

Abstract
In the US, newborn screening (NBS) is a unique health program that supports health equity and screens virtually every baby after birth, and has brought timely treatments to babies since the 1960's. With the decreasing cost of sequencing and the improving methods to interpret genetic data, there is an opportunity to add DNA sequencing as a screening method to facilitate the identification of babies with treatable conditions that cannot be identified in any other scalable way, including highly penetrant genetic neurodevelopmental disorders (NDD). However, the lack of effective dietary or drug‐based treatments has made it nearly impossible to consider NDDs in the current NBS framework, yet it is anticipated that any treatment will be maximally effective if started early. Hence there is a critical need for large scale pilot studies to assess if and how NDDs can be effectively screened at birth, if parents desire that information, and what impact early diagnosis may have. Here we attempt to provide an overview of the recent advances in NDD treatments, explore the possible framework of setting up a pilot study to genetically screen for NDDs, highlight key technical, practical, and ethical considerations and challenges, and examine the policy and health system implications.
1. INTRODUCTION
NBS provides the opportunity for equitable access to information that can prevent or decrease the burden of medical conditions and disabilities by enabling early pre‐symptomatic diagnosis, medical assessment, and intervention. Over time, there have been significant advances in technology platforms used for NBS including the adoption of tandem mass spectrometry for the screening of inborn errors of metabolism. Molecular genetic DNA analysis has also been integrated as either first or second tier tests with cystic fibrosis (CF), severe combined immunodeficiency (SCID), and spinal muscular atrophy (SMA). The next major advance in expanding the number of conditions addressable through NBS is likely to come via DNA sequencing and could include gene panels or more flexible exome/genome sequencing (ES/GS). Given the recent advances in emerging treatments for genetic diseases, an expandable sequencing‐based platform will allow for the flexibility to rapidly add genetic conditions in pilot NBS studies to gather the supporting evidence necessary prior to nomination to the Recommended Uniform Screening Panel (RUSP) (Kemper et al., 2014). Conditions on the RUSP are chosen based on the understanding of the conditions being screened, evidence that supports the ability to screen, the availability of effective treatments, and the potential net benefit of screening from early identification and treatment.
As an important class of conditions, there are currently over 500 genetic NDDs that could be readily identified with molecular genetic screening, affording the opportunity for presymptomatic diagnosis, early recognition and treatment of seizures when associated, and enrollment in Early Intervention Programs (EIP). EIP includes services and supports available to young children with developmental delays and disabilities by teaching them through additional support and repetition how to best adapt with their limitations and use their relative strengths. It has been shown to improve outcomes and minimize the burden of associated behavioral conditions including autism (Bonnier, 2008; Purpura et al., 2014; Wojcik, Stewart, Waisbren, & Litt, 2020). In a randomized clinical trial in infants age 9–14 months showing early behavioral signs of autism, EIP demonstrated reduced autism symptom severity and reduced likelihood of an eventual autism diagnosis (Whitehouse et al., 2021).
Previously, addition of some NDDs such as Fragile X have been explored in NBS pilot programs but have not been nominated to the RUSP due to the perceived ineffectiveness of the supportive treatment options available. However, nomination of NDDs warrants reconsideration. Rarely, there are effective treatments available such as the ketogenic diet for glucose transporter 1 deficiency syndrome (Klepper et al., 2020). While none of the supportive treatments available are curative for most NDDs, early intervention has been demonstrated to improve outcomes for Down Syndrome and other NDDs (Fidler et al., 2021; Guralnick, 2010, 2011; Pinero‐Pinto et al., 2020; Wojcik et al., 2020). Furthermore, in the conceptual framework established with SMA, the concurrent pilot NBS study and clinical trial with nusinersen were structured to co‐evolve, enabling early identification of newborns during a critical therapeutic window in which treatments were most efficacious. Importance of early intervention/therapy is not limited to SMA or the other RUSP conditions. Preclinical animal models suggest similar results with tuberous sclerosis complex (TSC), Angelman Syndrome, and Phelan‐McDermid Syndrome (Mei et al., 2016; Silva‐Santos et al., 2015; Tsai et al., 2018), such that treating early in life can be more effective than treating later. Therefore, there is a conundrum in which therapeutic advances will be catalyzed most rapidly if, and only if, there is NBS to identify individuals who will benefit most from early treatments, so that additional therapeutic development efforts will focus on these conditions.
NBS would enable identification of individuals before symptom onset, determination of the true population incidence, and understanding of the natural history from birth to prepare for future clinical trials. Given the seriousness of these NDDs, our current inability to cure them, and the impact they have on families, it is critically important to consider the complexities of population‐based pilot study of NDD screening and strategies to allow parents to make informed decisions within the context of a research study (Table 1). Here, we will review important considerations for NBS of NDDs and highlight the agenda that must be developed for a population level research pilot study aimed at providing the evidence needed to assess if and when these conditions should be included in NBS programs.
TABLE 1.
Reasons to add highly penetrant neurodevelopmental disorders to newborn screening
|
2. FRAMEWORK FOR INCLUDING NDDS IN NBS
Parents who previously participated in NBS research studies have shown interest in having their newborn screened for additional genetic conditions with strong desires to only know information about conditions with extremely high penetrance or near certainty of early childhood onset, even without curative treatments (Wynn et al., 2018). Traditionally, benefit of NBS is narrowly defined as cure or significant reduction of morbidity and mortality. The expanded benefits of early detection of NDDs may include prevention of secondary complications such as irreversible damage to the brain from untreated/inadequately treated seizures (Holmes, 2009), access to EIP leading to better long‐term health outcomes, avoidance of a prolonged “diagnostic odyssey” that often accompanies NDDs, as well as providing information and support for parents, empowering them to engage in their child's care more effectively. Note that we will discuss NDDs in the aggregate although there are clearly clinical and practical differences among the conditions which necessitate careful considerations in the context of each condition.
As an example of the benefit of early detection, TSC is often diagnosed prenatally based upon the associated cardiac rhabdomyomas (Davis et al., 2017). This early detection allows clinicians to provide anticipatory care, refer to EIP, and alert parents to the possibility of seizures, especially infantile spasms. Surveillance of and addressing associated hearing and vision issues could improve early development. Studies of the natural history of TSC from birth have identified pre‐symptomatic abnormal electroencephalograms as early as 4 months of age, followed by seizures 2–3 months later (Jozwiak, Kotulska, Wong, & Bebin, 2020; Wu et al., 2016). These natural history data were critical in designing the PReventing Epilepsy using Vigabatrin in Infants with TSC trial (PREVeNT, NCT02849457). Another example of an early detection benefit is Rett Syndrome which is associated with near normal development for the first year of life, followed by gradual but variable loss of skills. The average age of diagnosis is 3 years or older. Preclinical studies in mouse models of MECP2 variants (Achilly, Wang, & Zoghbi, 2021) have shown that repetitive training can help preserve function, which leads to the hypothesis that pre‐symptomatic task‐specific training may lessen symptom severity in females with Rett Syndrome. As we embark on these studies, it will be important for families and clinicians to define clinically meaningful differences and have robust outcome measures to assess the impact of earlier diagnosis and intervention.
Once newborns are identified to be at high risk for NDD conditions, a core set of follow ups should be defined, focusing on surveillance of neurodevelopment every 6 months or so depending on the condition and initiation of early intervention. For some conditions associated with epilepsy, referral to a pediatric neurologist, baseline electroencephalograms and monitoring for seizures will be appropriate. Detailed natural history studies and disease registries will be important to understand the emergence of symptoms (or lack thereof) in an unbiased cohort. There are economies of scale to use the same infrastructure such as that provided by the Newborn Screening Translational Research Network (NBSTRN) to study NDDs in the aggregate with tailoring of assessment modules based upon associations with specific features (epilepsy, autism) and ability level. Demonstrating efficacy of early diagnosis to improve outcomes for each condition individually could be challenging for ultra‐rare conditions, but aggregating individuals across conditions can help demonstrate an overall impact when compared with historical controls who were clinically ascertained. Importantly, as with other conditions ascertained through NBS, population‐based screening is likely to identify a wider phenotypic spectrum, and it may be difficult to disentangle ascertainment bias from efficacy of interventions unless the individual genes and variants are identical. A population‐based sampling method could also provide a strategy to identify genetic modifiers conferring resiliency since some individuals might not readily come to clinical attention if they are asymptomatic. Also important to the study is the assessment of the contribution to outcome differences by social equity factors such as access to a diagnosis and access to care including specialized treatment centers, therapists, and educators.
3. PILOT RESEARCH STUDY CONSIDERATIONS
For NBS, establishing the clinical validity and utility depends on the ability to identify the newborn with the NDD and to improve the outcome. Accumulating statistically robust outcome data during the pilot is critical, yet disease rarity often hampers the ability to acquire enough information in a typical follow up period of 2 years in research studies. Disease frequency for most NDDs is expected to range between 1 in 20,000 and 1 in 100,000 within the US population. Therefore, a pilot study would likely have to screen more than 100,000 newborns to identify at least one positive case for many NDDs. To help justify the large number needed to screen and maximize the yield, one strategy is to include NDDs that are relatively common with well‐established prevalence data (e.g., Fragile X Syndrome, Rett Syndrome, TSC) in addition to other rarer recessive (Martin et al., 2018) and de novo dominant conditions (Lopez‐Rivera et al., 2020).
The scope of the research pilot and its intended research question(s) should also be well matched. For a feasibility study focused on optimizing workflows, parental consent, and reporting procedures, a small study (500–1000 participants) and relatively short‐term follow‐up would be sufficient. In comparison, a study that intends to measure the real‐world yield and the downstream impact of positive findings must involve a much larger number of participants for >50 conditions given the relatively low prevalence of each NDD. Using ES/GS with analysis of a targeted gene set would allow for flexibility to assess and then add additional genes over time without the need for technical re‐validation of the same platform. Also, to assess clinical utility in terms of improved outcomes, the study would likely need to follow individuals who screen positive for many years and should provide the infrastructure to do so from the outset, which also provides the opportunity to better understand these diseases including penetrance, spectrum of disease severity, and natural history.
4. TECHNICAL AND IMPLEMENTATION CHALLENGES
Recent pilot studies such as the NIH funded Newborn Sequencing in Genomic Medicine and Public Health (NSIGHT) program (Adhikari et al., 2020; Ceyhan‐Birsoy et al., 2019; Roman et al., 2020) have demonstrated the feasibility and utility of genomic sequencing based NBS, including the use as a second‐tier test after biochemical analysis (Ruiz‐Schultz et al., 2021). For pilot studies using sequencing as the first‐tier screening test, the main challenges to address are variant detection, interpretation, data curation, documenting improved outcomes, and future implementation in state public health infrastructures.
The analytic validity of next generation sequencing methods for nucleotide substitutions and small insertions/deletions has been well established, though orthogonal confirmation methods of some reportable variants such as indels or copy number variants may still be necessary (Sanger sequencing or qPCR). Large copy number variants can be reliably detected with ES and GS, but single exon deletions or insertions in the range of 50–500 nucleotides can be difficult to reliably detect by short read sequencing technology. In addition, there are other specific technical challenges such as triplet repeat expansions (e.g., Fragile X) or imprinted loci (e.g., Angelman Syndrome).
For variant interpretation, it is important to distinguish between clinical diagnosis in a symptomatic individual and population‐based NBS since the priors in the two scenarios are different. NBS seeks to identify the infrequent newborns who require treatment among a large unaffected population, whereas diagnostic testing is focused on identifying a cause of symptoms in an affected individual. The distinction is important for setting thresholds for which genetic variants to report. ACMG/AMP guidelines provide a comprehensive set of rules to ensure the consistency of pathogenicity interpretation in the diagnostic setting. However, in the NBS setting, the criteria involving the individual's clinical phenotype cannot be used since it will likely not be available to the state screening program and rarely will the newborn be symptomatic. Some variants of unknown significance (VUS) may be reportable if second‐tier confirmatory testing (e.g., enzymatic activity, methylation analysis) or targeted inheritance testing of parents could consistently support variant reclassification. However, most NDDs cannot be confirmed biochemically, and in most cases, interpretation of the initial result can only be based on the individual baby's result without parental data. Therefore, pathogenicity will largely rely on reference population frequency, variant type (predicted loss of function), and previous reporting in public databases/publications. Some pathogenic variants may have minimal or modest impact, though we have no accurate estimates of the population frequency of such phenomena. Another significant initial challenge is variant interpretation in ancestrally diverse newborns due to less reference data, potentially leading to many VUS.
Therefore, given the reported parental desire of high diagnostic certainty (Wynn et al., 2018), to minimize anxiety and needless surveillance of newborns, it is prudent to initially favor specificity over sensitivity when interpreting variants for most NDD genes/conditions and only report curated pathogenic/likely pathogenic variants with high penetrance. These largely include previously reported highly recurrent missense variants and likely‐gene‐disrupting variants in well‐established NDD genes with consistent mode of action. This reporting strategy is especially relevant when the intervention is invasive (e.g., bone marrow transplantation), expensive, or experimental with significant risks. Some affected infants with novel variants will be missed due to the reduction of sensitivity, which is at odds with traditional NBS via mass spectrometry where most screened conditions have well‐defined confirmatory tests to reduce false positive findings and increase sensitivity. That being said, as variant interpretation improves over time and additional safe and effective treatments are developed, the reporting strategy can be adjusted to increase sensitivity (Figure 1). Based on the California genomic NBS pilot study for inborn errors of metabolism (Adhikari et al., 2020), this can be an accurate and scalable approach. Also note that decisions about variants reported may differ by gene based on disease severity, life expectancy, and likely effectiveness of treatment.
FIGURE 1.
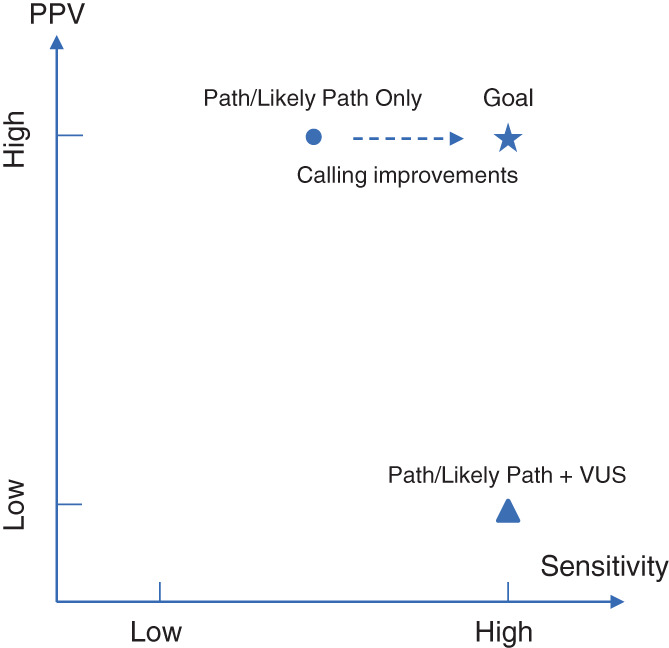
Trade‐off between sensitivity and PPV in reporting results. In individuals with monogenic cause of NDDs, the approach of reporting only Pathogenic and Likely Pathogenic variants will have high PPV but modest sensitivity, as some true positives are currently classified as VUSs. On the other hand, including VUS in the report will lead to high sensitivity but low PPV. Resolving VUS in the future, by improved computational and experimental tools and learning systems, is the key to improve sensitivity while maintaining high PPV.
Similarly, current variant databases, such as ClinVar, are largely based on diagnosed symptomatic individuals, and it will be essential to extend data curation to people ascertained in unbiased population studies to improve interpretation in asymptomatic people. In the longer term, the success of genomic sequencing based NBS hinges on iterative improvement of variant interpretation and understanding of natural history and genotype–phenotype relationships. Initially, there may be inequities in our ability to interpret genetic information for all ancestral groups. Eventually, the catalog of well‐established pathogenic and benign variants will become more comprehensive, enabling both higher sensitivity and specificity for genomic screening tests and more even performance across populations. One open question is to determine if there is parental interest in reanalysis and reinterpretation of previously reported results and permission to recontact within the context of a research study as knowledge changes over time, and if the labs have the capability to do so. While reanalysis of the data can potentially be automated, information technology systems would need to be developed to enable long term tracking of participants. Recent studies using advanced informatics and data mining tools such as machine learning have demonstrated the potential of such approaches (Baker et al., 2019; James et al., 2020; Robertson et al., 2022; Seo et al., 2022; Tan et al., 2020; Zaunseder et al., 2022).
Setting up sequencing‐based NBS in each state would be costly, technically challenging, and computationally intensive, but may not be necessary to replicate in each state. Practically, it would be more feasible and cost effective to have regional centers of expertise, rather than state‐by‐state programs. There should be a national platform that connects with individual NBS programs, provides a unified robust bioinformatics pipeline for alignment/variant calling/interpretation including gene specific guidelines, maintains a comprehensive database with annotated variants identified by NBS and associated clinical data, and houses disease registries to store follow‐up data. Such a platform will facilitate more state labs to implement genomic sequencing based NBS with rapid turnaround and high throughput and will minimize the workload for geneticists to interpret results. Furthermore, there could be a distributed model of gene specific experts to assist with interpretation of variants in particular genes (Figure 2).
FIGURE 2.
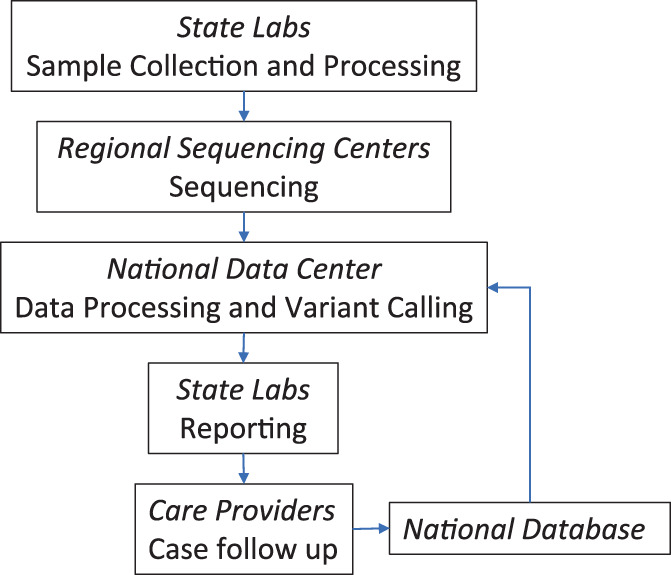
Infrastructure to support efficient implementation of sequencing based newborn screening nationwide. A small number of regional sequencing centers makes it easier to achieve uniform test performance. A national data center makes data curation and processing more efficient with the benefit of centralized continuous improvements from a more diverse and balanced representation of the US population while leveraging geographically scattered expertise.
5. PEDIATRIC ETHICAL, LEGAL, AND SOCIAL IMPLICATIONS (ELSI)
5.1. Informed consent
Informed consent is a critical issue for a pilot research study of expanded NBS focused on NDDs. State‐based NBS programs operate on an opt‐out model. Parents receive a pamphlet with brief information, and screening is performed unless the parents explicitly opt‐out. In contrast, opt‐in written informed consent is the default approach for interventional research, which requires infrastructure for research on NBS. Conducting the standard research informed consent process on a few thousand individuals is feasible. However, as described above, a large‐scale pilot study is necessary due to the rarity of NDDs, and the same consent process could become cost‐prohibitive, especially given that deliveries happen around the clock with typical postnatal hospital stays of <2 days. Additionally, opt‐in written consent is arguably the approach most likely to lead to participation bias which may significantly undermine the value of the resulting data. Research participation often requires parents to accept an element of risk for the child and a commitment to additional time and effort, which often pose a challenge for recruitment as seen in previous sequencing based NBS studies (Genetti et al., 2019), particularly for parents and communities that may harbor some distrust of the healthcare system.
One possible alternative is a waiver, granted by the IRB, to utilize verbal consent. Verbal consent is attractive from an ethical perspective because participation decisions are made after a more interactive, engaging discussion, which can better inform the participants and reflect their values than a consent process with a dense written document (Nishimura et al., 2013). As verbal consent conducted by research personnel might still be cost prohibitive, consideration should be given to using a concise verbal consent process conducted by clinical personnel and consider how electronic consent systems can complement the educational component of consent. This approach has proven successful with NBS public health research (Comeau & Levin, 2009).
The timing of recruitment and consent conversations is also important to consider as the newborn period can be chaotic. Approaching families first during the prenatal period could be an attractive approach since it permits families to consider research participation in a place and time more conducive to thoughtful decision‐making. However, since some parents do not seek prenatal care for a variety of reasons, it is likely that recruitment efforts that utilize prenatal recruitment approaches would also need to approach some families during the postnatal period. There are also substantial logistical hurdles in communicating prenatal consent decisions from the obstetrical clinics to birthing centers.
5.2. Selection of conditions
The success of NBS efforts hinges on the selection of conditions for which screening during the newborn period maximizes benefits while minimizing harms. From this perspective, the application of sequencing in the NBS process is simply the use of a different technology to achieve the same set of aims. However, there are several ways in which the use of sequencing creates novel challenges for selecting appropriate conditions for NBS, such as the lack of definitive genotype–phenotype correlation (e.g., Krabbe disease [Ross, 2015]), and the level of risk and benefit created by identifying and disclosing to the family of a child at risk for certain conditions. One important challenge is the ability to confirm a diagnosis. The ability to utilize a waiver of written consent hinges on a determination of minimal risk of the research, which is most applicable to screening for conditions that, when detected via sequencing, can be readily confirmed. The lack of confirmatory test methods for many of the NDDs would mean that for some of these cases, the only way to confirm the diagnosis is to wait until the symptoms emerge. In such cases, there are risks of potential harm to the parents due to the stress and anxiety from continued behavioral and developmental surveillance. Therefore, at least initially, written consent is appropriate for NDDs.
There are ethical reasons to consider the inclusion of a broader set of conditions. Chief among these is an interest in addressing health disparities. There is evidence, for example, that the introduction of SCID in state NBS panels provided greater benefit for newborns from non‐White families (Brosco, Grosse, & Ross, 2015). This is because universal screening helped partially alleviate disparities in early detection and treatment that were previously experienced by these groups. A pilot intended to examine the effects of universal screening for a broad range of conditions might offer an opportunity to address disparities in the detection and treatment of many of these conditions. A tiered approach is a way to include two groups of conditions and provide more clinically actionable information to more people: a group of readily confirmable and treatable conditions could be provided by default (i.e., with a waiver of written consent approach), and a second set of conditions including NDDs could be offered as an optional group (i.e., with a standard consent approach).
5.3. ELSI‐related study measures
Several ELSI issues raised by the application of sequencing in routine NBS could be addressed by collecting relevant data through carefully designed pilot studies. A document published by the Bioethics and Legal Workgroup of the NBSTRN detailed these issues (Goldenberg et al., 2019). One important concern is the effect of such expansion on the willingness of families with diverse backgrounds to participate, either in a research pilot or in the public health implementation contexts. For this reason, it would be important to design a pilot to include sites that are representative of the U.S. population and incorporate measures to help better understand the reasons behind non‐participation. It is also important to design process measures to capture the implications of participation such as when the results are received, when appropriate care (such as genetic counseling or evaluation by EIP therapists) is provided, and the impact of such information and services on the family. Since effects on parent–child bonding have been raised as a possible adverse effect of NBS, it would be helpful to explicitly survey both parents to help address such concerns. Finally, it would be helpful to consider outcomes for families participating in the study within all result categories of true positive, true negative, false positive, and false negative.
6. POLICY AND SYSTEMS
Core issues of cost‐effectiveness, benefits and harms, accessibility, and health equity have been extensively discussed regarding sequencing technologies and its application in healthcare generally and in NBS particularly. There are challenges with the lack of an appropriate public health framework and corresponding policies to enable NBS research, pilot studies, implementation, and adoption, and the following issues will require attention.
There are legislative and regulatory policy issues impeding the expansion of NDDs into NBS. Some arise from the distinctions between research and standard of care, and others from the challenges of screening for rare diseases. Strong scientific evidence often is hampered by low disease frequency. Expanded acceptance of innovative endpoints for population‐level pilot studies are needed. Thoughtful policies related to IT infrastructure for individual consent‐defined data sharing will be needed to ensure privacy and maximize benefit to participants and their communities. Further, the experiences of the NSIGHT programs (Berg et al., 2017) in interacting with the FDA highlight the importance of IRBs in assessing whether the Investigative Device Exemption (IDE) applies (Milko et al., 2019). When sequencing based NBS transitions from research to clinical care, there may be additional FDA regulatory oversight.
Additionally, recommendations to add sequencing‐based screening to the RUSP would require infrastructure that supports broad collaboration and funding at both the federal and state levels and from other stakeholders in genetic disease screening. Complementary efforts are occurring in NIH funded programs such as NSIGHT and private disease‐focused foundations are advocating for such collaborative efforts. Risk‐sharing strategies among stakeholders are increasingly being used to redistribute the intrinsically high costs of rare disease research, pilot studies, implementation, and treatment development. For the treatments for these rare diseases, a very small number of individuals will be identified in NBS that can result in very high costs of follow‐up care for these individuals (e.g., gene therapy) but can also lead to net fiscal benefits to the overall health care system and clear benefits to the families, and the development of reinsurance programs have potential in this area to distribute costs to society. Therefore, an emerging model of public‐private or multi‐institutional partnership of research, advocacy, industry, payers, and policy‐making stakeholders could potentially accelerate sequencing‐based NBS pilot studies to manage costs, increase efficiency, and develop data on potentially hundreds of conditions simultaneously.
Policies and systems related to NBS are largely under the control of state legislatures and public health programs while many genomic experts are in the academic medicine sector or clinical diagnostic laboratories. The goal of efficient translation of the sequencing technology and its implementation into NBS will benefit from broad participation in the development of this program. The current system of state‐by‐state adoption of conditions that have been reviewed by the Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC) and added to the RUSP has highlighted inequities and inconsistencies. Some children have access to public health NBS far in advance of others, often based solely on their state of birth. In practice, the evidence review process and decision‐making matrix may not require substantive changes to accommodate genetic sequencing based NBS. However, the ACHDNC is already resource‐challenged in keeping up with the number of conditions being nominated. Since each condition requires its own evidence base for inclusion on the RUSP, modifications to the review timeline and resource allocation would be required to accommodate the increased number of conditions that would likely be reviewed simultaneously. Continuing to bring together key stakeholders and consortia for groups of conditions will help advance the effort while working through critical policy issues that are still unidentified or under‐appreciated.
7. CONCLUSION
The challenges for genetic screening of neurogenetic developmental disorders are significant, but surmountable. It will require carefully designed pilot studies and additional empirical data to assess the key outcomes. Input from stakeholders including parents, health care providers, therapists, genomic researchers, ethicists, health economists, policy makers, and the newborn screen public health infrastructure are necessary to ensure responsible piloting of this strategy. The time is now to begin to assess the feasibility and impact of a genetically based NBS program for neurogenetic developmental disorders. Such a program will benefit from advances in sequencing based NBS that will surely be developing in parallel and will synergistically provide opportunities that we should not miss to equitably advance the support and treatment of these conditions which, although individually rare, are collectively common (at least 3% of all births across all racial and ethnic groups). Expansion of NBS to NDDs could have significant implications for equitable improvement in medical, cognitive, and behavioral outcomes for these children.
ACKNOWLEDGMENTS
We thank the Simons Foundation Autism Research Initiative for the generous support in organizing the SFARI Newborn Screening Workshop in September 2021. We thank Dr. Don Bailey, Mr. Jim Bialick, Dr. Joseph A. Bocchini, Jr, Dr Amy Brower, Dr. Michele Caggana, Dr. Pranesh Chakraborty, Dr. R. Rodney Howell, Ms. Terri Klein, Dr. Maximilian Muenke, Dr. Melissa A. Parisi, Dr. Andreas Rohrwasser, Dr. Bradford Therrell, and Dr. Huda Y. Zoghbi for their participation in the Workshop and contribution to the discussion that formed the basis of this work.
Chung, W. K. , Berg, J. S. , Botkin, J. R. , Brenner, S. E. , Brosco, J. P. , Brothers, K. B. , Currier, R. J. , Gaviglio, A. , Kowtoniuk, W. E. , Olson, C. , Lloyd‐Puryear, M. , Saarinen, A. , Sahin, M. , Shen, Y. , Sherr, E. H. , Watson, M. S. , & Hu, Z. (2022). Newborn screening for neurodevelopmental diseases: Are we there yet? American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 190C:222–230. 10.1002/ajmg.c.31988
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Achilly, N. P. , Wang, W. , & Zoghbi, H. Y. (2021). Presymptomatic training mitigates functional deficits in a mouse model of Rett syndrome. Nature, 592(7855), 596–600. 10.1038/s41586-021-03369-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari, A. N. , Gallagher, R. C. , Wang, Y. , Currier, R. J. , Amatuni, G. , Bassaganyas, L. , … Brenner, S. E. (2020). The role of exome sequencing in newborn screening for inborn errors of metabolism. Nature Medicine, 26(9), 1392–1397. 10.1038/s41591-020-0966-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, S. W. , Murrell, J. R. , Nesbitt, A. I. , Pechter, K. B. , Balciuniene, J. , Zhao, X. , … Santani, A. B. (2019). Automated clinical exome reanalysis reveals novel diagnoses. The Journal of Molecular Diagnostics, 21(1), 38–48. 10.1016/j.jmoldx.2018.07.008 [DOI] [PubMed] [Google Scholar]
- Berg, J. S. , Agrawal, P. B. , Bailey, D. B., Jr. , Beggs, A. H. , Brenner, S. E. , Brower, A. M. , … Wise, A. L. (2017). Newborn sequencing in genomic medicine and public health. Pediatrics, 139(2). 10.1542/peds.2016-2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnier, C. (2008). Evaluation of early stimulation programs for enhancing brain development. Acta Paediatrica, 97(7), 853–858. 10.1111/j.1651-2227.2008.00834.x [DOI] [PubMed] [Google Scholar]
- Brosco, J. P. , Grosse, S. D. , & Ross, L. F. (2015). Universal state newborn screening programs can reduce health disparities. JAMA Pediatrics, 169(1), 7–8. 10.1001/jamapediatrics.2014.2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceyhan‐Birsoy, O. , Murry, J. B. , Machini, K. , Lebo, M. S. , Yu, T. W. , Fayer, S. , … BabySeq Project, T . (2019). Interpretation of genomic sequencing results in healthy and ill newborns: Results from the BabySeq project. American Journal of Human Genetics, 104(1), 76–93. 10.1016/j.ajhg.2018.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeau, A. , & Levin, D. (2009, 01/01). Population‐based research within a public health system: Two models for common rule compliance in the Massachusetts newborn screening program. In Ethics and Newborn Genetics Screening: New Technologies, New Challenges (pp. 274–291). Baltimore, MD: Johns Hopkins Press. [Google Scholar]
- Davis, P. E. , Filip‐Dhima, R. , Sideridis, G. , Peters, J. M. , Au, K. S. , Northrup, H. , … Tuberous Sclerosis Complex Autism Center of Excellence Research, N . (2017). Presentation and diagnosis of tuberous sclerosis complex in infants. Pediatrics, 140(6). 10.1542/peds.2016-4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler, D. J. , Schworer, E. K. , Needham, A. , Prince, M. A. , Patel, L. , Will, E. A. , & Daunhauer, L. A. (2021). Feasibility of a syndrome‐informed micro‐intervention for infants with Down syndrome. Journal of Intellectual Disability Research, 65(4), 320–339. 10.1111/jir.12814 [DOI] [PubMed] [Google Scholar]
- Genetti, C. A. , Schwartz, T. S. , Robinson, J. O. , VanNoy, G. E. , Petersen, D. , Pereira, S. , … BabySeq Project, T . (2019). Parental interest in genomic sequencing of newborns: Enrollment experience from the BabySeq project. Genetics in Medicine, 21(3), 622–630. 10.1038/s41436-018-0105-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg, A. J. , Lloyd‐Puryear, M. , Brosco, J. P. , Therrell, B. , Bush, L. , Berry, S. , … Bioethics, & Legal Workgroup of the Newborn Screening Translational Research, N . (2019). Including ELSI research questions in newborn screening pilot studies. Genetics in Medicine, 21(3), 525–533. 10.1038/s41436-018-0101-x [DOI] [PubMed] [Google Scholar]
- Guralnick, M. J. (2010). Early intervention approaches to enhance the peer‐related social competence of young children with developmental delays: A historical perspective. Infants and Young Children, 23(2), 73–83. 10.1097/IYC.0b013e3181d22e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guralnick, M. J. (2011). Why early intervention works: A systems perspective. Infants and Young Children, 24(1), 6–28. 10.1097/IYC.0b013e3182002cfe [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, G. L. (2009). The long‐term effects of neonatal seizures. Clinics in Perinatology, 36(4), 901–914, vii‐viii. 10.1016/j.clp.2009.07.012 [DOI] [PubMed] [Google Scholar]
- James, K. N. , Clark, M. M. , Camp, B. , Kint, C. , Schols, P. , Batalov, S. , … Kingsmore, S. F. (2020). Partially automated whole‐genome sequencing reanalysis of previously undiagnosed pediatric patients can efficiently yield new diagnoses. NPJ Genomic Medicine, 5, 33. 10.1038/s41525-020-00140-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozwiak, S. , Kotulska, K. , Wong, M. , & Bebin, M. (2020). Modifying genetic epilepsies: Results from studies on tuberous sclerosis complex. Neuropharmacology, 166, 107908. 10.1016/j.neuropharm.2019.107908 [DOI] [PubMed] [Google Scholar]
- Kemper, A. R. , Green, N. S. , Calonge, N. , Lam, W. K. , Comeau, A. M. , Goldenberg, A. J. , … Bocchini, J. A., Jr. (2014). Decision‐making process for conditions nominated to the recommended uniform screening panel: Statement of the US Department of Health and Human Services Secretary's advisory committee on heritable Disorders in newborns and children. Genetics in Medicine, 16(2), 183–187. 10.1038/gim.2013.98 [DOI] [PubMed] [Google Scholar]
- Klepper, J. , Akman, C. , Armeno, M. , Auvin, S. , Cervenka, M. , Cross, H. J. , … de Vivo, D. C. (2020). Glut1 deficiency syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open, 5(3), 354–365. 10.1002/epi4.12414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Rivera, J. A. , Perez‐Palma, E. , Symonds, J. , Lindy, A. S. , McKnight, D. A. , Leu, C. , … Lal, D. (2020). A catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain, 143(4), 1099–1105. 10.1093/brain/awaa051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, H. C. , Jones, W. D. , McIntyre, R. , Sanchez‐Andrade, G. , Sanderson, M. , Stephenson, J. D. , … Deciphering Developmental Disorders, S. (2018). Quantifying the contribution of recessive coding variation to developmental disorders. Science, 362(6419), 1161–1164. 10.1126/science.aar6731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei, Y. , Monteiro, P. , Zhou, Y. , Kim, J. A. , Gao, X. , Fu, Z. , & Feng, G. (2016). Adult restoration of Shank3 expression rescues selective autistic‐like phenotypes. Nature, 530(7591), 481–484. 10.1038/nature16971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milko, L. V. , Chen, F. , Chan, K. , Brower, A. M. , Agrawal, P. B. , Beggs, A. H. , … Kingsmore, S. F. (2019). FDA oversight of NSIGHT genomic research: The need for an integrated systems approach to regulation. NPJ Genomic Medicine, 4, 32. 10.1038/s41525-019-0105-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura, A. , Carey, J. , Erwin, P. J. , Tilburt, J. C. , Murad, M. H. , & McCormick, J. B. (2013). Improving understanding in the research informed consent process: A systematic review of 54 interventions tested in randomized control trials. BMC Medical Ethics, 14, 28. 10.1186/1472-6939-14-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinero‐Pinto, E. , Benitez‐Lugo, M. L. , Chillon‐Martinez, R. , Rebollo‐Salas, M. , Bellido‐Fernandez, L. M. , & Jimenez‐Rejano, J. J. (2020). Effects of massage therapy on the development of babies born with down syndrome. Evidence‐Based Complementary and Alternative Medicine, 2020, 4912625. 10.1155/2020/4912625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purpura, G. , Tinelli, F. , Bargagna, S. , Bozza, M. , Bastiani, L. , & Cioni, G. (2014). Effect of early multisensory massage intervention on visual functions in infants with down syndrome. Early Human Development, 90(12), 809–813. 10.1016/j.earlhumdev.2014.08.016 [DOI] [PubMed] [Google Scholar]
- Robertson, A. J. , Tan, N. B. , Spurdle, A. B. , Metke‐Jimenez, A. , Sullivan, C. , & Waddell, N. (2022). Re‐analysis of genomic data: An overview of the mechanisms and complexities of clinical adoption. Genetics in Medicine, 24(4), 798–810. 10.1016/j.gim.2021.12.011 [DOI] [PubMed] [Google Scholar]
- Roman, T. S. , Crowley, S. B. , Roche, M. I. , Foreman, A. K. M. , O'Daniel, J. M. , Seifert, B. A. , … Berg, J. S. (2020). Genomic sequencing for newborn screening: Results of the NC NEXUS project. American Journal of Human Genetics, 107(4), 596–611. 10.1016/j.ajhg.2020.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, L. F. (2015). Ethical and policy issues in newborn screening of children for neurologic and developmental disorders. Pediatric Clinics of North America, 62(3), 787–798. 10.1016/j.pcl.2015.03.009 [DOI] [PubMed] [Google Scholar]
- Ruiz‐Schultz, N. , Sant, D. , Norcross, S. , Dansithong, W. , Hart, K. , Asay, B. , … Rohrwasser, A. (2021). Methods and feasibility study for exome sequencing as a universal second‐tier test in newborn screening. Genetics in Medicine, 23(4), 767–776. 10.1038/s41436-020-01058-w [DOI] [PubMed] [Google Scholar]
- Seo, G. H. , Lee, H. , Lee, J. , Han, H. , Cho, Y. K. , Kim, M. , … Eun, B. L. (2022). Diagnostic performance of automated, streamlined, daily updated exome analysis in patients with neurodevelopmental delay. Molecular Medicine, 28(1), 38. 10.1186/s10020-022-00464-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva‐Santos, S. , van Woerden, G. M. , Bruinsma, C. F. , Mientjes, E. , Jolfaei, M. A. , Distel, B. , … Elgersma, Y. (2015). Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. The Journal of Clinical Investigation, 125(5), 2069–2076. 10.1172/JCI80554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, N. B. , Stapleton, R. , Stark, Z. , Delatycki, M. B. , Yeung, A. , Hunter, M. F. , … Tan, T. Y. (2020). Evaluating systematic reanalysis of clinical genomic data in rare disease from single center experience and literature review. Molecular Genetics & Genomic Medicine, 8(11), e1508. 10.1002/mgg3.1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, P. T. , Rudolph, S. , Guo, C. , Ellegood, J. , Gibson, J. M. , Schaeffer, S. M. , … Sahin, M. (2018). Sensitive periods for cerebellar‐mediated autistic‐like behaviors. Cell Reports, 25(2), 357–367 e354. 10.1016/j.celrep.2018.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse, A. J. O. , Varcin, K. J. , Pillar, S. , Billingham, W. , Alvares, G. A. , Barbaro, J. , … Hudry, K. (2021). Effect of preemptive intervention on developmental outcomes among infants showing early signs of autism: A randomized clinical trial of outcomes to diagnosis. JAMA Pediatrics, 175(11), e213298. 10.1001/jamapediatrics.2021.3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik, M. H. , Stewart, J. E. , Waisbren, S. E. , & Litt, J. S. (2020). Developmental support for infants with genetic disorders. Pediatrics, 145(5). 10.1542/peds.2019-0629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. Y. , Peters, J. M. , Goyal, M. , Krueger, D. , Sahin, M. , Northrup, H. , … Bebin, E. M. (2016). Clinical electroencephalographic biomarker for impending epilepsy in asymptomatic tuberous sclerosis complex infants. Pediatric Neurology, 54, 29–34. 10.1016/j.pediatrneurol.2015.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn, J. , Ottman, R. , Duong, J. , Wilson, A. L. , Ahimaz, P. , Martinez, J. , … Chung, W. K. (2018). Diagnostic exome sequencing in children: A survey of parental understanding, experience and psychological impact. Clinical Genetics, 93(5), 1039–1048. 10.1111/cge.13200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaunseder, E. , Haupt, S. , Mutze, U. , Garbade, S. F. , Kolker, S. , & Heuveline, V. (2022). Opportunities and challenges in machine learning‐based newborn screening: A systematic literature review. JIMD Reports, 63(3), 250–261. 10.1002/jmd2.12285 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.