

Abstract
Claudins (Cldns) form a large family of protein homologs that are essential for the assembly of paracellular tight junctions (TJs), where they form channels or barriers with tissue‐specific selectivity for permeants. In contrast to several family members whose physiological role has been identified, the function of claudin 4 (Cldn4) remains elusive, despite experimental evidence suggesting that it can form anion‐selective TJ channels in the renal epithelium. Computational approaches have recently been employed to elucidate the molecular basis of Cldns’ function, and hence could help in clarifying the role of Cldn4. In this work, we use structural modeling and all‐atom molecular dynamics simulations to transfer two previously introduced structural models of Cldn‐based paracellular complexes to Cldn4 to reproduce a paracellular anion channel. Free energy calculations for ionic transport through the pores allow us to establish the thermodynamic properties driving the ion‐selectivity of the structures. While one model shows a cavity permeable to chloride and repulsive to cations, the other forms barrier to the passage of all the major physiological ions. Furthermore, our results confirm the charge selectivity role of the residue Lys65 in the first extracellular loop of the protein, rationalizing Cldn4 control of paracellular permeability.
Keywords: claudins, free energy calculations, ion‐selectivity, molecular dynamics, paracellular space, tight junctions
In this work, we use structural modeling and all‐atom molecular dynamics simulations to transfer two previously introduced structural models of claudin (Cldn)‐based paracellular complexes to Cldn4 to reproduce a paracellular anion channel. Our results confirm the charge selectivity role of the residue Lys65 in the first extracellular loop of the protein, rationalizing Cldn4 control of paracellular permeability.

INTRODUCTION
Claudins (Cldns) form a family of 27 homologous proteins with a pivotal role in endothelial and epithelial tight junction (TJ) 1 , 2 , 3 , 4 , 5 , 6 structure and function. Available structural information shows that Cldns fold in a four‐helices bundle (named from TM1 to TM4) that embeds in the cellular membrane and anchors two extracellular loops (ECL1–2) and a loop in the cytoplasmic region, where the two terminal domains are also contained. Cldns assemble in TJs via: (1) the formation of strands of protomers lining on the same cell membrane, held together by cis interactions, and (2) intercellular aggregates involving trans interactions between the ECLs belonging to the opposite protomers, which seal the thin layer separating two neighboring cells, named the paracellular space. The ECLs arrange in a β‐sheet layer and, typically, ECL1 affects the TJ permeability, while ECL2 determines the trans interactions between Cldns belonging to two adjacent cells. 7 These proteins regulate the paracellular transport of ions and molecules via highly selective mechanisms, 8 , 9 and for this reason, their dysfunction is directly associated with clinical disorders. 10 , 11 While the physiological role of few Cldns has been extensively characterized, 6 , 7 others are still to be completely understood. A remarkable example is the claudin 4 member (Cldn4), highly expressed in the kidney but also present in other tissues. 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 Recent investigations suggest that Cldn4 may play a relevant role in the dynamics of tumor growth, and its attitude to bind the Clostridium perfringens enterotoxin (CPE), as a receptor, could open new approaches for drug delivery strategies. 20 , 21 , 22 Several studies suggest that Cldn4 is responsible for anion reabsorption in the collecting ducts, forming highly selective channels for chloride, 23 , 24 , 25 , 26 , 27 most likely due to a positively charged residue, Lys65, belonging to the ECL1 domain. 23 , 28 , 29 However, the ability of Cldn4 to form TJ strands has been found to depend on the cellular system in which it is expressed. 26 , 30 , 31 , 32 While biochemical data reported by Hou et al. 23 suggest that heterotypic interactions between Cldn4 and Cldn8 are required for a functional localization of the TJ in the kidney epithelial cells, homotypic Cldn4 interactions have also been observed. 33
In the last years, structural modeling and molecular dynamics (MD) simulations have become instrumental to grasp the fine details of biophysical processes, such as protein–protein aggregation, protein conformational transitions, and selectivity of ion channels. 34 More recently, MD simulations allowed the atomic description of the structural and functional features of various Cldn‐based paracellular aggregates, including claudin 15 (Cldn15), claudin 5 (Cldn5), and claudin 2 (Cldn2). 35 , 36 , 37 , 38 , 39 , 40 A step toward a detailed description of TJ proteins was the introduction of the first model for Cldn15‐based channels in Ref. 41, often referred to as “Suzuki model.” The proposed arrangement is formed by multiple copies of the crystal structure of the isolated Cldn15 monomer (PDB ID: 4P79), 42 and it is consistent with cys‐scanning mutagenesis experiments and freeze‐fracture microscopy imaging. In this multimeric assembly, linear strands of neighboring Cldns are held together by cis interactions. Strands between two opposite cells seal the paracellular space and the ECLs of opposite cis dimers form β‐barrel super‐secondary structures that result in pores of radius smaller than 5 Å. Subsequent computational works contributed to refine and validate the original Cldn15‐based model, 37 , 38 , 40 , 43 also extending the pore configuration to channels of other members of the Cldn family and assessing its consistency with the tissue‐specific physiological properties. 36 A second model of a Cldn‐based pore was suggested in Refs. 35 and 39. Accordingly, two protomers belonging to the same cell assemble in a dimer via interactions between the TM2 and the TM3 helices. 44 Focusing on Cldn5, the authors emphasized that the structural stability of the dimer is due to the formation of a leucine zipper involving residues Leu83, Leu90, Leu124, and Leu131, supported by two homophilic π‐π interactions between the aromatic residues Phe127 and Trp138 on the opposing TM domains. This specific arrangement for the Cldn5 protomer matches the experimental results obtained by Rossa et al. 45 As described in the computational investigation of Refs. 35 and 39, two copies of these dimers, belonging to two opposing cells, can associate across the paracellular space to form another pore configuration. Coarse‐grained (CG) self‐assembly simulations suggested that this second model could be suitable for the assembly of Cldn4‐based TJs 35 as well, but the application to other homologs is highly questionable due to experimental evidence. 46
In this work, we present a computational investigation of the Cldn4 protein in the two pore arrangements, in order to assess their transferability to this member of the Cldn family. Consistently with the notation of Refs. 35, 39, and 44, we refer to the two models described above as Pore I and Pore II, respectively. The two tetramers are simulated in explicit double membrane bilayer and water environments, and free energy (FE) calculations are performed for single ion or water molecule permeation through the paracellular spaces. Results show that the two models are permeable to water with no relevant FE barriers for both the pores. Pore I is attractive for chloride, since its energetic profile displays a minimum of −2.5 kcal/mol in the inner portion of the cavity, where the Lys65 residues 23 belonging to the four protomers form a cage that functions as a selectivity filter. Indeed, the passage of cations is prevented by barriers of ∼4 and ∼8 kcal/mol for monovalent and divalent cations, respectively, with peaks framed by the same basic residues. In contrast, due to the drastically different distribution of charged pore‐lining residues, Pore II is impermeable to all the tested ions, showing double FE barriers of ∼3 and ∼6 kcal/mol for monovalent and divalent cations, respectively, and a single barrier of ∼5 kcal/mol for the anion.
These results reveal that the specific pore arrangement can strongly affect the selectivity properties of Cldn‐based TJ pores. In particular, the thermodynamic features of ionic permeation in the different Cldn4 pore models suggest that only the Pore I structure is consistent with the anionic selectivity of Cldn4 paracellular channels.
METHODS
Pore I assembly
Pore I was assembled with four Cldn4 monomers, following the protocol illustrated in Ref. 37 and matching the structure published by Suzuki et al. 41 The experimental Cldn4 structure (PDB ID: 5B2G) 47 is unlikely to be representative of the protein native conformation, because of its binding to the C‐terminal fragment of the CPE. For this reason, the Cldn4 monomers were homology modeled starting from the Cldn15 structure, available in the isolated form (PDB ID: 4P79). 42 The SWISS‐MODEL platform 48 , 49 was used to generate the starting Cldn4 configuration. Subsequently, the model was refined with ModRefiner. 50 The resulting protomer was replicated in four units and superimposed on the template of the Suzuki model 41 using the UCSF Chimera 51 Matchmaker tool. Afterward, a further refinement of the system was performed with GalaxyRefineComplex. 52 , 53 The cis interactions occurring between two protomers in the same cell are shown in Figure 1A, and the tetrameric structure sealing the paracellular space is introduced in Figure 2A,B.
FIGURE 1.
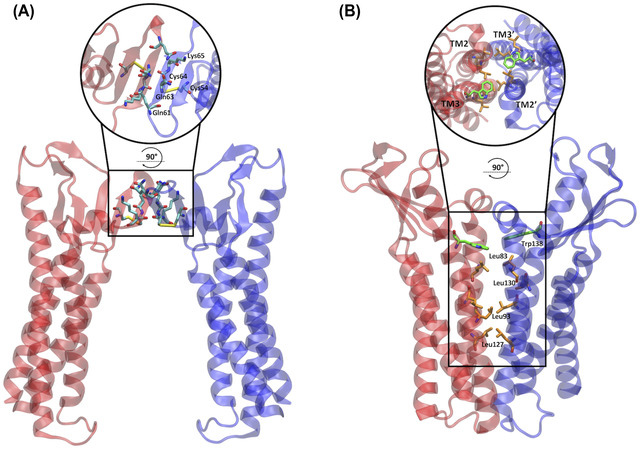
Dimeric cis interfaces of the two pore models. (A) In the Pore I model, two protomers of the same cell interact at the level of their ECLs, resulting in a hydrophilic interface. The apical zoomed view of the interface is shown in the circle. (B) In the Pore II model, two protomers of the same cell interact at the level of the TM domain forming a hydrophobic interface made by a leucine zipper involving the Leu83, Leu130, Leu93, and Leu127 residues and supported by the π–π interactions between the aromatic Trp138 sidechains. The apical view of the interface is shown in the circle.
FIGURE 2.
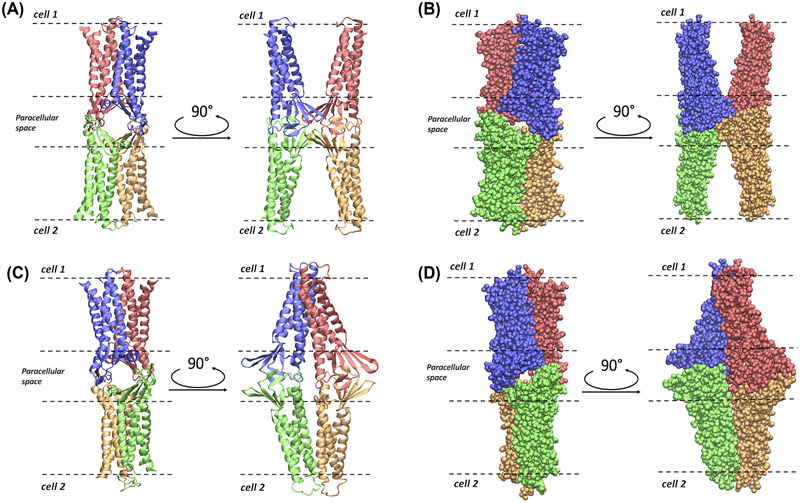
Representations of the Pore I and Pore II models. (A) Apical/basolateral and lateral view of the ribbon representation of the Pore I structure. (B) Apical/basolateral and lateral view of the Van der Waals representation of Pore I. (C) Apical/basolateral and lateral view of the ribbon representation of the Pore II structure. (D) Apical/basolateral and lateral view of the Van der Waals representation of Pore II. The four Cldn4 protomers are distinguished by their coloring. In all the panels, the dashed lines identify the boundaries of the membranes of two opposing cells separated by the paracellular space.
Pore II assembly
Because of the lack of a reference model based on experimental data of the pore, the strategy applied to reproduce the tetrameric architecture of the Pore II configuration was different from the one adopted for Pore I. First, we generated and equilibrated the cis dimer reproducing the leucine zipper (Figure 1B). A Cldn4 dimer with the specific protein–protein interface was obtained using MEMDOCK. 54 Following an additional refinement with the Rosetta DOCKING2 tool, 55 , 56 , 57 the structure was equilibrated in a homogeneous 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC) membrane, solvated with explicit three‐point (TIP3P) 58 water molecules, and neutralized with a physiological KCl concentration, and ∼250 ns of an all‐atom MD simulation was performed with the NAMD 3.0 program. 59 The CHARMM36m force field 60 was used, including the associated ionic parameters with the NBFIX corrections. 61 , 62 , 63 Then, ClusPro 64 , 65 , 66 , 67 , 68 was used to build the tetramer, starting from two copies of the equilibrated dimer. Positional restraints were included to keep the TM domains of the opposing dimers far from each other and to ensure the trans interactions of the ECL domains. Afterward, the structure was relaxed using the GalaxyRefineComplex server 52 , 53 (Figure 2C,D).
Double bilayer setup
Each tetrameric system was embedded in a double POPC bilayer, solvated with explicit three‐point (TIP3P) 58 water molecules, and neutralized with counterions, as described below. The pore axis was oriented along the VMD 69 y‐axis. The CHARMM PDB file of the protein complex was generated with the CHARMM‐GUI PDB manipulator tool. 70 , 71 A single hexagonal POPC bilayer, inscribed in a 128.0 × 128.0 Å square, was provided by the membrane builder tool of the same platform. 71 , 72 Two equilibrated copies of the membrane, based on NAMD simulations with the CHARMM36 force field, 73 were used to embed the pore transmembrane domains. After the removal of steric clashes between proteins and lipids, the resulting hexagonal box was filled with explicit TIP3P 58 water. When needed, counterions were added in both the cytosolic and the paracellular layers. The four disulfide bonds between the Cys54 and Cys64 residues were preserved.
Equilibration and unbiased MD simulation
After an initial short energy minimization, 30 ns of equilibration were performed by progressively releasing positional restraints on the heavy atoms. The system was simulated in the NPT ensemble at T = 310 K and P = 1 bar, maintained by a Langevin thermostat, with a damping coefficient of 1 ps, and a Nosé–Hoover Langevin piston, adopting an oscillation period of the piston of 50 fs and a damping time scale at 25 fs, respectively. 74 , 75 The NAMD 3.0 program 59 in combination with CHARMM36m force field 60 was used. The hexagonal box was inscribed in a square of about 128.0 × 128.0 Å and of height around 160.0 Å. Periodic boundary conditions were used to replicate the system and remove surface effects. Long‐range electrostatic interactions were computed using the particle‐mesh Ewald algorithm, 76 with an order 6 spline interpolation and a maximum space between grid points of 1.0 Å. Short range electrostatic and van der Waals interactions were calculated with a 12 Å cutoff and using a smoothing decay starting to take effect at 10 Å. A 16 Å pairlistdist was chosen for the neighbor list, and a 2 fs time‐step was employed. Chemical bonds involving hydrogen atoms and protein heavy atoms were constrained with SHAKE, 77 while those of the water molecules were kept fixed with SETTLE. 78
The transmembrane domain of each protomer was held fixed by harmonically restraining the C α atoms of residues 11, 14, 25, 28, 78, 81, 99, 102, 116, 119, 143, 146, 166, 169, 183, and 186 to their starting positions. Additional harmonic restraints on the Cα atoms of residues 30, 35, 40, 45, 50, 55, 60, 65, 70, 150, 153, 156, and 159, belonging to the ECLs, were added to Pore II during the production phase. The use of restraints on these atoms mimics the constriction exerted on the structure by the neighboring protomers in the TJ strand, that are not included in the single‐pore MD simulations. For each of the two systems, 250 ns of standard MD simulations were performed as additional equilibration, maintaining only the restraints of the C α atoms. The final configurations were adopted for the FE calculations.
Pore size analysis
The size of the paracellular channel was monitored along the trajectory with the HOLE program. 79 , 80 This algorithm maps the radius of a channel along a given axis (here, the paracellular channel was oriented along the VMD y‐axis) by fitting a spherical probe with the Amber Van der Waals radii 81 of the pore‐lining atoms. A threshold of 10 Å was chosen to define the boundaries of the channel.
Free energy calculations
Each FE profile was calculated with the umbrella sampling (US) method, 82 where a restraining potential term is added to the MD potential to confine a collective variable (CV) in selected regions, named windows, allowing proper sampling also of the high‐energy regions. Here, the CV is represented by the coordinate of the tagged permeating ion/water molecule along the pore axis, which was oriented along the VMD Cartesian y‐axis. The restraining potential Vi (y) in each window i is:
where yi 0 indicates the value in Å at which the CV is restrained in the window (called center) and k is a constant that is appropriately chosen in order to ensure a sufficient overlap of the CV distributions of adjacent windows (in this work, we used k = 2.0 kcal/(mol Å2) for all the simulations). In each window, the displacement of the ion orthogonal to the pore axis is confined within a disk of radius r 0 + δ, where r 0 is the pore radius as determined by the HOLE program 79 , 80 and δ = 2 Å. The equilibrated conformation of the system was used as the starting structure of all the US windows, and the ion/water molecule was manually positioned at each center yi 0. The Pore I channel axis was split into 60 windows spaced 1 Å from each other. After an initial minimization, 16 ns‐long trajectories were produced using the same setup and parameters of the unbiased MD simulation. The first 1 ns was excluded from the analysis for all the windows. Because of its elongated shape, sampling the Pore II model required 75 1‐Å‐spaced windows. The minimization, equilibration, and production procedures followed the same protocol of the Pore I MD run and up to 20 ns per window were simulated to achieve proper convergence of the FE profiles. The FE landscapes were obtained adopting the weighted histogram analysis method 83 , 84 , 85 and using the code available at http://membrane.urmc.rochester.edu/content/wham, 86 which calculates the statistical error associated with the FE estimation using the bootstrap method. The CV values were written every 10 ps.
Electrostatic potential surface
The electrostatic potential surface was computed with the adaptive Poisson–Boltzmann solver code, 87 using the default parameters set by the developers. Relative dielectric constants of 2 and 78.54 were used for the protein and the solvent, respectively, and the calculations were performed at a temperature of 298.15 K. The surface is shown with a red‐white‐blue color map ranging from −5 to +5 kT/e.
RESULTS
By comparing the two Cldn4‐based pore models, the position of the amino acids along the channel axis changes completely as a result of the opposite relative orientations of the monomers (Figure 3). In Pore I, we observe two pairs of ECL1 glutamine residues (Gln61 and Gln63) facing each other in the middle of the channel, with their sidechains pointing toward the lumen (Figure 3A,B). Moving away from the center along the axis, two pairs of Lys65 are found next to the glutamines, followed by other acidic (Asp48, Asp68, and Asp146) and basic (Arg31 and Arg158) residues. In Pore II, the positions of all these amino acids are inverted, with the glutamines at the mouths and the charged residues toward the central region (Figure 3C,D). In our MD simulation, the Pore I model revealed a remarkable stability of the paracellular β‐barrel super‐secondary structure. In particular, the sidechains of the pore lining residues maintained their relative orientation during the MD simulation. As illustrative examples, we report a set of representative distances between the amino C‐atom of Lys65 and the amide C‐atoms of Gln63, between the amino C‐atom of Lys65 and the amide C‐atoms of Gln61 sidechains, and between the amide C‐atoms of two neighboring Gln63 sidechains in Figure 4. Steady profiles around ∼5 and ∼6 Å are obtained for the distance between the Lys65 and the Gln63 sidechains and between the Lys65 and the Gln61 sidechains, respectively. Larger fluctuations are observed between the Gln63 residues from 50 to 150 ns of the simulated trajectory, but a stationary state around ∼4 Å is observed before and after that time window. Conversely, in Pore II, this network of interactions is not found due to the location of the Gln61, Gln63, and Lys65 residues of interacting monomers at opposing sides of the model.
FIGURE 3.
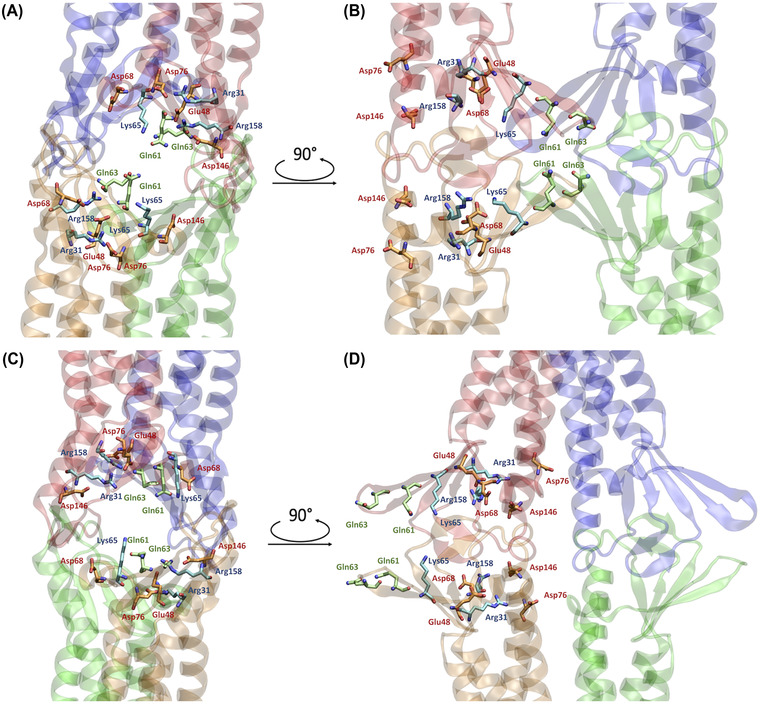
Representations of relevant residues in the two pore models. (A,B) Apical/basolateral A and lateral, B, views of the Pore I configuration. The β‐barrel arranged by the ECLs of the four protomers is visible. (C,D) Apical/basolateral, C, and lateral, D, views of the Pore II configuration. The reverse orientation of the pore‐lining residue compared to the Pore I is visible. The pore‐lining residues are indicated for two opposing protomers with respect to the paracellular plane. Acidic residues are depicted in orange, basic residues in light blue, and neutral residues in green. Oxygen and nitrogen atoms are shown in red and blue, respectively.
FIGURE 4.
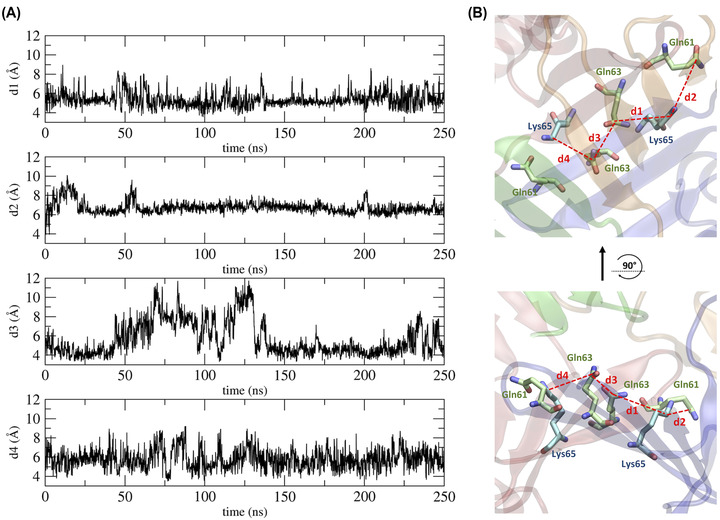
Distances between residues in the central region of the Pore I model. (A) Time evolution of distances between the amide C‐atom of the Gln residues and the amino C‐atom of the Lys residues. (B) 3D representation of the computed distances.
Free energy and electrostatic potential calculations
The physiological roles of Cldn‐based TJ pores are differentiated via their capacity to form channels or barriers to the passage of ions and molecules. In this work, we investigated the permeation of water and physiological ions through the two Cldn4 structures. FE profiles were calculated for both the systems using the US method. 82 Results are shown in Figures 5 and 6, for Pore I and Pore II, respectively. In these plots, the positions of the pore‐lining residues along the pore axis are overlaid to the FE profiles. In the Pore I model (Figure 5), a flat profile is obtained for the passage of the water molecule. On the contrary, barriers of ∼4 and 7–8 kcal/mol are observed for monovalent and divalent cations, respectively, all with a symmetrical shape with respect to the pore center, where the Gln61 and Gln63 sidechains are located. In this region, surrounded by the pore‐lining Lys65 sidechains pointing toward the pore lumen (Figure 3A,B), the pore radius reaches a minimum value (∼3 Å), as illustrated in Figure 7A. The four positively charged sidechains are symmetrically paired with respect to the cavity center, with two residues at ∼25 Å and two at ∼37 Å along the pore axis, generating a positively charged region in the inner section, as revealed by the electrostatic surface reported in Figure 8A. In contrast, the FE profile of the chloride is characterized by a minimum, ∼2.5 kcal/mol deep, with inflection points between the Arg31 and Arg158 residues, located close to the pore entrances with respect to the central glutamines (Figure 3A,B).
FIGURE 5.
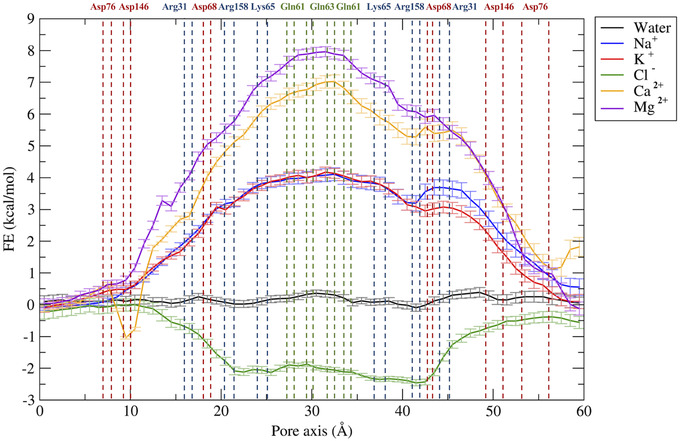
Free energy profiles for the permeation of water and ions through the Pore I model. The position of pore‐lining residues along the pore axis coordinates is indicated as dashed vertical lines. Acidic residues are colored in red, basic residues in blue, and neutral residues in green.
FIGURE 6.
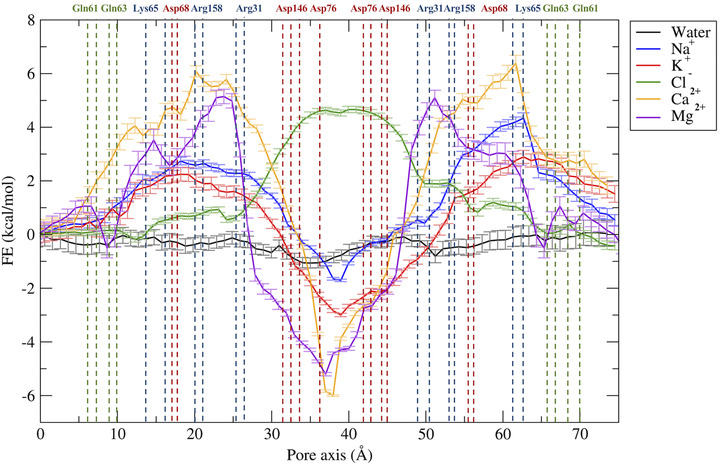
Free energy profiles for the permeation of water and ions through the Pore II model. The position of pore‐lining residues along the pore axis coordinate is indicated as dashed vertical lines. Acidic residues are colored in red, basic residues in blue, and neutral residues in green.
FIGURE 7.
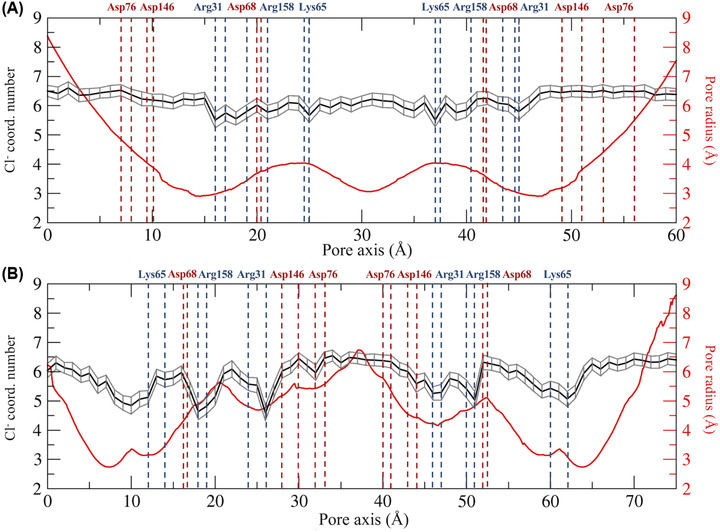
Hydration patterns of chloride and pore radius profile for the Pore I (A) and the Pore II (B) models. The position of the pore‐lining residues driving the ion selectivity is indicated. Acidic residues are shown in red and basic residues in blue.
FIGURE 8.
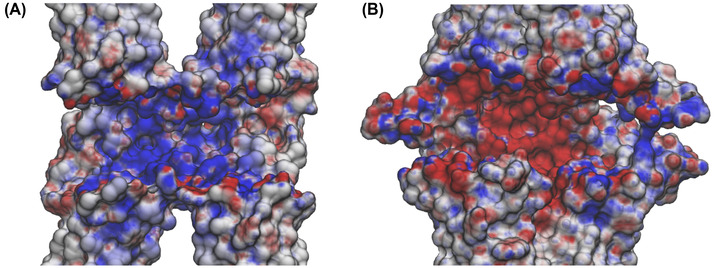
Electrostatic surface of the claudin 4 tetrameric models. Cross sections of the Pore I (A) and Pore II (B) electrostatic surfaces in the paracellular region computed with the adaptive Poisson–Boltzmann solver (APBS). Potentials are shown with a red‐white‐blue color map with values ranging from −5 to +5 kT/e.
In the Pore II structure, the FE profile for water permeation is also flat (Figure 6). Conversely, all the profiles for cations show two peaks localized at the opposite entrances. In comparison to the single peaks obtained for the Pore I model, smaller barriers oppose to the passage of both monovalent (∼2.5–3 kcal/mol) and divalent ions (∼5.5–6 kcal/mol). The two pairs of Lys65 residues are now close to the pore boundaries (Figure 3C,D), at ∼15 and ∼62 Å along the pore axis, but their position still correlates with the features of the FE profiles. Furthermore, the two FE peaks are located at the narrowest region of the channel, where the radius is at its minimum value (∼2.5 Å, Figure 7B). Between the two FE maxima, the profiles for the cations show minima at the cavity center due to the presence of a wide negatively charged chamber formed by the Asp146 and Asp76 residues, consistently with the electrostatic potential shown in Figure 8B. In this region, the Pore II configuration exhibits its maximum pore radius (∼6 Å, Figure 7B), and the electrostatic environment is defined by the acidic sidechains pointing toward the lumen of the channel and located between 32 and 45 Å along the pore axis (Figure 3C,D). The FE profile of the chloride shows a symmetrical barrier of ∼5 kcal/mol at the cavity center, correlating with the positions of the negatively charged sidechains of Asp76 and Asp146 (Figure 5). Remarkably, in Pore I, the acidic Asp76 and Asp146 residues do not obstacle the chloride flux because they are positioned close to the entrances, and thus have minimal interactions with the anion, as shown by the less extended acidic surface revealed by the electrostatic calculations (Figure 8A). In contrast, the anion passage is mainly driven by the inner, positively charged Arg31, Arg158, and Lys65 sidechains (Figure 3A,B), that are the major determinants of the Pore I cavity electrostatic potential.
Hydration scheme of the chloride ion
As a further investigation of the mechanisms associated with ion permeation through the two pore models, we computed the average coordination number of chloride in each US window, using a threshold radius of 3.5 Å. 88 , 89 , 90 Results are reported in Figure 7 as a function of the pore axis. The calculated hydration profiles reveal fluctuations in the number of coordinating water molecules surrounding the anion due to pore size variations and contacts with the charged pore‐lining residues in the two models. In Pore I (Figure 7A), whose radius varies smoothly and with limited differences in the inner region, minimal variations in the chloride hydration sphere are observed where, on average, the anion loses half water molecule in correspondence of the Arg31, Arg158, and Lys65 positions. The stabilizing interactions with the positively charged pore‐lining residues fill the slight depletion of the solvation sphere of the ion and favor its passage through the Pore I cavity, consistently with the energetic minima observed in Figure 5. Conversely, in the Pore II configuration, a more pronounced dehydration of the anion is observed. The chloride ion loses up to two water molecules in correspondence of the tight regions, where interactions with the sidechains of the positively charged residues (Lys65, Arg31, and Arg158) occur. On the contrary, in the inner part of the pore, the anion is fully hydrated, as a consequence of its passage through the widest region of the cavity and the occurrence of unfavorable interactions with the negatively charged pore‐lining residues Asp146 and Asp76 (Figure 7B), responsible for the FE barrier, hindering the permeation of the anion in the Pore II model (Figure 6).
DISCUSSION
The study of the ionic selectivity is a pivotal task to unravel the physiological function of biological channels. While in the literature there is a consolidated state‐of‐art for the computational investigation of transmembrane ion channel selectivity, 91 , 92 , 93 , 94 , 95 , 96 , 97 , 98 , 99 the study of paracellular channels is still limited to few works. 37 , 39 , 40 In this work, we compared the features of two different paracellular pore models, named as Pore I and Pore II, as putative TJ arrangements formed by Cldn4 proteins. In both the configurations, two copies of Cldn4 dimers, belonging to two neighboring cells, interact with each other in the paracellular space. The Pore I architecture was postulated in the work of Suzuki et al., 41 describing a homophilic strand of Cldn15‐based TJs starting from the crystal structure of the isolated protomer (PDB ID: 4P79). 42 Notably, it was also proposed independently by Irudayanathan et al. 39 for Cldns 3 and 5 from docking dimers spontaneously formed in CG MD simulations, first obtained for Cldn5. 44 This configuration was further investigated and refined in various studies for Cldn15, 37 , 38 , 40 , 43 Cldn2, 35 , 36 and Cldn5. 36 Conversely, the Pore II configuration was first introduced for Cldn5, 39 again by docking dimers assembled in CG MD simulations. 44 Comparisons of Pore I and II conformations were also made for Cldn10b and Cldn3, 46 and for Cldn2 and Cldn4, 100 based only on structural models, with no refinement by MD simulations. In particular, the study in Ref. 100 identified Lys65, Asp68, and Arg158 as key residues for Cldn4 anion selectivity.
Here, we used MD simulations and FE calculations to investigate the reliability of the two putative configurations as pore structures for Cldn4, and to further evaluate their transferability among different Cldn homologs in terms of preservation of tissue‐specific physiological functions, governed by the nonconserved ECL1 residues. 30 , 101 From a structural point of view, the two models exhibit significant differences at the intracellular, cis interfaces. In the Pore I architecture, two Cldn4 protomers of the same cell interact via a highly hydrophilic interface defined by the Gln61, Gln63, and Lys65 residues in the ECL1 domains (Figure 1A). Oppositely, the dimers originating the Pore II configuration are stabilized by a TM hydrophobic pattern, involving a leucine zipper supported by the π–π interactions provided by the TM3 Trp138 residues (Figure 1B). As previously described in Ref. 39 for Cldn5, although similar trans‐interactions are observed in both the models, they lead to opposite arrangements of the pore‐lining residues. The Pore I model is characterized by a central region formed by Gln61 and Gln63 pairs, surrounded by the two pairs of Lys65 residues symmetrically positioned with respect to the center, which contribute to the formation of a positively charged electrostatic surface (Figure 8A). Conversely, the Pore II residues arrangement generates an alternating charge environment with Lys65 close to the pore entrances, and two rings of negatively charged sidechains (Asp76 and Asp146) in the central region, resulting in a predominant acidic region in the inner section surrounded by a moderate basic potential at the pore entrances (Figure 8B). Consistently with the values proposed in Refs. 36 and 39 for the same pore models of other Cldns, the two configurations display a similar minimum pore radius between 2.5 and 3.0 Å (Figure 7). The Pore I duct shows a smooth profile, identifying two bottlenecks symmetrically paired at 15 and 45 Å on the pore axis, and a central constriction with a radius of 3 Å (Figure 7A). In contrast, the Pore II channel is tighter at the extremities and shows a central bulge with a radius of 6 Å (Figure 7B). In order to provide a quantitative assessment of the selectivity for the two configurations, we performed US simulations 82 to calculate the one‐dimensional FE for the permeation of water and physiological ions through the two pores. In the Pore I model, we observed an FE barrier to the passage of cations located at the cavity center with magnitude proportional to the ionic charge (Figure 5). The barriers encountered by the cations are located in the central region, where glutamines Gln61 and Gln63 are surrounded by the two pairs of Lys65. The overlap of the FE profiles of cations with same charge indicates a major role of the electrostatic interactions with respect to the steric effects in driving the passage of ions through the channel, similarly to what previously suggested by Alberini et al. 38 for the Cldn15 cation‐selectivity. In contrast, chloride is attracted through the channel by a ∼2.5 kcal/mol minimum spanning a region comprised between Arg31 and Arg158, and including the two pairs of Lys65 (Figure 5). Moreover, it was previously suggested that the passage of ions through the narrow region of cation selective Cldn15 38 , 102 and Cldn2 82 pores is associated with the loss of one or more hydrating water molecules. Here, analysis of the hydration state of the anion along the permeation pathway revealed a slight dehydration in correspondence of the positively charged pore‐lining residues, able to compensate for the missing water interactions (Figure 7A). On the contrary, the Pore II configuration shows barriers to the passage of all the ions. The pore‐lining residues sequence highlights a possible role of the Lys65 residues in reducing the permeation of cations at the pore entrance. The barriers associated with the cations in this pore arrangement are ∼1.5–2.0 kcal/mol, lower than those observed in Pore I, but symmetrically located at the two pore mouths. A pronounced minimum is found at the center of the FE profile corresponding to the position of the rings formed by the Asp76 and Asp146 residues that generate a negatively charged chamber with a diameter of almost 12 Å. Consistently, an FE barrier of 5 kcal/mol is obtained at the cavity center for the permeation of chloride (Figure 6). The hydration profile of the anion significantly varies along the cavity. In particular, the particle sheds almost two water molecules in correspondence of interactions between the ion and the positively charged sidechains of the Lys65, Arg31, and Arg158 residues and where the pore reveals major constrictions. Conversely, it preserves its hydration sphere in the central region of the cavity, where the unfavorable contact with the negatively charged residues takes place (Figure 7B) and the maximum in the FE profile is observed (Figure 6). This evidence confirms the driving role of electrostatic interactions in the formation of the FE barriers for the anionic permeation.
In conclusion, the two pore models suggest opposite behaviors for the Cldn4‐based TJ functionality, hence resulting mutually exclusive. Remarkably, the Pore I configuration provides the first atom‐detailed model recapitulating the observed anion‐selectivity of Cldn4‐based TJs. 23 , 24 , 25 , 27 In contrast, the Pore II model forms a barrier for both cations and anions. 28 , 32 , 101 , 103 , 104 The physiological role of Cldn4 as channel or barrier remains elusive, 23 , 24 , 25 , 26 , 27 , 28 , 30 , 32 , 33 , 101 , 103 , 104 , 105 and the factors modulating the expression of its functionality have not been fully clarified yet. 30 The capability to arrange in strands is also questioned due to the dependence on the cell expression system, 26 , 30 , 31 , 32 , 105 although experimental data supporting it have been reported. 106 Notwithstanding, our FE profiles for the Pore I configuration show higher barriers to the passage of the cations than Pore II. According to these considerations, we propose Pore I as the putative configuration to recapitulate the physiological properties of anion‐selective Cldn4‐based TJs. Further experimental characterization of Cldn4 physiology will ultimately provide the necessary knowledge to discriminate between different predicted configurations of Cldn4‐based TJs. Additional structural studies are also required to refine the peculiarities of the two architectures discussed in this work. In particular, the atom‐detailed characterization of Pore II model is currently less complete than Pore I model, and fewer experimental results supporting this arrangement are available to date. 45 Testing these models for different Cldns in terms of tissue‐specific TJ physiology will provide a fundamental contribution to their validation.
AUTHOR CONTRIBUTIONS
A.B. contributed to design the research, performed the simulations, analyzed the data, participated to the discussion for the data interpretation, and wrote the manuscript. G.A. shared his know‐how for the simulations setup, supervised the data production and analysis, participated to the discussion, and wrote the manuscript. L.M. and F.B. supervised the project, discussed the results, and revised the manuscript.
COMPETING INTERESTS
The authors declare no competing interests.
ACKNOWLEDGMENTS
We are grateful for the HPC infrastructure and the Support Team provided by the Fondazione Istituto Italiano di Tecnologia (IIT) that allowed us to run our MD simulations. We acknowledge Andrea L. Benfenati, Sergio Decherchi, and Diego Moruzzo for valuable help. We thank Jörg Piontek and Matteo Ceccarelli for useful discussions. The research was supported by IRCCS Ospedale Policlinico San Martino (Ricerca Corrente and 5×1000 grants to F.B. and L.M.) and by Telethon/Glut‐1 Onlus Foundations (GSP19002_PAsGlut009 and GSA22A002 to F.B.).
Open Access Funding provided by Universita Politecnica delle Marche within the CRUI‐CARE Agreement.
Berselli, A. , Alberini, G. , Benfenati, F. , & Maragliano, L. (2022). Computational study of ion permeation through claudin‐4 paracellular channels. Ann NY Acad Sci., 1516, 162–174. 10.1111/nyas.14856
Funding informationIRCCS Ospedale Policlinico San Martino; Telethon/Glut‐1 Onlus Foundations
Contributor Information
Fabio Benfenati, Email: fabio.benfenati@iit.it.
Luca Maragliano, Email: luca.maragliano@iit.it.
REFERENCES
- 1. Furuse, M. , Sasaki, H. , Fujimoto, K. , & Tsukita, S. (1998). A single gene product, claudin‐1 or ‐2, reconstitutes tight junction strands and recruits occludin in fibroblasts. Journal of Cell Biology, 143, 391–401. PMID: 9786950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morita, K. , Sasaki, H. , Furuse, M. , & Tsukita, S. (1999). Endothelial claudin. Journal of Cell Biology, 147, 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson, J. M. (2001). Molecular structure of tight junctions and their role in epithelial transport. News in Physiological Sciences, 16, 126–130. [DOI] [PubMed] [Google Scholar]
- 4. Farquhar, M. G. , & Palade, G. E. (1963). Junctional complexes in various epithelia. Journal of Cell Biology, 17, 375–412. PMID: 13944428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gumbiner, B. (1987). Structure, biochemistry, and assembly of epithelial tight junctions. American Journal of Physiology‐Cell Physiology, 253, C749–C758. [DOI] [PubMed] [Google Scholar]
- 6. Krause, G. , Winkler, L. , Mueller, S. L. , Haseloff, R. F. , Piontek, J. , & Blasig, I. E. (2008). Structure and function of claudins. Biochimica et Biophysica Acta (BBA) ‐ Biomembranes, 1778, 631–645. [DOI] [PubMed] [Google Scholar]
- 7. Krause, G. , Protze, J. , & Piontek, J. (2015). Assembly and function of claudins: Structure–function relationships based on homology models and crystal structures. Seminars in Cell & Developmental Biology, 42, 3–12. [DOI] [PubMed] [Google Scholar]
- 8. Powell, D. W. (1981). Barrier function of epithelia. American Journal of Physiology, 241, G275–G288. PMID: 7032321. [DOI] [PubMed] [Google Scholar]
- 9. Tang, V. W. , & Goodenough, D. A. (2003). Paracellular ion channel at the tight junction. Biophysical Journal, 84, 1660–1673. PMID: 12609869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martín De La Fuente, L. , Malander, S. , Hartman, L. , Jönsson, J.‐M. , Ebbesson, A. , Nilbert, M. , Måsbäck, A. , & Hedenfalk, I. (2018). Claudin‐4 expression is associated with survival in ovarian cancer but not with chemotherapy response. International Journal of Gynecological Pathology, 37, 101–109. PMID: 28481779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zihni, C. , Mills, C. , Matter, K. , & Balda, M. S. (2016). Tight junctions: From simple barriers to multifunctional molecular gates. Nature Reviews Molecular Cell Biology, 17, 564–580. PMID: 27353478. [DOI] [PubMed] [Google Scholar]
- 12. Soini, Y. (2005). Expression of claudins 1, 2, 3, 4, 5 and 7 in various types of tumours. Histopathology, 46, 551–560. PMID: 15842637. [DOI] [PubMed] [Google Scholar]
- 13. Nichols, L. S. , Ashfaq, R. , & Iacobuzio‐Donahue, C. A. (2004). Claudin 4 protein expression in primary and metastatic pancreatic cancer: Support for use as a therapeutic target. American Journal of Clinical Pathology, 121, 226–230. PMID: 14983936. [DOI] [PubMed] [Google Scholar]
- 14. Michl, P. , Buchholz, M. , Rolke, M. , Kunsch, S. , Löhr, M. , Mcclane, B. , Tsukita, S. , Leder, G. , Adler, G. , & Gress, T. M. (2001). Claudin‐4: A new target for pancreatic cancer treatment using Clostridium perfringens enterotoxin. Gastroenterology, 121, 678–684. PMID: 11522752. [DOI] [PubMed] [Google Scholar]
- 15. De Oliveira, S. S. , De Oliveira, I. M. , De Souza, W. , & Morgado‐Díaz, J. A. (2005). Claudins upregulation in human colorectal cancer. FEBS Letters, 579, 6179–6185. PMID: 16253248. [DOI] [PubMed] [Google Scholar]
- 16. Resnick, M. B. , Gavilanez, M. , Newton, E. , Konkin, T. , Bhattacharya, B. , Britt, D. E. , Sabo, E. , & Moss, S. F. (2005). Claudin expression in gastric adenocarcinomas: A tissue microarray study with prognostic correlation. Human Pathology, 36, 886–892. PMID: 16112005. [DOI] [PubMed] [Google Scholar]
- 17. Cunningham, S. C. , Kamangar, F. , Kim, M. P. , Hammoud, S. , Haque, R. , Iacobuzio‐Donahue, C. A. , Maitra, A. , Ashfaq, R. , Hustinx, S. , Heitmiller, R. E. , Choti, M. A. , Lillemoe, K. D. , Cameron, J. L. , Yeo, C. J. , Schulick, R. D. , & Montgomery, E. (2006). Claudin‐4, mitogen‐activated protein kinase kinase 4, and stratifin are markers of gastric adenocarcinoma precursor lesions. Cancer Epidemiology, Biomarkers & Prevention, 15, 281–287. [DOI] [PubMed] [Google Scholar]
- 18. Kominsky, S. L. , Vali, M. , Korz, D. , Gabig, T. G. , Weitzman, S. A. , Argani, P. , & Sukumar, S. (2004). Clostridium perfringens enterotoxin elicits rapid and specific cytolysis of breast carcinoma cells mediated through tight junction proteins claudin 3 and 4. American Journal of Pathology, 164, 1627–1633. PMID: 15111309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rangel, L. B. A. , Agarwal, R. , Souza, T. D. , Pizer, E. S. , Alò, P. L. , Lancaster, W. D. , Gregoire, L. , Schwartz, D. R. , Cho, K. R. , & Morin, P. J. (2003). Tight junction proteins claudin‐3 and claudin‐4 are frequently overexpressed in ovarian cancer but not in ovarian cystadenomas. Clinical Cancer Research, 9, 2567–2575. [PubMed] [Google Scholar]
- 20. Kondoh, M. , Takahashi, A. , Fujii, M. , Yagi, K. , & Watanabe, Y. (2006). A novel strategy for a drug delivery system using a claudin modulator. Biological & Pharmaceutical Bulletin, 29, 1783–1789. [DOI] [PubMed] [Google Scholar]
- 21. Suzuki, H. , Watari, A. , Hashimoto, E. , Yonemitsu, M. , Kiyono, H. , Yagi, K. , Kondoh, M. , & Kunisawa, J. (2015). C‐terminal Clostridium perfringens enterotoxin‐mediated antigen delivery for nasal pneumococcal vaccine. PLoS One, 10, e0126352. PMID: 26018248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shimizu, Y. , Isoda, K. , Taira, Y. , Taira, I. , Kondoh, M. , & Ishida, I. (2020). Anti‐tumor effect of a recombinant Bifidobacterium strain secreting a claudin‐targeting molecule in a mouse breast cancer model. European Journal of Pharmacology, 887, 173596. PMID: 32979353. [DOI] [PubMed] [Google Scholar]
- 23. Hou, J. , Renigunta, A. , Yang, J. , & Waldegger, S. (2010). Claudin‐4 forms paracellular chloride channel in the kidney and requires claudin‐8 for tight junction localization. Proceedings of the National Academy of Sciences of the United States of America, 107, 18010–18015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hou, J. , Rajagopal, M. , & Yu, A. S. L. (2013). Claudins and the kidney. Annual Review of Physiology, 75, 479–501. PMID: 23140368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hou, J. , Gomes, A. S. , Paul, D. L. , & Goodenough, D. A. (2006). Study of claudin function by RNA interference. Journal of Biological Chemistry, 281, 36117–36123. PMID: 17018523. [DOI] [PubMed] [Google Scholar]
- 26. Van Itallie, C. M. , Fanning, A. S. , & Anderson, J. M. (2003). Reversal of charge selectivity in cation or anion‐selective epithelial lines by expression of different claudins. American Journal of Physiology‐Renal Physiology, 285, F1078–F1084. [DOI] [PubMed] [Google Scholar]
- 27. Gong, Y. , Yu, M. , Yang, J. , Gonzales, E. , Perez, R. , Hou, M. , Tripathi, P. , Hering‐Smith, K. S. , Hamm, L. L. , & Hou, J. (2014). The Cap1–claudin‐4 regulatory pathway is important for renal chloride reabsorption and blood pressure regulation. Proceedings of the National Academy of Sciences of the United States of America, 111, E3766–E3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Borovac, J. , Barker, R. S. , Rievaj, J. , Rasmussen, A. , Pan, W. , Wevrick, R. , & Alexander, R. T. (2012). Claudin‐4 forms a paracellular barrier, revealing the interdependence of claudin expression in the loose epithelial cell culture model opossum kidney cells. American Journal of Physiology‐Cell Physiology, 303, C1278–C1291. PMID: 23076790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Colegio, O. R. , Van Itallie, C. M. , Mccrea, H. J. , Rahner, C. , & Anderson, J. M. (2002). Claudins create charge‐selective channels in the paracellular pathway between epithelial cells. American Journal of Physiology‐Cell Physiology, 283, C142–C147. PMID: 12055082. [DOI] [PubMed] [Google Scholar]
- 30. Piontek, J. , Krug, S. M. , Protze, J. , Krause, G. , & Fromm, M. (2020). Molecular architecture and assembly of the tight junction backbone. Biochimica et Biophysica Acta (BBA) ‐ Biomembranes, 1862, 183279. [DOI] [PubMed] [Google Scholar]
- 31. Günzel, D. , & Fromm, M. (2012). Claudins and other tight junction proteins. Comprehensive Physiology, 2, 1819–1852. PMID: 23723025. [DOI] [PubMed] [Google Scholar]
- 32. Krug, S. M. , Günzel, D. , Conrad, M. P. , Lee, I. N.‐F. M. , Amasheh, S. , Fromm, M. , & Yu, A. S. L. (2012). Charge‐selective claudin channels. Annals of the New York Academy of Sciences, 1257, 20–28. PMID: 22671585. [DOI] [PubMed] [Google Scholar]
- 33. Daugherty, B. L. , Ward, C. , Smith, T. , Ritzenthaler, J. D. , & Koval, M. (2007). Regulation of heterotypic claudin compatibility. Journal of Biological Chemistry, 282, 30005–30013. PMID: 17699514. [DOI] [PubMed] [Google Scholar]
- 34. Roux, B. (2017). Ion channels and ion selectivity. Essays in Biochemistry, 61, 201–209. PMID: 28487397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Irudayanathan, F. J. , Wang, X. , Wang, N. , Willsey, S. R. , Seddon, I. A. , & Nangia, S. (2018). Self‐assembly simulations of classic claudins—Insights into the pore structure, selectivity, and higher order complexes. Journal of Physical Chemistry B, 122, 7463–7474. PMID: 29869889. [DOI] [PubMed] [Google Scholar]
- 36. Irudayanathan, F. J. , & Nangia, S. (2020). Paracellular gatekeeping: What does it take for an ion to pass through a tight junction pore? Langmuir, 36, 6757–6764. PMID: 32450698. [DOI] [PubMed] [Google Scholar]
- 37. Alberini, G. , Benfenati, F. , & Maragliano, L. (2017). A refined model of claudin‐15 tight junction paracellular architecture by molecular dynamics simulations. PLoS One, 12, e0184190. PMID: 28863193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alberini, G. , Benfenati, F. , & Maragliano, L. (2018). Molecular dynamics simulations of ion selectivity in a claudin‐15 paracellular channel. Journal of Physical Chemistry B, 122, 10783–10792. PMID: 30372067. [DOI] [PubMed] [Google Scholar]
- 39. Irudayanathan, F. J. , Wang, N. , Wang, X. , & Nangia, S. (2017). Architecture of the paracellular channels formed by claudins of the blood–brain barrier tight junctions. Annals of the New York Academy of Sciences, 1405, 131–146. PMID: 28614588. [DOI] [PubMed] [Google Scholar]
- 40. Samanta, P. , Wang, Y. , Fuladi, S. , Zou, J. , Li, Y. E. , Shen, L. E. , Weber, C. , & Khalili‐Araghi, F. (2018). Molecular determination of claudin‐15 organization and channel selectivity. Journal of General Physiology, 150, 949–968. PMID: 29915162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suzuki, H. , Tani, K. , Tamura, A. , Tsukita, S. , & Fujiyoshi, Y. (2015). Model for the architecture of claudin‐based paracellular ion channels through tight junctions. Journal of Molecular Biology, 427, 291–297. PMID: 25451028. [DOI] [PubMed] [Google Scholar]
- 42. Suzuki, H. , Nishizawa, T. , Tani, K. , Yamazaki, Y. , Tamura, A. , Ishitani, R. , Dohmae, N. , Tsukita, S. , Nureki, O. , & Fujiyoshi, Y. (2014). Crystal structure of a claudin provides insight into the architecture of tight junctions. Science, 344, 304–307. PMID: 24744376. [DOI] [PubMed] [Google Scholar]
- 43. Zhao, J. , Krystofiak, E. S. , Ballesteros, A. , Cui, R. , Van Itallie, C. M. , Anderson, J. M. , Fenollar‐Ferrer, C. , & Kachar, B. (2018). Multiple claudin–claudin cis interfaces are required for tight junction strand formation and inherent flexibility. Communications Biology, 1, 50. PMID: 30271933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Irudayanathan, F. J. , Trasatti, J. P. , Karande, P. , & Nangia, S. (2016). Molecular architecture of the blood brain barrier tight junction proteins–A synergistic computational and in vitro approach. Journal of Physical Chemistry B, 120, 77–88. PMID: 26654362. [DOI] [PubMed] [Google Scholar]
- 45. Rossa, J. , Ploeger, C. , Vorreiter, F. , Saleh, T. , Protze, J. , Günzel, D. , Wolburg, H. , Krause, G. , & Piontek, J. (2014). Claudin‐3 and claudin‐5 protein folding and assembly into the tight junction are controlled by non‐conserved residues in the transmembrane 3 (TM3) and extracellular loop 2 (ECL2) segments. Journal of Biological Chemistry, 289, 7641–7653. PMID: 24478310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hempel, C. , Protze, J. , Altun, E. , Riebe, B. , Piontek, A. , Fromm, A. , Lee, I. M. , Saleh, T. , Günzel, D. , Krause, G. , & Piontek, J. (2020). Assembly of tight junction strands: Claudin‐10b and claudin‐3 form homo‐tetrameric building blocks that polymerise in a channel‐independent manner. Journal of Molecular Biology, 432, 2405–2427. PMID: 32142789. [DOI] [PubMed] [Google Scholar]
- 47. Shinoda, T. , Shinya, N. , Ito, K. , Ohsawa, N. , Terada, T. , Hirata, K. , Kawano, Y. , Yamamoto, M. , Kimura‐Someya, T. , Yokoyama, S. , & Shirouzu, M. (2016). Structural basis for disruption of claudin assembly in tight junctions by an enterotoxin. Science Reports, 6, 33632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Waterhouse, A. , Bertoni, M. , Bienert, S. , Studer, G. , Tauriello, G. , Gumienny, R. , Heer, F. T. , De Beer, T. A. P. , Rempfer, C. , Bordoli, L. , Lepore, R. , & Schwede, T. (2018). SWISS‐MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, 46, W296–W303. PMID: 29788355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Benkert, P. , Biasini, M. , & Schwede, T. (2011). Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics, 27, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu, D. , & Zhang, Y. (2011). Improving the physical realism and structural accuracy of protein models by a two‐step atomic‐level energy minimization. Biophysical Journal, 101, 2525–2534. PMID: 22098752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pettersen, E. F. , Goddard, T. D. , Huang, C. C. , Couch, G. S. , Greenblatt, D. M. , Meng, E. C. , & Ferrin, T. E. (2004). UCSF Chimera–A visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25, 1605–1612. PMID: 15264254. [DOI] [PubMed] [Google Scholar]
- 52. Heo, L. , Lee, H. , & Seok, C. (2016). GalaxyRefineComplex: Refinement of protein–protein complex model structures driven by interface repacking. Science Reports, 6, 32153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Heo, L. , Park, H. , & Seok, C. (2013). GalaxyRefine: Protein structure refinement driven by side‐chain repacking. Nucleic Acids Research, 41, W384–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hurwitz, N. , Schneidman‐Duhovny, D. , & Wolfson, H. J. (2016). Memdock: An α‐helical membrane protein docking algorithm. Bioinformatics, 32, 2444–2450. [DOI] [PubMed] [Google Scholar]
- 55. Lyskov, S. , & Gray, J. J. (2008). The RosettaDock server for local protein–protein docking. Nucleic Acids Research, 36, W233–W238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chaudhury, S. , Berrondo, M. , Weitzner, B. D. , Muthu, P. , Bergman, H. , & Gray, J. J. (2011). Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS One, 6, e22477. PMID: 21829626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lyskov, S. , Chou, F.‐C. , Conchúir, S. Ó. , Der, B. S. , Drew, K. , Kuroda, D. , Xu, J. , Weitzner, B. D. , Renfrew, P. D. , Sripakdeevong, P. , Borgo, B. , Havranek, J. J. , Kuhlman, B. , Kortemme, T. , Bonneau, R. , Gray, J. J. , & Das, R. (2013). Serverification of molecular modeling applications: The Rosetta Online Server that Includes Everyone (ROSIE). PLoS One, 8, e63906. PMID: 23717507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jorgensen, W. L. , Chandrasekhar, J. , Madura, J. D. , Impey, R. W. , & Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics, 79, 926–935. [Google Scholar]
- 59. Phillips, J. C. , Hardy, D. J. , Maia, J. D. C. , Stone, J. E. , Ribeiro, J. V. , Bernardi, R. C. , Buch, R. , Fiorin, G. , Hénin, J. , Jiang, W. , Mcgreevy, R. , Melo, M. C. R. , Radak, B. K. , Skeel, R. D. , Singharoy, A. , Wang, Y. I. , Roux, B. , Aksimentiev, A. , Luthey‐Schulten, Z. , … Tajkhorshid, E. (2020). Scalable molecular dynamics on CPU and GPU architectures with NAMD. Journal of Chemical Physics, 153, 044130. PMID: 32752662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huang, J. , Rauscher, S. , Nawrocki, G. , Ran, T. , Feig, M. , De Groot, B. L. , Grubmüller, H. , & Mackerell, A. D. (2017). CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nature Methods, 14, 71–73. PMID: 27819658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Noskov, S. Y. , & Roux, B. (2008). Control of ion selectivity in LeuT: Two Na+ binding sites with two different mechanisms. Journal of Molecular Biology, 377, 804–818. PMID: 18280500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Luo, Y. , & Roux, B. (2010). Simulation of osmotic pressure in concentrated aqueous salt solutions. Journal of Physical Chemistry Letters, 1, 183–189. [Google Scholar]
- 63. Venable, R. M. , Luo, Y. , Gawrisch, K. , Roux, B. , & Pastor, R. W. (2013). Simulations of anionic lipid membranes: Development of interaction‐specific ion parameters and validation using NMR data. Journal of Physical Chemistry B, 117, 10183–10192. PMID: 23924441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Desta, I. T. , Porter, K. A. , Xia, B. , Kozakov, D. , & Vajda, S. (2020). Performance and its limits in rigid body protein–protein docking. Structure, 28, 1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vajda, S. , Yueh, C. , Beglov, D. , Bohnuud, T. , Mottarella, S. E. , Xia, B. , Hall, D. R. , & Kozakov, D. (2017). New additions to the ClusPro server motivated by CAPRI. Proteins, 85, 435–444. PMID: 27936493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kozakov, D. , Hall, D. R. , Xia, B. , Porter, K. A. , Padhorny, D. , Yueh, C. , Beglov, D. , & Vajda, S. (2017). The ClusPro web server for protein–protein docking. Nature Protocols, 12, 255–278. PMID: 28079879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kozakov, D. , Beglov, D. , Bohnuud, T. , Mottarella, S. E. , Xia, B. , Hall, D. R. , & Vajda, S. (2013). How good is automated protein docking? Proteins, 81, 2159–2166. PMID: 23996272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xia, B. , Vajda, S. , & Kozakov, D. (2016). Accounting for pairwise distance restraints in FFT‐based protein–protein docking. Bioinformatics, 32, 3342–3344. PMID: 27357172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Humphrey, W. , Dalke, A. , & Schulten, K. (1996). VMD: Visual molecular dynamics. Journal of Molecular Graphics, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 70. Jo, S. , Cheng, X. , Islam, S. M. , Huang, L. , Rui, H. , Zhu, A. , Lee, H. S. , Qi, Y. , Han, W. , Vanommeslaeghe, K. , MacKerell Jr, A. D. , Roux, B. , & Im, W. (2014). CHARMM‐GUI PDB manipulator for advanced modeling and simulations of proteins containing nonstandard residues. Advances in Protein Chemistry and Structural Biology, 96, 235–265. PMID: 25443960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jo, S. , Kim, T. , Iyer, V. G. , & Im, W. (2008). CHARMM‐GUI: A web‐based graphical user interface for CHARMM. Journal of Computational Chemistry, 29, 1859–1865. PMID: 18351591. [DOI] [PubMed] [Google Scholar]
- 72. Wu, E. L. , Cheng, X. i , Jo, S. , Rui, H. , Song, K. C. , Dávila‐Contreras, E. M. , Qi, Y. , Lee, J. , Monje‐Galvan, V. , Venable, R. M. , Klauda, J. B. , & Im, W. (2014). CHARMM‐GUI Membrane Builder toward realistic biological membrane simulations. Journal of Computational Chemistry, 35, 1997–2004. PMID: 25130509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Klauda, J. B. , Venable, R. M. , Freites, J. A. , O'connor, J. W. , Tobias, D. J. , Mondragon‐Ramirez, C. , Vorobyov, I. , Mackerell, A. D. , & Pastor, R. W. (2010). Update of the CHARMM all‐atom additive force field for lipids: Validation on six lipid types. Journal of Physical Chemistry B, 114, 7830–7843. PMID: 20496934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Feller, S. E. , Zhang, Y. , Pastor, R. W. , & Brooks, B. R. (1995). Constant pressure molecular dynamics simulation: The Langevin piston method. Journal of Chemical Physics, 103, 4613–4621. [Google Scholar]
- 75. Martyna, G. J. , Tobias, D. J. , & Klein, M. L. (1994). Constant pressure molecular dynamics algorithms. Journal of Chemical Physics, 101, 4177–4189. [Google Scholar]
- 76. Darden, T. , York, D. , & Pedersen, L. (1993). Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. Journal of Chemical Physics, 98, 10089–10092. [Google Scholar]
- 77. Ryckaert, J.‐P. , Ciccotti, G. , & Berendsen, H. J. C (1977). Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n‐alkanes. Journal of Computational Physics, 23, 327–341. [Google Scholar]
- 78. Miyamoto, S. , & Kollman, P. A. (1992). SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. Journal of Computational Chemistry, 13, 952–962. [Google Scholar]
- 79. Smart, O. S. , Breed, J. , Smith, G. R. , & Sansom, M. S. (1997). A novel method for structure‐based prediction of ion channel conductance properties. Biophysical Journal, 72, 1109–1126. PMID: 9138559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Smart, O. S. , Neduvelil, J. G. , Wang, X. , Wallace, B. A. , & Sansom, M. S. P. (1996). HOLE: A program for the analysis of the pore dimensions of ion channel structural models. Journal of Molecular Graphics, 14, 354–360. PMID: 9195488. [DOI] [PubMed] [Google Scholar]
- 81. Smart, O. S. , Goodfellow, J. M. , & Wallace, B. A. (1993). The pore dimensions of gramicidin A. Biophysical Journal, 65, 2455–2460. PMID: 7508762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Torrie, G. M. , & Valleau, J. P. (1977). Nonphysical sampling distributions in Monte Carlo free‐energy estimation: Umbrella sampling. Journal of Computational Physics, 23, 187–199. [Google Scholar]
- 83. Kästner, J. (2011). Umbrella sampling. Wiley Interdisciplinary Reviews: Computational Molecular Science, 1, 932–942. [Google Scholar]
- 84. Kumar, S. , Rosenberg, J. M. , Bouzida, D. , Swendsen, R. H. , & Kollman, P. A. (1992). The weighted histogram analysis method for free‐energy calculations on biomolecules. I. The method. Journal of Computational Chemistry, 13, 1011–1021. [Google Scholar]
- 85. Souaille, M. , & Roux, B. (2001). Extension to the weighted histogram analysis method: Combining umbrella sampling with free energy calculations. Computer Physics Communications, 135, 40–57. [Google Scholar]
- 86. Grossfield, A. (2022) . WHAM: The weighted histogram analysis method.
- 87. Jurrus, E. , Engel, D. , Star, K. , Monson, K. , Brandi, J. , Felberg, L. E. , Brookes, D. H. , Wilson, L. , Chen, J. , Liles, K. , Chun, M. , Li, P. , Gohara, D. W. , Dolinsky, T. , Konecny, R. , Koes, D. R. , Nielsen, J. E. , Head‐Gordon, T. , Geng, W. , … Baker, N. A. (2018). Improvements to the APBS biomolecular solvation software suite. Protein Science, 27, 112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lamoureux, G. , & Roux, B. (2006). Absolute hydration free energy scale for alkali and halide ions established from simulations with a polarizable force field. Journal of Physical Chemistry B, 110, 3308–3322. PMID: 16494345. [DOI] [PubMed] [Google Scholar]
- 89. Horinek, D. , Mamatkulov, S. I. , & Netz, R. R. (2009). Rational design of ion force fields based on thermodynamic solvation properties. Journal of Chemical Physics, 130, 124507. PMID: 19334851. [DOI] [PubMed] [Google Scholar]
- 90. Lev, B. , Roux, B. , & Noskov, S. Y. U. (2013). Relative free energies for hydration of monovalent ions from QM and QM/MM simulations. Journal of Chemical Theory and Computation, 9, 4165–4175. PMID: 26592407. [DOI] [PubMed] [Google Scholar]
- 91. Allen, T. W. , Kuyucak, S. , & Chung, S.‐H. O (1999). Molecular dynamics study of the KcsA potassium channel. Biophysical Journal, 77, 2502–2516. PMID: 10545352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Khalili‐Araghi, F. , Tajkhorshid, E. , & Schulten, K. (2006). Dynamics of K+ ion conduction through Kv1.2. Biophysical Journal, 91, L72–L74. PMID: 16844753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bostick, D. L. , & Brooks, C. L. (2007). Selectivity in K+ channels is due to topological control of the permeant ion's coordinated state. Proceedings of the National Academy of Sciences of the United States of America, 104, 9260–9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Corry, B. , & Thomas, M. (2012). Mechanism of ion permeation and selectivity in a voltage gated sodium channel. Journal of the American Chemical Society, 134, 1840–1846. PMID: 22191670. [DOI] [PubMed] [Google Scholar]
- 95. Ulmschneider, M. B. , Bagnéris, C. , Mccusker, E. C. , Decaen, P. G. , Delling, M. , Clapham, D. E. , Ulmschneider, J. P. , & Wallace, B. A. (2013). Molecular dynamics of ion transport through the open conformation of a bacterial voltage‐gated sodium channel. Proceedings of the National Academy of Sciences of the United States of America, 110, 6364–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Boiteux, C. , Vorobyov, I. , & Allen, T. W. (2014). Ion conduction and conformational flexibility of a bacterial voltage‐gated sodium channel. Proceedings of the National Academy of Sciences of the United States of America, 111, 3454–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Domene, C. , Barbini, P. , & Furini, S. (2015). Bias‐exchange metadynamics simulations: An efficient strategy for the analysis of conduction and selectivity in ion channels. Journal of Chemical Theory and Computation, 11, 1896–1906. PMID: 26574394. [DOI] [PubMed] [Google Scholar]
- 98. Guardiani, C. , Rodger, P. M. , Fedorenko, O. A. , Roberts, S. K. , & Khovanov, I. A. (2017). Sodium binding sites and permeation mechanism in the NaChBac channel: A molecular dynamics study. Journal of Chemical Theory and Computation, 13, 1389–1400. PMID: 28024121. [DOI] [PubMed] [Google Scholar]
- 99. Noskov, S. Y. U. , & Roux, B. (2007). Importance of hydration and dynamics on the selectivity of the KcsA and NaK channels. Journal of General Physiology, 129, 135–143. PMID: 17227917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rajagopal, N. , Durand, A. J. , & Nangia, S. (2020). Predicting selectivity of paracellular pores for biomimetic applications. Molecular Systems Design & Engineering, 5, 686–696. [Google Scholar]
- 101. Van Itallie, C. , Rahner, C. , & Anderson, J. M. (2001). Regulated expression of claudin‐4 decreases paracellular conductance through a selective decrease in sodium permeability. Journal of Clinical Investigation, 107, 1319–1327. PMID: 11375422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yu, A. S. L. , Cheng, M. H. , Angelow, S. , GüNzel, D. , Kanzawa, S. A. , Schneeberger, E. E. , Fromm, M. , & Coalson, R. D. (2008). Molecular basis for cation selectivity in claudin‐2–based paracellular pores: Identification of an electrostatic interaction site. Journal of General Physiology, 133, 111–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lashhab, R. , Rumley, A. C. , Arutyunov, D. , Rizvi, M. , You, C. , Dimke, H. , Touret, N. , Zimmermann, R. , Jung, M. , Chen, X.‐Z. , Alexander, T. , & Cordat, E. (2019). The kidney anion exchanger 1 affects tight junction properties via claudin‐4. Science Reports, 9, 3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Acharya, P. , Beckel, J. , Ruiz, W. G. , Wang, E. , Rojas, R. , Birder, L. , & Apodaca, G. (2004). Distribution of the tight junction proteins ZO‐1, occludin, and claudin‐4, ‐8, and ‐12 in bladder epithelium. American Journal of Physiology‐Renal Physiology, 287, F305–F318. [DOI] [PubMed] [Google Scholar]
- 105. Veshnyakova, A. , Piontek, J. , Protze, J. , Waziri, N. , Heise, I. , & Krause, G. (2012). Mechanism of Clostridium perfringens enterotoxin interaction with claudin‐3/‐4 protein suggests structural modifications of the toxin to target specific claudins. Journal of Biological Chemistry, 287, 1698–1708. PMID: 22128179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sonoda, N. , Furuse, M. , Sasaki, H. , Yonemura, S. , Katahira, J. , Horiguchi, Y. , & Tsukita, S. (1999). Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: Evidence for direct involvement of claudins in tight junction barrier. Journal of Cell Biology, 147, 195–204. PMID: 10508866. [DOI] [PMC free article] [PubMed] [Google Scholar]