reported. [d] Two crystallographic independent molecules have crystallized in the asymmetric unit. Both molecules possess equal bond length within the standard deviation. Crystallographic data of the second molecule are located in the Supporting Information.
Further analysis methods like nuclear magnetic resonance (NMR) spectroscopy, infrared (IR) spectroscopy, and mass spectrometry (MS) also confirmed the structure of the various complexes C1 to C6.
Polymerization studies
To gain a first impression about the activity of the complexes C1–C6 in the ROP of lactide, the catalysts were tested with technical grade rac‐lactide in Schlenk tubes under solvent‐free conditions at 150 °C, which are desirable industrial conditions. All complexes exhibited activity in the melt polymerization, although huge differences in their polymerization rate were observed (Table 2): The four complexes with coordinating halide anions (C1, C2, C4, and C5) require hours to polymerize lactide to moderate conversion. By contrast, complexes C3 and C6, coordinated by two guanidine hydroquinoline ligands, reach nearly full conversions after a few minutes. With both complexes the production of atactic PLA was observed (P r=0.62–0.65; with P r = , with [sis] = integration of sis peak (s = syndiotactic and I = isotactic)) (Table 2, Figures S31 and S32). The slight heterotactic polymers produced with catalyst C3 and C6 could indicate stereoselective bias of the catalysts. By using l‐lactide, purely isotactic PLA was received, indicating the absence of epimerization (Figure S33). The enlarged dispersity of the polymer obtained with these two complexes after such a short time shows that transesterification reactions have already occurred (Table 2). This also explains the lower molar masses compared to the theoretical ones. On the basis of their activity with technical grade lactide, all complexes are found to be stable towards impurities in the non‐purified monomer at the tested monomer/initiator ([M]/[I]) ratio. The much higher activity of complex C3 and C6 points to the great potential of triflate complexes as catalysts in the ROP of lactide, which is supported by previous studies.[ 11g , 23 , 24 , 30 ]
Table 2.
Polymerization results of all complexes (C1–C6) under industrially relevant conditions.[a]
|
Complex |
t [min] |
Conv. [%] |
M n,theo [g mol−1] |
M n [b] [g mol−1] |
Ð [b] |
P r [c] |
|---|---|---|---|---|---|---|
|
C1 |
360 |
45 |
n. d. |
n. d. |
n. d. |
n. d. |
|
C2 |
360 |
45 |
n. d. |
n. d. |
n. d. |
n. d. |
|
C3 |
5 |
93 |
67000 |
61000 |
1.6 |
0.65 |
|
C4 |
360 |
44 |
n. d. |
n. d. |
n. d. |
n. d. |
|
C5 |
360 |
52 |
n. d. |
n. d. |
n. d. |
n. d. |
|
C6 |
5 |
95 |
68500 |
47800 |
1.7 |
0.62 |
[a] Polymerization conditions: technical grade rac‐lactide, [M]/[I] ratio=500 : 1, 150 °C, 260 rpm. n. d.=not determined. [b] Determined via gel permeation chromatography (GPC) analysis in THF. Mn : determined number‐average molar mass; M n,theo : theoretical number‐average molar mass calculated by conversion x molar mass x [M]/[I]; Ð: polydispersity obtained by Mw /Mn . [c] Determined via homonuclear decoupled NMR spectroscopy.
Density functional theory (DFT) studies followed by a natural bond orbital (NBO) [31] analysis provide insights into the electronic structure of the six complexes (C1–C6). In accordance with previous studies, we used the TPSSh [32] functional and the def2‐TZVP basis set [33] in combination with empirical dispersion correction with Becke–Johnson damping.[ 11g , 34 ] As described above, the zinc triflate complexes C3 and C6 possess a higher polymerization activity than the zinc halogenido complexes (C1, C2, C4, and C5). In complexes C1, C2, C4, and C5 the Lewis acidity of the zinc atom is diminished due to the coordination of the halogenido ligands. To quantify this, the NBO charges can be used. In the halogenido complexes the NBO charge of the zinc atom is +1.52 e units whereas the NBO charge of the zinc atom in the triflate complexes amounts to +1.65 e units.
In the publication of Börner et al. similar complexes were used for the ROP of lactide; the difference is a quinoline unit instead of a hydroquinoline backbone, which results in a much higher activity, where the polymerization reaction now needs minutes instead of hours.[ 24 , 25 ] For an explanation of the different catalytic activities enhanced by the slightly modified backbone, further computational chemistry studies were performed. In the molecular structures of the quinoline complexes one triflate anion coordinates to the zinc atom (Figure 2), whereas the zinc ion in complexes C3 and C6 is only N‐coordinated. Accordingly, the zinc atom is more shielded in the quinoline complexes and a lactide molecule has limited access to the zinc atom. In the hydroquinoline complexes the N donors possess a more negative NBO charge (Ngua and Npy) so the N donors are more basic than in the quinoline complexes (Table 3). Hence, the nucleophilic attack to the lactide in the hydroquinoline systems is easier than in the quinoline complexes, resulting in higher activity.
Figure 2.

Overlay of the cationic complex units in C3 with [Zn(DMEGqu)2OTf]+ (left), C6 with [Zn(TMGqu)2OTf]+ (center), and C3 with C6 (right). H‐atoms are omitted for clarity.
Table 3.
Selected NBO charges and charge transfer energies of the complex cations in C3, C6, [Zn(DMEGqu)2OTf]+ and [Zn(TMGqu)2OTf]+ (NBO6.0, TPSSh/def2‐TZVP GD3BJ).
|
Cation |
NBO charge [e units] |
Charge transfer energies [kcal mol−1] |
||
|---|---|---|---|---|
|
Npy |
Ngua |
Npy→Zn |
Ngua→Zn |
|
|
C3 |
−0.62 |
−0.79 |
35 |
37 |
|
[Zn(DMEGqu)2OTf]+ |
−0.57 |
−0.76 |
|
|
|
C6 |
−0.61 |
−0.80 |
33 |
40 |
|
[Zn(TMGqu)2OTf]+ |
−0.56 |
−0.76 |
|
|
A more detailed investigation about the complexes’ activity in the ROP of lactide was examined only with complexes C3 and C6 because they were showing the most promising results. Both complexes were first tested in solvent‐free conditions with different catalyst concentrations. The polymerizations were now performed in a reactor monitored by in situ Raman spectroscopic measurements. For each catalyst concentration an apparent polymerization rate constant k app was determined by a semilogarithmic linear plot (Figures S26–S29).
As shown in Tables 4 and 5, high k app values in the ROP of lactide were achieved with both complexes, with an even higher activity of the TMG complex (C6) compared to the DMEG one (C3). The effect of the guanidine unit on the activity is already known from literature.[ 11h , 24 ] As shown in Table 3, the Ngua donor in C6 is the stronger donor than the Npy donor, whereas in C3 both donor atoms donate similar amounts of charge density to the Zn atom. The stronger coordination of the TMG moiety (in complex C6) shortens the Zn−Ngua bond lengths, and the ZnN2−ZnN2 plane angle between both ligands is higher for C6 [C3: 71.0(1)°; C6: 75.6°]. Therefore, the nucleophilic attack of the lactide is facilitated in C6.
Table 4.
Polymerization of rac‐lactide in bulk using [Zn(DMEGhydroqu)2](OTf)2 (C3).[a]
|
Entry |
[M]/[I] |
k app [b] [s−1] |
t [s] |
Conv.[c] [%] |
M n,theo. [d] [g mol−1] |
M n [e] [g mol−1] |
Ð [e] |
|---|---|---|---|---|---|---|---|
|
1 |
1000 : 1 |
0.0379 |
166 |
80 |
115000 |
45500 |
1.4 |
|
2 |
1250 : 1 |
0.0245 |
225 |
79 |
142000 |
77000 |
1.5 |
|
3 |
1500 : 1 |
0.0126 |
285 |
64 |
138000 |
94400 |
1.5 |
|
4 |
2000 : 1 |
0.0061 |
405 |
53 |
153000 |
82100 |
1.4 |
|
5 |
2500 : 1 |
0.0017 |
582 |
48 |
173000 |
74200 |
1.4 |
|
6[f] |
2500 : 1 |
0.0069 |
285 |
49 |
177000 |
146000 |
1.4 |
[a] Conditions: non‐purified technical grade rac‐lactide, solvent‐free conditions, 150 °C, stirrer speed: 260 rpm. [b] The rate constant k app was determined by plotting ln([LA]0/[LA]t) vs. t. [c] Determined by 1H NMR spectroscopy. [d] Theoretical number‐average molar mass calculated by conversion×molar mass×[M]/[I]. [e] Determined via GPC analysis in THF. Mn : determined number‐average molar mass [f] Sublimated lactide. Ð: polydispersity obtained by Mw /Mn .
Table 5.
Polymerization of rac‐lactide in solution and bulk using [Zn(TMGhydroqu)2](OTf)2 (C6).[a]
|
Entry |
[M]/[I] |
kapp [b] [s−1] |
t [s] |
Conv.[c] [%] |
M n,theo. [d] [g mol−1] |
M n [e] [g mol−1] |
Ð [e] |
|---|---|---|---|---|---|---|---|
|
1 |
1250 : 1 |
0.0451 |
165 |
90 |
162000 |
58100 |
1.5 |
|
2 |
1500 : 1 |
0.0285 |
161 |
53 |
115000 |
89000 |
1.4 |
|
3 |
2000 : 1 |
0.0028 |
345 |
38 |
110000 |
72500 |
1.5 |
|
4 |
2500 : 1 |
0.0009 |
885 |
31 |
– |
– |
– |
|
5[f] |
2500 : 1 : 10 |
0.0178 |
285 |
95 |
34200 |
33000 |
1.3 |
|
6[g] |
2500 : 1 |
0.0326 |
285 |
94 |
339000 |
71400 |
1.8 |
|
7[f,g] |
2500 : 1 : 1 |
0.0551 |
285 |
96 |
346000 |
83500 |
1.4 |
|
8[h] |
500 : 1 |
0.0023 |
660 |
61 |
44000 |
54000 |
1.4 |
|
9[h] |
600 : 1 |
0.0014 |
660 |
37 |
32000 |
29000 |
1.5 |
|
10[h] |
750 : 1 |
0.0012 |
660 |
48 |
n. d. |
n. d. |
n. d. |
|
11[h] |
800 : 1 |
0.0010 |
660 |
41 |
n. d. |
n. d. |
n. d. |
[a] Conditions in bulk: non‐purified technical grade rac‐lactide, 150 °C, stirrer speed: 260 rpm. [b] The rate constant k app was determined by plotting ln([LA]0/[LA]t) vs. t. [c] Determined by 1H NMR spectroscopy. [d] Theoretical number‐average molar mass calculated by conversion×molar mass×[M]/[I]. [e] Determined via GPC analysis in THF. Mn : determined number‐average molar mass; Ð: polydispersity obtained by Mw /Mn . [f] Co‐initiator: benzyl alcohol. [g] Sublimated lactide. [h] Using c(lactide)=1.0 mol L−1 at 100 °C in toluene. n. d.=not determined.
By using the two complexes in the lactide polymerization, colorless polymers were obtained, which is important for industrial applications. For a classification of the complex activities, a comparison with the currently fastest robust zinc complex is useful. The literature‐known catalyst [Zn{(R,R)‐DMEG2(1,2)ch}2](OTf)2 ⋅ THF, which is a zinc triflate bisguanidine complex, resulted in a k app value of 0.023 s−1 ([M]/[I]=1250 : 1, 150 °C, rac‐lactide, 441 s, 89 %). [11g] While complex C3 showed a similar activity (k app=0.025 s−1) under the same conditions, complex C6 exhibited a rate constant nearly twice as high (k app=0.045 s−1) (Tables 4 and 5). The literature‐known zinc triflate hybridguanidine complex [Zn(TMGqu)2OTf](OTf) can also be used for comparison of the influence in the backbone design on the catalytic activity. However, a comparison of k app values is not feasible as only very high catalyst concentration ([M]/[I] ratios of 1000 : 1 or less) were tested in literature due to the lower activity of this quinoline complex. Therefore, the rate constant k p was used for comparison, showing a three orders of magnitude higher k p value for C6 compared to the quinoline complex {C3: 7.73 L mol−1 s−1; C6: 14.9 L mol−1 s−1; [Zn(TMGqu)2OTf](OTf): 2.63×10−3 L mol−1 s−1} (Figure S30). [25] However, this comparison is limited by signs of catalyst degradation at low concentrations for C6 and also C3 as described below. Nevertheless, all comparisons highlight the impressive activity of both triflate complexes at low [M]/[I] ratios.
Only at low catalyst loadings ([M]/[I] ratio of 2000 : 1 or higher) the activity decreases for both complexes C3 and C6. This hints at a sensitivity towards too many impurities in the technical grade rac‐lactide. Even so, molar masses with at least 70 000 g mol−1 were received despite decreasing rate constants at higher [M]/[I] ratios (Tables 4 and 5). Thermal deactivation can be excluded, as with thermogravimetric analysis (TGA) measurements the thermal stability at industrially conditions for complex C3 was identified (Figure S23).
The decrease in activity at higher [M]/[I] ratios makes it difficult to determine a useful k p value for a comparison of different catalyst systems. Due to the initially very high activity at low [M]/[I] ratios and the rapidly decreasing reactivity at higher [M]/[I] ratios the k p value appears to be much higher, which is an artefact of the catalyst deactivation at high [M]/[I] ratios (Figure S30). Indeed, both complexes demonstrated a high activity but are also more sensitive towards impurities of technical grade lactide. This sensitivity might be caused by the high Lewis acidity of the complexes. Especially for complex C6 a huge activity drop is apparent. The deactivation of these complexes due to impurities in the technical grade rac‐lactide was proved with the use of additional co‐initiator and the use of purified lactide (Table 5, entries 5–7). With both methods, a higher rate constant could be obtained through a better initiation phase. The addition of a high amount of co‐initiator resulted in a higher rate constant and a controlled polymerization with good agreement between theoretical molar mass and the measured one (Table 5, entry 5). By using sublimated lactide the rate constant increased rapidly, and notwithstanding a high [M]/[I] ratio, the polymerization was completed after less than 5 min. The strong difference between experimental and theoretical molar masses, as well as the high polydispersity, show that termination reactions like transesterifications occurred despite such a short polymerization time (Table 5, entry 6). This indicated again the enormously high catalyst activity. With matrix‐assisted laser desorption ionization time‐of‐flight (MALDI‐ToF)‐MS end group analyses were performed. We prepared therefore short‐chain polymers for analyses by using an excess of co‐initiator. The results for both complexes (C3 and C6) revealed end groups characteristic of single‐site catalysts (Table S8, Figures S34 and S35).
Differential scanning calorimetry (DSC) experiments show that the choice of catalyst does not affect the material properties: rac‐lactide samples derived from both complexes (Table 2) yielded amorphous materials (Figure S24). The material exhibited a glass transition temperature (T g) of 47 (C3) and 48 °C (C6), respectively, which is consistent with the literature. [11g]
In addition, the polymerization of lactide was performed in solution. Here, the focus was on complex C6 as its high activity seems to be more promising for solution polymerization. This hypothesis was tested by determining the activity of C6 in toluene at various [M]/[I] ratios, obtaining high k app values (Table 5, Figure S36). C6 showed again a higher activity than the reported zinc triflate bisguanidine complex. The k app value of complex C6 is at least twice the published complex one [C6: k app=1.2×10−3 s−1, 750 : 1, 11 min, 48 % (Table 5, entry 10) compared to [Zn{(R,R)‐DMEG2(1,2)ch}2](OTf)2 ⋅ THF: k app=0.46×10−3 s−1, 750 : 1, 15 min, 28 %]. [11g]
The reported new ligand system enabled the design of new highly active complexes for the ROP of lactide showing that the combination of hydroquinoline backbones and triflate counterions revealed complexes with excellent activity. This reported complex class produces colorless polymers with high activity, up to a certain catalyst concentration, and high molar masses in the melt and solution polymerization of lactide.
Degradation studies
For the circular economy, the end of life of the material plays a decisive role. Thus, chemical recycling studies have been accomplished with the two most promising complexes, C3 and C6, for industrial use. Both complexes were investigated in the degradation of PLA into methyl lactate (Me‐LA) in solution. Me‐LA obtained by alcoholysis is a commodity chemical that can be used as green solvent or converted to the monomer lactide, creating circularity for PLA. [20] For methanolysis, commercially available polylactide in form of a cup was used and degraded in THF. The degradation process was tracked via 1H NMR spectroscopy of the methine region (δ between 4.1 and 5.2 ppm). Quantification of the relative concentrations of the methine groups allows to define these as internal (Int), chain end (CE), and methyl lactate (Me‐LA). Wood and co‐workers have previously reported a two‐step process for Me‐LA production with CE methine groups as intermediate.[ 21b , 21c ] The different methine proton groups allow the calculation of internal methine conversion (X int), methyl lactate selectivity (S Me‐LA), and methyl lactate yield (Y Me‐LA) (Figure S38), which can be used to compare the catalysts’ activity. [21d]
It was previously shown that different degradation temperatures can be used for the transesterification reaction, with mild conditions being preferred.[ 11e , 21a , 21b , 35 ] All degradation studies and kinetics were performed in Schlenk tubes in an oil bath by using 1.0 mol % catalyst and THF/MeOH. For a first impression of the PLA degradation activity a temperature of 60 °C was used. Under these conditions both complexes showed good activity in the degradation process; with complex C3 PLA is preferably consumed (Figure 3). C3 achieved nearly full consumption of PLA after 120 min at 60 °C, with a few remaining chain end groups and 81 % yield of Me‐LA (Table 6, Figure S43).
Figure 3.
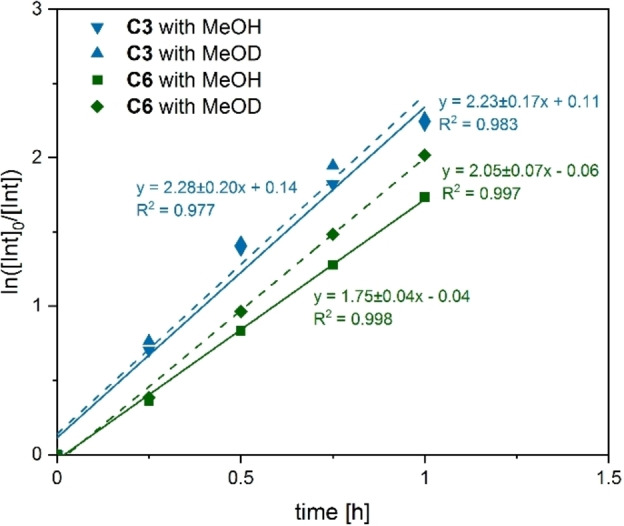
Pseudo‐first‐order semilogarithmic plots for the degradation of a PLA cup using C3 (blue) and C6 (green) as catalyst (1 mol % loading) in THF at 60 °C. Solid line: use of MeOH; dashed line: use of MeOD.
Table 6.
PLA cup degradation to methyl lactate using C3 and C6 in THF.[a]
|
Complex |
T [°C] |
t [h] |
X int [b] [%] |
S Me‐LA [b] [%] |
Y Me‐LA [b] [%] |
|---|---|---|---|---|---|
|
C3 |
40 |
6 |
91 |
78 |
71 |
|
60 |
2 |
97 |
83 |
81 |
|
|
60[c] |
2 |
97 |
83 |
81 |
|
|
C6 |
40 |
6 |
87 |
78 |
68 |
|
60 |
2 |
96 |
83 |
80 |
|
|
60[c] |
2 |
98 |
88 |
88 |
[a] Conditions: Mn (PLA cup)=54000 g mol−1; 1 mol % catalyst relative to ester linkages, V THF/V MeOH=4 : 1, n MeOH/n ester=7 : 1. [b] Determined by 1H NMR spectroscopy. [c] Use of methanol‐OD.
Milder reaction conditions at 40 °C demonstrated similar results after 6 h reaction time. Here, the Me‐LA conversion of C6 coincides with the one of C3 (Table 6, Figure S42). Kinetic plots of the PLA degradation at reaction conditions of 40 °C can be found in the Supporting Information (Figure S39) (C3: 0.680 h−1 and C6: 0.489 h−1). With these results the activity of both catalysts in the degradation of a post‐consumer PLA sample was proven, showing that complex C3 exhibited faster conversion from PLA to Me‐LA than C6 (Figures 3 and S39). This trend has been also observed for the chlorido complexes C1 and C4, although their degradation activity is considerably reduced compared to C3 and C6 (Table S9). A possible explanation could be different rate‐determining steps of the polymerization and depolymerization resulting in reverse trends. We performed, therefore, comparison experiments with methanol‐OD (MeOD) to analyze if the O−H bond cleavage is the rate‐determining step in the depolymerization reaction. As seen in Figure 3, the kinetic studies at 60 °C were plotted semilogarithmically, showing a linear plot for a pseudo first‐order kinetic process. For C3 and C6, no significant change in the reaction rate was observed by using deuterated methanol (Figure 3 and Figures S40 and S41). This indicates that no O−H bond scission or formation is involved in the rate‐determining step. The determination of a kinetic isotope effect (KIE) results in k H/k D=0.975±0.147 (C3) and 0.854±0.048 (C6). The KIE is identical for both complexes within the error range and is only marginally smaller than 1. This hints at a rate‐determining step that does not involve bond‐breaking or ‐formation processes for neither of the complexes. Therefore, the KIE experiments do not support a different rate‐determining step for the two complexes and rather suggest a similar one.
The kinetic activity of both zinc guanidine complexes demonstrates that this catalyst class can degrade PLA to Me‐LA faster than zinc complexes derived from bidentate Schiff‐base ligands. [15d] In comparison, state‐of‐the‐art zinc complexes based on tridentate Schiff base or {ONN} ligand systems showed roughly twice the activity in the methanolysis of PLA under similar mild reaction conditions.[ 21a , 21f ] In general, it is difficult to compare the results to other zinc‐mediated degradation outcomes as there are no standardized degradation conditions in literature (concerning the amount of catalyst, ratio of solvent to alcohol, etc.). A direct comparison with zinc guanidine carboxy complexes tested so far in chemical recycling shows an order of magnitude increased activity for C3 and C6 in the degradation process under same mild conditions. [22] This demonstrates the enormous potential of the presented zinc‐guanidine complexes for mediated alcoholysis of PLA. At the same time the results introduce zinc guanidine complexes with pure N donors as a new catalyst class for chemical recycling. Thus, C3 and C6 are attractive candidates for PLA formation and degradation, highlighting their potential to be used as catalysts in a circular economy.
Conclusion
A series of novel zinc guanidine hydroquinoline complexes was successfully synthesized and fully characterized. Their application in the ring‐opening polymerization (ROP) of non‐purified technical grade rac‐lactide under industrial conditions was investigated, showing high activity differences for various zinc salts. Two zinc guanidine complexes with triflate counterions proved to be the most active catalysts in the polymerization of lactide. The increased activity of the triflate complexes could be explained by density functional theory studies, as well as the influence of the modified ligand backbone. The aliphatic backbone results in higher natural bond orbital (NBO) charges of the guanidine N donor, facilitating the nucleophilic attack in the ROP.
Both complexes demonstrated high activity, yielding high conversions, molar masses up to 90000 g mol−1, and colorless polymers within a few minutes. With decreasing catalyst concentrations (at monomer/initiator ratios of around 2000 : 1), however, a massive decrease in activity was apparent. This deactivation is connected to the catalysts’ sensitivity towards too many impurities (which is again related to the high NBO charge of the guanidine N donor). Nevertheless, a polymerization in solution was possible with controlled molar masses and again high activities.
Both complexes were successfully applied in degradation studies of polylactide into methyl lactate under mild reaction conditions, although the complexes’ activity trend is reverse to the polymerization behavior.
Their efficiency in the methanolysis showed that zinc guanidine complexes with pure N donors are also excellent depolymerization catalysts, using them in a fully sustainable process in the circular approach.
This study emphasizes the importance of a sophisticated complex design to develop highly active zinc guanidine catalysts for a circular economy.
Experimental Section
Ligand synthesis and characterization
The ligand synthesis was carried out starting from rac‐5,6,7,8‐tetrahydroquinolin‐8‐ol (1). All intermediates as well as the ligands were therefore present as a mixture of two enantiomers.
8‐azido‐5,6,7,8‐tetrahydroquinoline (2): The title compound was prepared following partly the protocol of Uenishi and Hamada [26] and Bruns et al.: [27] 5,6,7,8‐tetrahydroquinolin‐8‐ol (1, 1.491 g, 10.00 mmol, 1.0 equiv.), DMAP (3.665 g, 30.00 mmol, 3.0 equiv.), and sodium azide were dissolved in dichloromethane (abs., 80 mL), and methanesulfonyl chloride (1.55 mL, 2.291 g, 20.00 mmol, 2.0 equiv.) was added at 0 °C. The reaction was stirred for 60 min at 0 °C. After warming up to RT, DMF (abs. 20 mL) was added, and the mixture was stirred overnight. The mixture was quenched with a sodium hydroxide solution (1 m, 50 mL) and extracted with hexane (3×50 mL). The combined organic layers were washed with brine (50 mL), dried over magnesium sulfate, filtered, and concentrated. The crude product was purified by column chromatography using hexane/EtOAc (7 : 3) (R f value=0.5) and additional 3 % of methanol as mobile phase to obtain the title compound as yellow oil (1.389 g, 80 %). 1H NMR (400 MHz, CDCl3): δ=8.50–8.48 (m, 1H), 7.46–7.44 (m, 1H), 7.17 (dd, 3 J=4.7 Hz, 3 J=7.8 Hz, 1H), 4.71 (t, 3 J=4.6 Hz, 1H), 2.87–2.69 (m, 2H). 2.10–2.00 (m, 2H), 1.98–1.90 (m, 1H), 1.85–1.79 ppm (m, 1H). The NMR spectroscopic data is in agreement with the reported literature. [26]
5,6,7,8‐tetrahydroquinolin‐8‐amine (3): The synthesis was performed following the description of Uenishi and Hamada. [26] To a solution of 8‐azido‐5,6,7,8‐tetrahydroquinoline (2, 1.342 g, 7.70 mmol, 1.0 equiv.) in methanol (abs. 60 mL), Pd/C (10 wt %, 0.410 g, 3.85 mmol, 0.5 equiv.) was added. The mixture was stirred under H2 atmosphere at RT overnight. Pd/C was filtered through a Celite® pad, washed with methanol, and the filtrate was concentrated afterwards. The product was obtained without further purification as yellow oil (1.007 g, 88 %). 1H NMR (400 MHz, CDCl3): δ=8.41–8.40 (m, 1H), 7.38–7.35 (m, 1H), 7.07 (dd, 3 J=4.7 Hz, 3 J=7.7 Hz, 1H), 4.01 (t, 3 J=5.4 Hz, 1H), 2.86–2.72 (m, 2H), 2.55 (br s, 2H, NH2), 2.25–2.19 (m, 1H), 2.01–1.92 (m, 1H), 1.84–1.67 ppm (m, 2H). The NMR spectroscopic data is in agreement with the reported literature.[ 26 , 27 ]
General synthesis of guanidine‐hydroquinoline ligands (L1 and L2): To an ice‐cooled solution of 5,6,7,8‐tetrahydroquinolin‐8‐amine (L1: 0.800 g, 5.40 mmol, 1.0 equiv.; L2: 1.380 g, 9.31 mmol, 1.0 equiv.) and triethylamine (1.1 equiv.) in acetonitrile (abs. 30 mL) a solution of chloroformamidinium chloride [DMEG‐VS (1.00 g, 5.94 mmol, 1.1 equiv.) or TMG‐VS (1.752 g, 10.2 mmol, 1.1 equiv.)] in acetonitrile (abs., 20 mL) was added dropwise. After 3 h at reflux, the mixture was cooled down to RT and an aqueous solution of NaOH (1.1 equiv. in 5 mL H2O) was added. The solvent and triethylamine were removed under reduced pressure. The remaining guanidine hydrochloride was deprotonated by KOH (50 wt %, in 10 mL H2O) and extracted with acetonitrile (3×20 mL). The organic phase was dried with Na2SO4 over activated carbon. After filtration over Celite®, the solvent was evaporated under reduced pressure and the product was dried under high vacuum.
DMEGhydroqu (L1): Yellow oil, 1.174 g (4.805 mmol, 89 %). Both stereoisomers of R‐ and S‐configuration were identified in the molecular structure in the solid state through XRD analysis. Therefore, in the liquid state (in the NMR analysis) a racemic mixture of the two isomers was present. 1H NMR (400 MHz, CDCl3): δ=8.42 (ddt, J=0.7 Hz, 4 J=1.8 Hz, 3 J=4.7 Hz, 1H, a), 7.32 (ddt, J=0.9 Hz, 4 J=1.9 Hz, 3 J=7.6 Hz, 1H, c), 6.99 (dd, 3 J=4.7 Hz, 3 J=7.6 Hz, 1H, b), 4.94 (t, 3 J=5.8 Hz, 1H, h), 3.26–3.14 (m, 4H, l), 2.98–2.84 (m, 6H, k), 2.78–2.69 (m, 2H, e), 2.18–2.09 (m, 1H, f), 2.03–1.89 (m, 2H, g/g’), 1.80–1.70 ppm (m, 1H, f’). 13C NMR (100 MHz, CDCl3): δ=160.3 (j), 156.8 (i), 147.3 (a), 136.6 (c), 132.2 (d), 121.2 (b), 55.5 (h), 49.8 (l), 37.4 (k), 33.6 (g), 29.3 (e), 19.7 ppm (f). IR (ATR): ṽ=3043 [vw, ṽ(CHarom)], 2954 [m, ṽ(CHaliph)], 2935 [m, ṽ(CHaliph)], 2833 [s, ṽ(CHaliph)], 1702 (vw), 1647 [vs, ṽ(C=Ngua)], 1572 (m), 1561 (w), 1477 (w), 1441 (m), 1423 (m), 1419 (m), 1378 (m), 1371 (m), 1353 (vw), 1343 (vw), 1330 (vw), 1266 (s), 1256 (s), 1231 (m), 1206 (w), 1199 (m), 1185 (w), 1179 (w), 1156 (vw), 1140 (vw), 1118 (w), 1110 (w), 1095 (w), 1087 (w), 1065 (w), 1051 (w), 1040 (m), 1010 (w), 994 (vw), 987 (vw), 981 (vw), 951 (s), 930 (vw), 881 (m), 863 (m), 854 (w), 831 (w), 810 (s), 787 (m), 740 (w), 719 (m), 637 (m), 595 (m), 582 (w), 564 (m), 545 cm–1 (w). MS EI: m/z [%]: 243.9 (4) [C14H20N4]+, 147.9 (3) [C9H12N2]+, 132.9 (3) [C9H11N]+, 113.9 (100) [C5H11N3]+, 98.9 (2) [C6H13N]+. HRMS ESI(+): m/z [%] calculated 244.1688 (100) [C14H20N4]+, found 244.1677 [C14H20N4]+ (100). Elemental analysis calculated [%] for C14H20N4: C 68.82, H 8.25, N 22.93; found: C 68.40, H 8.11, N 23.30. Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://doi.org/10.14272/reaction/SA‐FUHFF‐UHFFFADPSC‐BWTQRWNXOS‐UHFFFADPSC‐NUHFF‐NUHFF‐NUHFF‐ZZZ.
TMGhydroqu (L2): Yellow oil, 2.129 g (8.642 mmol, 93 %). Both stereoisomers of R‐ and S‐configuration were identified in the molecular structure in the solid state through XRD analysis. Therefore, in the liquid state (in the NMR analysis) a racemic mixture of the two isomers was present. 1H NMR (400 MHz, CDCl3): δ=8.38 (ddt, J=0.8 Hz, 4 J=1.7 Hz, 3 J=4.7 Hz, 1H, a), 7.30 (ddt, J=1.0 Hz, 4 J=1.9 Hz, 3 J=7.7 Hz, 1H, c), 6.97 (dd, 3 J=4.7 Hz, 3 J=7.6 Hz, 1H, b), 4.54 (t, 3 J=6.0 Hz, 1H, h), 2.89 (s, 6H, k), 2.87–2.73 (m, 2H, e), 2.70 (s, 6H, k), 2.15–2.06 (m, 1H, f), 1.97–1.83 (m, 2H, g/g’), 1.80–1.71 ppm (m, 1H, f’). 13C NMR (100 MHz, CDCl3): δ=160.6 (j), 147.3 (a), 136.8 (i), 136.5 (c), 132.2 (d), 121.1 (b), 58.0 (h), 40.5 (k), 38.8 (k), 33.1 (g), 29.4 (e), 20.3 (f) ppm. IR (ATR): ṽ=3144 [vw, ṽ(CHarom)], 3046 [vw, ṽ(CHarom)], 2938 [m, ṽ(CHaliph)], 2888 [m, ṽ(CHaliph)], 2798 [w, ṽ(CHaliph)], 1613 [vs, ṽ(C=Ngua)], 1584 (vs), 1573 (vs), 1473 (w), 1441 (m), 1425 (m), 1404 (s), 1363 (m), 1319 (w), 1234 (w), 1194 (w), 1171 (w), 1149 (w), 1128 (w), 1114 (w), 1088 (vw), 1067 (w), 1019 (m), 1008 (m), 995 (w), 946 (w), 934 (w), 901 (s), 894 (w), 859 (m), 838 (w), 827 (w), 804 (m), 780 (m), 755 (m), 721 (w), 700 (w), 685 (w), 646 (w), 614 (w), 573 (w), 563 (w), 506 cm–1 (vw). MS EI: m/z [%]: 245.9 (72) [C14H22N4]+, 200.9 (100) [C11H13N4]+, 185.9 (96) [C10H10N4]+, 132.9 (51) [C9H11N]+, 114.9 (4) [C5H13N3]+, 100.0 (22) [C6H14N]+, 78.0 (4) [C6H6]+. HRMS ESI(+): m/z [%] calculated 246.1845 (100) [C14H22N4]+, found 246.1839 (42) [C14H22N4]+. Elemental analysis calculated [%] for C14H20N4: C 68.26, H 9.00, N 22.74; found: C 68.67, H 8.67, N 23.05. Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://doi.org/10.14272/reaction/SA‐FUHFF‐UHFFFADPSC‐QXXJFVVUCP‐UHFFFADPSC‐NUHFF‐NUHFF‐NUHFF‐ZZZ.
General synthesis of zinc complexes
Both, ligand (0.500 mmol, 1 equiv. for complex C1, C2, C4, C5; 0.300 mmol, 1 equiv. for complex C3 and C6) and zinc salt (0.500 mmol, 1 equiv. for complex C1, C2, C4, C5; 0.120 mmol, 0.4 equiv. for complex C3 and C6) were each dissolved in THF (abs., 4 mL each) under heating. The warm solution of the zinc salt was added afterwards to the warm ligand solution. Single crystals were obtained.
[ZnCl2(DMEGhydroqu)] (C1): Yellow crystals, yield 0.121 g (0.318 mmol, 64 %). Both stereoisomers of R‐ and S‐configuration were identified in the molecular structure in solid state through XRD analysis. Therefore, in the liquid state (in the NMR analysis) a racemic mixture of the two isomers was present. 1H NMR (400 MHz, CDCl3): δ=8.49 (d, 3 J=5.1 Hz, 1H, a), 7.64 (d, 3 J=7.8 Hz, 1H, c), 7.36 (dd, 3 J=5.1 Hz, 3 J=7.7 Hz, 1H, b), 4.99 (dd, 3,4 J=5.1, 11.4 Hz, 1H, h), 3.76–3.72 (m, 2H, l), 3.40–3.30 (m, 2H, l), 3.08 (s, 6H, k), 2.96–2.83 (m, 2H, e), 2.40–2.34 (m, 1H, g), 2.08–2.01 (m, 1H, f), 1.98–1.83 (m, 1H, f’), 1.52–1.42 ppm (m, 1H, g’). 13C NMR (100 MHz, CDCl3): δ=166.5 (j), 157.2 (i), 145.6 (a), 139.9 (c), 133.5 (d), 123.8 (b), 56.1 (h), 49.6/49.5 (l), 36.5 (k), 29.2 (g), 27.7 (e), 20.6 ppm (f). IR (ATR): ṽ=2946 [w, ṽ(CHaliph)], 2876 [w, ṽ(CHaliph)], 1591 [s, ṽ(C=Ngua)], 1571 [vs, ṽ(C=Ngua)], 1506 (m), 1478 (m), 1456 (m), 1427 (m), 1417 (m), 1398 (m), 1379 (w), 1340 (w), 1329 (vw), 1287 (s), 1218 (vw), 1135 (w), 1099 (w), 1079 (vw), 1051 (w), 1036 (w), 1010 (m), 970 (w), 902 (m), 864 (m), 833 (m), 805 (s), 771 (s), 750 (m), 719 (m), 660 (m), 631 (m), 604 (w), 592 (m), 571 cm–1 (m). HRMS ESI(+): m/z [%] calculated 343.0668 (100) [C14H20ClN4Zn]+, 345.0637 (92) [C14H20 35ClN4 66Zn]+, 347.0625 (58) [C14H20 35ClN4 68Zn]+; found: 343.0659 (4) [C14H20ClN4Zn]+, 345.0628 (3) [C14H20 35ClN4 66Zn]+, 347.0613 (<1) [C14H20 35ClN4 68Zn]+. Elemental analysis calculated [%] for C14H20Cl2N4Zn: C 44.18, H 5.30, N 14.72; found: C 44.57, H 5.31, N 14.40. Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://doi.org/10.14272/reaction/SA‐FUHFF‐UHFFFADPSC‐VHSSPBJSZZ‐UHFFFADPSC‐NUHFF‐LUHFF‐NUHFF‐ZZZ.
[ZnBr2(DMEGhydroqu)] (C2): Yellow crystals, yield 0.186 g (0.396 mmol, 79 %). Both stereoisomers of R‐ and S‐configuration were identified in the molecular structure in the solid state through XRD analysis. Therefore, in the liquid state (in the NMR analysis) a racemic mixture of the two isomers was present. 1H NMR (400 MHz, CDCl3): δ=8.52 (d, 3 J=5.1 Hz, 1H, a), 7.64 (d, 3 J=7.7 Hz, 1H, c), 7.37 (dd, 3 J=5.2 Hz, 3 J=7.7 Hz, 1H, b), 5.01 (dd, 3,4 J=5.0, 11.4 Hz, 1H, h), 3.77–3.73 (m, 2H, l), 3.41–3.32 (m, 2H, l), 3.11 (s, 6H, k), 2.97–2.85 (m, 2H, e), 2.39–2.33 (m, 1H, g), 2.09–2.01 (m, 1H, f), 1.98–1.83 (m, 1H, f’), 1.54–1.44 ppm (m, 1H, g’). 13C NMR (100 MHz, CDCl3): δ=166.3 (j), 156.7 (i), 145.2 (a), 139.6 (c), 133.1 (d), 123.5 (b), 55.8 (h), 49.3 (l), 36.7 (k), 29.0 (g), 27.4 (e), 20.3 ppm (f). IR (ATR): ṽ=3076 (vw), 2941 [m, ṽ(CHaliph)], 2872 [w, ṽ(CHaliph)], 2840 [w, ṽ(CHaliph)], 1589 (m), 1568 [vs, ṽ(C=Ngua)], 1504 (m), 1478 (m), 1462 (m), 1454 (m), 1425 (m), 1415 (m), 1397 (w), 1379 (w), 1356 (vw), 1339 (m), 1328 (w), 1286 (s), 1273 (m), 1240 (m), 1217 (w), 1195 (w), 1187 (w), 1156 (vw), 1133 (m), 1119 (w), 1098 (w), 1077 (w), 1069 (w), 1051 (w), 1034 (w), 1010 (m), 988 (w), 970 (m), 944 (w), 915 (w), 901 (m), 863 (m), 831 (m), 811 (w), 801 (s), 770 (s), 749 (m), 719 (m), 660 (m), 630 (s), 604 (w), 591 (m), 570 cm–1 (m). HRMS ESI(+): m/z [%] calculated 387.0163 (65) [C14H20 79BrN4 64Zn]+, 389.0142 (100) [C14H20 81BrN4 64Zn]+, 391.0111 (63) [C14H20 81BrN4 66Zn]+ ; found 387.0149 (15) [C14H20 79BrN4 64Zn]+, 389.0124 (22) [C14H20 81BrN4 64Zn]+, 391.0100 (14) [C14H20 81BrN4 66Zn]+. Elemental analysis calculated [%] for C14H20Br2N4Zn: C 35.81, H 4.29, N 11.93; found: C 36.14, H 4.29, N 11.74. Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://doi.org/10.14272/reaction/SA‐FUHFF‐UHFFFADPSC‐DTLOYKSPTO‐UHFFFADPSC‐NUHFF‐LUHFF‐NUHFF‐ZZZ.
[Zn(DMEGhydroqu)2](OTf)2 (C3): Colorless crystals, yield 0.093 g (0.109 mmol, 91 %). For better splitting of diasteromeric signals NMR measurements were performed at higher temperatures: 1H NMR (400 MHz, CDCl3, 67 °C): δ=8.22 (d, 3 J=4.9 Hz, 2H, a), 7.86 (d, 3 J=4.7 Hz, 1H, a), 7.79 (d, 3 J=7.5 Hz, 2H, c), 7.72 (d, 3 J=7.8 Hz, 1H, c), 7.47 (dd, 3 J=5.4 Hz, 3 J=7.4 Hz, 2H, b), 7.33 (dd, 3 J=5.2 Hz, 3 J=7.6 Hz, 1H, b), 5.13–5.09 (m, 3H, h), 3.90–3.85 (m, 2H, l), 3.69–3.60 (m, 4H, l), 3.47–3.42 (m, 2H, l), 3.41–3.34 (m, 4H, l), 3.02–2.99 (m, 6H, e), 2.90 (s, 6H, k), 2.83 (s, 12H, k) 2.46–2.39 (m, 3H, g), 2.12–2.02 (m, 6H, f/f’), 1.58–1.46 ppm (m, 3H, g’). 13C NMR (100 MHz, CDCl3, 67 °C): δ=166.6 (j), 157.8/157.5 (i), 145.1/143.9 (a), 140.7/140.4 (c), 134.7/134.6 (d), 124.3/123.5 (b), 57.2/56.2 (h), 49.2/49.1 (l), 36.0/35.4 (k), 29.7/29.6 (g), 27.6/27.4 (e), 20.2 ppm (f). IR (ATR): ṽ=2952 [w, ṽ(CHaliph)], 2923 [m, ṽ(CHaliph)], 2855 [m, ṽ(CHaliph)], 1591 (m), 1574 [m, ṽ(C=Ngua)], 1508 (w), 1488 (vw), 1451 (m), 1424 (w), 1405 (w), 1378 (w), 1341 (w), 1271 [m, ṽ(SO3)], 1259 (s), 1223 (m), 1149 [m, ṽ(CF3)], 1076 (w), 1028 [s, ṽ(SO3)], 1015 (m), 971 (w), 901 (w), 868 (w), 831 (w), 800 (w), 772 (w), 748 (w), 723 (w), 664 (w), 637 [vs, δ(SO3)], 572 [m, δ(CF3)], 516 cm–1 [m, δ(SO3)]. HRMS (APCI+): m/z [%] calculated 457.0499 (100) [C15H20F3N4O3S64Zn]+ (ZnL1+OTf), 459.0469 (65) [C15H20F3N4O3S66Zn]+, 461.0457 (44) [C15H20F3N4O3S68Zn]+; found 457.0493 (73) [C15H20F3N4O3S64Zn]+, 459.0461 (46) [C15H20F3N4O3S66Zn]+, 461.0450 (31) [C15H20F3N4O3S68Zn]+. Elemental analysis calculated [%] for C30H40F6N8O6S2Zn: C 42.28, H 4.73, N 13.15; found: C 42.22, H 4.63, N 12.97. Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://doi.org/10.14272/reaction/SA‐FUHFF‐UHFFFADPSC‐QZYBMJJXWB‐UHFFFADPSC‐NUHFF‐LUHFF‐NUHFF‐ZZZ.
[ZnCl2(TMGhydroqu)] (C4): Yellow crystals, yield 0.114 g (0.298 mmol, 60 %). Both stereoisomers of R‐ and S‐configuration were identified in the molecular structure in the solid state through XRD analysis. Therefore, in the liquid state (in the NMR analysis) a racemic mixture of the two isomers was present. 1H NMR (400 MHz, CDCl3): δ=8.47 (d, 3 J=5.2 Hz, 1H, a), 7.64 (d, 3 J=7.8 Hz, 1H, c), 7.35 (dd, 3 J=5.2 Hz, 3 J=7.7 Hz, 1H, b), 4.59 (dd, 3,4 J=5.0, 11.2 Hz, 1H, h), 3.13 (s, 3H, k), 2.95–2.87 (m, 11H, k/e), 2.04–1.91 (m, 3H, f/f’/g), 1.51–1.41 ppm (m, 1H, g’). 13C NMR (100 MHz, CDCl3): δ=166.8 (j), 157.4 (i), 145.7 (a), 139.6 (c), 133.5 (d), 123.7 (b), 56.8 (h), 40.8/40.2/39.5/39.4 (k), 29.8 (g), 27.5 (e), 20.7 ppm (f). IR (ATR): ṽ=2945 [w, ṽ(CHaliph)], 2891 [w, ṽ(CHaliph)], 1592 (s), 1549 [vs, ṽ(C=Ngua)], 1532 [vs, ṽ(C=Ngua)], 1474 (w), 1458 (m), 1452 (m), 1445 (m), 1422 (m), 1404 (w), 1390 (s), 1353 (w), 1339 (w), 1303 (w), 1273 (w), 1237 (w), 1186 (w), 1161 (m), 1133 (m), 1110 (w), 1080 (w), 1067 (w), 1054 (w), 1034 (w), 1004 (m), 990 (w), 926 (w), 899 (m), 860 (w), 830 (w), 816 (m), 797 (m), 771 (m), 742 (w), 724 (w), 637 (m), 579 cm–1 (w). HRMS ESI(+): m/z [%] calculated 345.0824 (100) [C14H22 35ClN4 64Zn]+, 347.0796 (92) [C14H22 35ClN4 66Zn]+, 349.0782 (58) [C14H22 35ClN4 68Zn]+; found 345.0.827 (5) [C14H22 35ClN4 64Zn]+, 347.0796 (4) [C14H22 35ClN4 66Zn]+, 349.0783 (<1) [C14H22 35ClN4 68Zn]+. Elemental analysis calculated [%] for C14H22Cl2N4Zn: C 43.95, H 5.80, N 14.64; found: C 43.54, H 5.50, N 14.35. Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://doi.org/10.14272/reaction/SA‐FUHFF‐UHFFFADPSC‐PUPMHNBPAO‐UHFFFADPSC‐NUHFF‐LUHFF‐NUHFF‐ZZZ.
[ZnBr2(TMGhydroqu)] (C5): Yellow crystals, yield 0.181 g (0.384 mmol, 77 %). Both stereoisomers of R‐ and S‐configuration were identified in the molecular structure in the solid state through XRD analysis. Therefore, in the liquid state (in the NMR analysis) a racemic mixture of the two isomers was present. 1H NMR (400 MHz, CDCl3): δ=8.49 (d, 3 J=5.2 Hz, 1H, a), 7.64 (d, 3 J=7.8 Hz, 1H, c), 7.37 (dd, 3 J=5.2 Hz, 3 J=7.6 Hz, 1H, b), 4.60 (dd, 3,4 J=4.6, 11.4 Hz, 1H, h), 3.16 (s, 3H, k), 2.98–2.88 (m, 11H, k/e), 2.04–1.90 (m, 3H, f/f’/g), 1.53–1.43 ppm (m, 1H, g’). 13C NMR (100 MHz, CDCl3): δ=166.8 (j), 157.2 (i), 145.7 (a), 139.6 (c), 133.5 (d), 123.8 (b), 56.9 (h), 41.5/40.3/39.5/39.3 (k), 29.9 (g), 27.5 (e), 20.7 ppm (f). IR (ATR): ṽ=2941 [m, ṽ(CHaliph)], 2890 [w, ṽ(CHaliph)], 1592 (w), 1548 [vs, ṽ(C=Ngua)], 1531 [vs, ṽ(C=Ngua)], 1471 (m), 1457 (m), 1451 (m), 1444 (m), 1420 (m), 1403 (m), 1389 (s), 1351 (m), 1337 (m), 1305 (w), 1272 (w), 1236 (m), 1216 (vw), 1185 (w), 1161 (m), 1142 (w), 1132 (m), 1110 (vw), 1080 (w), 1066 (w), 1052 (w), 1034 (w), 1003 (m), 988 (w), 944 (w), 925 (w), 898 (m), 859 (w), 830 (m), 816 (m), 794 (m), 769 (m), 741 (w), 723 (m), 636 (m), 579 (m), 564 cm–1 (w). HRMS ESI(+): m/z [%] calculated 389.0319 (64) [C14H22 79BrN4 64Zn]+, 391.0299 (100) [C14H22 81BrN4 64Zn]+, 393.0268 (63) [C14H22 81BrN4 66Zn]+; found 389.0318 (<1) [C14H22 79BrN4 64Zn]+, 391.0292 (<1) [C14H22 81BrN4 64Zn]+, 393.0271 (<1) [C14H22 81BrN4 66Zn]+. Elemental analysis calculated [%] for C14H22Br2N4Zn: C 35.66, H 4.70, N 11.88; found: C 35.94, H 4.62, N 11.84. Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://doi.org/10.14272/reaction/SA‐FUHFF‐UHFFFADPSC‐QSTVHJCKUO‐UHFFFADPSC‐NUHFF‐LUHFF‐NUHFF‐ZZZ.
[Zn(TMGhydroqu)2] (OTf)2 (C6): Colorless crystals, yield 0.101 g (0.118 mmol, 98 %). For better splitting of diasteromeric signals NMR measurements were performed at higher temperatures: 1H NMR (400 MHz, CDCl3, 67 °C): δ=8.13–8.14 (m, 2H, a), 7.78–7.68 (m, 4H, a/c), 7.43 (dd, 3 J=5.4 Hz, 3 J=7.5 Hz, 2H, b), 7.30–7.26 (m, 1H, b), 4.82–4.78 (m, 1H, h), 4.73–4.70 (m, 2H, h), 3.01–2.71 (m, 42H, k/e), 2.06–2.04 (m, 6H, f/f’), 2.00–1.96 (m, 3H, g), 1.64–1.54 ppm (m, 3H, g’). 13C NMR (100 MHz, CDCl3, 67 °C): δ=167.3 (j), 157.5/157.1 (i), 144.9/143.9 (a), 140.4/139.8 (c), 134.6/134.5 (d), 124.0/123.5 (b), 57.8/56.7 (h), 40.6/40.3/40.2/39.9 (k), 30.3/29.8 (g), 27.5/27.4 (e), 20.8/20.7 ppm (f). IR (ATR): ṽ=2949 [vw, ṽ(CHaliph)], 2917 [vw, ṽ(CHaliph)], 2866 [vw, ṽ(CHaliph)], 1585 (vw), 1560 (m), 1540 [m, ṽ(C=Ngua)], 1481 (w), 1466 (w), 1452 (w), 1429 (w), 1413 (w), 1403 (m), 1356 (w), 1340 (w), 1262 [s, ṽ(CF3)], 1224 (m), 1168 (w), 1139 (m), 1082 (w), 1067 (vw), 1051 (vw), 1031 [s, ṽ(SO3)], 1004 (w), 928 (vw), 904 (vw), 888 (vw), 869 (vw), 831 (vw), 822 (w), 812 (w), 789 (vw), 775 (w), 752 (w), 740 (w), 722 (w), 636 [vs, δ(SO3)], 592 (vw), 584 (vw), 572 (w), 517 (m), 503 cm–1 (w). HRMS (APCI+): m/z [%] calculated 459.0656 (100) [C15H22F3N4O3S64Zn]+ (ZnL2+OTf), 461.0625 (65) [C15H22F3N4O3S66Zn]+, 463.0613 (43) [C15H22F3N4O3S68Zn]+; found 459.0646 (13) [C15H22F3N4O3S64Zn]+, 461.0614 (8) [C15H22F3N4O3S66Zn]+, 463.0629 (5) [C15H22F3N4O3S68Zn]+. Elemental analysis calculated [%] for C30H44F6N8O6S2Zn: C 42.08, H 5.18, N 13.09; found: C 42.26, H 5.14, N 12.90. Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://doi.org/10.14272/reaction/SA‐FUHFF‐UHFFFADPSC‐LGSDFUJUBG‐UHFFFADPSC‐NUHFF‐LUHFF‐NUHFF‐ZZZ.
Bulk polymerization
Method A (in Schlenk tubes): Polymerization in Schlenk tubes has been performed at a [M]/[I] ratio of 500 : 1, therefore technical rac‐lactide (1.00 g, 6.94 mmol) and the corresponding amount of catalyst C1–C6 (0.0139 mmol) were weighed in and homogenized in a mortar. The reaction mixture was added to Schlenk tubes with stirring bars, closed air‐proofed with a Young closure afterwards, and removed out of a nitrogen‐filled glovebox. The Schlenk tubes were fixed in a preheated oil bath of 150 °C (260 rpm). After 5 min or 6 h, the Schlenk tubes were removed from the oil bath and cooled down under running water. The samples were solved in DCM (2 mL); an aliquot was taken, cleared of DCM, and analyzed with 1H NMR spectroscopy (CDCl3). The dissolved polymer was than precipitated in ethanol (200 mL) at room temperature, dried under vacuum, and characterized via GPC.
Polymerization with technical grade l‐lactide (1.00 g, 6.94 mmol) has been performed at a [M]/[I] ratio of 1500 : 1 with complex C6 (4.0 mg, 4.63 10–3 mmol). The homogenized reaction mixture was added to Schlenk tubes with stirring bars, closed air‐proofed with a Young closure afterwards, and removed out of a nitrogen‐filled glovebox. The Schlenk tubes were fixed in a preheated oil bath of 150 °C (260 rpm), and after less than 3 min the reaction was terminated under running water. The further treatment of the polymer sample was done as described above.
Method B (in a reactor): A homogenous mixture of non‐purified technical grade rac‐lactide (8.00 g, 55.5 mmol), catalyst (according to the respective [M]/[I] ratio between 1000 : 1 and 2500 : 1), and in some cases benzyl alcohol were added into an argon‐flashed, preheated reactor at 150 °C. The in situ Raman measurement started directly after the reaction mixture insertion (150 °C, 260 rpm), as soon as the reactor was closed, and the reaction time was adjusted to the [M]/[I] ratio and the viscosity of the polymer. After the desired reaction time the reaction was stopped, and the crude product was analyzed by 1H NMR spectroscopy (CDCl3). The crude polymer was dissolved in an appropriate amount of DCM, precipitated in ethanol (200 mL) at room temperature, and dried under high vacuum. The molar mass of the polymer was characterized via GPC.
Solution polymerization
Non‐purified technical grade rac‐lactide (1.153 g, 8.00 mmol) and catalyst (according to the respective [M]/[I] ratio between 500 : 1 and 800 : 1) were added into Schlenk tubes in a nitrogen‐filled glovebox. Toluene (8 mL) was inserted in nitrogen counterflow to the Schlenk tube, which was fixed in an oil bath of 100 °C (260 rpm) afterwards. Aliquots were taken after defined reaction times and analyzed by 1H NMR spectroscopy (CDCl3). The reaction was terminated by cooling down under cold running water. Samples with high polymer conversion were dissolved in an appropriate amount of DCM, precipitated in ethanol (100 mL) at room temperature, and dried under vacuum. The molar mass of the polymer was characterized via GPC.
Depolymerization procedure
A Schlenk tube was loaded with catalyst (1 mol % related to ester linkages) and THF (2 mL, abs.) in a nitrogen‐filled glovebox. PLA (0.127 g, Huhtamäki, PLLA cup, M n=54000 g mol−1) was added under nitrogen counterflow to the Schlenk tube. The polymer was then dissolved in the solvent with heating. The Schlenk tube was immersed in a preheated oil bath (60 or 40 °C), to which MeOH (0.5 mL) or MeOD (0.51 mL) were added. The amount of methanol was defined as n MeOH/n ester=7 : 1. Aliquots were taken for 1H NMR spectroscopy (CDCl3) of the methine region.
Descriptions of all used materials and methods and further experimental details and information are detailed in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors thank the European Commission under H2020 Grant Agreement No. 953073 (acronym: UPLIFT) for funding, as well as Total Corbion PLA for lactide donations. Moreover, the authors acknowledge the financial support of the Bioeconomy Science Center as part of the project R2HPBio. The scientific activities of the Bioeconomy Science Center were financially supported by the Ministry of Innovation, Science and Research within the framework of the NRW Strategieprojekt BioSC (no. 313/323‐400‐002 13). We thank H. Hüppe and M. Paul for XRD measurements. We furthermore thank the Regional Computing Center of the University of Cologne (RRZK) for providing computing time on the DFG‐funded High Performance Computing (HPC) system CHEOPS as well as support. Thanks to B. Pütz for mass spectroscopic measurements, N. Avraham‐Radermacher for TGA measurements, S. Buschmann for DSC measurements and Dr. G. Fink for NMR measurements. We thank NFDI4Chem for support and funding of the repository RADAR4Chem. Open Access funding enabled and organized by Projekt DEAL.
A. Hermann, T. Becker, M. A. Schäfer, A. Hoffmann, S. Herres-Pawlis, ChemSusChem 2022, 15, e202201075.
Data Availability Statement
Additional information on the synthesis of the target compounds and original analysis data files are available via the Chemotion Repository (for corresponding links see Experimental Section). The MALDI‐ToF‐MS data as csv files are deposited in the repository RADAR4Chem by FIZ Karlsruhe ‐ Leibniz‐Institut für Informationsinfrastruktur and are published under an Open Access model (CC BY‐NC‐SA 4.0 Attribution‐NonCommercial‐ShareAlike; (DOI) 10.22000/693).
References
- 1.
- 1a. Geyer R., Jambeck J. R., Law K. L., Sci. Adv. 2017, 3, e1700782; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Heidbreder L. M., Bablok I., Drews S., Menzel C., Sci. Total Environ. 2019, 668, 1077–1093. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Jambeck J. R., Geyer R., Wilcox C., Siegler T. R., Perryman M., Andrady A., Narayan R., Law K. L., Science 2015, 347, 768–771; [DOI] [PubMed] [Google Scholar]
- 2b. Chamas A., Moon H., Zheng J., Qiu Y., Tabassum T., Jang J. H., Abu-Omar M., Scott S. L., Suh S., ACS Sustainable Chem. Eng. 2020, 8, 3494–3511. [Google Scholar]
- 3.
- 3a.Ellen MacArthur Foundation, in The New Plastic Economy: Rethinking the Future of Plastics, 2016, https://ellenmacarthurfoundation.org/the-new-plastics-economy-rethinking-the-future-of-plastics;
- 3b. Lambert S., Wagner M., Chem. Soc. Rev. 2017, 46, 6855–6871. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a.Ellen MacArthur Foundation, in The New Plastics Economy: Catalysing Action, 2017, https://ellenmacarthurfoundation.org/the-new-plastics-economy-rethinking-the-future-of-plastics-and-catalysing;
- 4b. Keijer T., Bakker V., Slootweg J. C., Nat. Chem. 2019, 11, 190–195. [DOI] [PubMed] [Google Scholar]
- 5. Gregory G. L., López-Vidal E. M., Buchard A., Chem. Commun. 2017, 53, 2198–2217. [DOI] [PubMed] [Google Scholar]
- 6. Payne J., McKeown P., Jones M. D., Polym. Degrad. Stab. 2019, 165, 170–181. [Google Scholar]
- 7.
- 7a. Vink E. T. H., Rábago K. R., Glassner D. A., Gruber P. R., Polym. Degrad. Stab. 2003, 80, 403–419; [Google Scholar]
- 7b. Groot W. J., Borén T., Int. J. Life Cycle Assess. 2010, 15, 970–984; [Google Scholar]
- 7c. Rezvani Ghomi E., Khosravi F., Saedi Ardahaei A., Dai Y., Neisiany R. E., Foroughi F., Wu M., Das O., Ramakrishna S., Polymer 2021, 13, 1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Drumright R. E., Gruber P. R., Henton D. E., Adv. Mater. 2000, 12, 1841–1846; [Google Scholar]
- 8b. Lasprilla A. J. R., Martinez G. A. R., Lunelli B. H., Jardini A. L., Filho R. M., Biotechnol. Adv. 2012, 30, 321–328; [DOI] [PubMed] [Google Scholar]
- 8c. Rabnawaz M., Wyman I., Auras R., Cheng S., Green Chem. 2017, 19, 4737–4753. [Google Scholar]
- 9. Linder M., Green Chem. Lett. Rev. 2017, 10, 428–435. [Google Scholar]
- 10.
- 10a. Leenslag J. W., Pennings A. J., Makromol. Chem. 1987, 188, 1809–1814; [Google Scholar]
- 10b. Zhang X., MacDonald D. A., Goosen M. F. A., McAuley K. B., J. Polym. Sci. Part A 1994, 32, 2965–2970; [Google Scholar]
- 10c. Stjerndahl A., Finne-Wistrand A., Albertsson A.-C., Bäckesjö C. M., Lindgren U., J. Biomed. Mater. Res. Part A 2008, 87A, 1086–1091; [DOI] [PubMed] [Google Scholar]
- 10d. Kricheldorf H. R., Chemosphere 2001, 43, 49–54; [DOI] [PubMed] [Google Scholar]
- 10e. Jones M. D., Wu X., Chaudhuri J., Davidson M. G., Ellis M. J., Mater. Sci. Eng. C 2017, 80, 69–74; [DOI] [PubMed] [Google Scholar]
- 10f. Lewis R. J., Sax N., in Sax's dangerous properties of industrial materials, Vol. 12, Van Nostrand Reinhold Company, New York, 1996. [Google Scholar]
- 11.
- 11a. Chamberlain B. M., Cheng M., Moore D. R., Ovitt T. M., Lobkovsky E. B., Coates G. W., J. Am. Chem. Soc. 2001, 123, 3229–3238; [DOI] [PubMed] [Google Scholar]
- 11b. Abbina S., Du G., ACS Macro Lett. 2014, 3, 689–692; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Thevenon A., Romain C., Bennington M. S., White A. J., Davidson H. J., Brooker S., Williams C. K., Angew. Chem. Int. Ed. 2016, 55, 8680–8685; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8822–8827; [Google Scholar]
- 11d. Schäfer P. M., McKeown P., Fuchs M., Rittinghaus R. D., Hermann A., Henkel J., Seidel S., Roitzheim C., Ksiazkiewicz A. N., Hoffmann A., Pich A., Jones M. D., Herres-Pawlis S., Dalton Trans. 2019, 48, 6071–6082; [DOI] [PubMed] [Google Scholar]
- 11e. McKeown P., Román-Ramírez L. A., Bates S., Wood J., Jones M. D., ChemSusChem 2019, 12, 5233–5238; [DOI] [PubMed] [Google Scholar]
- 11f. Yang R., Xu G., Lv C., Dong B., Zhou L., Wang Q., ACS Sustainable Chem. Eng. 2020, 8, 18347–18353; [Google Scholar]
- 11g. Hermann A., Hill S., Metz A., Heck J., Hoffmann A., Hartmann L., Herres-Pawlis S., Angew. Chem. Int. Ed. 2020, 59, 21778–21784; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 21962–21968; [Google Scholar]
- 11h. Schäfer P. M., Fuchs M., Ohligschläger A., Rittinghaus R., McKeown P., Akin E., Schmidt M., Hoffmann A., Liauw M. A., Jones M. D., Herres-Pawlis S., ChemSusChem 2017, 10, 3547–3556. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Rosen T., Goldberg I., Navarra W., Venditto V., Kol M., Angew. Chem. Int. Ed. 2018, 57, 7191–7195; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7309–7313; [Google Scholar]
- 12b. Sánchez-Barba L. F., Garcés A., Fajardo M., Alonso-Moreno C., Fernández-Baeza J., Otero A., Antiñolo A., Tejeda J., Lara-Sánchez A., López-Solera M. I., Organometallics 2007, 26, 6403–6411; [Google Scholar]
- 12c. Wang L., Ma H., Macromolecules 2010, 43, 6535–6537; [Google Scholar]
- 12d. Payne J., McKeown P., Driscoll O., Kociok-Köhn G., Emanuelsson E. A. C., Jones M. D., Polym. Chem. 2021, 12, 1086–1096; [Google Scholar]
- 12e. Rosen T., Rajpurohit J., Lipstman S., Venditto V., Kol M., Chem. Eur. J. 2020, 26, 17183–17189. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Rittinghaus R. D., Schäfer P. M., Albrecht P., Conrads C., Hoffmann A., Ksiazkiewicz A. N., Bienemann O., Pich A., Herres-Pawlis S., ChemSusChem 2019, 12, 2161–2165; [DOI] [PubMed] [Google Scholar]
- 13b. Marin P., Tschan M. J.-L., Isnard F., Robert C., Haquette P., Trivelli X., Chamoreau L.-M., Guérineau V., del Rosal I., Maron L., Venditto V., Thomas C. M., Angew. Chem. Int. Ed. 2019, 58, 12585–12589; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 12715–12719; [Google Scholar]
- 13c. Stewart J. A., McKeown P., Driscoll O. J., Mahon M. F., Ward B. D., Jones M. D., Macromolecules 2019, 52, 5977–5984; [Google Scholar]
- 13d. Rittinghaus R. D., Zenner J., Pich A., Kol M., Herres-Pawlis S., Angew. Chem. Int. Ed. 2022, 61, e202112853; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e2021128; [Google Scholar]
- 13e. Rittinghaus R. D., Karabulut A., Hoffmann A., Herres-Pawlis S., Angew. Chem. Int. Ed. 2021, 60, 21795–21800; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 21965–21971. [Google Scholar]
- 14.
- 14a. Mezzasalma L., Dove A. P., Coulembier O., Eur. Polym. J. 2017, 95, 628–634; [Google Scholar]
- 14b. Lim J. Y. C., Yuntawattana N., Beer P. D., Williams C. K., Angew. Chem. Int. Ed. 2019, 58, 6007–6011; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6068–6072. [Google Scholar]
- 15.
- 15a. McKeown P., McCormick S. N., Mahon M. F., Jones M. D., Polym. Chem. 2018, 9, 5339–5347; [Google Scholar]
- 15b. Schäfer P. M., Dankhoff K., Rothemund M., Ksiazkiewicz A. N., Pich A., Schobert R., Weber B., Herres-Pawlis S., ChemistryOpen 2019, 8, 1020–1026; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Fuchs M., Schmitz S., Schäfer P. M., Secker T., Metz A., Ksiazkiewicz A. N., Pich A., Kögerler P., Monakhov K. Y., Herres-Pawlis S., Eur. Polym. J. 2020, 122, 109302; [Google Scholar]
- 15d. Payne J., McKeown P., Mahon M. F., Emanuelsson E. A., Jones M. D., Polym. Chem. 2020, 11, 2381–2389; [Google Scholar]
- 15e. dos Santos Vieira I., Herres-Pawlis S., Eur. J. Inorg. Chem. 2012, 2012, 765–774; [Google Scholar]
- 15f. Schäfer P. M., Herres-Pawlis S., ChemPlusChem 2020, 85, 1044–1052. [DOI] [PubMed] [Google Scholar]
- 16. Lackmann C., Brendt J., Seiler T.-B., Hermann A., Metz A., Schäfer P. M., Herres-Pawlis S., Hollert H., J. Hazard. Mater. 2021, 416, 125889. [DOI] [PubMed] [Google Scholar]
- 17. Hong M., Chen E. Y. X., Green Chem. 2017, 19, 3692–3706. [Google Scholar]
- 18.
- 18a. McKeown P., Jones M. D., Sustain. Chem. 2020, 1, 1–22; [Google Scholar]
- 18b. Rahimi A., García J. M., Nat. Chem. Rev. 2017, 1, 0046. [Google Scholar]
- 19. Fan Y., Zhou C., Zhu X., Catal. Rev. Sci. Eng. 2009, 51, 293–324. [Google Scholar]
- 20.
- 20a. Pereira C. S. M., Silva V. M. T. M., Rodrigues A. E., Green Chem. 2011, 13, 2658–2671; [Google Scholar]
- 20b. Upare P. P., Hwang Y. K., Chang J.-S., Hwang D. W., Ind. Eng. Chem. Res. 2012, 51, 4837–4842. [Google Scholar]
- 21.
- 21a. Payne J. M., Kociok-Köhn G., Emanuelsson E. A. C., Jones M. D., Macromolecules 2021, 54, 8453–8469; [Google Scholar]
- 21b. Román-Ramírez L. A., McKeown P., Jones M. D., Wood J., ACS Catal. 2019, 9, 409–416; [Google Scholar]
- 21c. Román-Ramírez L. A., McKeown P., Jones M. D., Wood J., ACS Omega 2020, 5, 5556–5564; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21d. Román-Ramírez L. A., McKeown P., Shah C., Abraham J., Jones M. D., Wood J., Ind. Eng. Chem. Res. 2020, 59, 11149–11156; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21e. English L. E., Jones M. D., Liptrot D. J., ChemCatChem 2022, 14, e202101904; [Google Scholar]
- 21f. Santulli F., Lamberti M., Mazzeo M., ChemSusChem 2021, 14, 5470–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fuchs M., Walbeck M., Jagla E., Hoffmann A., Herres-Pawlis S., ChemPlusChem 2022, 87, e202200029. [DOI] [PubMed] [Google Scholar]
- 23. Metz A., McKeown P., Esser B., Gohlke C., Kröckert K., Laurini L., Scheckenbach M., McCormick S. N., Oswald M., Hoffmann A., Jones M. D., Herres-Pawlis S., Eur. J. Inorg. Chem. 2017, 2017, 5557–5570. [Google Scholar]
- 24. Börner J., Flörke U., Huber K., Döring A., Kuckling D., Herres-Pawlis S., Chem. Eur. J. 2009, 15, 2362–2376. [DOI] [PubMed] [Google Scholar]
- 25. Börner J., dos Santos Vieira I., Pawlis A., Döring A., Kuckling D., Herres-Pawlis S., Chem. Eur. J. 2011, 17, 4507–4512. [DOI] [PubMed] [Google Scholar]
- 26. Uenishi J., Hamada M., Synthesis 2002, 5, 0625–0630. [Google Scholar]
- 27. Bruns D., Merk D., Santhana Kumar K., Baumgartner M., Schneider G., ChemistryOpen 2019, 8, 1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raab V., Harms K., Sundermeyer J., Kovačević B., Maksić Z. B., J. Org. Chem. 2003, 68, 8790–8797. [DOI] [PubMed] [Google Scholar]
- 29. Yang L., Powell D. R., Houser R. P., Dalton Trans. 2007, 955–964. [DOI] [PubMed] [Google Scholar]
- 30. Börner J., Herres-Pawlis S., Flörke U., Huber K., Eur. J. Inorg. Chem. 2007, 2007, 5645–5651. [Google Scholar]
- 31.
- 31a. Weinhold F., Landis C. R., in Valency and bonding: a natural bond orbital donor-acceptor perspective, Cambridge University Press, New York, 2005; [Google Scholar]
- 31b.E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis, F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013;
- 31c. Glendening E. D., Landis C. R., Weinhold F., J. Comput. Chem. 2013, 34, 1429–1437. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Tao J., Perdew J. P., Staroverov V. N., Scuseria G. E., Phys. Rev. Lett. 2003, 91, 146401; [DOI] [PubMed] [Google Scholar]
- 32b. Staroverov V. N., Scuseria G. E., Tao J., Perdew J. P., J. Chem. Phys. 2003, 119, 12129–12137; [Google Scholar]
- 32c. Staroverov V. N., Scuseria G. E., Tao J., Perdew J. P., J. Chem. Phys. 2004, 121, 11507–11507. [Google Scholar]
- 33.
- 33a. Schäfer A., Huber C., Ahlrichs R., J. Chem. Phys. 1994, 100, 5829–5835; [Google Scholar]
- 33b. Eichkorn K., Weigend F., Treutler O., Ahlrichs R., Theor. Chem. Acc. 1997, 97, 119–124; [Google Scholar]
- 33c. Weigend F., Häser M., Patzelt H., Ahlrichs R., Chem. Phys. Lett. 1998, 294, 143–152; [Google Scholar]
- 33d. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456–1465; [DOI] [PubMed] [Google Scholar]
- 34b. Goerigk L., Grimme S., Phys. Chem. Chem. Phys. 2011, 13, 6670–6688; [DOI] [PubMed] [Google Scholar]
- 34c.For TPSSh, the values of the original paper have been substituted by the corrected values kindly provided by S. Grimme as private communication and published in;
- 34d. Hoffmann A., Grunzke R., Herres-Pawlis S., J. Comput. Chem. 2014, 35, 1943–1950. [DOI] [PubMed] [Google Scholar]
- 35. Fliedel C., Vila-Viçosa D., Calhorda M. J., Dagorne S., Avilés T., ChemCatChem 2014, 6, 1357–1367. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
Additional information on the synthesis of the target compounds and original analysis data files are available via the Chemotion Repository (for corresponding links see Experimental Section). The MALDI‐ToF‐MS data as csv files are deposited in the repository RADAR4Chem by FIZ Karlsruhe ‐ Leibniz‐Institut für Informationsinfrastruktur and are published under an Open Access model (CC BY‐NC‐SA 4.0 Attribution‐NonCommercial‐ShareAlike; (DOI) 10.22000/693).