

Abstract
Three‐dimensional (3D) human organotypic skin cultures provide a physiologically relevant model that recapitulates in vivo skin features. Most commonly, organotypic skin cultures are created by seeding isolated epidermal keratinocytes onto a collagen/fibroblast plug and lifting to an air liquid interface. These conditions are sufficient to drive stratification and differentiation of the keratinocytes to form an epidermal‐like sheet with remarkable similarities to human epidermis. Coupled with genetic or pharmacological treatments, these cultures provide a powerful tool for elucidating keratinocyte biology. Recent focus has been placed on increasing the utility of organotypic skin cultures by incorporating other cell types that are present in the skin, such as melanocytes, immune cells, and other cells. Here we describe a step‐by‐step protocol for the isolation of neonatal human epidermal keratinocytes and melanocytes from foreskins, and the creation of organotypic skin cultures that include both cell types. We also describe methods that can be used to assess melanocyte behavior in these organotypic cultures, including methods for whole mount staining, measurement of melanocyte dendricity, staining for pigment, and 5‐bromo‐2′‐deoxyuridine (BrdU) labeling for identification of proliferating cells. © 2022 The Authors. Current Protocols published by Wiley Periodicals LLC.
Basic Protocol 1: Isolation of primary cells
Alternate Protocol: Isolation of primary cells using differential trypsinization
Basic Protocol 2: Organotypic culture protocol
Support Protocol 1: Culture and maintenance of NHEKs and melanocytes
Support Protocol 2: Lentiviral transduction of melanocytes
Support Protocol 3: Retroviral transduction of NHEKs
Support Protocol 4: Whole mount immunostaining protocol
Support Protocol 5: Measuring melanocyte dendricity
Support Protocol 6: Fontana‐Masson staining protocol
Support Protocol 7: BrdU labeling and staining
Keywords: keratinocytes, melanocytes, organotypic skin, organotypic human skin cultures
INTRODUCTION
This protocol describes methods for adapting one of the most well‐defined human organotypic models, organotypic skin cultures, to include incorporation of the pigment producing cells of the skin, melanocytes. When compared to traditional 2D cell culture models, where cells are grown as a monolayer submerged in medium, 3D organotypic models more faithfully recapitulate tissue architecture and cell differentiation patterns. Organotypic skin cultures were developed to model the outermost layer of the skin, the epidermis, and this model faithfully reestablishes much of the organization observed in skin epidermis in vivo (Arnette, Koetsier, Hoover, Getsios, & Green, 2016). These cultures can also be modified to incorporate other skin cell types, including inflammatory cells such as monocytes and T cells, neurons, endothelial cells, melanocytes, and melanoma cells (Arnette et al., 2020; Enjalbert et al., 2020; Ghosh et al., 2005; Li, Fukunaga‐Kalabis, & Herlyn, 2011; Lorthois, Simard, Morin, & Pouliot, 2019; Marino, Luginbühl, Scola, Meuli, & Reichmann, 2014; Martorina, Casale, Urciuolo, Netti, & Imparato, 2017; Michielon et al., 2020; Müller & Kulms, 2018; Shin et al., 2020). This protocol describes methods for adapting the organotypic skin culture protocol to include melanocytes in the culture (Fig. 1A‐C). Organotypic cultures incorporating melanocytes have been used to address mechanisms regulating pigmentation and melanoma pathogenesis (Ghosh et al., 2005; Li et al., 2011; Michielon et al., 2020). The methods described here can be used to address mechanisms leading to activation of the tanning response, including methods for measuring pigment production and melanocyte dendricity, as well as endpoints that are relevant for melanomagenesis, including pagetoid movement, retraction of dendrites, and proliferation.
Figure 1.
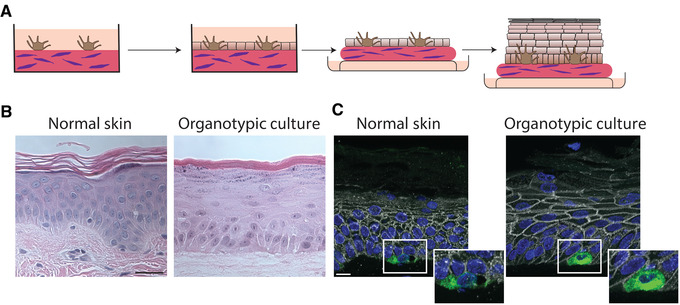
Organotypic skin cultures model normal skin. (A) Schematic representing the general formation of 3D organotypic skin cultures incorporating melanocytes. A collagen‐fibroblast plug is created on which melanocytes are seeded and allowed to adhere for 24 hr. NHEKs are seeded onto the plug and allowed to adhere and reach confluence for 48 hr. The culture is then lifted to an air liquid interface inducing stratification and differentiation. (B) H and E stained FFPE samples from normal skin compared to an organotypic skin culture with incorporated melanocytes after lifting to an air liquid interface for 6 days. Organotypic skin cultures retain the layers normally present in skin, including a cornified layer and the presence of keratohyalin granules in the granular layer. Scale bar = 50 μm. (C) FFPE tissue sections from normal skin or organotypic skin cultures stained for Melan A to label melanocytes, plakoglobin to label cell borders, and DAPI. Melanocytes in the organotypic skin cultures are localized to the basal layer like normal skin. Scale bar = 20 μm. Abbreviations: NHEKs, neonatal human epidermal keratinocytes; H and E, hematoxylin and eosin; FFPE, formalin fixed paraffin embedded.
This article will outline steps for isolating and maintaining primary neonatal human epidermal keratinocytes (NHEKs) and melanocytes needed to generate the organotypic cultures (Basic Protocol 1, Alternate Protocol), and the methods for generating the organotypic cultures (Basic Protocol 2). Support protocols describe methods to maintain primary NHEKs and melanocytes in vitro (Support Protocol 1), methods to genetically manipulate the melanocytes and NHEKs that are used to incorporate in the organotypic culture (Support Protocols 2 and 3), methods for whole mount staining of epidermal sheets from organotypic cultures (Support Protocol 4), methods for analyzing melanocyte dendricity (Support Protocol 5), staining for pigment using Fontana‐Masson stain (Support Protocol 6), and labeling and staining for 5‐bromo‐2′‐deoxyuridine (BrdU; Support Protocol 7).
STRATEGIC PLANNING
The investigator must decide whether matched NHEKs and melanocytes, as in cells isolated from the same donor, are needed for the experiment. While not necessary for generating organotypic co‐cultures, an advantage of using matched cells is that it simplifies the statistical analysis. There are some situations where using matched cells may not be possible, for instance if cells are purchased commercially instead of isolated in the laboratory. Isolating both cell types from the same tissue sample also reduces yields of both, which may be limiting for some experiments. If it is not possible to use melanocytes and NHEKs isolated from the same donor, the cultures will still form normally. Also, for NHEKs to properly stratify and differentiate in organotypic skin cultures, the cells must be used 12 to 18 days after the initial harvest. In our experience cells used outside of this window do not stratify and differentiate properly. The timing of experiments must be carefully planned, especially if using cells that have been genetically manipulated, so that all cells and equipment are ready when needed. Another factor to consider is the type of pigment produced by the melanocytes. If using melanocytes isolated from individuals with skin type 5 or 6 from the Fitzpatrick scale then pigment will be visible in the culture, including in whole mount images and in hematoxylin and eosin (H and E) stained sections (Fitzpatrick, 1988). Melanocytes isolated from individuals with skin types 1 to 4 will still produce pigment but it will not be visible unless labeled, such as with Fontana‐Masson stain. Finally, typically four biological replicates (cells isolated from four different individuals) are enough to determine statistical differences between groups in our hands, though this will vary depending on the specific experiment and will need to be experimentally determined by the investigator.
Basic Protocol 1. ISOLATION OF PRIMARY CELLS
This protocol describes the method used to isolate both NHEKs and melanocytes from the same tissue sample. Primary cells are isolated from neonatal foreskin, though primary cells can also be isolated from adult skin (e.g., samples can be derived from abdominoplasty), if desired. Tissue samples can be stored in Hank's balanced salt solution (HBSS) for up to 72 hr.
NOTE: Federal regulations require that research projects involving human subjects be reviewed by an Institutional Review Board (IRB). The IRB must approve or determine the project to be exempt prior to the start of any research activities.
NOTE: This protocol involves working with human tissue. Follow all institutional biosafety procedures. All steps should be performed inside a Biosafety Level 2 (BSL‐2) cabinet.
NOTE: Both NHEKs and melanocytes can be isolated from the same tissue sample, allowing for the use of matched cells when making the organotypic culture. This protocol describes isolating melanocytes and NHEKs from the same sample. This is achieved by dividing the solution containing the isolated single cells, followed by isolation of melanocytes from one portion and the keratinocytes from the other portion. This strategy results in the best purity of each cell type when performing the isolation. However, this does reduce the overall yield of cells. If a high yield of cells is required, all cells can be isolated from the same sample using differential trypsinization. The Alternate Protocol describes this method.
Materials
Human neonatal foreskins (can be acquired under IRB approval through one's institution; neonatal foreskins used in this work were provided by the Northwestern University Skin Biology and Diseases Resource‐based Center): Collect and place in transport medium, HBSS without calcium or magnesium, and store at 4°C until ready to perform the isolation; foreskins can be stored for up to 72 hr before beginning the isolation protocol with minimal loss of viability
PBS containing no Ca2+ or Mg2+
M154 supplemented medium (see recipe)
FBS (MilliporeSigma, F0926‐500ML)
Basic fibroblast growth factor (bFGF) medium (see recipe)
Dispase solution, 1.4 U/ml: Reconstitute dispase powder (Roche, 04942078001) at 105 U/ml following the manufacturer's instructions; for dispase solution, dilute to 1.4 U/ml in PBS without calcium and magnesium and sterile filter; store at 4°C for up to 3 days
0.25% trypsin (Gibco, 25200072)
Sterile Forceps
Sterile Scissors
100‐mm TC‐treated Culture Dish (Corning, 430167)
60‐mm TC‐treated Culture Dish (Corning, 430166)
40‐μm nylon cell strainers (Corning, 352340)
70‐μm nylon cell strainers (Corning, 431751)
Day 1: Prepare foreskin for isolation
This step is best performed at the end of the day; extended incubation in dispase solution can lead to a small reduction in the number of viable NHEKs and melanocytes isolated from the foreskin.
-
1
Add 3 ml dispase solution to a 60‐mm cell culture dish. Set aside while preparing the foreskin.
-
2
Remove transport solution from the tube containing the foreskin and wash two times in PBS containing no Ca2+ or Mg2+.
All solutions that touch the foreskin should be placed in a beaker containing bleach.
-
3
Move foreskin to a clean 100‐mm dish and using sterile forceps and scissors, cut foreskin so that it will lay flat in the dish, remove excess fat and vessels, and cut into smaller (roughly 8 mm × 8 mm) pieces.
-
4
Carefully place pieces, dermis side down, into the 60‐mm dish containing dispase solution. Place this dish inside a 100‐mm culture dish and place in 4°C refrigerator overnight.
Day 2: Prepare single cells for NHEK and melanocyte isolation
This step is best performed early in the day following the day 1 protocol, as leaving the foreskin in dispase for too long can lead to a small decrease in cell viability.
-
5
Add 3 ml of 0.25% trypsin solution to a 100‐mm dish, with the dish propped up on a lid so the solution collects on one side. Set aside while working with the foreskin in dispase solution.
-
6
Collect the 100‐mm dish containing the foreskin in dispase solution from 4°C refrigerator and bring to the laminar flow hood.
-
7
Using two pairs of sterile forceps, separate epidermis from the dermis from each piece of the foreskin and float epidermis on trypsin solution.
Incubation overnight in dispase allows for the epidermis and dermis to be separated but the NHEKs together with the melanocytes will remain in an intact sheet of cells in the epidermis. Incubating in trypsin allows disassociation of the sheet into single cells, though leaving the cells in trypsin for too long can reduce yield of viable cells.
-
8
Incubate at 37°C for 15 min.
-
9
Inactivate trypsin by adding 0.5 ml FBS to the dish and release single cells from the epidermis by scraping the epidermal sheets along the bottom of the dish using sterile forceps.
The sheets will not dissociate during this process, as the upper layers of the epidermis will remain intact; however, cells will be released from the lower layers of the epidermis.
Scrape each chunk of tissue for ∼1 min to release all the cells. At this point the single cells present in the solution will include both NHEKs and melanocytes. When scraping cells, it is easiest to use a set of curved forceps, with the curved end of the forceps to hold the tissue down while scraping (Fig. 2A).
Trypan blue can be used to check cell viability at this step. Typically, viability should be >90%.
Figure 2.
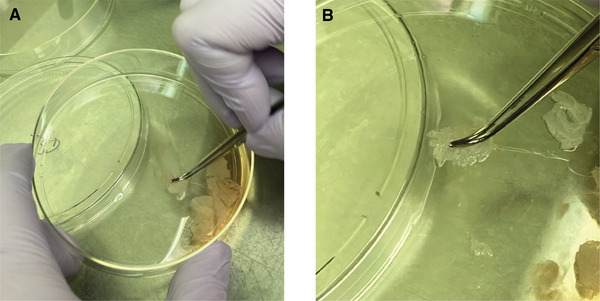
Neonatal human epidermal keratinocyte (NHEK) and melanocyte isolation protocol. When scraping the tissue along the bottom of the dish to dislodge cells from the epidermal sheet it is easiest to use curved forceps to hold the tissue flat on the bottom of the dish. (A) Note that the dish is held at an angle to allow the solution to pool at the bottom of the dish, allowing the epidermal sheets to be scraped on an area of the plate not covered in solution. (B) Zoom in of the tissue being held to the bottom of the plate using curved forceps.
-
10
To isolate both NHEKs and melanocytes from the same tissue sample, divide sample into two portions for further isolation.
Typically, melanocytes will expand to a greater extent after isolation than keratinocytes, thus we isolate NHEKs from three‐quarters of the solution and melanocytes from the remaining one‐quarter. The method for isolating NHEKs and melanocytes are described separately below.
-
11
For the isolation of NHEKs, pass the single cell suspension through the 40‐μm nylon cell strainer into a 50‐ml conical tube. Rinse plate with 5 ml PBS and pass this rinse solution through the 40‐μm strainer into the 50‐ml conical tube.
-
12
For isolation of melanocytes, pass the single cell suspension through the 70‐μm nylon cell strainer into a separate 50‐ml conical tube.
-
13
Centrifuge tubes containing the single cell suspensions at 300 × g for 5 min to pellet the cells.
-
14
Resuspend NHEKs in 7 ml M154 medium and plate in a 100‐mm dish.
-
15
Resuspend melanocytes in 3 ml bFGF medium and plate in a 60‐mm dish.
ISOLATION OF PRIMARY CELLS USING DIFFERENTIAL TRYPSINIZATION
This protocol describes methods to isolate melanocytes and NHEKs from the same tissue sample using differential trypsinization. This allows for higher yields of each cell type from a given tissue sample, noting that cells cannot be perfectly separated by differential trypsinization and there will be some contamination of both cell types in each culture.
Materials
Single cell suspension (see Basic Protocol 1)
M154 supplemented medium (see recipe)
bFGF medium (see recipe)
0.25% trypsin (Gibco, 25200072)
0.05% trypsin/1 mM EDTA (Thermo Fisher Scientific, 25300054)
DMEM with 10% FBS
PBS
Sterile Forceps
Sterile Scissors
100‐mm TC‐treated Culture Dish (Corning, 430167)
60‐mm TC‐treated Culture Dish (Corning, 430166)
70‐μM nylon cell strainers (Corning, 431751)
Day 2: Isolate primary cells
The day 1 isolation protocol is the same as in Basic Protocol 1, steps 1 to 4 and this protocol will start just after Day 2, step 5 of Basic Protocol 1.
-
1
Pass single cell suspension through the 70‐μM nylon cell strainer into a separate 50‐ml conical tube.
-
2
Wash dish with 5 ml PBS and pass this solution through the 70‐μM strainer.
-
3
Centrifuge tubes containing single cell suspension at 300 × g for 5 min to pellet the cells.
-
4
Resuspend cells in 7 ml M154 medium and plate in a 100‐mm dish.
-
5
Change medium every day until the cells are roughly 60%‐70% confluent.
At this point there will be both melanocytes and NHEKs present in the culture.
Differential trypsinization to separate melanocytes and NHEKs
This step takes advantage of the fact that melanocytes will release from the culture dish much more quickly than NHEKs in the presence of trypsin. The exact timing will vary between batches of trypsin, so it is necessary to check the culture under a microscope and inhibit the trypsin once the melanocytes have lifted from the dish while the NHEKS are still attached.
-
6
Remove medium from cell culture dish and wash with PBS.
-
7
Add 3 ml 0.05% trypsin/1 mM EDTA to the culture well and incubate 2 min at 37°C. After 2 min, check cells under a microscope.
At this point the melanocytes should begin to retract their dendrites and lift off the bottom of the cell culture dish while the NHEKs will remain attached.
-
8
Inactivate trypsin by adding 3 ml DMEM with 10% FBS and collect this solution in a 50‐ml conical tube.
This solution contains a majority of melanocytes.
-
9
Wash plate with PBS and add this solution to the 50‐ml conical tube containing melanocytes.
-
10
Add 3 ml 0.05% trypsin/1 mM EDTA to the culture well and incubate for 3‐5 min at 37°C. Check cells under a microscope to determine if the cells have lifted off the bottom of the culture dish.
-
11
Inactivate trypsin by adding 3 ml DMEM with 10% FBS and collect this solution in a 50‐ml conical tube.
This solution contains a majority of NHEKs.
-
12
Centrifuge tubes containing single cell suspension at 300 × g for 5 min to pellet the cells.
-
13
Resuspend NHEKs in 7 ml M154 medium and plate in a 100‐mm dish.
-
14
Resuspend melanocytes in 7 ml bFGF medium and plate in a 100‐mm dish.
Basic Protocol 2. ORGANOTYPIC CULTURE PROTOCOL
This protocol describes the generation of organotypic cultures. To generate a final organotypic culture that has melanocytes and keratinocytes in a similar ratio to normal skin, we plate at a ratio of 1:5 melanocytes/keratinocytes. This ratio is much higher than in normal skin, where the ratio is 1:32 melanocytes/keratinocytes. Plating at the 1:5 ratio is necessary as keratinocytes proliferate at a higher rate than melanocytes in the organotypic cultures.
NOTE: The J2 subclone of 3T3 cells is incorporated into the collagen plug to support the growth of the organotypic skin culture. J2 3T3 cells are available through ATCC (CCL‐92), which also provides instructions for propagation and maintenance.
NOTE: Collagen plugs must be prepared 2 to 3 days prior to seeding of the NHEKs and melanocytes onto the surface. The number of collagen plugs needed will determine the number of J2 cells and volumes of all solutions that are needed. Final concentrations of J2 cells should be 2.66 × 105 cells/ml, and the final collagen concentration should be 4 mg/ml. The following protocol will describe the final concentrations of each component and will give volumes/cell numbers required to make ten collagen plugs as an example.
NOTE: This protocol lifts the organotypic skin cultures to an air liquid interface by moving the cultures to a metal grid that lifts the cultures above the medium below. Other methods for achieving this include growing the cultures in transwell inserts that can be “lifted” to the air liquid interface by removing the medium in the top of the transwell and lowering the medium level in the bottom well. Using transwell inserts is required for some experiments, such as measuring transepithelial electrical resistance. Using transwells to create organotypic skin cultures requires a larger number of cells for each culture, which can be limiting in some instances. For methods on creating organotypic skin cultures using transwells see Arnette et al. (2016).
Materials
J2 3T3 fibroblasts (ATCC, CCL‐92)
0.05% trypsin/1mM EDTA (Thermo Fisher Scientific, 25300054)
Rat Tail Collagen Type 1, High Concentration (Corning, 354249)
Reconstitution buffer (see recipe)
10× DMEM (MilliporeSigma, D2429‐100ML)
DMEM with 10% FBS
Molecular grade water (Corning, 46‐000‐CM)
0.5 N sodium hydroxide (NaOH; sterile filtered)
bFGF medium (see recipe)
E‐medium (see recipe)
12‐well Flat Bottom TC‐treated Cell Culture Plate (Corning, 353225)
1.5 in. Stainless Steel Mesh Grids (cleaned and autoclaved; McMaster‐Carr, 9317T145; mesh grids will arrive as flat grids; prepare for use by bending three small legs equidistant from each other so grids are raised ∼3 mm from the bottom of the dish; ensure grids sit flat on the bottom of the dish, see Fig. 3)
60‐mm TC‐treated Culture Dish (Corning, 430166)
Laboratory spatula (cleaned and autoclaved)
Figure 3.
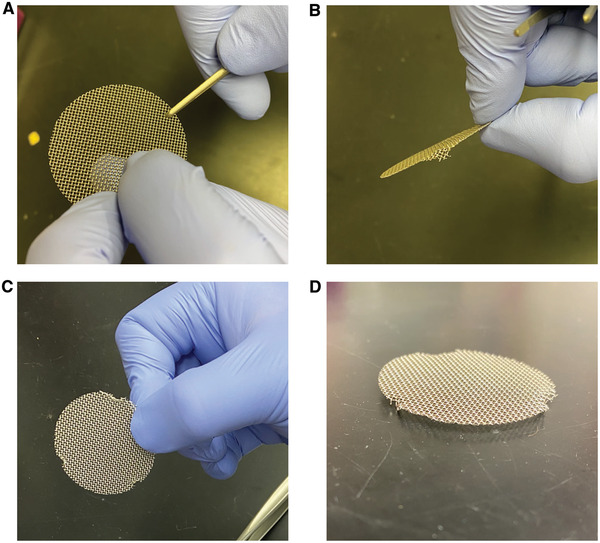
Creating grids for lifting cultures to an air liquid interface. (A) Grab close to the edge with a pair of forceps and (B) bend the edge down to create a leg to hold the grid above the bottom of the dish. (C) Repeat this process two more times around the circumference of the grid. (D) The final grid should stand above the surface it is resting on and should be level with the surface below.
Prepare collagen plugs
Keep all solutions on ice while creating the collagen plug to keep the collagen from forming a gel before it is placed into the culture wells.
-
1
Incubate J2 3T3 cells in 3 ml 0.05% trypsin/1mM EDTA to dissociate the cells. After cells have dissociated and released from the bottom of the culture dish, inactivate trypsin by adding serum containing medium.
-
2
Count the number of J2 3T3 cells and calculate the number needed to generate collagen plugs.
For the grid system, plugs are made in volumes of 1.5 ml and J2 cells should be at a final concentration of 2.66 × 105 cells/ml (for example, if ten collagen plugs are needed, then pellet 4 × 106 cells).
Pellet the number of J2 cells need by spinning at 300 × g for 5 min.
-
3
Resuspend J2 cells in 10× reconstitution buffer (for ten collagen plugs this would mean resuspending in 1.5 ml of reconstitution buffer, leading to a final concentration of 1× in the collagen plug). Also add 10× DMEM to the cell suspension (add 1.5 ml if making ten collagen plugs). Add molecular grade water and collagen to a final concentration of 4 mg/ml of collagen in the final solution. Mix solution by carefully inverting, trying to minimize the amount of air bubbles that are added into the mixture.
The concentration of purchased collagen changes between batches so the exact volumes of water and collagen that need to be added will have to be calculated for each experiment.
-
4
Adjust pH to ∼7.4 with 0.5 N NaOH and mix by inverting, again trying to minimize the addition of air bubbles to the mixture.
This adjustment is based on the color of the phenol red indicator in the medium. Add NaOH until the medium corresponds to a pH of ∼7.4 using the color of the phenol red as a rough measure of pH (an example of the proper color would be the color of phenol red containing DMEM in an unopened bottle). Typically, this will involve adding μl volumes to adjust; we recommend starting with 50 μl volumes. Do not allow the pH of the collagen solution to increase above ∼7.4 as a proper gel will not form if this occurs.
-
5
Pipet 1.5 ml collagen/cell mixture into each well of a 12‐well plate and allow collagen to polymerize for 30 min at 37°C in a cell culture incubator.
If the collagen does not polymerize to form a collagen plug, potentially the pH of the mixture was not correct.
-
6
After 30 min, add 2 ml supplemented DMEM to each well and incubate 48 hr at 37°C in cell culture incubator.
Seeding melanocytes and NHEKs onto collagen plugs
For these cultures, we plate NHEKs and melanocytes in a 5:1 ratio. Melanocytes are grown in bFGF medium originally developed by Ruth Halaban, which we have found supports pigment production in melanocytes (Halaban, 2003; Zhang et al., 2013) and are seeded onto the collagen plugs in this medium for 24 hr. Once the NHEKs are seeded on the plugs, the medium is changed to E‐medium supplemented with EGF.
-
7
Trypsinize melanocytes and count the number of cells using a hemocytometer. Pellet the required number of melanocytes by centrifuging at 300 × g for 5 min; 1.2 × 105 melanocytes are plated on each collagen plug (pellet 1.2 × 106 cells if seeding onto ten collagen plugs).
-
8
Resuspend melanocytes in bFGF medium at a concentration of 6 × 104 cell/ml. Carefully remove medium from the collagen plugs and add 2 ml melanocyte cell suspension to each plug. Evenly distribute melanocytes by moving the plate in a figure eight motion and incubate the organotypic cultures 24 hr at 37°C in a cell culture incubator.
This step allows the melanocytes to adhere to the collagen plug prior to adding NHEKs. Take care not to swirl the plate in a circle, as this will cause the melanocytes to cluster in the center of the well.
-
9
The following day, trypsinize NHEKs and count cells. Each plug will get 6 × 105 NHEKs (pellet 6 × 106 NHEKs if seeding ten collagen plugs). Pellet the required number of cells by centrifuging at 300 × g for 5 min and resuspend cells in E‐medium supplemented with EGF (5 ng/ml) to a final concentration of 3 × 105 cells/ml.
-
10
Carefully remove bFGF medium from the collagen plugs and replace it with 2 ml NHEK suspension. Evenly distribute NHEKs by moving the plate in a figure eight motion and incubate plug at 37°C in a cell culture incubator for 24 hr.
-
11
Change E‐medium by carefully aspirating the medium in the well without disturbing the collagen plug or the cells on top. Add another 2 ml E‐medium supplemented with EGF (5 ng/ml). Incubate at 37°C in a cell culture incubator for 24 hr.
Incubating the cells for 48 hr after seeding ensures that the NHEKs will be 100% confluent before lifting to an air liquid interface, which is required for proper stratification and differentiation.
Lifting organotypic cultures to air liquid interface
Cultures will be lifted to an air liquid interface by removing the collagen plugs and placing them on a metal grid that will raise them slightly above the bottom of the dish, allowing the plug to sit on the surface of the medium in the dish (Fig. 4). To prepare for lifting the cultures to the air liquid interface, the metal grids must be cleaned and autoclaved. To lift the collagen plugs from the 12‐well plate to place on the grids, use a cleaned and autoclaved laboratory spatula.
Figure 4.
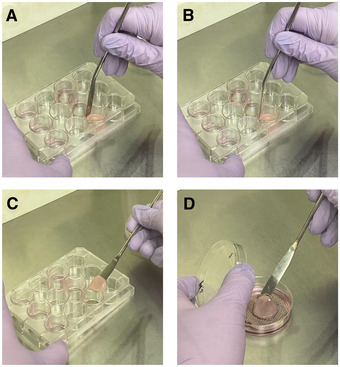
Lifting organotypic cultures to an air liquid interface. (A and B) Carefully break the connection between the collagen plug and the side of the well by pressing a sterile lab spatula up and down around the edge. Do not run the spatula around the edge once pressed down. This can cause the cells on the plug to pull from the plug itself. (C and D) Using the sterile lab spatula, scoop underneath the collagen plug and place the collagen plug onto the metal grid with E‐medium underneath the collagen plug.
-
12
Begin the process by placing one of the autoclaved grids in a 60‐mm culture dish (see Fig. 3 for the preparation of the metal grids).
-
13
Remove medium from the organotypic culture in the 12‐well plates and break the attachment between the collagen plug and the wall of the 12‐well plate using a laboratory spatula. To achieve this, press spatula between the edge of the collagen plug and the side of the culture well until the spatula hits the bottom of the well. Pull the spatula back out vertically and repeat this around the entire circumference of the plug.
Do not run the spatula around the periphery of the plug, as this can cause the cells on the collagen plug to separate from it.
Carefully, using the spatula, lift the collagen plug from the culture well and place it on the metal grid in a 60‐mm culture dish (Fig. 4). See Movie 1 for a demonstration of the lifting technique.
-
14
Carefully add E‐medium to the culture dish until the bottom of the metal grid is in contact with the medium. Make sure that there are no air bubbles between the metal grid, collagen plug, and medium, and incubate plug at 37°C in a cell culture incubator.
If there are air bubbles underneath the collagen plug, the cultures above that point will not be fed by the medium and will not properly form. It is also important at this point to ensure that the top of the organotypic cultures stay free of medium, otherwise the organotypic cultures will not form.
Movie 1.
Demonstration of the lifting technique used for removing the collagen plug from the culture well and placing it on the metal grid.
-
15
To maintain the organotypic cultures, change medium every other day with fresh E‐medium. To change the medium, carefully remove the exhausted medium and add fresh medium until it comes into contact with the bottom of the metal grid. Follow the same guidelines as above.
The cultures can be maintained for 12 days after lifting to an air liquid interface following this protocol. The organotypic cultures will begin to stratify and differentiate quickly after lifting to an air liquid interface and by day 3 multiple layers of cells will be present, granular markers such as keratohyalin granules will be visible by day 4‐5, and by day 6 the cornified layer will be present.
Support Protocol 1. CULTURE AND MAINTENANCE OF NHEKs AND MELANOCYTES
NHEKs and melanocytes must be expanded in cell culture prior to genetic manipulation or creating organotypic skin cultures. The following protocols describe methods for propagating these cells in culture.
Additional Materials (see also Basic Protocols 1 and 2)
Passage of NHEKs
For culture of NHEKs: The first day after isolating NHEKs, the cells will have attached to the bottom of the culture dish but will still be rounded and are only weakly attached to the bottom of the dish. Carefully remove the medium from the cells and replace with fresh M154 with 0.07 mM Ca2+.
For the first 7 days after NHEK isolation, we recommend changing medium daily with M154 medium with 0.07 mM Ca2+ to remove floating cells. After 7 days, the medium can be changed every other day.
Do not allow NHEKs to become 100% confluent when maintaining in vitro; passage cells at ∼80% confluence.
NHEKs can be sustained in cell culture for ∼30 days, or six to eight passages. In our experience, the days since isolation offer a more consistent view of the lifespan of NHEKs than passage number.
-
1
Remove medium from the culture dish and wash with PBS.
-
2
Add 3 ml 0.05% trypsin/1 mM EDTA and incubate at 37°C for 5‐8 min.
The exact time will vary between NHEK isolations; it is best to check the cells every couple of minutes to determine when cells have been released from the cell culture dish. Tapping the side of the dish can help detach some cells if there are a small number still attached.
-
3
Neutralize trypsin by addition of 7 ml DMEM with 10% FBS and pellet cells by spinning at 300 × g for 5 min.
-
4
Resuspend cells in M154 medium and re‐plate cells in a cell culture dish.
Passage protocol for melanocytes
Culture of melanocytes: The first day after isolating melanocytes, the cells will have attached to the cell culture dish and will have flattened out and projected dendrites into the surrounding area. There will also be contaminating NHEKs and fibroblasts attached to the dish as well. Most of these contaminating cells will be removed during the first passage and do not need to be addressed immediately. Change the bFGF medium with fresh medium every other day as the cells divide. The cells can be grown on a coverslip and stained to check for cell purity. Labels for specific cell types include: Melan‐a for melanocytes, keratin 14 or keratin 5 for keratinocytes, and alpha smooth muscle actin to label fibroblasts.
This protocol uses bFGF medium that in our hands is effective at supporting pigment production by melanocytes. However, melanocyte behavior is heavily influenced by medium composition and it will benefit the investigator to identify a medium that works best for the specific use.
Do not allow melanocyte cultures to reach 100% confluence; passage the cells before reaching 80% confluence.
-
5
Remove medium from the culture dish and wash with PBS.
-
6
Add 3 ml 0.05% trypsin/1 mM EDTA and incubate at 37°C for 3‐5 min.
The exact time will vary between melanocyte isolations and it is best to check the cells every couple of minutes to determine when the cells have released from the cell culture dish.
When passaging melanocytes for the first time, pay close attention to when they release from the bottom of the dish, as they will release much more quickly than other cells. Neutralizing the trypsin and collecting the cells as soon as the melanocytes release will aid in removing many of the contaminating cells.
-
7
Neutralize trypsin by addition of 7 ml DMEM with 10% FBS, and pellet cells by spinning at 300 × g for 5 min.
-
8
Resuspend cells in bFGF medium and re‐plate.
Support Protocol 2. LENTIVIRAL TRANSDUCTION OF MELANOCYTES
Organotypic cultures provide a platform to genetically manipulate cells in a more complex model of skin. This protocol will describe the method used in the results presented here to genetically manipulate melanocytes to be used in organotypic cultures. Once the melanocytes are expressing the plasmid, they can be used for incorporating into organotypic cultures.
Additional Materials (see also Basic Protocols 1 and 2)
Melanocyte infection
-
1
Plate melanocytes at ∼30% confluence or less into a 6‐well plate. Incubate overnight at 37°C in a cell culture incubator to allow cells to attach to the plate.
-
2
The next day, change medium with bFGF medium with 1 µg/ml Polybrene added.
-
3
Add lentivirus to the top of the cells and gently swirl to spread virus; incubate at 37°C for 4‐8 hr.
The volume of lentivirus added to the cells will depend on how concentrated the virus is. This should be experimentally determined prior to performing infections to be used in organotypic cultures.
-
4
Remove medium and replace with normal growth medium.
Cells should express the constructs several days later.
Support Protocol 3. RETROVIRAL TRANSDUCTION OF NHEKs
It is also possible to genetically manipulate NHEKs prior to incorporating into organotypic cultures. There are many ways to successfully genetically manipulate NHEKs for use in organotypic cultures that each have their advantages and disadvantages. Electroporating small interfering RNAs (siRNAs) into the NHEKs can be used to knockdown genes of interest before creating organotypic cultures. siRNA is much easier to create compared to other methods and is often commercially available to knockdown a gene of interest. However the duration of knockdown can be short, limiting the timepoints that can be used. NHEKs can also be transduced using lentiviruses and retroviruses. In our experience using a retroviral system provides more effective transduction with less toxicity when compared to lentivirus but both can be used effectively. Viral transduction can be used to either knockdown or overexpress a gene of interest. Viral transductions also provide more stable genetic modification of the NHEKs than other methods, allowing for sustained knockdown or overexpression. Viral systems do take more time to develop and test, which can be a disadvantage of this system. The following protocol describes the method to transduce NHEKs using a retrovirus containing supernatant. See Simpson, Kojima, and Getsios (2010) for detailed methods for creating retroviral supernatants.
Additional Materials (see also Basic Protocols 1 and 2)
NHEK infection
-
1
Plate NHEKs at ∼20%‐30% confluence in 100‐mm dish. Incubate overnight to allow the cells to attach to the plate.
-
2
Make transduction solution by adding viral supernatant and 4 μl Polybrene to 4 ml fresh M154 medium.
-
3
Remove medium from NHEK culture and replace with transduction solution. Incubate plate at 32°C for 1 hr.
-
4
Wash cells two times in PBS, discarding wash into 10% bleach solution.
-
5
Remove medium and replace with normal growth medium.
Cells can be maintained as described above and should express the next day.
Support Protocol 4. WHOLE MOUNT IMMUNOSTAINING PROTOCOL
Whole mount staining of organotypic cultures allows for the analysis of three‐dimensional structures present in the cultures that are difficult to visualize using tissue sections. This includes melanocyte dendrite morphology, which is difficult to observe in tissue sections but is easily observed and quantified in whole mount staining. It is recommended that antibodies, or other labels such the F‐actin label phalloidin or DAPI, are tested to determine if they work in a pilot experiment before beginning a large experiment, as not all antibodies or labels work well in whole mount staining. We have successfully used ProLong Gold as a mounting medium for prolonging fluorescence staining in whole mount samples. If another mounting medium is used, we recommend testing first in a pilot experiment.
To image stained whole mount samples, it is best to use a fluorescent microscope that can image through thicker tissues. These would include confocal microscopes, multi‐photon microscopes, or light sheet microscopes as examples. Grid‐based optical sectioning techniques, such as those used by the Zeiss Apotome, do not work well for imaging whole mounts as the tissue is too thick. See Figure 5A for example images of whole mount stained samples taken on a confocal microscope. Another option is to whole mount unstained organotypic skin cultures and image with a brightfield microscope. This allows visualization of the pigmented melanocytes and any pigment released into the culture. To be able to visualize the pigment without prior staining, melanocytes isolated from type 5 or 6 skin from the Fitzpatrick scale are required, as melanocytes isolated from type 1‐4 skin do not produce easily visible pigment. See Figure 5B for example images of organotypic skin culture whole mounts taken on a brightfield microscope.
Figure 5.
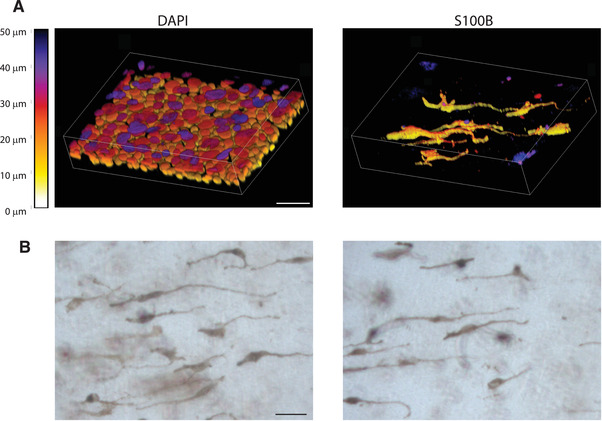
Whole mount staining of organotypic cultures. (A) Example images showing whole mount stained organotypic cultures stained with DAPI and S100B to label melanocytes. Images were collected using a Nikon A1R confocal laser scanning microscope equipped with GaAsP detectors and 20× Plan‐Apochromat objective with an NA of 0.75. NIS‐Elements were used to generate 3D images of z stacks using the alpha‐blend method with z‐depth color coding. Lookup table represents z depth based on voxel color, scale bar = 50 μm. (B) Example images of whole mount samples taken with a brightfield microscope. These samples were not stained allowing visualization of the pigmented melanocytes and any pigment released into the culture. Scale bar = 50 μm.
Additional Materials (see also Basic Protocols 1 and 2)
Organotypic skin culture (see Basic Protocol 2)
4% formaldehyde solution: Dilute 16% formaldehyde (Thermo Fisher Scientific, PI28908) to 4% formaldehyde in PBS and keep on ice while using
Blocking and permeabilizing solution (PBS, 5% Normal Goat Serum, 1% Triton X‐100)
Primary and secondary antibodies, e.g., in Figure 5A Anti‐S100B primary antibody (Abcam, ab52642) and Goat Anti‐Rabbit IgG, Alexa Flour 568 secondary antibody (Thermo Fisher Scientific, A‐11011) were used
ProLong Gold Antifade mounting medium (Thermo Fisher Scientific, P36930)
60‐mm TC‐treated Culture Dish (Corning, 430166)
12‐well Flat Bottom TC‐treated Cell Culture Plate (Corning, 353225)
Forceps
Fix and wash the epidermal sheet
-
1
Remove medium from the well and flood with PBS until the top of the organotypic culture is covered.
-
2
Carefully remove the organotypic culture from the metal grid.
Sometimes the epidermal sheet will have grown around the edge of the plug and is attached to the metal grid. These areas can be lifted off the grid by grabbing with forceps and pulling up carefully.
-
3
Remove epidermal sheet from the collagen plug by grabbing the sheet with forceps at an edge and peel away from the collagen plug. Place epidermal sheet with the cornified layer (the topmost layer of the sheet) down onto a 60‐mm culture dish and place it on ice.
-
4
Fix epidermal sheet by adding 3 ml ice cold 4% formaldehyde and incubating 15 min on ice.
-
5
Wash epidermal sheet three times in PBS for 5 min each time.
Whole mount staining
-
6
Move each tissue to a well in a 12‐well tissue culture plate, ensuring that the tissue is placed with the stratum corneum up (flipping the tissue over from the direction placed when fixing the tissue).
-
7
Add 1 ml blocking and permeabilizing solution, Parafilm the plate closed, and incubate at 37°C for 1 hr.
-
8
Remove solution and incubate tissue in blocking and permeabilizing solution containing primary antibody. Parafilm the plate closed, and incubate overnight at 37°C.
-
9
After incubating overnight, wash tissue three times in PBS for 10 min each wash.
-
10
Add blocking and permeabilization solution containing secondary antibody and a nuclear stain such as DAPI, if desired. Incubate overnight at 37°C.
-
11
Wash tissue three times in PBS for 10 min each wash.
-
12
To mount the samples, place tissue on a slide, add 30 μl Prolong Gold, and place coverslip on top of tissue. Allow to harden overnight at room temperature.
Support Protocol 5. MEASURING MELANOCYTE DENDRICITY
To facilitate transfer of pigment between melanocytes and surrounding keratinocytes, melanocytes extend dendrites into the surrounding tissue. This allows a relatively smaller number of melanocytes to transfer pigment to a large number of surrounding keratinocytes. During the normal tanning response, melanocytes will increase the length and branching of dendrites, which is thought to further facilitate pigment transfer. The opposite, retraction of melanocyte dendrites, is observed during melanomagenesis. An advantage of the organotypic culture models, combined with whole mount staining and imaging, allows for quantification of 3D melanocyte dendricity, including total dendrite length and number of branchpoints. To measure changes in melanocyte dendrites, we use a tool originally developed for the analysis of neuron dendrites, SNT, available in the free image analysis software FIJI (Arshadi, Günther, Eddison, Harrington, & Ferreira, 2021). Below we describe a method using SNT to model and measure changes in melanocyte morphology in 3D confocal images. General instructions about all functions of SNT and FIJI are also available through the FIJI website (https://imagej.net). To see an example of images used for mapping and data generated from this method see Figure 6.
Figure 6.
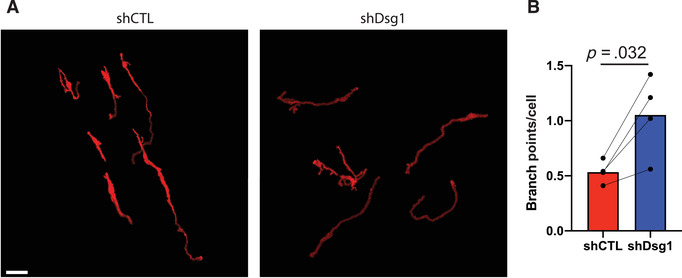
Measuring changes in melanocyte dendricity in organotypic skin cultures. Activation of the tanning response in melanocytes results in both an increase in pigment production, as well as an increase in melanocyte dendricity to promote transfer of pigment to the surrounding keratinocytes. Activation of the tanning response can be mediated by loss of keratinocyte desmoglein 1 (Arnette et al., 2020). To measure melanocyte morphology the melanocytes were transduced with a construct expressing tdTomato allowing visualization of the cells in the organotypic culture. To whole mount stain the organotypic cultures the epidermal equivalent was removed from the collagen plug and fixed in 4% paraformaldehyde and stained with DAPI before mounting to a slide. The entire thickness of the organotypic culture was then imaged using a Nikon A1R confocal microscope equipped with GaAsP detectors and 20× Plan‐Apochromat objective with a NA of 0.75. To measure changes in melanocyte morphology the SNT plugin for FIJI was used to generate 3D models of the melanocytes, and these were used to determine dendrite length and branches of the melanocyte dendrites. (A) Representative images of melanocytes in organotypic cultures with control (shCTL) or desmoglein 1 knockdown (shDsg1) NHEKs. Scale bar = 50 μm. (B) Quantification of the average number of branchpoints per melanocyte under each condition. NHEKs, neonatal human epidermal keratinocytes.
Materials
Image analysis software (FIJI, https://imagej.net)
Fluorescent images
Open image in SNT and create model of single dendrite
-
1
Open the image using FIJI.
It is easiest if the channel that will be used to generate the model is pseudo colored gray; the colors used by SNT to represent the models are most easily visible if the image is black and white. To change the color of the channel, select Image > Look Up Table > Greys.
-
2
Open SNT by going to Plugins > Neuroanatomy > SNT and select the desired image in the prompt.
If the Neuroanatomy selection is not available in the plugins menu then FIJI will have to be updated to include these tools. To start, open the FIJI updater by going to Help > Update… and allow FIJI to update. If prompted to restart FIJI, restart the program, then go back to updater by going to Help > Update… again. After the update runs, an ImageJ Updater dialog box will open; click the Manage Update Sites button on the bottom left of the dialog box. This will open another dialog box showing plugin resources that are freely available for ImageJ. Scroll down the list and click the check box next to Neuroanatomy. Close the Manage Update Sites dialog box by clicking the Close button on the bottom right of the dialog box. This will bring up the ImageJ Updater dialog box again; click the Apply Changes button in the bottom right and this will download the Neuroanatomy plugins, including SNT. Restart ImageJ and check to make sure SNT is available in the plugins dropdown menu.
-
3
Pick a starting point by selecting a point in the cell body of one of the melanocytes in the image that you wish to model. This will lay a point that represents the start of the model.
-
4
Select another point along the dendrite that is being modeled and SNT will identify and draw a path connecting these points, following the staining of the dendrite. The path drawn between the points will be cyan indicating that this is a temporary path until it is approved by the user. To approve the path, click yes in the SNT dialog box or click the Y key on the keyboard. Once approved, the path will turn red, indicating that the path is the current working path.
-
5
Repeat by clicking a point further along the current dendrite, approve the connecting path, and repeat this until the entire length of the dendrite has been modeled.
-
6
Finish the path by clicking Finish in the SNT dialog box or by clicking F on the keyboard. This will cause the path to turn pink representing a completed unselected path. This will also place a path in the Path Manager.
Create a branching path off an existing path
-
7
To create a branchpoint off an existing path, first select the path in the Path Manager. This will cause the selected path to turn green. Hold the Alt key; this will cause the selection point to be along the currently selected path and click at the point where the dendrite branches. This will drop a starting point along the current path, creating a branchpoint. Extend the path as described above and finish the path.
-
8
Clicking finish will add a daughter path in the Path Manager representing the branched path just created.
-
9
Repeat creating branched paths until all of the dendrites of the current melanocyte have been modeled.
Output model analysis
-
10
To output data from the model, including dendrite lengths and number of branchpoints, select Analyze > Measurements > Measure Path(s). This will open a dialog box.
-
11
To get data from each individual path, select Measure Individual Paths check box, select any boxes representing the desired output data, and select OK.
-
12
This will open a table with the selected data that can be copied and pasted into an Excel document or any other analysis software desired.
Support Protocol 6. FONTANA‐MASSON STAINING PROTOCOL
In general, analysis that can be performed on normal skin can also be performed on organotypic cultures. The cultures can be formalin fixed and paraffin embedded (FFPE), and sectioned like normal skin, and pigment can be stained using the Fontana‐Masson stain protocol as would be used for normal skin. Fontana‐Masson stain will stain melanin black and the kit used here uses a nuclear fast red stain to label the tissue. The intensity of the nuclear fast red stain can be modified by increasing or decreasing the time incubated in the stain, and if quantifying Fontana‐Masson stain, reducing it is best to reduce the nuclear red fast stain as much as possible. For quantification of Fontana‐Masson stain, the images can be taken in brightfield and areas that are positive for stain can be quantified. For an example of Fontana‐Masson stained organotypic skin cultures used for quantification see Figure 7.
Figure 7.
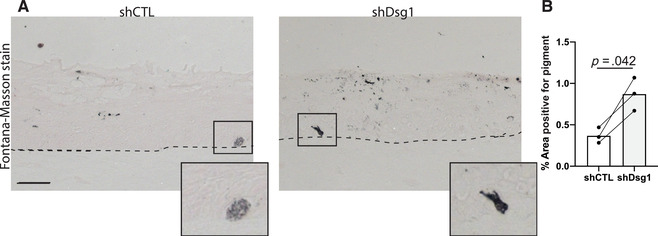
Measuring changes in pigment in organotypic skin cultures. Loss of keratinocytes desmoglein 1 results in an increase in pigment production and release from melanocytes through paracrine signaling. Fontana‐Masson stain can be used to visualize pigment present in sections from organotypic cultures to demonstrate changes in pigment levels in different conditions. (A) Representative images of organotypic culture sections stained with the Fontana‐Masson stain. Organotypic cultures were created using either control NHEKs (shCTL) or desmoglein 1 knockdown keratinocytes (shDsg1). Scale bar = 20 μm. (B) Quantification of area positively stained with Fontana‐Masson stain. NHEKs, neonatal human epidermal keratinocytes.
Materials
Prepare organotypic culture sections
-
1
Begin to deparaffinize sections by baking the slides overnight in 60°C oven.
-
2
The following day, incubate sections three times in xylenes for 3 min each time to remove the remaining paraffin.
-
3
Rehydrate sections by incubating three times in 100% ethanol for 3 min each time, one time in 70% ethanol for 3 min, and two times in distilled water for 3 min each time.
Prepare ammoniacal silver solution
-
4
In new glassware, mix 27 ml distilled or deionized water with a vial of the silver nitrate solution from the Fontana‐Masson staining kit and blend completely.
-
5
Add ammonium hydroxide to the solution one drop at a time. Each drop will cause the solution to turn dark brown and fade to clear with a layer of sediment on the bottom of the glassware. Keep adding drops of ammonium hydroxide until the sediment is fully dissolved.
-
6
Place ammoniacal silver solution in 60°C water bath and allow to equilibrate.
Stain tissue sections
-
7
Incubate rehydrated slides in warmed ammoniacal silver solution for 30 min.
-
8
Wash slides by placing three times in distilled water for 3 min each time.
-
9
Incubate slides in gold chloride solution (from Fontana‐Masson staining kit) for 30 s at room temperature.
-
10
Wash slides by placing three times in distilled water for 3 min each time.
-
11
Incubate slides in sodium thiosulfate solution (from Fontana‐Masson staining kit) for 1 min at room temperature.
-
12
Rinse for 2 min in running tap water followed by two washes in distilled water for 3 min each time.
-
13
Incubate slide in nuclear fast red solution (from Fontana‐Masson staining kit) for 5 min.
-
14
Wash three times in distilled water for 5 min each time.
-
15
Dehydrate by incubating three times in 100% ethanol for 3 min each time.
-
16
Mount slides using the ProLong Gold mounting medium.
The nuclear red fast stain will fade quickly so image the slides as soon as the mounting medium has hardened.
Support Protocol 7. BrdU LABELING AND STAINING
Identifying changes in proliferation rates within any cell type in an organotypic culture can be quantified by labeling cells with the thymidine analog BrdU. BrdU functions by incorporating into DNA during DNA synthesis and will be maintained in the genome over time, thus labeling any cells which have replicated their DNA while the BrdU was present. Figure 8 shows the results of BrdU staining in control organotypic cultures. In this experiment the organotypic cultures were lifted to an air liquid interface for 6 days, and labeled with BrdU for the last 24 hr, from day 5 to day 6. Under these conditions it is rare to find a BrdU labeled melanocyte (<5% of the melanocytes will label with BrdU). Figure 8 shows two melanocytes, one that has BrdU labeling in the nucleus and one that is negative for BrdU.
Figure 8.
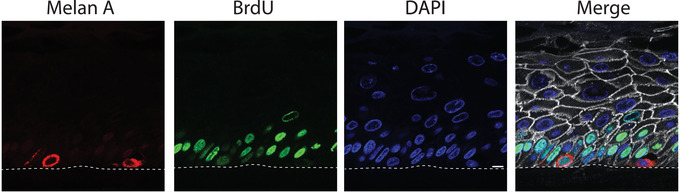
BrdU incorporation and staining in organotypic cultures. Adding BrdU to the medium of organotypic cultures allows the labeling of any cells that replicate their genome during the time BrdU is present. Staining for BrdU and for other cell markers to allow identification of cell types which transition through the cell cycle. Representative images of organotypic cultures, which were labeled for BrdU for 24 hr prior to harvesting and fixing the samples, and stained for BrdU, Plakoglobin to label cell borders, Melan A to label melanocytes, and DAPI. Merge shows overlay of all images. Basal keratinocytes label with BrdU at this timepoint and can be used as a positive control for BrdU staining. The image contains two melanocytes, one labeling positive for BrdU and one negative for BrdU. Scale bar = 10 μm.
Materials
Organotypic skin cultures (see Basic Protocol 2)
5‐Bromo‐2′‐deoxyuridine (BrdU; MilliporeSigma, B9285) stock (10 mM BrdU in PBS): Add 3 mg BrdU to 1 ml PBS, sterile filter; if BrdU does not dissolve, heat in a 37°C water bath; store at −20°C for up to 6 months
E‐medium (see recipe)
10% neutral buffered formalin
DAPI (MilliporeSigma, D9542)
Xylenes
100% ethanol
70% ethanol
Anti‐BrdU antibody (Abcam, ab6326)
Goat Anti‐Rat IgG, Alexa Flour 568 (Thermo Fisher Scientific, A‐11077)
Permeabilization solution (0.05% Triton X‐100 in PBS)
Citrate buffer (0.01 M citrate, 0.05% Tween 20, pH 6.0)
Blocking solution (1% BSA, 2% normal goat serum in PBS)
ProLong Gold Antifade mounting medium (Thermo Fisher Scientific, P36930)
ImmEdge Pen (Vector Laboratories, H‐4000)
Label dividing cells with BrdU
BrdU will be incorporated into the DNA in any cell that replicates its DNA while present. Thus, the investigator must identify a time span when identifying dividing cells is desired. For experiments performed here, BrdU was added to the medium for the last 24 hr, from day 5 to day 6, to label cells which replicate their DNA in this window.
-
1
Dilute BrdU to a final concentration of 10 μM in E‐medium.
-
2
Remove exhausted medium from organotypic cultures and add E‐medium containing BrdU.
-
3
Remove BrdU‐containing medium and replace with fresh E‐medium when done labeling cells.
Stain for BrdU in sections
-
4
Deparaffinize sections by baking overnight in a 60°C oven.
-
5
The following day incubate sections three times in xylenes for 3 min each time to remove the remaining paraffin.
-
6
Rehydrate sections by incubating three times in 100% ethanol for 3 min each time, one time in 70% ethanol for 3 min, and three times in PBS for 3 min each time.
-
7
Permeabilize tissue by incubating in permeabilization solution for 10 min.
-
8
Wash three times in PBS for 3 min each time.
-
9
Perform antigen retrieval by transferring slides to a metal carrier placed in a dish containing the citrate buffer.
-
10
Heat citrate buffer to 95°C and incubate 15 min.
-
11
Remove dish from heat source and allow to cool to room temperature on benchtop.
-
12
Wash three times in PBS for 3 min each time.
-
13
Use the ImmEdge pen to circle the organotypic culture sections on the slide.
-
14
Add 30‐100 μl blocking solution, so that the entire section of the organotypic culture is covered. Place slides into a humidity chamber and incubate 30 min in 37°C incubator.
-
15
Create primary antibody solution by adding 1:5000 BrdU antibody/blocking solution.
If co‐staining with other antibodies also add these to the primary antibody solution at this time.
-
16
Remove block solution from the slides and add primary antibody solution. Place slides in humidity chamber and incubate overnight at 4°C.
-
17
The following day, wash slides three times in PBS for 3 min each time.
-
18
Create secondary antibody solution by diluting goat anti‐rat IgG 1:300 and DAPI 1:500 in blocking solution.
-
19
Add secondary antibody solution to the tissue sections and incubate 1 hr at 37°C.
-
20
Wash slides three times in PBS for 3 min each time.
-
21
Dry slides and mount by adding 30 μl Prolong Gold to each section and cover with coverslip. Allow slides to dry overnight at room temperature protected from light before imaging.
REAGENTS AND SOLUTIONS
Basic fibroblast growth factor (bFGF) medium
500 ml OptiMem Medium (Thermo Fisher Scientific, 11058021)
25 ml FBS (MilliporeSigma, F0926‐500ML)
5 ml penicillin/streptomycin solution (Corning, B003L42)
50 μl bFGF stock (see recipe)
500 μl dbcAMP stock (see recipe)
50 μl heparin stock (see recipe)
9 ml 3‐isobutyl‐1‐methylxanthine stock (see recipe)
Combine all ingredients and store at 4°C for up to 1 month.
Basic fibroblast growth factor (bFGF) stock
Dissolve 10 μg of bFGF (ConnStem, F1004) in 100 μl water.
Store at −20°C for up to 1 month.
Dibutyryl cyclic‐AMP sodium salt (dbcAMP) stock
Dissolve 0.1 g dbcAMP (MilliporeSigma, D0627) in 2.03 ml water.
Store at −20°C for up to 1 year.
E‐cocktail
20 ml adenine solution: Dissolve 486 mg adenine in 20 ml of deionized water. Add 500 μl 6 N HCl (MilliporeSigma, A2786)
20 ml human recombinant insulin solution: Dissolve 100 mg in 20 ml 0.1 N HCl (MilliporeSigma, 91077C)
20 ml human apo‐transferrin solution: Dissolve 100 mg in 20 ml PBS (MilliporeSigma, T1147)
20 ml triiodothyronine solution (see recipe)
Combine all solutions above and bring to 200 ml with PBS. Make 10‐ml aliquots and store at −20°C for up to 1 year.
E‐medium
500 ml DMEM/F12 (Corning, 10‐090‐CV)
500 ml DMEM (Corning, 10‐013‐CV)
10 ml gentamycin/amphotericin B (Thermo Fisher Scientific, R01510)
10 ml E‐cocktail (see recipe)
Hydrocortisone (Final concentration 0.4 μg/ml, MilliporeSigma, H0888)
Cholera toxin (Final concentration 10 ng/ml, MilliporeSigma, C8052)
Combine all ingredients and sterile filter.
Store up to 1 month at 4°C.
Heparin stock
Dissolve 100 mg heparin (MilliporeSigma, 3393) in 10 ml water.
Store at 4°C for up to 2 years.
3‐Isobutyl‐1‐methylxanthine stock
Dilute 0.1 g 3‐isobutyl‐1‐methylxanthine (MilliporeSigma, I5879) in 1 ml DMSO. Add 44 ml H2O and 45 ml Ham's F12 medium.
Store at −20°C for up to 1 year.
M154 supplemented medium
500 ml calcium free M154 medium (from Medium 154CF kit: Thermo Fisher Scientific, M154CF500)
175 μl CaCl2 (from Medium 154CF kit: Thermo Fisher Scientific, M154CF500)
5 ml human keratinocyte growth supplement (Thermo Fisher Scientific, S0015)
-
Combine and store at 4°C for up to 1 month.
Final concentration: 0.07 mM calcium.
Reconstitution buffer
1.1 g NaHCO3
2.3 g HEPES
Resuspend in 50 ml of 0.05 N NaOH and sterile filter.
Store at 4°C for up to 2 years.
Triiodothyronine solution
Dissolve 6.8 mg triiodothyronine (MilliporeSigma, T6397) in 50 ml 0.02 N NaOH. Then dilute 0.1 ml of this solution in 9.9 ml of PBS, and further dilute by taking 0.2 ml and adding to 19.8 ml of PBS.
Store at 4°C for up to 30 days.
COMMENTARY
Background Information
Organotypic cultures have significant advantages when compared to traditional 2D submerged cultures grown in culture dishes. Organotypic cultures more faithfully model in vivo skin architecture as they contain the same structural layers present in normal skin, including a cornified outer layer. Other components of normal skin are also present in the correct locations in organotypic cultures, such as tight junctions, which are localized to the stratum granulosum 2, and proteins like keratin 1, keratin 10, and loricrin, which are correctly localized in the spinous and granular layers (Broussard, Koetsier, Hegazy, & Green, 2021; Getsios et al., 2009). Other skin properties, such as an increase in tension in superficial layers are also faithfully recapitulated in organotypic skin cultures. Organotypic cultures can be fixed and embedded in paraffin for sectioning and staining, and the epidermal sheets can be stained and whole mounted to analyze 3D structure of the entire sheet. Because organotypic skin cultures are created using in vitro expanded cells, they are also easily manipulable, either pharmacologically or genetically, in a way in vivo skin is not. These attributes allow for the study of complex in vivo like behaviors in an in vitro setting.
Traditional organotypic skin cultures, which only incorporate keratinocytes into the epidermal layer of the culture, faithfully recapitulate many aspects of in vivo epidermis; however, they lack other cell types present in the epidermis. Using the methods described here, melanocytes incorporated into organotypic cultures also replicate key in vivo behaviors, such as localizing to the basal layer (Fig. 1).
A limitation of organotypic cultures specific to models which incorporate melanocytes is a lack of efficient uptake of pigment into the keratinocytes. This limitation has been a consistent challenge in the field. While pigment is not efficiently taken up by keratinocytes in these cultures, the melanocytes produce pigment which is released into the surrounding culture (Fig. 7). More generally, the keratinocytes in organotypic skin cultures express wound keratins such as keratin 6 and keratin 16, suggesting they are in a more activated state than in normal skin. Many of these limitations also exist in 2D cell cultures (pigment is also not efficiently taken up by keratinocytes in 2D cultures and the wound keratins are expressed).
Critical Parameters
Isolation and maintenance of primary cells
It is important that the NHEKs used for organotypic raft cultures are used within an optimal window after isolation. This is typically 12 to 18 days after isolation and will often correspond with passage numbers between 3 and 5, though we find that days since isolation is a better determinant of when cells can be used for organotypic cultures. When maintaining all cells in culture, NHEKs, melanocytes, and J2 3T3 cells, ensure that the cells never reach 100% confluence.
Preparation of collagen plugs
Establishing the correct pH of the collagen matrix solution is critical for correct formation of the collagen plug. The pH can be determined by using the phenol red indicator in the solution. The solution should be a light pink color before pipetting into the culture plate and allowing to polymerize. When adding the collagen solution to the culture wells, try to prevent the introduction of air bubbles into the solution, as NHEKs above the air bubbles may not differentiate properly.
Lifting organotypic cultures to air liquid interface
When transitioning the collagen plugs to an air liquid interface it is important to ensure there are no air bubbles between medium and the metal grid. Air bubbles under the collagen plug prevent even feeding of the organotypic culture and can result in areas of the culture not differentiating properly.
Viral transduction of melanocytes or NHEKs
Before creating an organotypic culture with transduced melanocytes, the viral construct should be tested for expression in primary melanocytes. For the results shown here the pLVX‐tdTomato‐C1 vector was used. It is also important to plan the viral infection of melanocytes so that they are ready when needed for seeding onto the collagen plug. Often this requires infecting the melanocytes up to 1 week before seeding to allow the cells to expand to the needed number for the experiment. The same is also true when transducing NHEKs with retrovirus. The virus should be tested prior to use, and timing of the viral infection planned so that cells are healthy and within the optimal timeframe of 12 to 18 days after isolation.
Troubleshooting
Possible problems, causes, and suggested solutions can be found in Table 1.
Table 1.
Troubleshooting Commonly Encountered Problems
| Problem | Possible cause | Solution |
|---|---|---|
| Collagen gel does not polymerize | pH of the fibroblast/collagen solution is not correct | Check pH of the collagen solution and verify it is ∼pH 7.4 |
| Neonatal human epidermal keratinocytes (NHEKs) do not stratify and differentiate | There are several possible causes of NHEKs not stratifying in organotypic cultures, including:
|
Solutions for these problems include:
|
| Melanocytes do not incorporate into the organotypic culture | Melanocytes from some donors do not incorporate into rafts well and will lift off the basement membrane | Use melanocytes from a different donor |
Statistical Analysis
The statistical tests used when performing experiments using organotypic skin cultures will depend on the specific experimental design. We recommend that experiments using organotypic cultures be performed with cells isolated from independently isolated unique donors. We consider these biological replicates. For experiments where single cell level data is generated, e.g., fluorescent intensity of individual cells for imaging experiments, individual cells are akin to technical replicates, with biological replicates again being cultures created from different donors. When graphing data with large numbers of technical repeats, we recommend using “super plots,” which present both technical and biologic repeats on a single graph, allowing visualization of data from individual experiments to be shown and aiding in interpretation. A web browser based tool to create super plots is available (see https://huygens.science.uva.nl/SuperPlotsOfData/; Goedhart, 2021).
When designing organotypic culture experiments, the investigator must also account for significant clonal heterogeneity between cells isolated from different individuals. One method to account for this variability to design experiments that allows the use of paired statistical tests, such as a paired t‐test. These tests determine whether the average of differences between paired samples across a control and treatment group are different from 0. This allows the statistical test to ignore variability within a treatment or group and test if the treatment causes the same change across all samples. This method can be helpful when there is significant clonal variability. Importantly, the decision whether a paired statistical test is going to be used must be made during the design stage of the experiment and the experiment must be designed to use a paired statistical test. For organotypic skin cultures, we would pair a control and treated sample if the cultures are created from cells from the same donor. Typically, enough cells can be isolated from an individual to use replicates for each group of an experiment, allowing pairing of the samples with the control from the same clone and the use of paired statistical tests when analyzing the data. When graphing paired data, we recommend using super plots that have lines connecting the paired samples.
Understanding Results
Figure 1B and C illustrate the results of creating organotypic skin cultures with incorporated melanocytes. When analyzed by hematoxylin and eosin the epidermal sheet in organotypic skin cultures will look remarkably like normal skin, with keratohyalin granules in the granular layer, a cornified layer, and melanocytes localized to the basal layer. The images show an organotypic culture after being lifted to an air liquid interface for 6 days. The organotypic skin cultures will form a cornified layer as early as 3 days after lifting to an air liquid interface, but other layers will be less defined at this time point, and the cultures can be allowed to form for 12 days, after which the cells will be exhausted. Cultures can be extended beyond 12 days by altering the underlying matrix, for instance with the use of de‐epidermalized human skin or incorporating human fibroblasts.
Figures 5, 6, and 7 show examples of types of analyses that can be performed using organotypic skin cultures with incorporated melanocytes. For additional details on the data shown here see Arnette et al. (2020). Figure 5 shows an example of whole mount staining of organotypic skin cultures. Whole mount staining of organotypic skin cultures can be a powerful tool when looking at complex cellular morphologies that are difficult to assess using sagittal tissue sections. Figure 6 shows an example of analyses that can be done using whole mount staining, specifically measuring melanocyte dendricity. The irregular morphology of melanocyte dendrites makes it impossible to accurately measure changes in length and branching in sagittal sections but can be easily visualized with whole mount staining. In Figure 6, we tested the role of keratinocyte desmoglein 1 in controlling melanocyte dendricity associated with activation of the tanning response. We note that the loss of keratinocyte desmoglein 1 caused an increase in melanocyte dendrite branchpoints. Pigment released by melanocytes into the organotypic culture can also be measured using the Fontana‐Masson stain protocol. Figure 7 shows the results of Fontana‐Masson stain in sections from organotypic skin cultures, with released pigment stained in black. Here we test the hypothesis that loss of keratinocyte desmoglein 1 would increase pigment release into the culture, noting that in organotypic cultures lacking desmoglein 1 there is an increase in area stained with pigment.
Time Considerations
If starting from the time point of isolating the primary cells from skin samples, the entire protocol can take up to 25 days. The exact length of time can vary depending on exactly how many days after isolating the NHEKs and melanocytes they are used, and how long the organotypic culture is lifted to an air liquid interface. Typically, cultures that have been maintained at the air liquid interface for between 3 and 12 days are used for the assessments described in this protocol, with greater amounts of stratification and differentiation occurring at later time points.
Author Contributions
Quinn R. Roth‐Carter: Conceptualization, Methodology, Formal analysis, Investigation, Writing original draft, Visualization. Jennifer L. Koetsier: Conceptualization, Methodology, Investigation, Writing editing and review. Joshua A. Broussard: Conceptualization, Methodology, Investigation, Formal analysis, Writing editing and review. Kathleen J. Green: Conceptualization, Writing review and editing, Supervision, Funding acquisition.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases R01 AR041836, National Institutes of Health R37 AR43380, and National Cancer Institute R01 CA228196 to KJG. Additional support was provided by the J.L. Mayberry Endowment to KJG. QRRC was supported by National Institutes of Health T32 Training Grant (T32 CA009560), and National Institute of Arthritis and Musculoskeletal and Skin Diseases F32 AR078645. JAB was supported by National Institutes of Health K01 AR075087.
Roth‐Carter, Q. R. , Koetsier, J. L. , Broussard, J. A. , & Green, K. J. (2022). Organotypic human skin cultures incorporating primary melanocytes. Current Protocols, 2, e536. doi: 10.1002/cpz1.536
Published in the Cell Biology section
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Literature Cited
- Arnette, C. , Koetsier, J. L. , Hoover, P. , Getsios, S. , & Green, K. J. (2016). In vitro model of the epidermis: Connecting protein function to 3D structure. Methods in Enzymology, 569, 287–308. doi: 10.1016/bs.mie.2015.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnette, C. R. , Roth‐Carter, Q. R. , Koetsier, J. L. , Broussard, J. A. , Burks, H. E. , Cheng, K. , … Green, K. J. (2020). Keratinocyte cadherin desmoglein 1 controls melanocyte behavior through paracrine signaling. Pigment Cell & Melanoma Research, 33(2), 305–317. doi: 10.1111/pcmr.12826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arshadi, C. , Günther, U. , Eddison, M. , Harrington, K. I. S. , & Ferreira, T. A. (2021). SNT: A unifying toolbox for quantification of neuronal anatomy. Nature Methods, 18(4), 374–377. doi: 10.1038/s41592-021-01105-7 [DOI] [PubMed] [Google Scholar]
- Broussard, J. A. , Koetsier, J. L. , Hegazy, M. , & Green, K. J. (2021). Desmosomes polarize and integrate chemical and mechanical signaling to govern epidermal tissue form and function. Current Biology, 31(15), 3275–3291.e3275. doi: 10.1016/j.cub.2021.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enjalbert, F. , Dewan, P. , Caley, M. P. , Jones, E. M. , Morse, M. A. , Kelsell, D. P. , … O'Toole, E. A. (2020). 3D model of harlequin ichthyosis reveals inflammatory therapeutic targets. The Journal of Clinical Investigation, 130(9), 4798–4810. doi: 10.1172/JCI132987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick, T. B. (1988). The validity and practicality of sun‐reactive skin types I through VI. Archives of Dermatology, 124(6), 869–871. doi: 10.1001/archderm.1988.01670060015008 [DOI] [PubMed] [Google Scholar]
- Getsios, S. , Simpson, C. L. , Kojima, S.‐i. , Harmon, R. , Sheu, L. J. , Dusek, R. L. , … Green, K. J. (2009). Desmoglein 1–dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. Journal of Cell Biology, 185(7), 1243–1258. doi: 10.1083/jcb.200809044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, S. , Spagnoli, G. C. , Martin, I. , Ploegert, S. , Demougin, P. , Heberer, M. , & Reschner, A. (2005). Three‐dimensional culture of melanoma cells profoundly affects gene expression profile: A high density oligonucleotide array study. Journal of Cellular Physiology, 204(2), 522–531. doi: 10.1002/jcp.20320 [DOI] [PubMed] [Google Scholar]
- Goedhart, J. (2021). SuperPlotsOfData‐a web app for the transparent display and quantitative comparison of continuous data from different conditions. Molecular Biology of the Cell, 32(6), 470–474. doi: 10.1091/mbc.E20-09-0583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaban, R. (2003). Culture of melanocytes from normal, benign and malignant lesions. In R. Pfragner & R. I. Freshney (Eds.), Culture of human tumor cells, (pp. 289‐318). doi: 10.1002/0471722782.ch12. Hoboken, NJ: John Wiley & Sons [DOI] [Google Scholar]
- Li, L. , Fukunaga‐Kalabis, M. , & Herlyn, M. (2011). The three‐dimensional human skin reconstruct model: A tool to study normal skin and melanoma progression. Journal of Visualized Experiments, 54, doi: 10.3791/2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorthois, I. , Simard, M. , Morin, S. , & Pouliot, R. (2019). Infiltration of T cells into a three‐dimensional psoriatic skin model mimics pathological key features. International Journal of Molecular Sciences, 20(7), 1670. doi: 10.3390/ijms20071670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino, D. , Luginbühl, J. , Scola, S. , Meuli, M. , & Reichmann, E. (2014). Bioengineering dermo‐epidermal skin grafts with blood and lymphatic capillaries. Science Translational Medicine, 6(221), 221ra214. doi: 10.1126/scitranslmed.3006894 [DOI] [PubMed] [Google Scholar]
- Martorina, F. , Casale, C. , Urciuolo, F. , Netti, P. A. , & Imparato, G. (2017). In vitro activation of the neuro‐transduction mechanism in sensitive organotypic human skin model. Biomaterials, 113, 217–229. doi: 10.1016/j.biomaterials.2016.10.051 [DOI] [PubMed] [Google Scholar]
- Michielon, E. , López González, M. , Burm, J. L. A. , Waaijman, T. , Jordanova, E. S. , de Gruijl, T. D. , & Gibbs, S. (2020). Micro‐environmental cross‐talk in an organotypic human melanoma‐in‐skin model directs M2‐like monocyte differentiation via IL‐10. Cancer Immunology, Immunotherapy: CII, 69(11), 2319–2331. doi: 10.1007/s00262-020-02626-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, I. , & Kulms, D. (2018). A 3D organotypic melanoma spheroid skin model. Journal of Visualized Experiments, 135, 57500. doi: 10.3791/57500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, J. U. , Abaci, H. E. , Herron, L. , Guo, Z. , Sallee, B. , Pappalardo, A. , … Christiano, A. M. (2020). Recapitulating T cell infiltration in 3D psoriatic skin models for patient‐specific drug testing. Scientific Reports, 10(1), 4123. doi: 10.1038/s41598-020-60275-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, C. L. , Kojima, S.‐i. , & Getsios, S. (2010). RNA interference in keratinocytes and an organotypic model of human epidermis. In Turksen K. (Ed.), Epidermal cells: Methods and protocols (pp. 127–146). doi: 10.1007/978-1-60761-380-0_10. New York: Humana Press [DOI] [PubMed] [Google Scholar]
- Zhang, R. , Premi, S. , Kilic, S. S. , Bacchiocchi, A. , Halaban, R. , & Brash, D. E. (2013). Clonal growth of human melanocytes using cell‐free extracellular matrix. Pigment Cell & Melanoma Research, 26(6), 925–927. doi: 10.1111/pcmr.12159 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.