

Abstract
Small‐molecular‐weight (MW) additives can strongly impact amorphous calcium carbonate (ACC), playing an elusive role in biogenic, geologic, and industrial calcification. Here, we present molecular mechanisms by which these additives regulate stability and composition of both CaCO3 solutions and solid ACC. Potent antiscalants inhibit ACC precipitation by interacting with prenucleation clusters (PNCs); they specifically trigger and integrate into PNCs or feed PNC growth actively. Only PNC‐interacting additives are traceable in ACC, considerably stabilizing it against crystallization. The selective incorporation of potent additives in PNCs is a reliable chemical label that provides conclusive chemical evidence that ACC is a molecular PNC‐derived precipitate. Our results reveal additive‐cluster interactions beyond established mechanistic conceptions. They reassess the role of small‐MW molecules in crystallization and biomineralization while breaking grounds for new sustainable antiscalants.
Keywords: Antiscalants, Crystal Engineering, Green Chemistry, Reaction Mechanisms, Solid-State REDOR NMR
Small but effective: only few small additives are potent antiscalants, extremely solubilizing CaCO3. These underlying molecular mechanisms act beyond standard concepts of nucleation inhibition: they trigger, stabilize, and feed PNCs, presetting key properties of amorphous calcium carbonate (ACC). This study reveals how small molecules may co‐regulate biomineralization and breaks grounds for sustainable antiscalants.
Introduction
Amorphous calcium carbonate (ACC) is a ubiquitous precursor to biominerals, pivotal in the biocrystallization of calcifying organisms.[ 1 , 2 , 3 , 4 ] Organisms meticulously control ACC formation and transformation by a complex molecular machinery whose mechanistic foundations still require further elucidation.[ 5 , 6 , 7 ] Since the detection of nanometric prenucleation clusters (PNCs) in non‐saturated calcium carbonate solutions, it has been argued that both formation and polyamorphism of ACC are linked to PNCs.[ 8 , 9 , 10 , 11 ] It was assumed that the correlation between PNC stability, PNCs formation rates, ACC solubility, and ACC near‐range order as a function of solution conditions implies that these multinuclear carbonato‐calcium complexes [12] are precursors of solid ACC.[ 9 , 10 , 11 ] Molecular simulations of Demichelis et al. describe PNCs as dynamic, thus constantly reorganizing networks, [10] and their stability appears as a balance between ionic coordination and ion hydration with entropic contributions.[ 13 , 14 ] THz spectroscopy showed ACC formation coincides with a change in the solution's dynamic hydrogen‐bond network. [15] The notion of “nonclassical nucleation” induced by reduced cluster dynamics [16] was repeatedly challenged.[ 17 , 18 ] As of yet, it is impossible to chemically trace the evolution of PNCs from the solute to the solid state due to the lack of accountable chemical labeling techniques.
ACC acts as a transient precursor to crystalline calcium carbonates;[ 19 , 20 , 21 , 22 ] controlling ACC formation by additives is thus central for chemistry, biology, geochemistry, and industry. Consequently, the disclosure of PNCs provokes the question of whether additives interact with these carbonato‐calcium complexes and, thereby, impact calcium carbonate formation.[ 23 , 24 ] A refined molecular understanding of how cluster‐additive interactions impact ACC formation would spur the development of sustainable antiscalants. Eutrophic phosphorous antiscalants are still used since a molecular understanding for their impressive antiscaling efficacy and a equipotent but sustainable alternative are lacking. [25] Cölfen et al. have listed nine different additives traits, five of which can be qualitatively discriminated by in situ mineralization profiles:[ 9 , 24 , 26 ] additives can I) sequester cations from solution, II) impact prenucleation‐stage cluster equilibria, III) inhibit nucleation and precipitation of a specific secondary phase, IV) adsorb to and stabilize a secondary phase, or V) favor formation of a specific poly(a)morph.[ 24 , 26 ] Meanwhile, various compounds have been screened with this assay, e.g., carbohydrates, [27] amino acids, [28] polyelectrolytes. [29] These profiling experiments revealed a remarkable complexity and raised further questions. For instance, citrate (CIT) was described as a Type I/II/V additive which stabilizes PNCs by still unidentified mechanisms.[ 26 , 30 ] Another unsettled case is tripolyphosphate (TPP) that was classified as Type I/III/IV/V with dominating Type III/V action. Initially, TPP was supposed to inhibit precipitation by surface adsorption to PNCs. [24] Later, it was suggested that TPP inhibits precipitation via induction of a PILP[ 31 , 32 ]‐like phase [26] and not by steric shielding because TPP is only “weakly involved” [26] in PNCs. These unresolved issues betoken our current lack of a deeper, molecular‐level understanding. Consequently, simplified models inveterately persist in applied industrial studies dictating that additives interfere by ion sequestration or growth inhibition. [33]
Herein, we assess the modes by which small‐molecular‐weight (MW) additives molecularly interact with calcium carbonate solutes, both ions and PNCs. We identify three distinct classes of molecular interaction. The first is the classically expected Ca binding, which reduces Ca activity and leaves ACC stability and composition unaffected. The remaining two novel classes are molecular interaction modes that operate beyond the framework of classical models. The first of these novel modes is the association of a few carbonate ions via Ca2+ bridges, thus, in the second coordination sphere of the additive. This trait allows additives to bind to the surface of PNCs, with pronounced consequences for selected additives: CIT enhances PNC growth by incorporating its ionic freight into PNCs during temporary contact. The second novel class of molecular interaction characterizes the most potent inhibitory agents against ACC precipitation. These additives actively trigger PNC formation by associating a higher number of both calcium and carbonate ions from the solution. Due to subsequent cluster overgrowth, these PNC‐triggering additives molecularly integrate into PNCs. Additives of this novel class considerably enhance the solution stability against ACC precipitation and distinctly alter ACC composition and stability. We found that only these additives can be traced in the ACC precipitate in significant amounts. PNC‐incorporating additives are embedded into the amorphous precipitate as individual molecules in close contact with the mineral matrix. These co‐precipitated additive molecules that are deeply integrated into the ACC matrix considerably stabilize the amorphous phase against crystallization, both spontaneous and thermally induced in wet and dry conditions. Our results demonstrate that additive‐cluster interactions in solution regulate solution speciation (i.e., cluster formation) and solution stability but also later the stability and chemical composition of the forming precipitate.
Results and Discussion
This study screens six different additives (Figure 1), all of which are antiscalants and some relevant to biomineralization.[ 7 , 34 , 35 , 36 ] Ethylenediaminetetraacetic acid (EDTA) is the common cation masking agent but shows unfavorably slow biodegradation rates. Methylglycinediacetic acid (MGDA, also known as α‐ADA) is an EDTA derivative with enhanced biodegradability. Further, citric acid (CIT) and cyclopentane‐1,2,3,4‐tetracarboxylic acid (CPTC) were analyzed. Besides these four carboxylate‐bearing compounds, we further analyzed the bisphosphonate etidronic acid (1‐hydroxyethane 1,1‐diphosphonic acid, HEDP) and sodium tripolyphosphate (TPP). All additives are largely deprotonated under the employed experimental conditions; Tables S2A and S2B in the Supporting Information give dissociation constants and theoretical speciation.
Figure 1.

Top—Molecular structures of the employed small‐MW additives (For clarity, the fully protonated version is shown; theoretical speciation at pH 9.75 is given in Table S2B; note that additives undergo further deprotonation upon cation‐binding[ 41 , 42 ]). Middle—SEM images of the products after fast precipitation in the presence of additives. All powders were amorphous. Bottom—Products retrieved after 15 min of ripening. Powders generated with EDTA, MGDA, and CIT transformed into mixtures of calcite and vaterite. Only in the presence of CPTC, HEDP, and TPP the powders remained amorphous.
As a first assessment, we precipitated ACC in their presence (and in absence as a reference) by mixing solutions of calcium chloride (3.5 mL, 80 mM) and sodium carbonate (50 mL, 80 mM) at pH 9.75, immediately retrieving the precipitate without further reaction time. These precipitation experiments were conducted at a fixed additive concentration of 800 μM. In all cases, ACC with similar particle morphologies formed (see Figure 1 and Figures S1, S2 A in the Supporting Information).
To examine the stability of the initially formed ACC against crystallization, we conducted ripening experiments by expanding the reaction time by 15 min. Additive‐free ACC, EDTA‐, MGDA‐, and CIT‐ACC transformed to varying mixtures of crystalline vaterite and calcite (Figure S2B). In stark contrast, CPTC‐, HEDP‐, and TPP‐ACC withstood phase transformation, demonstrating a remarkable stabilization of the amorphous state. A comparable phase transformation behavior is found in aging experiments, re‐suspending dry ACC powders in pure water for 15 min (Figure S2C). Then, only CPTC‐, HEDP‐, and TPP‐ACC withstood phase transformation.
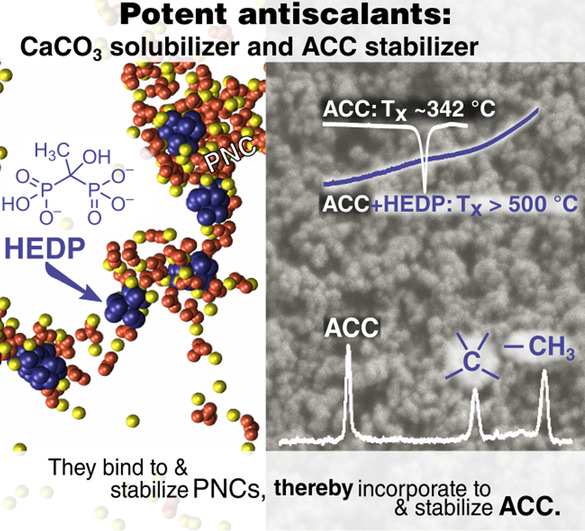
DSC under N2 atmosphere allowed to quantify the additives’ stabilization effects on ACC powders (Figure S3). EDTA and MGDA left the crystallization temperature mostly unaffected (approx. 340 °C, matching additive‐free ACC). CIT‐ACC showed a minor increase to about 360 °C. Again, in striking contrast, the additive triad of CPTC, TPP, and HEDP increased the crystallization temperature considerably. CPTC led to a crystallization temperature of 411 °C, whereas the most extreme cases were HEDP and TPP, for which no pronounced endo‐ or exothermic reaction was observed up to 600 °C. In both cases, no crystalline phases could be detected after annealing to 450 °C, and only traces of crystalline phases after 600 °C.
TGA under N2 atmosphere gave the co‐precipitated additive mass fraction (Figure S4). EDTA and MGDA were present at insignificant levels if at all (<0.4 w %, at the method's resolution limit), while CIT was found at an intermediate level of 1.3 w %. In contrast, the additive triad CPTC, HEDP, and TPP was incorporated at significantly higher fractions (3.7 w %, 4.5 w %, and 4.7 w %, respectively).
SS‐NMR measurements of the additive‐ACC powders enabled us to identify which additives co‐precipitated with ACC, their incorporation levels, and that they are molecularly dispersed in the calcium carbonate matrix. Furthermore, SS‐NMR served to assess the temporal stability of these precipitates under dry conditions.
The notable additive peaks in the 13C and/or 31P cross‐polarization (CP) MAS spectra of the precipitates (Figure 2, left and Figures S5a, b) show that only HEDP, TPP, CPTC co‐precipitated with ACC at levels of approx. 2–3 mol % (see below), conforming with the TGA analyses perfectly. The much weaker CIT peaks indicate its co‐precipitation occurred at a much lower level. Cross‐polarization selects and enhances peaks of carbons (or phosphorous) proximate to hydrogen atoms, hence straightforwardly exposed the hydrogen bearing carbons and phosphorus species of each of the co‐precipitated additives. ACC's intrinsic structural water molecules (ACC: H2O≈1 : 1) detected by 1H MAS NMR (not shown) render the amorphous carbonates visible in the CP MAS spectra as fingerprinted by the dominant 168.5(δν≈4.0) ppm peak (Figure 2A).
Figure 2.

75.4 MHz 13C CP (left) and DE (right) MAS SS‐NMR spectra of the ACC samples. The molecular structures of the additives given on the left are the most abundant at pH 9.75 (Table S2B); note that further deprotonation occurs upon cation‐binding.[ 41 , 42 ] The CP MAS technique (A) enhances peaks of carbons proximate to hydrogen atoms and, as such, selectively exposes environments near water and/or the organic additive molecules—the hydrated ACC carbonates and the occluded additives. The DE MAS technique (B) directly excites all 13C species under quantitative conditions (less sensitive), which allows identifying the occurring crystalline polymorphs and the disordered forms (ACC). The CP MAS spectra for all precipitates (B) were re‐acquired after one to two weeks (after recording the DE spectra in A). While for CPTC, HEDP, and TPP, the spectra remained unchanged (not shown), for EDTA, MGDA, and CIT, the carbonate peak vanished from the CP spectra, indicating complete dehydration (overlayed red spectra), as a result of concomitant partial crystallization seen in the DE spectra.
To identify that the co‐precipitated additives have become occluded within the ACC matrix, we probed interatomic proximities between the matrix carbonates 13C's and the 31P's of HEDP and TPP using 13C{31P} CP‐REDOR measurements (Supporting Information and Figure S5d–f).[ 37 , 38 ] Specifically, the extensive attenuation of the ACC peak (Figure S5e) due to carbonate⋅⋅⋅phosphorus dipolar interactions, identifies that carbonates within<1 nm from P‐atoms of HEDP or TPP constitute a fraction larger than 25 %. A more accurate assessment is obtained by fitting the increasing attenuation of the carbonate vs. on‐time of the dipolar interaction—REDOR evolution (Supporting Information and Figure S5f). The fitted evolution indicates that these additives are molecularly dispersed throughout the entire ACC matrix, with ≈6 mol % P/Ca concentration. For P2(HEDP)/Ca and P3(TPP)/Ca this translates into ≈3 and ≈2 mol %, respectively. [7] In turn, these concentrations amount, on the average, to having an additive molecule in every ACC sphere of 7–8 Å radius. Importantly, occlusion in such dimensions can be realized only if it occurs when PNCs form and grow, as is also shown by our MD simulations (see below). The broad additive peaks (13C peaks of HEDP, CPTC, and CIT (δν≈6 ppm); 31P peaks of TPP and HEDP, Figures 2 and S5) represent disordered local environments consistent with these four additives being molecularly dispersed in an amorphous matrix (ACC). [37] Our CP MAS spectra suggest that citrate incorporation level is up to an order of magnitude lower than those of TPP, HEDP, and CPTC; the incorporation level of EDTA and MGDA, if any, is at least two orders of magnitude lower. As structural water molecules (and to a lesser extent the molecularly dispersed additives) render the amorphous carbonates peak visible in the CP MAS spectra, this peak serves as a marker of the temporal stability of these ACC variants under ambient conditions.
Indeed, this peak persisted intact for CPTC‐, HEDP‐, and TPP‐ACC, however, after one to two weeks it vanished from the CP MAS spectra of EDTA‐, MGDA‐ and CIT‐ACC, evidencing they have spontaneously dehydrated (Figure 2A, red spectra). Moreover, the 13C DE MAS spectra (Figure 2B), a complimentary quantitative technique, further identify that EDTA‐ and MGDA‐ACC precipitates underwent partial crystallization, resulting in anhydrous disordered vaterite (≈85 %) and disordered calcite (≈15 %) (Figure 2B; excessively broadened vaterite and calcite peaks). [37] CIT‐ACC transformation advanced further, yielding more ordered (narrower peaks) vaterite and calcite mixture. The additive‐free ACC precipitate, our reference sample, had transformed almost completely to vaterite within 4 days, apart from a small ACC remainder (≈2 %) and residual hydrated disordered vaterite (Figure S5c). The extent of amorphous‐to‐crystalline transformation of the ACC samples seen in the NMR experiments is like that observed for ripened samples (XRD; Figure S2). After longer waiting times (months), calcite also appeared in the control sample (not shown), an observation in line with Ostwald's rule of stages. The high abundance of vaterite and its persistence suggests it is stabilized due to our precipitation conditions in line with earlier reports.[ 26 , 37 ]
Of the six additives, only CPTC and the two phosphorous‐bearing additives, HEDP and TPP, yielded stable, hydrated ACC, as attested by the persistence of the ACC peak in both the CP and DE MAS spectra. These are also the additives found to be effectively occluded within the amorphous matrix. Their 2–3 mol % concentration implies occlusion occurred during the early stages of cluster formation, when strong additive‐solute interactions are formed and maintained throughout growth and precipitation, and so also stabilize the solution against precipitation. ACCs generated in the presence of EDTA, MGDA, CIT, and the additive‐free, have undergone spontaneous dehydration and amorphous‐to‐crystalline transformation to vaterite and calcite mixtures reaching various degrees of completeness and disorder.[ 26 , 37 ]
Mineralization profiles monitor the solution's calcium activity during pH‐constant Ca titrations of carbonate buffers; they provide detailed information[ 24 , 26 ] about how our selected additives impact calcium carbonate formation (Figure 3) when compared to additive‐free mineralization profiles and carbonate‐free Ca‐binding profiles. To be comparable with earlier studies and to attain workable profiling of the additives, ion and additive concentrations were reduced by a factor of 8 relative to the initial precipitation procedures, maintaining their concentration ratio.
Figure 3.

Mineralization profiles (thus, in presence of carbonate; two left columns) and Ca‐binding profiles (thus without carbonate; right, framed column) of the individual additives. These profiles show the measured Ca activity as a function of the Ca concentration. The middle column is an enlarged view of the left. The dashed black line in mineralization profiles gives the median of reference experiments (without additives). Its gray envelope shows the scatter of reference profiles around the black dotted median, which is insignificant in the beginning and only becomes significant in the nucleation regime due to stochastic nature of nucleation. The reference profile with standard deviation is given in Figure S7. The dashed gray line in Ca‐binding profiles displays the total dosed Ca concentration, whereas the continuous gray line shows the initial slope of the additive‐free reference mineralization profiles. All additives were used at a concentration of 100 μM; both mineralization and Ca‐binding profiles were recorded at a constant pH of 9.75.
The mineralization profiles in presence of EDTA and MGDA show efficient Ca‐masking capabilities as the mineralization profiles are shifted to the right; they stoichiometrically remove free Ca ions from the solution. After reaching their Ca‐binding capacity, their slopes equal the reference; thus, Ca‐saturated masking agents give way for Ca‐binding by carbonate/clusters, and Ca‐binding proceeds as in the absence of additives. Both EDTA and MGDA allow for a higher maximal prenucleation Ca activity; they also lead to elevated post‐nucleation plateau levels, indicating a change in phase selectivity in favor of more soluble phases.
In contrast, CIT is not a Ca‐masking but only a mediocre Ca‐binding agent on par with carbonate under the given conditions, as evidenced by its Ca‐binding profile. The initial slope is only slightly lower than the additive‐free reference. Later, the slope steepens without reaching the slope of the reference, an effect that is more prominent at higher CIT concentrations (Figure S6). The nucleation point is right‐shifted by 30–50 μM, less than half the additive nominal concentration. These changes suggest a two‐fold action of CIT: Ca‐binding and—as proposed earlier [26] —PNC stabilization by a mechanism indefinable by mere in situ profiling. CIT is the only additive without impact on the maximal prenucleation Ca activity.
The remaining triad of additives, i.e., TPP, HEDP, and CPTC, forms a class of its own. They efficiently suppress precipitation by allowing significantly higher maximal Ca activities, especially the phosphorous‐bearing agents TPP and HEDP. Thus, they act as solution stabilizers (solubilizers), shifting the point of nucleation to higher Ca dosages. This inhibitory action is independent of a strong Ca‐binding: CPTC is almost as mediocre as CIT in its Ca‐binding affinity in contrast to TPP, which is a Ca‐masking agent as strong as MGDA. Turning to their mineralization profiles, their initial stages resemble their respective Ca‐binding curves suggesting that Ca‐binding by additives initially outcompetes Ca‐binding by carbonate/clusters. In contrast to CIT, the slopes of the individual mineralization profiles approach the reference slope at later stages. Because of this behavior, it was supposed that TPP is “only weakly involved in these [prenucleation] clusters”. [26]
Reviewing these results, we can group the inspected additives into two sets of end‐members, with CIT as a transitional case. The first set is exemplified by the masking agents EDTA and MDGA. These Ca‐binding agents delay precipitation in a stoichiometric manner by effectively reducing the solution's Ca activity. However, they hardly stabilize the solution against precipitation. The triad of solution stabilizers CPTC, HEDP, and TPP form the other set of end members. Their presence exquisitely stabilizes the solution against precipitation. The particular case of CPTC shows that neither strong Ca‐binding affinity [26] nor specific chemical moieties such as phosphate or phosphonate groups are necessary preconditions for the solubilizing action of this additive triad.
This grouping corresponds to our compositional analyses (TGA, SS‐NMR) as well as to our ACC reactivity (ripening/ageing) tests and DSC analyses. EDTA and MDGA are hardly traceable in the amorphous precipitates, leaving the crystallization temperature of ACC unaffected and exerting no stabilization of ACC against crystallization in wet and dry conditions. The solution stabilizers CPTC, HEDP, and TPP are dispersed as individual molecules in the amorphous precipitate in significant amounts, and they are closely associated with the surrounding mineral matrix. They stabilize ACC both in wet and dry conditions, increasing the crystallization temperature of ACC considerably. CIT is the intermediate case: it is sparsely traceable in the precipitate, and it marginally stabilizes ACC according to DSC measurements to an extent too little to considerably delay Ostwald step ripening under dry and wet conditions.
MD simulations allow us to suggest different modes of molecular action of the additives and, thereby, link experimental observations summarized above with molecular‐scale behavior patterns. We adopted the seminal approach of Gale and co‐workers. [10] We performed a series of simulations, exploring the behavior of each additive for 50 ns in an aqueous solution of 1.5 M calcium carbonate at pH≈10 and ambient conditions. By comparing MD simulations of all six additives, three dynamic interaction modes with PNCs—Classes A to C—can be distinguished that originate from distinct ion‐association motifs. These three interaction modes are summarized in Figure 4A; close‐up snapshots of MD simulation provided in Figure S8 further visualize the different ion association motifs: (a) Additives such as EDTA and MGDA tend to remain dispersed in solution (see label a in Figure 4). Whilst associating several calcium ions, they hardly associate carbonates from solution. Thus, these additives basically associate calcium ions but do not associate to PNCs. (b) For additives such as CIT (but to some extent also MGDA) we find the additive associates both calcium and carbonate ions (label b). Moreover, they feature Ca2+‐mediated association to terminal carbonates that reside at PNC surfaces (label b*). (c) The additive triad of CPTC, HEDP, and TPP associates large numbers of both calcium and carbonate ions. This behavior triggers PNC formation, and, in the course of cluster growth, these additives integrate into the interior of PNCs (label c).
Figure 4.

MD simulations in the presence of additives. Additives are blue, CO3 2− ions red, Ca2+ ions yellow. Water molecules are not shown. Snapshots are representative top views of the simulation box. A) MD simulations provide an additive classification based on the nearest‐neighbor numbers of associated Ca2+ ions in the first and CO3 2− in the second coordination shell of the additive. While all additives bind Ca2+ ions, three distinct modes of CO3 2− association are apparent. Chelating additives such as EDTA bind Ca2+ ions while hardly associating CO3 2− ions. They bind to PNC surfaces only infrequently. The coordination occurrence diagram of EDTA reflects this behavior, showing limited CO3 2− association numbers (in the center top, ). The occurrence diagram of CIT differs in this respect (in the middle center, ), highlighting it as an intermediate case. CIT features several calcium Ca2+ ions in its near vicinity (see label a in the upper right snapshot of CIT) but can also associate CO3 2− ions via Ca2+ bridges (see label b). CIT's average carbonate association number has a further contribution since CIT preferentially associates to PNC surfaces but does not entirely incorporate into PNCs (see label b*). Additives such as HEDP attract larger numbers of both Ca2+ and CO3 2− ( ) and incorporate into PNCs (label c in the lower snapshot of HEDP). Occasionally, they reside at the PNC interface (label b* in the lower snapshot). Further simulations show that these additives actively trigger PNC formation: PNCs form around the additives, and the additive is overgrown by the forming cluster (Figure S12). B) Consecutive snapshots taken after 15 ns of initial equilibration show a single CIT molecule (in green) laden with three Ca2+ ions and one CO3 2− ion. It attaches to a PNC, while some other CIT molecules remain at the surface of the PNC during the sampled time frame. Upon attachment, the PNC incorporates the CIT‐associated ions. Later, CIT was found to detach from the PNC surface.
For all additives, three‐dimensional plots of occurrence densities (Figure S9) illustrate the complex and dynamic behavior of binding and association of Ca2+ ions from solution. These plots demonstrate that, although Ca2+ ions are preferentially coordinated in specific domains in the surrounding of a given additive, neither the Ca2+ positions nor the terminal oxygen atoms of the acidic groups of the additives are spatially well‐defined.
For further quantification and classification of the additive's interaction modes, we evaluated statistics of the nearest‐neighbor numbers of Ca2+ ions in the first coordination shell of the additive and the number of nearby carbonate ions in the additive's second coordination shell, indirectly bound to the additives via Ca‐bridges. For that purpose, we defined distance delimiters (Table S3) based on the interatomic distance distribution functions between Ca2+ and carboxylate‐C or phosphate/phosphonate‐P atoms and between carbonates‐C and terminal additive oxygens (Figure S10). The variety of complexes observed is reflected by the statistics sampled for the number of Ca2+ ions in the first coordination shell of the additive and the number of CO3 2− ions in the second coordination shell, respectively (Figure S11 and Figure 4A, middle). Based on average nearest neighbor numbers ) a standard Class A agent is observed to accord with , thus binding calcium ions from the solution but only a few carbonates. Additives of type B, which preferably reside at PNC surfaces, must associate about two carbonate ions, whereas additive incorporation into the bulk of the PNCs, Class C, is found for >3. We therefore attribute and as characteristics to additive types B and C, respectively.
Along their time course, additives may span more than one interaction mode; however, the average coordination numbers provide a mean of classification for their preferential mode of action. The coordination number statistics of EDTA, CIT, and HEDP are shown as representative cases in Figure 4A, whereas all profiles are provided in Figure S11. These profiles, along with simulation snapshots (Figure 4A, right), demonstrate that each of the explored small‐MW additives exploits two of these three possible interaction modes. For example, MGDA acts both as a Ca‐binding agent but occasionally resides at the PNC‐surface. Contrastingly, HEDP, TPP, and CPTC are predominantly found in the bulk volume of PNCs (label c in Figure 4) but also, less frequently, at their surface (label b* in Figure 4). Indeed, the members of this additive triad are not found to be individually dispersed in the solution but stay associated with clusters. Simulation snapshots revealed that these three additives actively triggered PNC formation and were subsequently overgrown by PNCs (Figure S12). This classification based on coordination statistics (Table S4) aligns with the grouping of additives suggested by our experimental evidence on ACC composition and stability. Together, they highlight the key role of strong additive‐cluster interactions in producing a potent additive. These interactions provoke additive integration in the PNC and, in turn, co‐precipitation within ACC. As such, they play dual roles: solution stabilization and precipitated ACC stabilization.
MD simulations furthermore showed an unforeseen concomitant of the capability of CIT to attach to PNC surfaces. These CIT interactions with PNCs are found to be a dynamic equilibrium of association and dissociation events. Despite the short time scales of MD runs (50 ns), we consistently find that the number of calcium and carbonate ions associated with CIT additives is larger during CIT attachment than upon detachment from PNC surfaces (see snapshot series in Figure 4B), hence demonstrating that CIT unloads some of its ionic cargo at the PNC surface. Thus, CIT can be described as a carrier molecule that actively feeds the growth of PNCs by transporting both calcium and carbonate ions to PNCs.
SANS allowed us to probe the ultrastructure of the non‐ripened ACC and showed a consistent multilevel structure and comparable specific surface areas for all of these powders, be it in the presence or absence of additives (Figure S13–S15). A multilevel Beaucage fit, with the assumption of dense and non‐porous building units, allowed extraction of comparable gyradii of the smallest detectable entities (Table S5). Throughout, these gyradii range from 18.2±0.2 Å to 29.8±0.9 Å, evidencing a persistent nanoscopic organization that correlates well with the estimated size of prenucleation clusters.[ 8 , 9 , 39 , 40 ]
Conclusion
Our experimental findings show that only selected additives such as CPTC, HEDP, and TPP significantly stabilize the solution against precipitation (mineralization profiles). These additives also lead to a remarkable stabilization of ACC against spontaneous crystallization in dry and wet conditions and against thermally induced transformation (ripening/ageing experiments, DSC). Only these solution‐stabilizing additives incorporate into ACC considerably upon precipitation (TGA, SS‐NMR), in form of individually dispersed molecules closely associated with the amorphous mineral matrix (SS‐NMR). Common Ca‐masking additives such as EDTA and MGDA neither impact ACC stability (ripening/ageing experiments, DSC) nor can they be traced in the precipitate in significant amounts (TGA, SS‐NMR). MD simulations on calcium carbonate solutions disclose that only these solution‐stabilizing additives induce PNC formation and incorporate into PNCs, while additives such as EDTA and MGDA hardly interact with PNCs but stay dispersed in solution.
The strong correlation between (a) PNC incorporation of solution‐stabilizing additives in simulation, with (b) their abundance in the resulting ACC and its stabilization, and with (c) the pronounced calcium carbonate solubilization, reveals for the first time, a unified multidisciplinary view on the path of action of high‐potency additives. The fact that precipitation is strongly suppressed only for PNC‐incorporating additives demonstrates that solution stability against precipitation and ACC stability against crystallization are intimately connected via the stability/composition of PNCs. These findings experimentally evidence that ACC is derived from aggregating PNCs, a conclusion fully in line with our recent insights into the ultra‐ and mesostructure of ACC. [40] SANS data provide further corroboration, suggesting that ACC forms by the agglomeration of nanometer‐sized units that do not coalesce but preserve their nanoparticular character upon assembly, an assumption coherent with our recent report on the ultrastructure of ACC. [40]
The additive CIT exhibits an intermediate mode of action. Only trace amounts of CIT occur in the precipitates (SS‐NMR, TGA). It marginally stabilizes ACC against thermally induced transformation (DSC), and no stabilization was observed against transformation in solution (ripening/ageing experiments). CIT is the only additive that does not significantly increase the maximally attainable Ca2+ activity but alters the solution's Ca speciation (mineralization profiles). By relying on mineralization profiles, it has been assumed earlier that CIT exerts a distinct stabilization effect on PNCs.[ 24 , 26 ] Our results show that CIT promotes PNC formation from ion solutions (MD simulations, mineralization profiling) by feeding PNC growth (MD simulations), a transport mechanism that corresponds well with the characteristic slope observed in mineralization profiling. However, MD simulations imply that the main effect exhibited by CIT is PNC surface binding. We additionally infer that CIT acts as a “surfactant” that reduces PNC‐PNC interactions and hinders PNC agglomeration, leading to decelerated precipitation while only weakly affecting ACC stability.
In conclusion, we classify the modes of action of additives based on their molecular interactions with PNCs (Classes A to C). As additives typically show not only one but two of these traits (Figure 4A, snapshots on the right), our classification highlights the additive's predominant role. It is the weighted contributions of mechanisms that lead to all our experimentally observed properties.
The apparent mode of action is the sequestration of cations from solution, i.e., Ca‐binding. This trait simply reduces the apparent activity product of the parental solution. Ca‐masking agents such as EDTA and MGDA are relatively clean representatives of this class, masking individual Ca2+ ions. In MD simulations, they also weakly associate further calcium ions in their first coordination shell but less than two carbonate ions in their second coordination shell ( ).
The preferential affinity of additives to PNC surfaces typifies the second molecular interaction mode. In this mode, additives associate a small number of calcium and carbonate ions ( ). Then, the additive can bind to PNCs surfaces without fully integrating into the PNC bulk volume. CIT is the additive that mainly takes this mode of molecular interaction. As a concomitant effect, CIT increases the cluster growth rate by transporting Ca2+ and CO3 2− ions from the solution to the PNCs.
The third mode of action is shown by additives that generously associate both Ca2+ and CO3 2− ions from solution, thereby provoking PNC formation , e.g., the triad of CPTC, HEDP, and TPP). Their incorporation into clusters leads to a notable suppression of precipitation. They also provide stabilization of ACC against transformation as these additives are incorporated into the precipitate as individually dispersed molecules.
Our results show that PNCs regulate their chemical composition by binding/integrating only selected foreign components. Thereby, PNCs can be seen as gatekeepers that govern the composition of ACC and, thereby, fundamental macroscopic properties of the resulting ACC. The selective incorporation of Class C additives in PNCs renders these additives accountable for chemical labels that provide conclusive chemical evidence that ACC is a molecular precipitate derived from PNCs.
We expect the above interaction modes to be valid for other precipitation systems that are prone to amorphous precursor phases and PNCs. Our work builds a basis for future understanding and for potential designing high potency additives once further molecular physicochemical factors—e.g., the additive's molecular and conformational dynamics and binding geometries (e.g., Figure S9) or ion residence times—are scrutinized. Thereby, our study will provide new starting points for non‐eutrophic, sustainable mineralization inhibitors and novel drug design concepts against nephrolithiasis and other atopic biomineralization processes. Since small‐MW molecules are involved in biomineralization processes,[ 7 , 34 , 35 , 36 ] our findings also bear substantial implications for these systems, including other bio‐ and ACC‐mediated mineralization processes (e.g., in geosciences). As these processes typically occur at near‐neutral conditions, future studies will have to address whether an additive's modes of action are regulated by pH (or other factors), e.g., due to a change in the additive's protonation state.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
P.D., D.Z., and S.E.W. acknowledge financial support from Procter & Gamble Inc. S.E.W. received an Emmy Noether grant from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, grant number 251939425) and gratefully acknowledges financial support by JCNS to perform the neutron scattering measurements at the Heinz Maier‐Leibnitz Zentrum (MLZ), Garching, Germany. A.S. and S.K. acknowledge the Israel Science Foundation grant 2001/17 for financial support. We thank Dr. N. Khansur and S. Fiedler for support in XRD, TGA, and DSC. Open Access funding enabled and organized by Projekt DEAL.
P. Duchstein, P. I. Schodder, S. Leupold, T. Q. N. Dao, S. Kababya, M. R. Cicconi, D. de Ligny, V. Pipich, D. Eike, A. Schmidt, D. Zahn, S. E. Wolf, Angew. Chem. Int. Ed. 2022, 61, e202208475; Angew. Chem. 2022, 134, e202208475.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.33774/chemrxiv‐2021‐fvvwb).
Contributor Information
Prof. Asher Schmidt, Email: asher@ch.technion.ac.il.
Prof. Dr. Dirk Zahn, Email: dirk.zahn@fau.de.
Priv.‐Doz. Dr. Stephan E. Wolf, Email: stephan.e.wolf@fau.de.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Addadi L., Raz S., Weinfurter H., Weiner S., Adv. Mater. 2003, 15, 959–970. [Google Scholar]
- 2. Gower L. B., Chem. Rev. 2008, 108, 4551–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jo C., Hwang J., Lim W. G., Lim J., Hur K., Lee J., Adv. Mater. 2018, 30, 1703829. [DOI] [PubMed] [Google Scholar]
- 4. Politi Y., Arad T., Klein E., Weiner S., Addadi L., Science 2004, 306, 1161–4. [DOI] [PubMed] [Google Scholar]
- 5. New Perspectives on Mineral Nucleation and Growth (Eds.: Van Driessche A. E. S., Kellermeier M., Benning L. G., Gebauer D.), Springer Nature, Cham, 2017. [Google Scholar]
- 6. Zou Z., Yang X., Albéric M., Heil T., Wang Q., Pokroy B., Politi Y., Bertinetti L., Adv. Funct. Mater. 2020, 30, 2000003. [Google Scholar]
- 7. Akiva-Tal A., Kababya S., Balazs Y. S., Glazer L., Berman A., Sagi A., Schmidt A., Proc. Natl. Acad. Sci. USA 2011, 108, 14763–14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pouget E. M., Bomans P. H. H., Goos J. A. C. M., Frederik P. M., de With G., Sommerdijk N. A. J. M., Bomans P. H. H., Goos J. A. C. M., Frederik P. M., de With G., Sommerdijk N. A. J. M., Science 2009, 323, 1455–1458. [DOI] [PubMed] [Google Scholar]
- 9. Gebauer D., Völkel A., Cölfen H., Science 2008, 322, 1819–22. [DOI] [PubMed] [Google Scholar]
- 10. Demichelis R., Raiteri P., Gale J. D., Quigley D., Gebauer D., Nat. Commun. 2011, 2, 590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cartwright J. H. E., Checa A. G., Gale J. D., Gebauer D., Sainz-Díaz C. I., Angew. Chem. Int. Ed. 2012, 51, 11960–11970; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12126–12137. [Google Scholar]
- 12. Wolf S. E., Müller L., Barrea R., Kampf C. J., Leiterer J., Panne U., Hoffmann T., Emmerling F., Tremel W., Nanoscale 2011, 3, 1158–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Finney A. R., Rodger P. M., Faraday Discuss. 2012, 159, 47. [Google Scholar]
- 14. Kellermeier M., Raiteri P., Berg J. K., Kempter A., Gale J. D., Gebauer D., ChemPhysChem 2016, 17, 3535–3541. [DOI] [PubMed] [Google Scholar]
- 15. Sebastiani F., Wolf S. L. P., Born B., Luong T. Q., Cölfen H., Gebauer D., Havenith M., Angew. Chem. Int. Ed. 2017, 56, 490–495; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 504–509. [Google Scholar]
- 16. Gebauer D., Kellermeier M., Gale J. D., Bergström L., Cölfen H., Chem. Soc. Rev. 2014, 43, 2348–71. [DOI] [PubMed] [Google Scholar]
- 17. Smeets P. J. M., Finney A. R., Habraken W., Nudelman F., Friedrich H., Laven J., De Yoreo J. J., Rodger P. M., Sommerdijk N. A. J. M., Proc. Natl. Acad. Sci. USA 2017, 114, E7882–E7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Henzler K., Fetisov E. O., Galib M., Baer M. D., Legg B. A., Borca C., Xto J. M., Pin S., Fulton J. L., Schenter G. K., Govind N., Siepmann J. I., Mundy C. J., Huthwelker T., De Yoreo J. J., Sci. Adv. 2018, 4, eaao6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walker J. M., Marzec B., Nudelman F., Angew. Chem. Int. Ed. 2017, 56, 11740–11743; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11902–11905. [Google Scholar]
- 20. Mass T., Giuffre A. J., Sun C.-Y., Stifler C. A., Frazier M. J., Neder M., Tamura N., Stan C. V., Marcus M. A., Gilbert P. U. P. A., Proc. Natl. Acad. Sci. USA 2017, 114, E7670–E7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DeVol R. T., Sun C.-Y., Marcus M. A., Coppersmith S. N., Myneni S. C. B., Gilbert P., J. Am. Chem. Soc. 2015, 137, 13325–13333. [DOI] [PubMed] [Google Scholar]
- 22. Kababya S., Gal A., Kahil K., Weiner S., Addadi L., Schmidt A., J. Am. Chem. Soc. 2015, 137, 990–998. [DOI] [PubMed] [Google Scholar]
- 23. Gebauer D., Minerals 2018, 8, 179. [Google Scholar]
- 24. Gebauer D., Cölfen H., Verch A., Antonietti M., Adv. Mater. 2009, 21, 435–439. [Google Scholar]
- 25.2004, Regulation (EC) N°648/2004 of the European Parliament and of the Council on Detergents, 2004.
- 26. Verch A., Gebauer D., Antonietti M., Cölfen H., Phys. Chem. Chem. Phys. 2011, 13, 16811–20. [DOI] [PubMed] [Google Scholar]
- 27. Rao A., Berg J. K., Kellermeier M., Gebauer D., Eur. J. Mineral. 2014, 26, 537–552. [Google Scholar]
- 28. Picker A., Kellermeier M., Seto J., Gebauer D., Co H., Z. Kristallogr. 2012, 227, 744–757. [Google Scholar]
- 29. Wolf S. L. P., Jähme K., Gebauer D., CrystEngComm 2015, 17, 6857–6862. [Google Scholar]
- 30. Raiteri P., Demichelis R., Gale J. D., Kellermeier M., Gebauer D., Quigley D., Wright L. B., Walsh T. R., Faraday Discuss. 2012, 159, 61. [Google Scholar]
- 31. Gower L. B., Tirrell D., J. Cryst. Growth 1998, 191, 153–160. [Google Scholar]
- 32. Harris J., Mey I. P., Böhm C. F., Trinh T. T. H., Leupold S., Prinz C., Tripal P., Palmisano R., Wolf S. E., Nanoscale Horiz. 2019, 4, 1388–1393. [Google Scholar]
- 33. Oshchepkov M. S., Rudakova G. Y., Tkachenko S. V., Larchenko V. E., Popov K. I., Tusheva M. A., Therm. Eng. 2021, 68, 370–380. [Google Scholar]
- 34. Davies E., Muller K. H., Wong W. C., Pickard C. J., Reid D. G., Skepper J. N., Duer M. J., Proc. Natl. Acad. Sci. USA 2014, 111, E1354–E1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu Y.-Y., Rawal A., Schmidt-Rohr K., Proc. Natl. Acad. Sci. USA 2010, 107, 22425–22429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Omelon S. J., Grynpas M. D., Chem. Rev. 2008, 108, 4694–4715. [DOI] [PubMed] [Google Scholar]
- 37. Ben Shir I., Kababya S., Zax D. B., Schmidt A., J. Am. Chem. Soc. 2020, 142, 13743–13755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gullion T., Schaefer J., J. Magn. Reson. 1989, 81, 196–200. [Google Scholar]
- 39. Gebauer D., Wolf S. E., J. Am. Chem. Soc. 2019, 141, 4490–4504. [DOI] [PubMed] [Google Scholar]
- 40. Clark S. M., Colas B., Jacob D. E., Neuefeind J. C., Wang H.-W., Page K. L., Soper A. K., Schodder P. I., Duchstein P., Zubiri B. A., Yokosawa T., Pipich V., Zahn D., Spiecker E., Wolf S. E., Sci. Rep. 2022, 12, 6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jones F., Jones P., Ogden M. I., Richmond W. R., Rohl A. L., Saunders M., J. Colloid Interface Sci. 2007, 316, 553–561. [DOI] [PubMed] [Google Scholar]
- 42. Danil de Namor A. F., Alfredo Pacheco Tanaka D., J. Chem. Soc. Faraday Trans. 1998, 94, 3105–3110. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.