

Abstract
Background
Aquaporin‐4 IgG seropositive neuromyelitis optica spectrum disorder (AQP4‐IgG NMOSD) might occur in association with cancer. According to diagnostic criteria, a probable paraneoplastic NMOSD can be diagnosed only in patients with isolated myelitis and adenocarcinoma or tumors expressing AQP4. The aim of this study was to explore the features of paraneoplastic NMOSD through a data‐driven approach.
Methods
A systematic literature review was performed. Patients with AQP4‐IgG positivity in association with tumor in the absence of history of checkpoint inhibitors administration/central nervous system metastases were included. Demographic, clinical, and oncological data were collected. A hierarchical cluster analysis (HCA) was performed and data were compared between resulting clusters.
Results
A total of 1333 records were screened; 46 studies (72 patients) fulfilled inclusion criteria. Median age was 54 (14–87) years; adenocarcinoma occurred in 41.7% of patients, and 44% of cases had multifocal index events. Cancer and NMOSD usually co‐occurred. HCA classified patients in three clusters that differed in terms of isolated/multifocal attacks, optic neuritis, pediatric onset, and type of underlying tumor. Age, time from neoplasm to NMOSD onset, and tumor AQP4 staining did not differ between clusters.
Conclusions
Our data‐driven approach reveals that paraneoplastic NMOSD does not present a homogeneous phenotype nor peculiar features. Accordingly, cancer screening may be useful in AQP4‐IgG NMOSD regardless of age and clinical presentation.
Keywords: AQP4, cancer, NMOSD, paraneoplastic, tumor
Aquaporin‐4‐IgG‐seropositive neuromyelitis optica spectrum disorder (AQP4‐IgG NMOSD) may seldom present in association with neoplasms. This systematic review of the literature, including 72 patients from 46 studies, provides evidence that paraneoplastic AQP4‐IgG NMOSD does not have a homogeneous phenotype and should be considered regardless of age or clinical presentation. Three main clusters were identified through a hierarchical cluster analysis, and oncological accompaniments were the most relevant discriminative factor.
INTRODUCTION
Neuromyelitis optica spectrum disorder (NMOSD) is a heterogeneous syndrome characterized by longitudinally extensive transverse myelitis (LETM), optic neuritis, and brainstem, cortical, or diencephalic involvement. Most patients with NMOSD have serum antibodies directed against the astrocytic water channel aquaporin‐4 (AQP4‐IgG), which simultaneously represents both the disease biomarker and the pathogenic agent [1].
Since a minority of patients with AQP4‐IgG‐related NMOSD have a neoplasm, the correlation between cancer and NMOSD is still a matter of debate. The evolving diagnostic criteria for paraneoplastic neurological syndromes (PNS) have reshaped the approach to this unclear association [2, 3, 4, 5].
The purpose of the present study was to describe the features of paraneoplastic NMOSD and explore the association between cancer and AQP4‐IgG‐related NMOSD using a data‐driven approach through a systematic literature analysis.
MATERIALS AND METHODS
We performed a systematic review to identify reported patients with AQP4‐IgG‐related NMOSD and any concomitant neoplasms. Clinical and demographic data were collected. Then, a hierarchical cluster analysis (HCA) was performed, and data were compared between resulting clusters.
Systematic review
An independent search of PubMed/Medline was performed by three of the authors (AD, GUB, GC), with no temporal restrictions, including the following research queries: “paraneoplastic neuromyelitis optica”, “Aquaporin‐4 and cancer”, “Aquaporin‐4 and paraneoplastic”, “Aquaporin‐4 and tumor” (last update: February 4, 2022).
Inclusion criteria were (i) diagnosis of AQP4‐IgG‐related NMOSD according to current diagnostic criteria [1, 6] and (ii) any concomitant onset or relapse of a neoplasm (if multiple neoplasms were reported, we included only the closest to the index event), regardless of the timing between NMOSD onset and neoplasm. Exclusion criteria were: (i) history of exposure to immune checkpoint inhibitors prior to NMOSD onset, since cases of AQP4‐IgG‐positive NMOSD have been reported as drugs immune‐related adverse effects [7] and (ii) history or evidence of active central nervous system metastasis at the index event.
Data collection
Demographic data, clinical features of NMOSD onset attack (LETM, optic neuritis, brainstem syndrome, cortical syndrome), features of underlying neoplasm, and timing between neoplasm and disease onset were independently collected in an electronic database by the same three authors. Any discrepancy was collectively discussed and resolved.
Statistical analysis
Data were reported as median (range) or number (percentage), as appropriate. For the HCA, data collected through the systematic review were categorized in the following binary categorial variables (presence/absence): male sex, age at onset <18 years (pediatric NMOSD), age at onset >50 years (late‐onset NMOSD), LETM, optic neuritis, brainstem syndrome, cortical syndrome, multifocal attack, any adenocarcinoma (defined as neoplasms originating from epithelial glandular cells), any other solid neoplasm, hematological neoplasms, and cancer within 2 years from onset.
Agglomerative HCA with Ward's method, using Euclidean squared distance, was performed and clusters were identified through the visual inspection of the resulting dendrogram. A dendrogram is a tree‐like figure that shows the hierarchical relationship between different clusters. The distance represents the similarity between clusters (the shorter the distance, the closer the similarity) and it could be read at any node in the diagram.
A comparison of data among clusters was performed with Chi‐square test, Fisher's Test, and Kruskal–Wallis test, as appropriate. Values of p < 0.05 were considered statistically significant. Statistical analysis was performed with IBM SPSS 26.
RESULTS
We identified 1333 records through a systematic literature search and a further five records through cross‐referencing. After removing duplicates, abstracts of 781 records were screened for eligibility. The full text of 72 compatible papers were then assessed and 46 of them fulfilled the study inclusion criteria and were included in the qualitative synthesis. Included studies are reported in Appendix S1. The PRISMA flow chart, summarizing the systematic search and included papers, is reported in Figure 1.
FIGURE 1.
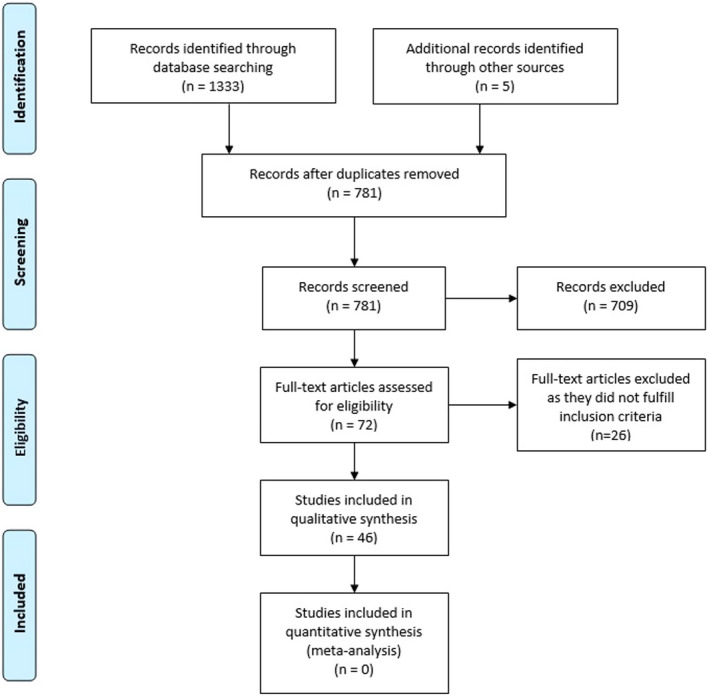
PRISMA flow chart of the study selection process [Colour figure can be viewed at wileyonlinelibrary.com]
Seventy‐two patients were included. Median age at onset was 54 (range 14–87) years and 57 (79.2%) were female. LETM was the most frequent clinical presentation (66.7%), followed by optic neuritis (44.4%), brainstem syndrome (27.8%), and cortical syndromes (5.6%). Multifocal onset attacks were reported in 44% of cases. Adenocarcinoma and other solid neoplasms equally accounted for about 80% of all neoplasms (41.7% and 40.3%, respectively), while the remaining patients presented with hematological neoplasms. The analysis of AQP4 expression on tumor tissue was tested in only 19 patients and showed positive results in 17 (89.5%). Given this small sample, this feature was not included in further analyses.
Median time from neoplasms to NMOSD onset was 0 months, indicating the co‐occurrence of the two conditions, even though neoplasms were diagnosed over a wide time range (between 96 months before and 180 months after NMOSD onset). Despite this variability, cancer occurred within 2 years from NMOSD onset in about 90% of included cases. Clinical, demographic, and oncological findings of the analyzed cohort are summarized in Table 1 and in the supplementary material (Table S1).
TABLE 1.
Demographic and clinical features of included patients (n = 72)
| Demographic and clinical features | |
|---|---|
| Age at disease onset (years) a | 54 (14–87) |
| Sex | 15 (20.8%) Male |
| 57 (79.2%) Female | |
| Time from neoplasm to NMOSD onset (months) | 0 (−96 to 180) b |
| Associated neoplasm c | 30 (41.7%) Adenocarcinoma |
| 29 (40.3%) Other solid neoplasms | |
| 13 (18%) Hematological neoplasms | |
| AQP4 expression in cancer tissue (n = 19) | 2 (10.5%) Absent |
| 17 (89.5%) Present | |
| Clinical features d | 48 (66.7%) LETM |
| 31 (43.1%) Optic neuritis | |
| 19 (26.4%) Brainstem syndrome | |
| 4 (5.6%) Cortical syndrome | |
Note: Data are represented as median (range), number (percentage), as appropriate.
Abbreviations: AQP4, aquaporin 4; LETM, longitudinally extensive transverse myelitis; NMOSD, neuromyelitis optica spectrum disorder.
5 (6.9%) pediatric‐onset and 43 (59.7%) late‐onset NMOSD were included.
Negative values indicate that cancer preceded NMOSD onset, while positive values indicate that NMOSD preceded cancer. NMOSD and cancer occurred within 2 years in 64/72 (88.9%) patients.
Individual data including cancer histotypes are reported in the supplementary material (Table S1).
Multifocal attacks were observed in 32 (44%) patients.
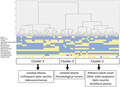
HCA defined three clusters including, respectively, 31 (43.1%), 30 (41.6%), and 11 (15.3%) patients. The resulting dendrogram with a heat map representing the clinical features of individual patients and clusters identified through the HCA is reported in Figure 2.
FIGURE 2.
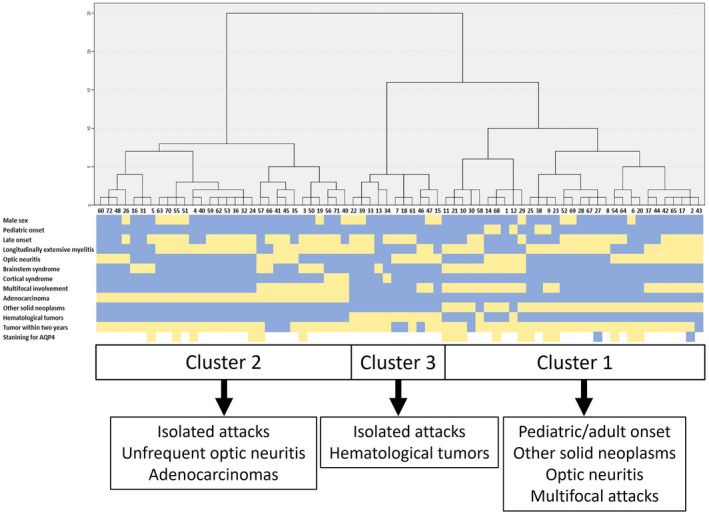
Dendrogram, heatmap, and peculiar features of each cluster. In the upper part of the figure the dendrogram obtained through a hierarchical cluster analysis is represented. Each number at the bottom of the dendrogram represents the identity (ID) of an individual patient (refer to the [Table S1] for references and extracted data [Appendix S1])) and each column under ID represents the most relevant clinical and oncological accompaniments of each included patient (middle part of the figure). The heatmap summarizes the presence (yellow) or absence (light blue) of each clinical and oncological feature. Finally, in the lower part of the figure, the boundaries of each cluster and their peculiar features are summarized [Colour figure can be viewed at wileyonlinelibrary.com]
Cluster 1 was characterized by the presence of both adult and pediatric patients, optic neuritis, and more frequent multifocal attacks in association with other solid neoplasms (non‐adenocarcinoma).
Cluster 2 was defined by the predominant presence of adenocarcinomas, isolated attacks and less frequent optic neuritis. Finally, hematological tumors and isolated clinical phenotypes were characterized in Cluster 3.
The frequency of LETM, brainstem and cortical syndrome, as well as median age, tumor staining for AQP4 (excluding patients with hematological tumors), and time from cancer to NMOSD diagnosis did not differ among clusters. Data from individual clusters and their comparison are reported in Table 2.
TABLE 2.
Comparison of demographic and clinical features between clusters
| Demographic and clinical features | Cluster 1 (n = 31) | Cluster 2 (n = 30) | Cluster 3 (n = 11) | P value Cluster 1 and 2 | P ‐value Cluster 1 and 3 | P value Cluster 2 and 3 |
|---|---|---|---|---|---|---|
| Age at disease onset (years) | 50 (14–76) | 57 (29–87) | 55 (28–76) | 0.192 | 0.192 | 0.192 |
| Pediatric‐onset NMOSD | 5 (18.5%) | 0 (0%) | 0 (0%) | 0.029 | 0.200 | NA |
| Late‐onset NMOSD | 16 (51.6%) | 20 (66.7%) | 7 (63.6%) | 0.175 | 0.371 | 0.568 |
| Sex (male) | 4 (12.9%) | 7 (23.3%) | 4 (36.4%) | 0.234 | 0.107 | 0.323 |
| Time from neoplasm to NMOSD onset (months) | 0 (−96 to 48) | 0 (−36 to 180) | 0 (−96 to 55) | 0.391 | 0.391 | 0.391 |
| Neoplasm within 2 years from NMOSD | 29 (93.5%) | 27 (90%) | 19 (86.4%) | 0.484 | 0.103 | 0.184 |
| Adenocarcinoma | 0 (0%) | 30 (100%) | 0 (0%) | <0.001 | NA | <0.001 |
| Other solid neoplasms | 29 (93.5%) | 0 (0%) | 0 (0%) | <0.001 | <0.001 | NA |
| Hematological neoplasms | 2 (6.5%) | 0 (0%) | 11 (100%) | 0.254 | <0.001 | <0.001 |
| Tumor staining for AQP4 | 2 (15.4%) Absence 11 (84.6%) Presence | 6 (100%) Presence | NA | 0.456 | NA | NA |
| LETM | 22 (71%) | 21 (70%) | 5 (45.5%) | 0.578 | 0.126 | 0.140 |
| Optic neuritis | 16 (51.6%) | 9 (30%) | 6 (54.5%) | 0.072 | 0.574 | 0.140 |
| Brainstem syndrome | 10 (32.3%) | 8 (26.7%) | 1 (9.1%) | 0.422 | 0.134 | 0.225 |
| Cortical syndrome | 0 (0%) | 3 (10%) | 1 (9.1%) | 0.113 | 0.262 | 0.712 |
| Multifocal attacks | 19 (61.3%) | 11 (36.7%) | 2 (18.2%) | 0.047 | 0.016 | 0.231 |
Note: Data are expressed as median (range) or number (%), as appropriate. Statistical comparison was performed with Chi‐square test, Fisher's test, and Kruskal–Wallis test, as appropriate. Bold and italic indicates statistically significant (P < 0.05). Italic indicates trend (P < 0.1).
Abbreviations: AQP4, aquaporin‐4; LETM, longitudinally extensive transverse myelitis; NA, not available; NMOSD, neuromyelitis optica spectrum disorder.
DISCUSSION
The most recent PNS diagnostic criteria questioned the significance of AQP4‐IgG in a paraneoplastic setting, given the unclear association between NMOSD and cancer [5]. Previous reports described paraneoplastic AQP4‐IgG‐related NMOSD cases mainly associated with adenocarcinoma, in particular in patients with brainstem involvement (mainly area postrema syndrome [APS]) or in older (>45 years old) male patients presenting with LETM [2].
However, according to established criteria, AQP4‐related NMOSD is considered to have a lower risk of being paraneoplastic (<5% associated with cancer). Adenocarcinomas, older age, male sex, and clinical presentation with severe nausea/vomiting are described as major risks factors for a paraneoplastic origin [5]. According to the recently proposed score, none of the AQP4‐related NMOSD cases satisfy the criteria of definite PNS (i.e., PNS‐Care Score ≥ 8). Only patients with isolated myelitis (an intermediate risk phenotype, PNS‐Care Score = 2) with concomitant adenocarcinoma occurring within 2 years (the only cancer considered consistent with the antibody) or with demonstrated tumor antigen expression (PNS‐Care Score = 4) can be considered of probable paraneoplastic origin.
In this context, our data‐driven approach, which analyzed the association between neoplasms and AQP4‐IgG‐related NMOSD, provides some interesting findings.
First, in accordance with previous studies [2], the median age of included patients was 54 years, even though a minority of pediatric and younger patients were also observed [8, 9, 10, 11]. This finding suggests that a paraneoplastic origin of NMOSD should be considered independently of age at onset. Of note, cancer screening may be relevant even in pediatric patients, where an association with ovarian teratoma has been most frequently reported [12].
Second, median time from NMOSD onset to cancer was 0 months, indicating that the two conditions are usually concomitant.
Third, we found no clear differences in terms of clinical phenotypes of the index attacks, with the notable exception of patients with non‐adenocarcinoma solid neoplasms, who more frequently had multifocal attacks at onset when compared to patients with hematological tumors and more commonly optic neuritis in comparison to patients with adenocarcinoma. Overall, the categorization into clusters was determined mainly by the underlying neoplasms rather than the presence of any peculiar clinical feature, challenging the concept of isolated myelitis or APS as predominant manifestations of paraneoplastic NMOSD.
A critical point for the application of the PNS criteria to NMOSD is the demonstration of AQP4‐IgG binding to tumor tissue, since discordant findings have been reported in the literature, also influenced by the different methods used to analyze AQP4 staining [13, 14, 15, 16]. In the absence of adenocarcinoma, AQP4 expression in tumor tissue has paramount importance in determining the PNS‐Care Score, although it is rarely tested. According to the literature, AQP4 tumor expression predominantly occurs in adenocarcinoma [13, 17] and ovarian teratoma [8], while it has been rarely reported in other solid neoplasms [14, 18]. Our results show that AQP4 expression in the neoplastic tissue did not differ among various solid neoplasms, suggesting that AQP4 may be found in cancer tissue beyond adenocarcinomas. However, most of the included studies did not account for AQP4 expression in tumor tissue, as they were published before the update of the PNS diagnostic criteria, so definitive conclusions on this relevant topic cannot be drawn. Another remarkable point is that at variance with other antibodies associated with PNS, AQP4‐IgG requires three‐dimensional binding with its conformational antigen, which is determined by the ratio of the M1 and M23 isoforms. The M1/M23 ratio within the neoplasms is mandatory for AQP4‐IgG binding and to elicit an immune response against cancer [19]. The different M1/M23 ratio in cancer tissue may explain the lack of serum reactivity for AQP4‐IgG in asymptomatic patients with non‐small cell lung cancer, which usually expresses AQP4 [20]. More studies are required to identify the specific factors which determine AQP4‐IgG production according to the presence or absence of AQP4 expression within the neoplasms and to clarify the role of AQP4‐IgG in the cancer immune response.
According to the latest diagnostic criteria, most of the AQP4‐IgG‐seropositive NMOSD patients included in our study would have been classified as “non‐PNS” and therefore excluded from cancer screening, with relevant clinical and treatment consequences. We herein provide evidence that patients with paraneoplastic NMOSD have heterogeneous demographic, clinical features, and tumor associations, which extend beyond the presence of myelitis and adenocarcinoma.
Of note, currently there are no specific recommendations for cancer screening in patients with NMOSD. Taking into account the cost‐effectiveness and the benefit–risk ratio related to radiation, magnetic resonance imaging and pelvis ultrasound studies should be used in pediatric patients to rule out the presence of teratomas and to avoid exposure to radiation. Conversely, total‐body computed tomography (CT) scans may be a reasonable approach for adult patients, while 18F‐fluorodeoxyglucose (FDG) positron emission tomography (PET) should be reserved for selected cases or patients with suspected lymphoma. Finally, full blood count, flow cytometry, and protein electrophoresis may be performed as first‐line screening to exclude hematological tumors.
Coexistent antibodies and some clinical clues might also guide cancer screening. For example, the concomitance of antibodies directed against N‐methyl‐D‐aspartate receptor or glial fibrillary acid protein may suggest the presence of an underlying teratoma [8, 21]. Furthermore, treatment of antecedent autoimmune comorbidities (such as systemic lupus erythematous or Sjögren’s syndrome) with chronic immunosuppression has to be considered as a potential additional factor increasing the risk of cancer, in particular hematological neoplasms.
Our study has some limitations including: (i) the possible reporting bias of the systematic review; (ii) the small sample size, despite the large number of articles screened; (iii) the paucity of cases reporting AQP4 expression in tumor tissue, which have hindered a proper application of PNS diagnostic criteria to the whole cohort and thus limited a proper definition of paraneoplastic NMOSD; (iv) the classification of solid tumors in adenocarcinoma versus other solid tumors on the basis of the current PNS diagnostic criteria, which resulted in a widely heterogenous cluster (Cluster 1); and (v) the lack of a control group of non‐paraneoplastic AQP4‐IgG NMOSD. The latter limitation hindered a proper comparison of patients with paraneoplastic versus non‐paraneoplastic NMOSD, which might be analyzed in future studies focusing on the identification of the peculiar clinical and paraclinical features of paraneoplastic NMOSD.
Our study suggests that the association of neoplasms and AQP4‐IgG‐related NMOSD should not be overlooked, and that cancer screening should be performed at baseline, regardless of age and clinical presentation. Future studies are urgently needed to determine the association of cancer and AQP4‐IgG‐seropositive NMOSD in large cohorts of patients and to analyze the expression of AQP4 in cancer tissue and cells, given the prognostic and therapeutic relevance of these features.
AUTHOR CONTRIBUTIONS
Alessandro Dinoto: Conceptualization (equal); data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); visualization (equal); writing – original draft (equal). Giovanni Umberto Borin: Investigation (equal); methodology (equal); writing – review and editing (equal). Giulia Campana: Investigation (equal); methodology (equal); writing – review and editing (equal). Sara Carta: Methodology (equal); writing – review and editing (equal). Sergio Ferrari: Methodology (equal); writing – review and editing (equal). Sara Mariotto: Conceptualization (equal); data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); visualization (equal); writing – original draft (equal).
CONFLICT OF INTEREST
The authors have nothing to disclose.
Supporting information
ACKNOWLEDGEMENT
Open Access Funding provided by Universita degli Studi di Verona within the CRUI‐CARE Agreement.
Dinoto A, Borin GU, Campana G, Carta S, Ferrari S, Mariotto S. Investigating paraneoplastic aquaporin‐4‐IgG‐seropositive neuromyelitis optica spectrum disorder through a data‐driven approach. Eur J Neurol. 2022;29:3466‐3472. doi: 10.1111/ene.15479
DATA AVAILABILITY STATEMENT
Data are available for sharing and further examination from the corresponding author on reasonable request by qualified investigators.
REFERENCES
- 1. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177‐189. doi: 10.1212/WNL.0000000000001729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sepúlveda M, Sola‐Valls N, Escudero D, et al. Clinical profile of patients with paraneoplastic neuromyelitis optica spectrum disorder and aquaporin‐4 antibodies. Mult Scler J. 2018;24(13):1753‐1759. doi: 10.1177/1352458517731914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dinoto A, Bosco A, Sartori A, et al. Hiccups, severe vomiting and longitudinally extensive transverse myelitis in a patient with prostatic adenocarcinoma and aquaporin‐4 antibodies. J Neuroimmunol. 2021;352:577488. doi: 10.1016/j.jneuroim.2021.577488 [DOI] [PubMed] [Google Scholar]
- 4. Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75(8):1135‐1140. doi: 10.1136/jnnp.2003.034447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Graus F, Vogrig A, Muñiz‐Castrillo S, et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm. 2021;8(4):e1014. doi: 10.1212/NXI.0000000000001014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66(10):1485‐1489. doi: 10.1212/01.wnl.0000216139.44259.74 [DOI] [PubMed] [Google Scholar]
- 7. Weiss D, Cantré D, Zettl UK, Storch A, Prudlo J. Lethal form of a late‐onset aquaporin‐4 antibody‐positive NMOSD related to the immune checkpoint inhibitor nivolumab. J Neurol. 2022;269(5):2778‐2780. doi: 10.1007/s00415-021-10913-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bernard‐Valnet R, Cobo‐Calvo A, Siegfried A, et al. Paraneoplastic neuromyelitis optica and ovarian teratoma: a case series. Mult Scler Relat Disord. 2019;31:97‐100. doi: 10.1016/j.msard.2019.03.031 [DOI] [PubMed] [Google Scholar]
- 9. Cai G, He D, Chu L, Dai Q, Xu Z, Zhang Y. Paraneoplastic neuromyelitis optica spectrum disorders: three new cases and a review of the literature. Int J Neurosci. 2016;126(7):660‐668. doi: 10.3109/00207454.2015.1054481 [DOI] [PubMed] [Google Scholar]
- 10. Pittock SJ, Lennon VA. Aquaporin‐4 autoantibodies in a paraneoplastic context. Arch Neurol. 2008;65(5):629‐632. doi: 10.1001/archneur.65.5.629 [DOI] [PubMed] [Google Scholar]
- 11. Tardo L, Wang C, Rajaram V, Greenberg BM. Pediatric paraneoplastic neuromyelitis optica spectrum disorder associated with ovarian teratoma. Mult Scler J. 2022;28(1):160‐163. doi: 10.1177/13524585211037582 [DOI] [PubMed] [Google Scholar]
- 12. Abdel‐Mannan O, Hacohen Y. Aquaporin‐4 antibody neuromyelitis optica spectrum disorder: a paraneoplastic disease? Mult Scler J. 2022;28(1):163‐164. doi: 10.1177/13524585211039755 [DOI] [PubMed] [Google Scholar]
- 13. Iorio R, Rindi G, Erra C, Damato V, Ferilli M, Sabatelli M. Neuromyelitis optica spectrum disorder as a paraneoplastic manifestation of lung adenocarcinoma expressing aquaporin‐4. Mult Scler. 2015;21(6):791‐794. doi: 10.1177/1352458515572241 [DOI] [PubMed] [Google Scholar]
- 14. Figueroa M, Guo Y, Tselis A, et al. Paraneoplastic neuromyelitis optica spectrum disorder associated with metastatic carcinoid expressing aquaporin‐4. JAMA Neurol. 2014;71(4):495‐498. doi: 10.1001/jamaneurol.2013.6331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kon T, Ueno T, Suzuki C, et al. Aquaporin‐4 antibody positive neuromyelitis optica spectrum disorder associated with esophageal cancer. J Neuroimmunol. 2017;309:38‐40. doi: 10.1016/j.jneuroim.2017.05.009 [DOI] [PubMed] [Google Scholar]
- 16. Beauchemin P, Iorio R, Traboulsee AL, Field T, Tinker AV, Carruthers RL. Paraneoplastic neuromyelitis optica spectrum disorder: a single center cohort description with two cases of histological validation. Mult Scler Relat Disord. 2018;20:37‐42. doi: 10.1016/j.msard.2017.12.012 [DOI] [PubMed] [Google Scholar]
- 17. Baik KW, Kim SH, Shin HY. Paraneoplastic neuromyelitis optica associated with lung adenocarcinoma in a young woman. J Clin Neurol. 2018;14(2):246‐247. doi: 10.3988/jcn.2018.14.2.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mitsui S, Tanaka Y, Kimura K, Jimbo N, Chihara N, Maniwa Y. Paraneoplastic neuromyelitis optica spectrum disorder associated with atypical thymic carcinoid: a case report. Ann Thorac Cardiovasc Surg. 2021;2‐5. doi: 10.5761/atcs.cr.20-00354. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin‐4 M1/M23 isoforms and orthogonal arrays. J Biol Chem. 2011;286(18):16516‐16524. doi: 10.1074/jbc.M111.227298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jarius S, Warth A, Wandinger KP, et al. Antibodies to aquaporin‐4 in non‐small cell lung cancer: a study on 50 patients. Neurol Sci. 2010;31(6):871‐872. doi: 10.1007/s10072-010-0290-9 [DOI] [PubMed] [Google Scholar]
- 21. Zoccarato M, Saddi MV, Serra G, et al. Aquaporin‐4 antibody neuromyelitis optica following anti‐NMDA receptor encephalitis. J Neurol. 2013;260(12):3185‐3187. doi: 10.1007/s00415-013-7182-x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available for sharing and further examination from the corresponding author on reasonable request by qualified investigators.