

Abstract
Background
Breast cancer results in a three‐ to four‐fold increased risk of venous thromboembolism (VTE), which is associated with reduced patient survival. Despite this, the mechanisms underpinning breast cancer‐associated thrombosis remain poorly defined. Tumor cells can trigger endothelial cell (EC) activation resulting in increased von Willebrand factor (VWF) secretion. Importantly, elevated plasma VWF levels constitute an independent biomarker for VTE risk. Moreover, in a model of melanoma, treatment with low molecular weight heparin (LMWH) negatively regulated VWF secretion and attenuated tumor metastasis.
Objective
To investigate the role of VWF in breast cancer metastasis and examine the effect of LMWH in modulating EC activation and breast tumor transmigration.
Methods
von Willebrand factor levels were measured by ELISA. Primary ECs were used to assess tumor‐induced activation, angiogenesis, tumor adhesion, and transendothelial migration.
Results and Conclusion
Patients with metastatic breast cancer have markedly elevated plasma VWF:Ag levels that also correlate with poorer survival. MDA‐MB‐231 and MCF‐7 breast cancer cells induce secretion of VWF, angiopoietin‐2, and osteoprotegerin from ECs, which is further enhanced by the presence of platelets. Vascular endothelial growth factor‐A (VEGF‐A) plays an important role in modulating breast cancer‐induced VWF release. Moreover, VEGF‐A from breast tumor cells also contributes to a pro‐angiogenic effect on ECs. VWF multimers secreted from ECs, in response to tumor‐VEGF‐A, mediate adhesion of breast tumor cells along the endothelium. LMWH inhibits VWF‐breast tumor adhesion and transendothelial migration. Our findings highlight the significant crosstalk between tumor cells and the endothelium including increased VWF secretion which may contribute to tumor metastasis.
Keywords: endothelium, LMWH, metastasis, thrombosis, von Willebrand factor
Essentials
Breast cancer cells trigger secretion of VWF through VEGF‐A‐mediated endothelial cell activation.
Breast cancer induced VWF secretion is enhanced in the presence of platelets.
LMWH ablates adhesion of circulating breast cancer cells to endothelial VWF under shear stress.
Increased vascular permeability and tumor transendothelial migration is attenuated by LMWH.
1. INTRODUCTION
Breast cancer is associated with a three‐ to four‐fold increased risk of venous thromboembolism (VTE). 1 , 2 This VTE risk increases further in patients with metastatic breast cancer and in those receiving chemotherapy, with thrombosis rates of up to 17.5% reported in some studies. 3 , 4 Although the relative risk of VTE in breast cancer is lower than with some other tumors (e.g., pancreatic cancer), the high prevalence of breast cancer means that it is actually the commonest malignancy associated with thrombosis. 5 Importantly, cancer‐associated thrombosis is associated with significant morbidity and mortality. 6 For example, cancer patients diagnosed with VTE had a three‐fold reduction in 1‐year survival rate (12% vs. 36%; p < .001) compared with cancer patients without VTE. 7 Similarly, multivariate analysis has demonstrated that VTE is associated with reduced survival in breast cancer patients (hazard ratio 2.3; 95% confidence interval, 2.1–2.6). 3 However, the biological mechanisms underpinning breast cancer‐associated thrombosis remain poorly defined.
von Willebrand factor (VWF) is a large multimeric plasma glycoprotein that plays important roles in normal hemostasis. Under normal conditions, in vivo biosynthesis of VWF is limited to endothelial cells (ECs) and megakaryocytes only. 8 VWF synthesized within megakaryocytes is stored within the alpha‐granules of platelets. Consequently, plasma VWF is predominantly derived from EC secretion. Beyond its importance in coagulation, recent studies have identified additional novel roles for VWF in regulating inflammation and angiogenesis. 9 , 10 Furthermore, accumulating evidence suggests that VWF also impacts cancer cell metastasis. 11 Significantly elevated plasma VWF antigen (VWF:Ag) levels have been reported in various cancer types, including glioblastoma, colorectal, lung, and gastric cancer. 11 Interestingly, significantly higher VWF:Ag levels have been reported in the presence of metastatic disease and have been shown to correlate with reduced survival. Elevated plasma VWF:Ag levels have also been shown to constitute an independent biomarker for VTE risk in breast cancer patients. 12 , 13
The EC wall serves as both the entry and exit point for metastasizing tumor cells. Disseminating cancer cells secrete factors that directly induce EC activation, leading to upregulated expression of specific adhesion receptors (including P‐selectin, ICAM‐1, and β2 integrins) and enhanced vessel permeability. 14 , 15 , 16 , 17 , 18 In keeping with this concept, Bauer et al. recently reported that malignant melanoma cells directly trigger EC activation in vitro and in vivo. This EC activation not only resulted in increased VWF secretion, but also in the accumulation of VWF ultra‐large multimer (UL‐VWF) strings along the EC vessel wall. 19 These UL‐VWF multimer strings bound to platelets and induced aggregation, thus contributing to local microvascular occlusion in vivo. A significant positive correlation was observed between metastatic burden and local VWF secretion of tumor‐bearing mice. 16 This finding is consistent with other studies that have shown that VWF secreted from activated ECs can facilitate adhesion of a variety of tumor cells (including melanoma and colon carcinoma) under shear stress conditions. 20 , 21 , 22 Based on these data, the authors proposed that (1) VWF is important in the pathogenesis of melanoma‐induced VTE and (2) melanoma cells trigger EC activation with VWF secretion, which in turn facilitates melanoma metastasis. 16 Treatment with low molecular weight heparin (LMWH) ablated the melanoma‐induced upregulation in VWF secretion, reduced platelet aggregation, and attenuated tumor metastasis in this murine melanoma model. 16 These findings are consistent with other studies that have reported antimetastatic properties for LMWH in animal models of pancreatic, colon, and breast cancer, respectively. 23 , 24 , 25 LMWH has also been reported to reduce breast cancer metastasis in a murine model. 25 Critically, however, the pathobiology underlying tumor cell–EC interactions and the downstream effects including EC activation remain poorly understood. In particular, the roles played by secreted UL‐VWF multimers in enabling tumor tethering and extravasation have yet to be defined. In this study, we have investigated the specific roles of VWF and LMWH in modulating breast cancer metastasis.
2. MATERIALS AND METHODS
Detailed methodology is available in Appendix S1.
2.1. Patient cohort
Plasma samples were obtained from patients with metastatic breast cancer via a prospective cohort of patients (n = 44) recruited at The Christie NHS Foundation Trust Hospital (Manchester, UK) between February 2013 and June 2015 as part of the TuFClot (Tumor Fragments and Clotting) study. 26 All had either a new diagnosis of metastatic disease or new evidence of clinical or radiological disease progression. Healthy control plasma samples were obtained from gender‐matched patients attending with benign disease as part of the CHAMPion (Cancer‐induced Hypercoagulability As a Marker of Prognosis) Study, 27 as well as established historical control samples (median age 50 years; range 45–69 years; n = 11). 28 Use of patient materials was approved by UK National Research Ethics Service Committee North West‐Greater Manchester Central Ethics Committee.
2.2. Cell lines
A number of human breast cancer cell lines were included in this study; mesenchymal‐like, highly metastatic triple‐negative MDA‐MB‐231, epithelial‐like, low metastatic hormone receptor‐positive MCF‐7, invasive lobular‐type MM‐134‐VI (MDA‐MD‐134‐VI), non‐tumorigenic cell MCF‐10A, and control primary mammary epithelial cells (HMEC). All cells were obtained from the American Type Culture Collection and tested negative for Mycoplasma throughout study duration. Specific details on the culture conditions are provided in Appendix S1. Pooled primary human umbilical vein endothelial cells (HUVEC) were purchased from PromoCell Germany and cultured in endothelial cell growth medium (PromoCell) supplemented with growth media kit (PromoCell), 18% fetal bovine serum, and 1% penicillin–streptomycin. HUVECs were maintained at 37°C, 5% CO2. HUVECs were used a maximum of four passages.
2.3. Platelet preparation
Blood was drawn into sodium citrate tubes (Sarstedt) and centrifuged at 170g for 10 min at room temperature to obtain platelet‐rich plasma. Platelet‐rich plasma was supplemented with prostaglandin E1 (1 μM; Sigma Aldrich) and centrifuged at 900g for 5 min. The platelet pellet was resuspended in wash buffer (10 mM sodium citrate, 1% w/v dextrose, 150 mM NaCl, pH 7.4). Subsequently, platelets were centrifuged for 5 min at 720g, supernatant was removed, and the platelet pellet was resuspended in Tyrode's buffer (134 mM NaCl, 12 mM NaHCO3, 2.9 mM KCl, 0.34 mM Na2HPO4, 1 mM MgCl2, 10 mM HEPES, pH 7.4) containing 5 mM dextrose and 0.5 U/ml apyrase. Platelets were recalcified to 1.8 mM CaCl2 after 15–30 min.
2.4. Generation of breast cancer cell‐derived supernatants
To generate supernatants, 5 × 105 breast cells were grown to confluence in T75 cm2 flasks for 48 h. After aspiration of the culture medium, the cells were rinsed twice with HEPES‐buffered Ringer's solution (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM glucose, and 10 mM HEPES) and incubated in 7 ml of OptiMEM (Gibco) with or without LMWH Tinzaparin (100 IU/ml) at 37°C and 5% CO2. After 16 h of incubation, the supernatants were collected, centrifuged to remove cellular debris, and stored at −20°C until further use. For conditions with platelets, platelets (2 × 108/ml) were co‐cultured with breast cancer cells (3 × 106/ml) for 16 h in OptiMEM with or without LMWH Tinzaparin (100 IU/ml), platelets alone were seeded as a control. Supernatant from co‐culture was collected, centrifuged to remove cellular debris, and stored at −20°C until used.
2.5. Endothelial cell stimulation
HUVECs were grown on gelatin‐coated standard 12‐well culture plates. At confluency, culture medium was removed, and cells were rinsed twice with prewarmed HEPES‐buffered Ringer's solution. Subsequently, different stimuli were applied: breast cancer cell supernatants, breast cancer‐platelet co‐culture supernatant, recombinant human vascular endothelial growth factor‐A (VEGF165; 10 ng/ml, Peprotech) or phorbol 12‐myristate 13‐acetate (PMA) (100 nM; Sigma‐Aldrich). Where indicated, the VEGF‐A neutralizing antibody bevacizumab (100 ng/ml; R&D Systems) was incubated with the cells. Prewarmed serum free medium (OptiMEM) served as control. The supernatants of endothelial cells were collected after 15, 30, and 60 min of incubation at 37°C, centrifuged at 300g for 5 min to remove cell debris, and kept at −20°C for VWF ELISA.
2.6. ELISAs
VWF was measured as previous. 29 Briefly, the release of endothelial VWF was measured by a sandwich ELISA technique using a polyclonal rabbit anti‐VWF antibody (Dako) and a polyclonal rabbit horseradish peroxidase‐labeled anti‐human VWF antibody (Dako). A standard curve was generated using standard human reference plasma (Behring) with a defined VWF content. VWF secretion was normalized as a percentage of control untreated HUVECs across the same time course for consistency. The absolute normal range of VWF release in our assays is typically in the 40 to 100 ng/ml range depending on stimulus used. Plasma VWF:Ag levels in our patients cohort were quantified as previously 30 and expressed as IU/dl. VWF propeptide (VWFpp) was measured by ELISA using CLB‐Pro 35 and CLB‐Pro 14.3‐horseradish peroxidase monoclonal antibodies (Sanquin), as previously described 29 and expressed as U/ml. VEGF‐A, angiopoietin (Ang 2), osteoprotegerin (OPG), and TGF‐β1 levels were assessed using VEGF, Human Angiopoietin‐2,Human Osteoprotegerin/TNFRSF11B and Human transforming growth factor‐ β1 (TGF‐β1) Duoset ELISA kits (R&D Systems) respectively according to the manufacturer's instructions. Data were analyzed by generating a four‐parametric logistic curve‐fit on GraphPad Prism.
2.7. Endothelial tube formation assay
In vitro endothelial cell tube formation assay was performed using Ibidi μ‐slides (IBIDI GmbH). Briefly, 3 × 105 HUVECs were suspended in 1 ml of OptiMEM serum free medium (Gibco). Stimulants were supplemented with or without LMWH tinzaparin (100 IU/ml) or bevacizumab (100 ng/ml). Cell suspension (50 μl) was applied to the growth factor reduced matrigel (R&D Systems) coated well of each μ‐Slide and incubated in a humidified chamber at 37°C for 8 h. Tube formation was assessed using an Andor iXon3 888 emCCD camera on a Zeiss Axiovert 200 microscope using Metamorph v7.8.2 software. The total tube length was quantified using Angiogenesis analyzer on ImageJ. 31
2.8. Data presentation and statistical analysis
Experimental data were analyzed with GraphPad Prism version 9.0 (GraphPad Software). Data were expressed as mean values ± standard error of the mean. Data were analyzed with Student's unpaired two‐tailed t‐ test, and p values <.05 were considered to be significant. For multiple comparisons, data were analyzed by one‐way anova followed by Bonferroni adjustment t‐ test and p values <.05 were considered to be significant. Non‐normally distributed quantitative data were compared using the Mann–Whitney U test and the Spearman correlation coefficient where appropriate with p values <.05 considered to be significant.
3. RESULTS
3.1. Breast cancer cells promote VWF secretion from endothelial cells through platelet‐dependent and platelet‐independent pathways
Given reports of elevated plasma VWF:Ag levels in various cancer types, we assessed VWF plasma levels in patients with breast cancer (n = 44). The median age of the female patient cohort was 60 years (range 36–82 years). All patients had evidence of metastasis to liver, bone, lungs, or brain. Moreover, 77.5% of the cohort had breast cancer metastasis at multiple sites. The clinicopathological data is supplied in Table S1. Patients with breast cancer had markedly elevated VWF:Ag levels compared to healthy controls (217 ± 13 IU/dl vs. 89.1 ± 8.8 IU/dl, p < .0001) (Figure 1A). Two‐year follow‐up data demonstrated a significant inverse correlation between VWF:Ag levels and overall survival (r = −.47, p = .01) (Figure 1B).
FIGURE 1.
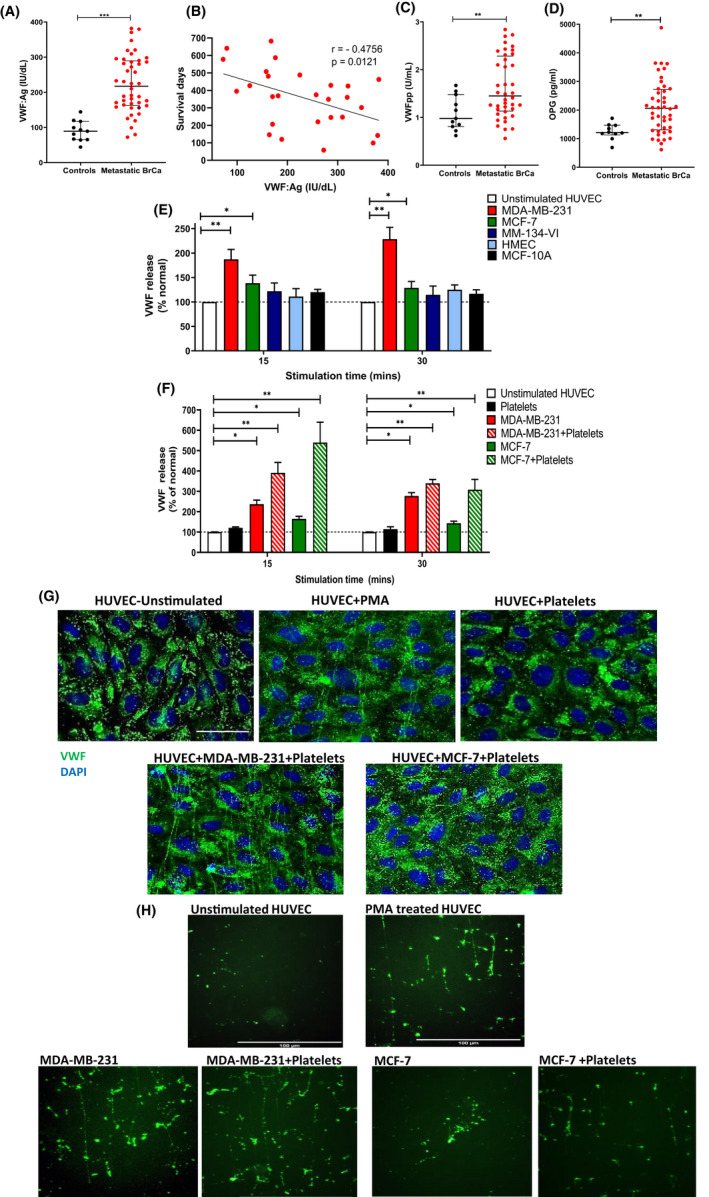
Breast cancer cells promote VWF secretion from EC through platelet‐dependent and platelet‐independent pathways. (A) Plasma VWF:Ag levels were quantified in patients with advanced metastatic breast cancer and compared to sex‐matched healthy controls. Data are graphed as median and interquartile range. (B) Plasma VWF:Ag levels were correlated with patient survival over a 2‐year period. (C) Plasma VWFpp levels and (D) OPG levels were quantified in patients with advanced metastatic breast cancer and compared with sex‐matched healthy controls. Data are graphed as median and interquartile range. Patient data were analyzed with using the Mann–Whitney U test for unpaired two‐tailed t‐ test and p values < .05 were considered to be significant. (E) Breast tumor‐induced endothelial cell activation was assessed using specific VWF:Ag ELISA and percentage VWF release was calculated by comparing the VWF:Ag levels to unstimulated HUVEC controls. (F) Tumor cell supernatant from co‐culture of MDA‐MB‐231 or MCF‐7 with platelets was assessed for EC activating potential. (G) HUVEC stimulation by either MDA‐MB‐231+platelets, MCF‐7+platelets or PMA as a positive control was visualized by immunofluorescence VWF staining (green). Scale bar represents 50 μm. (H) HUVEC were perfused with supernatant from MDA‐MB‐231 or MCF‐7 cells alone or supernatant from breast tumor‐platelet co‐cultures. VWF staining was visualized by immunofluorescence VWF staining (green) under conditions of venous shear stress. Scale bar represents 100 μm. Data are presented as the mean ± SEM of at least three independent experiments. Data were analyzed with Student unpaired two‐tailed t‐ test versus control conditions and p values <.05 were considered to be significant (*p <0 .05, **p < .005, ***p < .001).
We observed that plasma VWFpp levels were also markedly elevated in metastatic breast cancer (1.45 ± 0.1 U/ml vs. 0.98 ± 0.1 U/ml, p < .01, Figure 1C). VWFpp is a surrogate marker for acute endothelial cell activation. 28 Previous studies have reported VWFpp levels to correlate with risk of venous thrombosis within the general population 32 and reinforces the concept that high VWF levels observed in breast cancer patients are largely mediated by increased VWF secretion from the endothelium. Given the marked increases in both VWF:Ag and VWFpp plasma levels, we next investigated whether EC activation in breast cancer patients may also be associated with increased secretion of other proteins stored within Weibel Palade bodies (WPB). Plasma osteoprotegerin (OPG) levels are significantly increased in patients with metastatic breast cancer (2062 ± 140 pg/ml vs. 1214 ± 89 pg/ml, p < .005, Figure 1D). Importantly in the context of cancer, OPG has been shown to directly trigger upregulation in endothelial adhesion receptors including ICAM‐1 and E‐selectin and to have direct effects on VWF function by modulating platelet recruitment to VWF multimers. 33 , 34 Consistent with the concept that metastatic breast cancer is associated with acute EC activation, we observed significant correlations between plasma levels of all the WPB cargo proteins (VWF:Ag, VWFpp, and OPG) in our patient cohort (Figure S1A,B).
To investigate how breast cancer causes increased plasma VWF levels, we next examined whether human breast cancer cells could trigger WPB exocytosis and VWF secretion in vitro. Incubation of HUVECs with supernatant from MDA‐MB‐231 breast cancer cells induced a significant increase in VWF secretion compared with unstimulated control HUVECs (228 ± 21% vs. 100 ± 10%, p < .05) (Figure 1E). Similarly, treatment with supernatant from MCF‐7 breast cancer cells also resulted in increased VWF secretion, although levels were lower than those seen with MDA‐MB‐231 (129 ± 11%, p < .05) (Figure 1E). In contrast, incubation with supernatant from MM‐134‐VI breast cancer cells and nontumorigenic breast cells (HMECs and MCF‐10A) had no significant effect on VWF secretion (Figure 1E).
Breast cancer cells have previously been shown to activate platelets leading to secretion of a variety of factors that promote metastasis (including cytokines, growth factors, and matrix metalloproteases). 35 , 36 , 37 Consequently, we next investigated whether supernatants from breast tumor cells incubated with platelets impacted endothelial VWF secretion. Supernatants were isolated from breast tumor cells co‐cultured with washed human platelets. These tumor‐platelet supernatants were subsequently used to stimulate HUVEC and VWF release quantified. Importantly, baseline levels of VWF were measured in tumor‐platelets supernatants before incubation with HUVEC to exclude contribute of platelet‐derived VWF in the assay. Moreover, supernatant from washing platelets alone did not significantly stimulate VWF secretion from HUVEC. Interestingly, co‐culture of platelets with MDA‐MB‐231 and MCF‐7 cells markedly increased the ability of breast tumor cells to induce EC activation, with four‐ and five‐fold increased VWF levels observed, respectively, compared with unstimulated HUVECs or HUVECs exposed to human platelets alone (Figure 1F). In further support for tumor‐induced EC activation, increased secretion of both OPG and angiopoietin‐2 (Ang‐2) were also observed following treatment of HUVEC with supernatant from co‐cultures of platelets with MDA‐MB‐231 or MCF‐7 cells (Figure S1C–F). Immunofluorescent staining under static conditions demonstrated UL‐VWF multimer strings on the EC surface following treatment with supernatant derived from MDA‐MB‐231 or MCF‐7 tumor‐platelet co‐cultures that were similar to PMA control‐treated HUVECs (Figure 1G). In contrast, no such multimer strings were observed on the surface of untreated HUVECs, or HUVECs incubated with platelet supernatant alone under static conditions (Figure 1G). Similarly, perfusion of tumor cell supernatant from MDA‐MB‐231 cells or supernatant from platelet co‐culture with MDA‐MB‐231 and MCF‐7 cells across coated HUVEC cells under conditions of venous shear stress revealed significant VWF multimer accumulation along the endothelium (Figure 1H). In keeping with our previous findings, no VWF multimers were observed in HUVEC perfused with supernatant from MCF‐7 cells. Collectively, these data demonstrate that specific types of breast cancer cells trigger acute EC activation under both static and shear stress conditions and thereby enhanced VWF secretion, as well as the release of other WPB constituents including OPG and Ang‐2. Moreover, this increase in VWF secretion is further increased in the presence of tumor‐induced platelet releasate.
3.2. VEGF‐A plays a key role in modulating breast cancer‐induced VWF secretion
VEGF‐A is a potent EC activator expressed by a variety of tumor cells (including melanoma, urothelial bladder carcinoma, breast and ovarian cancer). 14 , 17 , 38 , 39 In particular, elevated plasma levels of VEGF‐A have been reported in patients with metastatic breast cancer versus those with benign disease. 40 We examined VWF release from HUVEC in response to recombinant VEGF‐A and observed that VEGF‐A levels of 300 pg/ml and greater induced significant VWF release (Figure S2A). Consistent with the effect of MDA‐MB‐231 in stimulating VWF secretion, VEGF‐A levels were markedly elevated in MDA‐MB‐231 supernatant compared with other breast cancer cells and non‐cancer breast cell lines that had no effect on VWF secretion (1207 ± 114 pg/ml vs. 8.7 ± 1.0 pg/ml for MM134‐VI cells and 178 ± 77 pg/ml for HMEC) (Figure 2A). VEGF‐A levels in MCF‐7 supernatant were also significantly increased but remained lower than those seen with MDA‐MB‐231 cells (418 ± 43 pg/ml) (Figure 2A). VEGF‐A levels were markedly increased following co‐culture of both breast tumor cells with platelets (Figure 2A).
FIGURE 2.
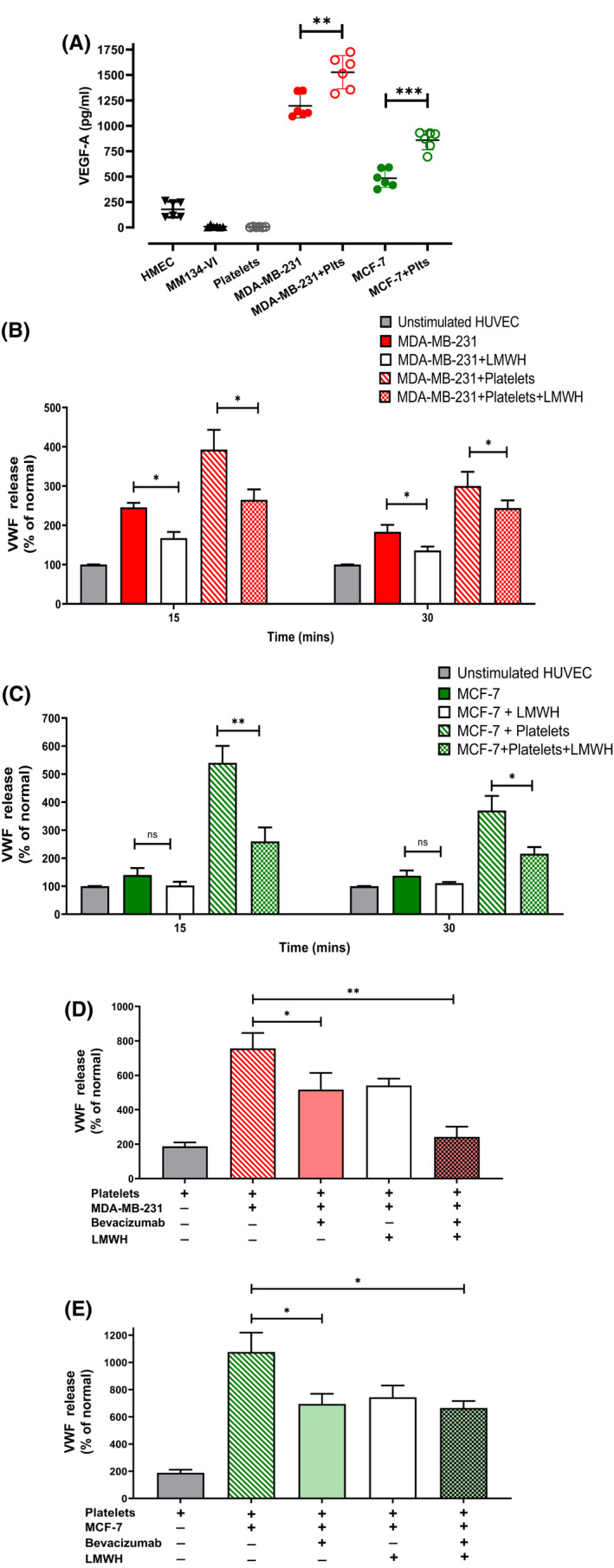
VEGF‐A plays a key role in modulating breast cancer‐induced VWF secretion. (A) VEGF‐A levels in supernatant from breast tumor cell supernatants cultured with and without platelets (plts) were measured by ELISA. (B) MDA‐MB‐231 and (C) MCF‐7 cells were cultured with or without platelets in the presence and absence of low molecular weight heparin (LMWH; Tinzaparin 100 IU/ml) and supernatant subsequently applied to HUVEC monolayer. VWF release was measured by ELISA following 15 and 30 min of stimulation. (D) MDA‐MB‐231 and (E) MCF‐7 cells were co‐cultured with platelets and treated with LMWH (Tinzaparin‐100 IU/ml), or anti‐VEGF antibody (bevacizumab 100 ng/ml) or a combination of both. This supernatant was subsequently applied to HUVEC monolayer. VWF release was measured by ELISA following 15 and 30 min of stimulation. Data are presented as the mean ± SEM of at least three independent experiments Data were analyzed with anova followed by Bonferroni t ‐test and p values < .05 were considered to be significant (*p < .05, **p < .005, ***p < .001).
Previous studies have demonstrated that LMWH binds and inhibits VEGF‐A activity. 19 , 41 In addition, LMWH was also reported to significantly reduce melanoma‐induced VWF secretion. 19 In keeping with those data, we observed that the ability of MDA‐MB‐231 supernatant to trigger VWF exocytosis from HUVECs was significantly attenuated in the presence of LMWH (245 ± 11% vs. 167 ± 10%, p < .05) (Figure 2B). Similarly, an LMWH inhibitory effect was also observed in the presence of co‐culture with platelets (392 ± 52% vs. 264 ± 26%, p < .05). LMWH also attenuated the ability of MCF‐7 supernatant to trigger VWF exocytosis from HUVECs in the presence or absence of platelets (Figure 2C). LMWH also resulted in significantly reduced secretion of other endothelial activation markers, OPG, and Ang‐2 (Figure S1C–F). In keeping with an important role for VEGF‐A in modulating endothelial cell activation, significantly decreased VEGF‐A levels were observed following treatment of breast tumor‐platelet co‐cultures with LMWH (Figure S2B,C).
To examine whether VEGF‐A plays a role in modulating breast cancer‐induced VWF release, HUVEC activation experiments were repeated in the presence or absence of the inhibitory VEGF‐A antibody (bevacizumab). Addition of bevacizumab to MDA‐MB‐231‐platelet co‐culture significantly attenuated VWF release (Figure 2D). Moreover, combined treatment with LMWH and bevacizumab resulted in a significantly more marked reduction in VWF secretion. Similarly, bevacizumab also significantly reduced VWF secretion from HUVECs in response to MCF‐7‐platelet co‐culture supernatant (1076 ± 90% vs. 649 ± 50%, p < .05) (Figure 2E). Conversely however, no further reduction in VWF secretion was observed following combined treatment with LMWH and bevacizumab, suggesting other VEGF‐A independent mechanisms through which MCF‐7 cells may also induce VWF secretion in the presence of platelets. Proteome microarray analysis of supernatant from breast tumor cells alone versus that co‐cultured with platelets revealed elevated expression of a number of cytokines previously reported to induce endothelial cell activation including IL‐8, platelet factor 4, PDGF‐AA, and TGF‐β1 (Figure 3A). Moreover, direct contact between MCF‐7 tumor cells and platelets has previously reported to induce the release of TGF‐β1 from platelets, which contributes to metastasis. 42 We thus quantified TGF‐β1 within MCF‐7 breast tumor supernatant and observed markedly elevated levels in supernatant from MCF‐7 cells co‐cultured with platelets versus MCF‐7 supernatant (2081 ± 82 pg/ml vs. 135 ± 6 pg/ml, Figure 3B). TGF‐β1 levels were significantly reduced upon treatment with LMWH. To determine whether these high levels of TGF‐β1 may contribute to MCF‐7 platelet‐induced endothelial cell activation, we assessed VWF release from HUVEC dose‐dependent exposure to recombinant TGF‐β1 and observed a dose‐dependent increase in VWF secretion (Figure S1C). Taken together, these data highlight a key role for VEGF‐A in modulating breast cancer‐induced VWF secretion from EC, but there are likely a number of other secreted factors that may play additional roles such as TGF‐β1, and these factors may be breast tumor‐type specific.
FIGURE 3.
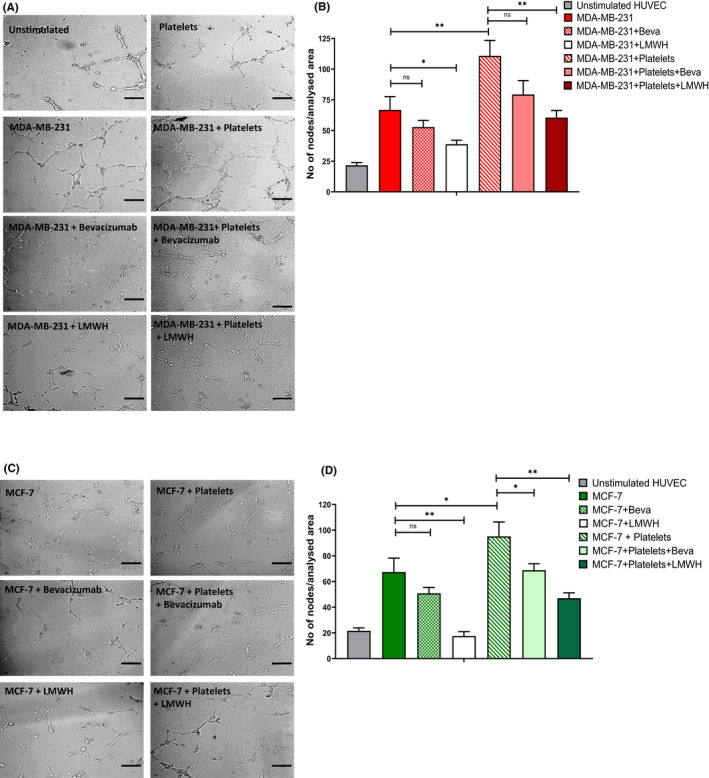
Breast cancer cell‐induced angiogenesis is inhibited by LMWH. MDA‐MB‐231 and MCF‐7 cells were cultured alone or with platelets (plts), in the presence and absence of anti‐VEGF‐A antibody bevacizumab (100 ng/ml) or LMWH (100 IU/ml). Tumor supernatant was then used for a HUVEC angiogenesis assay using growth factor reduced matrigel. HUVECs were cultured with the tumor supernatant from (A, B) MDA‐MB‐231 or (C, D) MCF‐7 and representative images of capillary tube formation generated were taken with Zeiss Axiovert microscope. Quantification of number of EC nodes formed following exposure to tumor supernatant from (B) MDA‐MB‐231 and (D) MCF‐7. Data are presented as the mean ± SEM of at least three independent experiments. Data were analyzed with anova followed by Bonferroni t‐ test (*p < .05, **p < .005, ***p < .001). Scale bar 100 μm.
3.3. Breast cancer cells induce endothelial cell angiogenesis that can be inhibited by LMWH
VEGF‐A plays a key role in regulating in vivo angiogenesis. 43 Recent studies have demonstrated that VWF and other proteins stored within WPB (including Ang‐2 and osteoprotegerin) also influence angiogenesis. 44 Given the high levels of VEGF‐A and EC activation associated with breast tumor‐platelet co‐cultures, we next investigated the effects of breast cancer cells on EC angiogenesis. In the presence of tumor supernatant from either MDA‐MB‐231 or MCF‐7, significantly increased EC capillary tube network and node formation were observed (66.7 ± 10.8 nodes and 67.4 ± 10.9 nodes versus 21.6 ± 2.2 nodes for unstimulated EC, p < .0001) (Figure 3A–D). Interestingly, the pro‐angiogenic effects of the breast tumor‐platelet co‐culture supernatants were only partial inhibited by anti‐VEGF‐A antibody (Figure 3B,D). For both MDA‐MB‐231 and MCF‐7 cell lines, LMWH was significantly more effective in attenuating capillary tube network formation. These data suggest that the interactions between malignant breast tumor cells and platelets trigger acute EC activation and a local pro‐angiogenic effect that is modulated at least in part through VEGF‐A. LMWH inhibits this pro‐angiogenic effect, likely through a number of different pathways.
3.4. VWF multimer strings recruit breast cancer cells to EC surfaces under shear stress
Previous studies have suggested that UL‐VWF strings on EC surfaces play a role in tethering circulating cancer cells and thereby promote tumor metastases. Triple‐negative MDA‐MB‐231 cells rapidly undergo bloodborne metastasis in vivo. In a parallel flow perfusion assay, fluorescently labeled MDA‐MB‐231 cells adhered to PMA‐activated HUVECs. MDA‐MD‐231 cells adhered to activated EC in a linear arrangement consistent with the appearance of UL‐VWF strings (Figure 4A). In support of this putative VWF involvement, breast tumor‐EC adhesion under shear was ablated in the presence of a polyclonal anti‐VWF antibody (2.5 ± 0.3 cells/mm2 vs. 42.3 ± 4.2 cells/mm2 surface coverage for untreated endothelium, p < .001) (Figure 4B,D, Video S1). Interestingly, MDA‐MB‐231 tethering was also significantly inhibited by LMWH (7.3 ± 1.7 cells/mm2, p < .001, Figure 4C,D). In addition to quantifying adherent breast cancer cells, we further examined transient adhesion events between EC and tumor cells and observed significantly increased detachment rates (K off rate) in the presence of either anti‐VWF antibody or LMWH, respectively (0.43 ± 0.09 s−1 and 0.55 ± 0.09 s−1 vs. 0.04 ± 0.01 s−1 for control conditions, p < .001, Figure 4E). Collectively, these data demonstrate that VWF multimers secreted from activated ECs can facilitate adhesion of circulating breast tumor cells under shear stress conditions and thereby potentially play a role in promoting metastasis. Next, we examined whether VEGF‐A in tumor supernatant could induce the release of VWF under shear and thus promote breast tumor tethering along the endothelium. HUVEC were perfused with supernatant from MDA‐MB‐231 cells or MDA‐MB‐231 cells co‐cultured with platelets. As a positive control, HUVECs were perfused with media containing PMA or VEGF‐A. The adhesion of fluorescently labeled MDA‐MB‐231 cells to the endothelial cells was quantified. We observed similar surface coverage of breast tumor cells to the endothelium in response to PMA, VEGF‐A, and MDA‐MB‐231 supernatant (Figure 4F). In keeping with previously observed increased VWF release, supernatant from MDA‐MB‐231‐platelet co‐culture significantly enhanced breast tumor adhesion to the endothelium (86.3 ± 8.4 vs. 45.2 ± 7.6 cells/mm2, p < .001, Figure 4F). Reinforcing the key role for VEGF‐A derived from breast tumor cells and breast tumor activated platelets in mediating VWF release, addition of bevacizumab to the perfusion conditions ablated MDA‐MB‐231 endothelial adhesion and significantly increased the K off rate for circulating breast tumor cells (Figure 4F,G).
FIGURE 4.
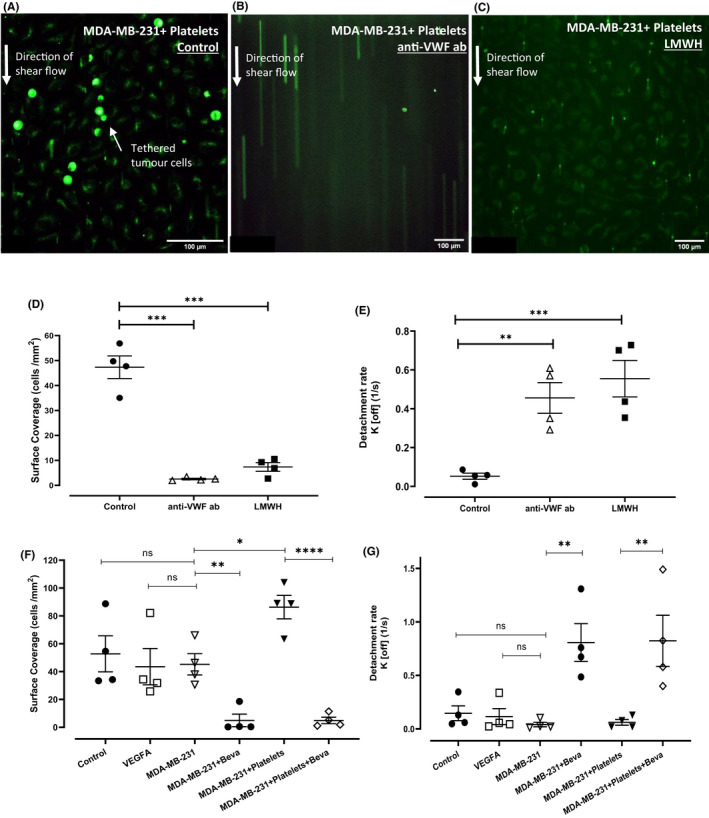
VWF multimer strings recruit breast cancer cells to EC surfaces under shear stress. HUVEC monolayers were cultured on Ibidi channel slides and stimulated with PMA to allow maximum release of VWF. MDA‐MB‐231, fluorescently labeled with Cell Tracker green were flown over the HUVEC layer at flow rate of 0.2 ml/min resulting in venous shear stress of 0.25 dyn/cm2. (A) MDA‐MB‐231 cells binds to VWF strings under shear flow. (B) The specificity of this interaction was confirmed using a blocking anti‐VWF antibody. (C) LMWH significantly decreased the number of binding events observed. (D) Equilibrium surface coverage (cells per mm2) and (E) detachment rates (K off) were calculated for each treatment condition. Perfusion assays were performed on HUVEC monolayers stimulated with supernatant from MDA‐MB‐231 cells or supernatant from MDA‐MB‐231+platelets and adhesion of labeled MDA‐MB‐231 cells were quantified (F and G). Data are presented as the mean ± SEM of at least three independent experiments. Data were analyzed with analysis of variance (anova) followed by Bonferroni t ‐test and p values < .05 were considered to be significant (*p < .05, **p < .005, ***p < .001). Scale bar 100 μm.
3.5. Breast cancer cells induce enhanced endothelial cell permeability and transendothelial migration
To disseminate, circulating breast cancer cells not only need to adhere to ECs, but must infiltrate through the vessel wall. We hypothesized that breast cancer cells may not only activate ECs, but also alter permeability of the EC monolayer. To examine this, HUVEC were cultured on a transwell insert to form an intact monolayer, which was then treated with tumor supernatant from MDA‐MB‐231 or MCF‐7. Permeability was assessed using fluorescein isothiocyanate (FITC)‐dextran leakage across the EC barrier. Treatment with breast tumor supernatants markedly increased EC permeability compared with untreated HUVEC monolayer (MDA‐MB‐231; 47 ± 7.6% FITC‐dextran leakage; MCF‐7; 40 ± 7.8% FITC‐dextran leakage, p < .05, Figure 5A,B). Treatment of the monolayer with supernatants from breast tumor‐platelet co‐culture further enhanced the endothelial barrier permeability. Importantly, breast cancer cell‐induced endothelial barrier breakdown was significantly attenuated in the presence of LMWH and bevacizumab to a similar extent (Figure 5A,B), highlighting a key role for VEGF‐A derived from both tumor‐ and tumor‐activated platelets in contributing to endothelial barrier permeability. Similar findings were also observed using impedance technology to assess endothelial integrity in a label‐free environment in real‐time (Figure S4A,B). Staining of control HUVECs for tight junction protein VE‐cadherin revealed characteristic jagged pattern of tight junctions across adjacent cell membranes. In contrast, following breast tumor supernatant exposure, HUVEC barrier integrity was reduced (Figure 5C, arrows). Once again, treatment with LMWH, significantly increased VE‐cadherin staining in HUVECs treated with breast tumor supernatant and supernatant from breast tumor‐platelet co‐cultures (Figure 5C). Finally, enhanced permeability induced by breast tumor cells or breast tumor‐platelets co‐cultures results in increased transendothelial migration for both MDA‐MB‐231 and MCF‐7 cells (Figure 6A). In keeping with its barrier protective function, LMWH significantly attenuated transendothelial migration of breast tumor cells (Figure 6A). Taken together, these findings demonstrate that LMWH inhibits breast tumor cell‐mediated vascular permeability and transendothelial migration in vitro, this protective effect is likely to be, at least in part, mediated by inhibiting tumor‐derived VEGF‐A.
FIGURE 5.
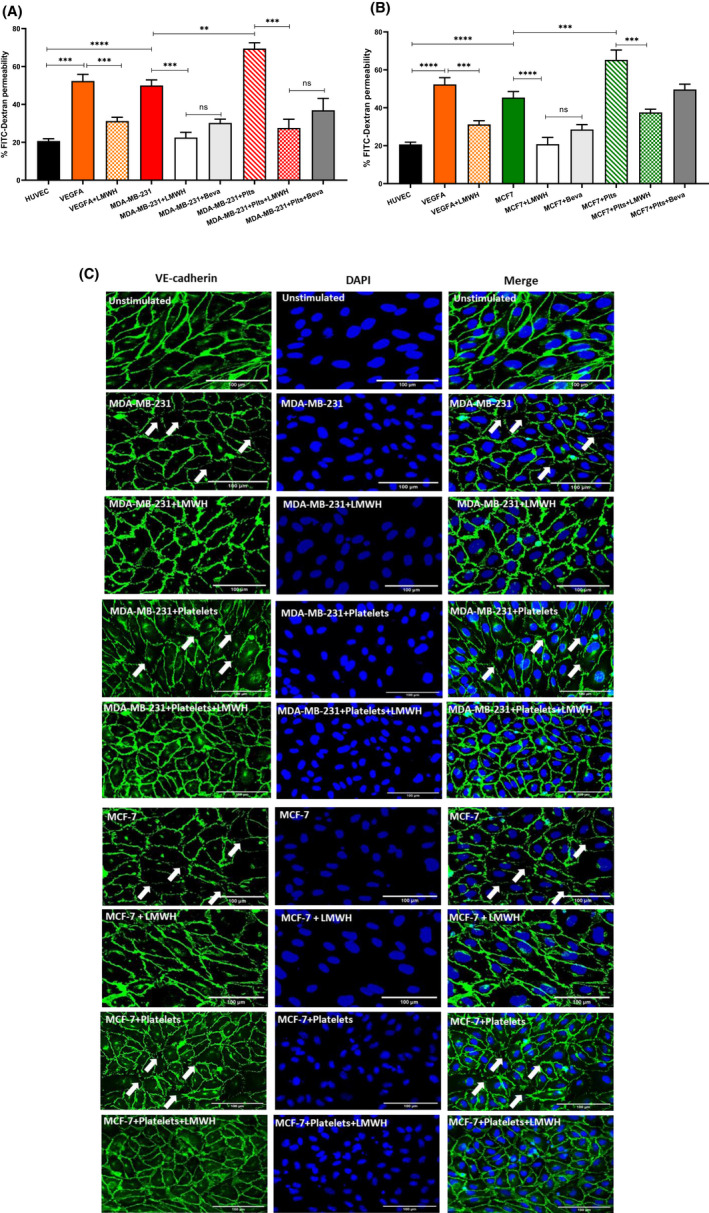
Breast cancer cells induce enhanced EC permeability and transendothelial migration. Breast tumor supernatant from (A) MDA‐MB‐231 and (B) MCF‐7 cells either alone or co‐cultured with platelets was collected and applied to a confluent HUVEC monolayer on a transwell insert. EC permeability was assessed by leakage of 10 kDa FITC‐Dextran from the top chamber after stimulation. (C) Immunofluorescence staining for VE‐cadherin was performed on HUVEC after tumor stimulation with and without LMWH pretreatment. Arrows indicate areas with reduced HUVEC barrier integrity. Scale bar 100 μm. Data were analyzed with anova followed by Bonferroni t ‐test and p values < .05 were considered to be significant (*p < .05, **p < .005, ***p < .001).
FIGURE 6.
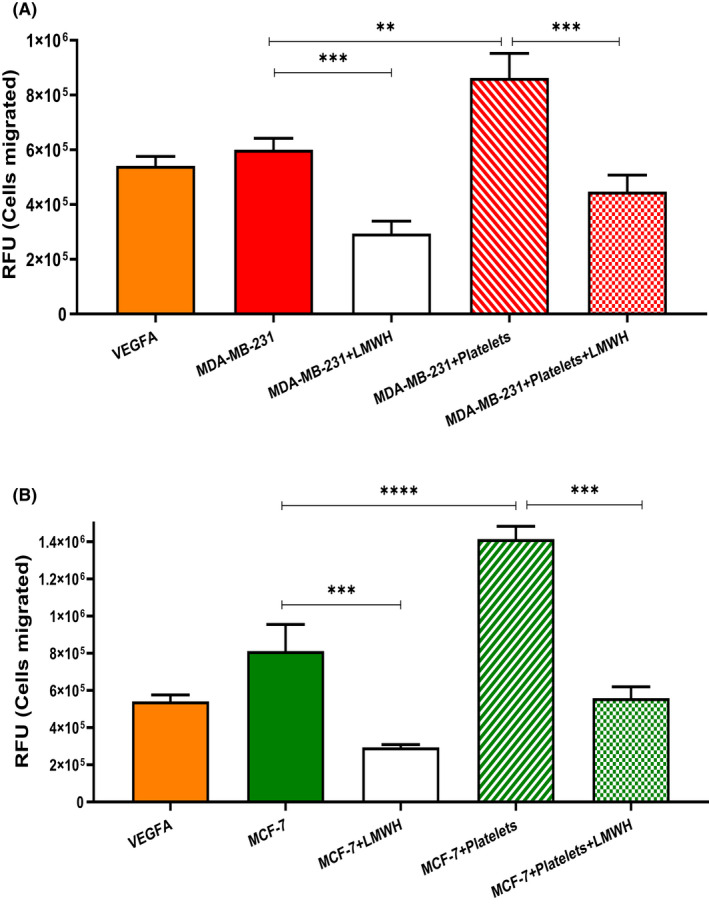
LMWH attenuates breast tumor transendothelial migration. Transendothelial migration of (A) MDA‐MB‐231 and (B) MCF‐7 breast tumor cells was assessed using fluorescently labeled tumor cells seeded on top of HUVEC confluent monolayer. Various stimuli were used to alter endothelial integrity including VEGF‐A, supernatant from breast tumor cell alone of following their co‐culture with platelets. After 24 h, tumor cells that had migrated into bottom chamber were lysed to measure the relative fluorescence units (RFU). Data are presented as the mean ± SEM of at least three independent experiments. Data were analyzed with anova followed by Bonferroni t‐test and p values < .05 were considered to be significant (*p < .05, **p < .005, ***p < .001).
4. DISCUSSION
A number of studies have reported that elevated plasma VWF:Ag levels in cancer cohorts are associated with disease progression and poorer patient outcomes, highlighting VWF:Ag as an independent prognostic marker in specific cancer subtypes. Our findings now also demonstrate significantly increased VWF:Ag levels in patients with metastatic breast cancer as well as VWFpp and OPG levels, suggestive of an underlying endothelial cell activation in these patients. Moreover, plasma VWF:Ag in this cohort inversely correlated with patient survival. These novel findings raise the intriguing possibility that VWF may play a role in directly contributing to breast cancer progression rather than merely being an epiphenomenon of a tumor acute phase response. However, the mechanisms underpinning increased VWF levels in cancer patients remains poorly defined.
In this study, we now report for the first time that breast cancer cells promote EC activation, through both platelet‐dependent and platelet‐independent mechanisms. EC activation results in acute VWF release in vitro. Our findings confirm a key role of tumor secreted VEGF‐A in promoting VWF release. In particular, MDA‐MB‐231 triple‐negative breast cancer cells were potent mediators of EC activation and VWF release via direct secretion of VEGF‐A. In contrast, hormone receptor‐positive MCF‐7 breast cancer cells were largely dependent on platelets to promote significant VWF release. In agreement with this, levels of VEGF‐A secreted directly by MCF‐7 cells were significantly lower than MDA‐MB‐231 cells. However, co‐culture of MCF‐7 cells with human platelets, resulted in markedly elevated VEGF‐A levels as well as TGF‐β1 in the co‐culture supernatant, which induced acute VWF release from ECs. In breast cancer patients, platelets have been identified as a very significant source of circulating VEGF. 36 , 45 Furthermore, Battinelli et al. 36 has previously reported that secretion of thrombin from MCF‐7 breast cancer cells directly activates platelets via PAR1 cleavage resulting in secretion of proangiogenic VEGF‐A from platelets. Collectively, our data suggest a key role of breast tumor‐derived VEGF‐A, as well as VEGF‐A derived from tumor‐activated platelets, in promoting release of VWF from ECs. However, given the increased expression of a number of other mediators in breast tumor‐platelet releasate, it is likely that other factors, independent of VEGF‐A, may also play a role. This also highlights the distinct biological mechanisms through which breast cancer cells may promote elevated VWF levels in vivo.
LMWH treatment significantly attenuated breast cancer‐mediated VWF release from ECs. This effect was likely mediated by significant reductions in VEGF‐A levels following LMWH treatment of breast cancer cells alone and in co‐culture with platelets. A number of studies have previously reported high affinity of LMWH for VEGF‐A. LMWH‐VEGF‐A binding inducing conformational changes in VEGF‐A inhibiting its activity. 19 , 41 Moreover, platelets from patients on LMWH anticoagulant treatment have decreased VEGF levels and reduced angiogenic potential following activation by MCF‐7 breast cancer cells ex vivo. 36 Similarly, we observed that the pro‐angiogenic potential of breast tumor‐platelet co‐cultures was markedly decreased following treatment with LMWH. This aligns with reports that LMWH is a potent inhibitor of VEGF‐mediated human microvascular endothelial cell proliferation. 46 However, because this inhibitory effect of LMWH was more potent than treatment with anti‐VEGF‐A antibody, bevacizumab, it suggests that LMWH may also modulate other pro‐ or anti‐angiogenic signaling pathways in the setting of breast cancer‐mediated angiogenesis. Notably, LMWH also served to attenuate expression of TGF‐β1 from tumor‐platelet co‐cultures. This may be important given the well‐defined role of TGF‐β1 in regulating endothelial angiogenesis in vivo. 47 Nevertheless, these findings may point to a dual clinical benefit of LMWH, first by reducing the risk of cancer‐associated thrombosis; and second, by attenuating tumor progression via reduced proangiogenic effects on EC and reduced VWF release from the endothelium.
Importantly, intraluminal accumulation of VWF within the tumor vasculature contributes to cancer progression and metastasis in vivo. 17 , 19 In tumor‐bearing mice, VWF was deposited along the endothelium in tumor‐free distal organs, including lung, liver, and brain, which are frequent sites of metastasis. 16 A strong positive correlation was observed between VWF release and pulmonary metastatic burden in these tumor models in vivo. 16 More recently, Feinauer et al. 48 provided direct evidence of the pro‐metastatic role of VWF, whereby intravital imaging of tumor‐bearing mice demonstrated that VWF release contributes to tumor cell arrest within brain capillaries. Accordingly, formation of brain macro‐metastasis was significantly reduced in mice treated with blocking anti‐VWF antibody. We now report that VWF multimers can directly tether circulating MDA‐MB‐231 breast tumor cells along the endothelium under conditions of shear stress in vitro. This effect was ablated by addition of LMWH to the circulating MDA‐MB‐231 cells. Because the major heparin binding domain of VWF resides within the A1 domain, this may indicate a key role for this region in mediating interaction between VWF and MDA‐MB‐231 tumor cells, further studies will be required to determine the role of the VWF A1 domain. 49 Nevertheless, the data provide evidence of multimeric VWF, released in response to elevated breast tumor VEGF‐A levels, as a novel adhesive mediator for triple negative breast tumor‐endothelial interactions.
Previous studies using EA.hy926, an immortalized endothelial cell line, have reported a protective effect of LMWH on endothelial barrier function, although the biological mechanism of this effect remained unclear. 50 Our data now provide further evidence that LMWH alters VE‐cadherin expression in primary EC, which contributes to enhanced barrier protective function. This effect may be partially mediated through its inhibition of VEGF‐A because a similar inhibitory effect was observed with bevacizumab. Increased endothelial barrier integrity served to inhibit transendothelial migration of breast tumor cells MDA‐MB‐231 and MCF‐7. These findings reveal novel antimetastatic properties of LMWH in the setting of breast cancer. Interestingly, this cytoprotective role of LMWH was achieved at very low doses, below LMWH doses currently used for thromboprophylaxis clinically.
In conclusion, this study provides novel insights into the crosstalk that exists between circulating breast tumor cells and the endothelium that is known to promote thrombosis and metastasis. 51 , 52 , 53 Our findings highlight increased VWF secretion as a novel feature in this crosstalk (Figure 7). Marked release of VWF from the EC is likely to contribute to a prothrombotic milieu within the circulation. Moreover, endothelial VWF may also serve as an anchor point in mediating breast tumor cell adhesion to the endothelium, a key prerequisite step in the metastatic pathway. Importantly, both VWF‐mediated adhesion of circulating breast tumor cells and transendothelial migration of breast cancer cells were attenuated with LMWH. These findings may have significant clinical implications given that triple‐negative breast cancer is an aggressive form of breast cancer with higher risk of early metastasis with a proclivity for bloodborne dissemination. 54 Thus, there is an unmet clinical need to identify and develop novel treatment strategies for patients to attenuate metastasis and reduce thrombotic complications. In this context, improved understanding of the interplay between breast cancer cells, the endothelium, and VWF will be essential in the development of these novel therapeutic strategies.
FIGURE 7.
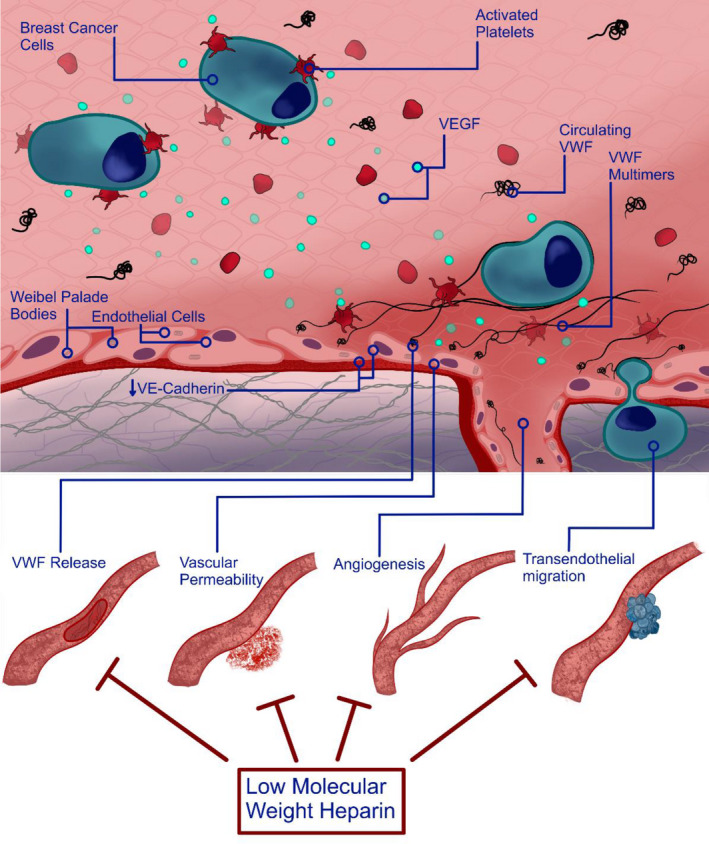
Schematic summary. Proposed model of breast tumor‐mediated EC activation. Breast cancer cells promote EC activation, which is further enhanced by the presence of platelets. Tumor‐secreted VEGF‐A, as well as other potential mediators including TGF‐β1, play an important role in modulating breast cancer‐induced VWF secretion, which is negatively regulated by LMWH. Elevated VEGF‐A from breast tumor cells also contributes to a pro‐angiogenic effect on EC. VWF multimers released from activated EC in response to increased VEGF‐A levels can tether circulating breast tumor cells, which is inhibited by LMWH. Increased vascular permeability and tumor transendothelial migration is attenuated by LMWH.
AUTHOR CONTRIBUTIONS
S.P.S.D. performed experiments and analyzed data; S.P., C.C., C.M.B., M.K., I.S., and B.C., assisted with experiments; I.S. analyzed flow data; J.C. and C.C.K. provided clinical samples and clinical data; J.S.O.D. and J.M.O.S. designed the research and analyzed the data. All authors were involved in writing and reviewing the manuscript.
CONFLICT OF INTEREST
S.P.S.D., S.P., C.C., C.M.B., M.K., I.S., B.C., J.C., C.C.K., and J.S.O.D. have no conflicts of interest related to this research. J.M.O.S. has received research grant funding awards from LEO Pharma.
Supporting information
Video S1
Figure S1
Figure S2
Figure S3
Figure S4
Table S1
Appendix S1
ACKNOWLEDGMENT
The research conducted in this publication was funded by a RCSI‐Industry partnership with LEO Pharma and the Irish Research Council, under grant number (EPSPD/2021/67) to S.P.S.D. The authors would like to acknowledge Dr Dishon Hiebner for his assistance with generating washed platelets. Open access funding provided by IReL.
Dhami SPS, Patmore S, Comerford C, et al. Breast cancer cells mediate endothelial cell activation, promoting von Willebrand factor release, tumor adhesion, and transendothelial migration. J Thromb Haemost. 2022;20:2350‐2365. doi: 10.1111/jth.15794
Manuscript Handled by: David Lillicrap
Final decision: David Lillicrap, 13 June 2022
Contributor Information
Sukhraj Pal Singh Dhami, @sps_dhami.
Jamie M. O'Sullivan, Email: jamieosullivan@rcsi.ie, @Jme_os.
REFERENCES
- 1. Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer‐associated venous thrombosis. Blood. 2013;122(10):1712‐1723. [DOI] [PubMed] [Google Scholar]
- 2. Walker AJ, Card TR, West J, Crooks C, Grainge MJ. Incidence of venous thromboembolism in patients with cancer‐a cohort study using linked United Kingdom databases. Eur J Cancer. 2013;49(6):1404‐1413. [DOI] [PubMed] [Google Scholar]
- 3. Chew HK, Wun T, Harvey D, Zhou H, White RH. Incidence of venous thromboembolism and its effect on survival among patients with common cancers. Arch Intern Med. 2006;166(4):458‐464. [DOI] [PubMed] [Google Scholar]
- 4. Goodnough LT, Saito H, Manni A, Jones PK, Pearson OH. Increased incidence of thromboembolism in stage IV breast cancer patients treated with a five‐drug chemotherapy regimen. A study of 159 patients. Cancer. 1984;54(7):1264‐1268. [DOI] [PubMed] [Google Scholar]
- 5. Stein PD, Beemath A, Meyers FA, Skaf E, Sanchez J, Olson RE. Incidence of venous thromboembolism in patients hospitalized with cancer. Am J Med. 2006;119(1):60‐68. [DOI] [PubMed] [Google Scholar]
- 6. Dhami SPS, Patmore S, O'Sullivan JM. Advances in the management of cancer‐associated thrombosis. Semin Thromb Hemost. 2021;47(2):139‐149. [DOI] [PubMed] [Google Scholar]
- 7. Sørensen HT, Mellemkjaer L, Olsen JH, Baron JA. Prognosis of cancers associated with venous thromboembolism. N Engl J Med. 2000;343(25):1846‐1850. [DOI] [PubMed] [Google Scholar]
- 8. Lenting PJ, Christophe OD, Denis CV. von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood. 2015;125(13):2019‐2028. [DOI] [PubMed] [Google Scholar]
- 9. Kawecki C, Lenting PJ, Denis CV. von Willebrand factor and inflammation. J Thromb Haemost. 2017;15(7):1285‐1294. [DOI] [PubMed] [Google Scholar]
- 10. Ishihara J, Ishihara A, Starke RD, et al. The heparin binding domain of von Willebrand factor binds to growth factors and promotes angiogenesis in wound healing. Blood. 2019;133(24):2559‐2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Patmore S, Dhami SPS, O'Sullivan JM. Von Willebrand factor and cancer; metastasis and coagulopathies. J Thromb Haemost. 2020;18(10):2444‐2456. [DOI] [PubMed] [Google Scholar]
- 12. Pépin M, Kleinjan A, Hajage D, et al. ADAMTS‐13 and von Willebrand factor predict venous thromboembolism in patients with cancer. J Thromb Haemost. 2016;14(2):306‐315. [DOI] [PubMed] [Google Scholar]
- 13. Obermeier HL, Riedl J, Ay C, et al. The role of ADAMTS‐13 and von Willebrand factor in cancer patients: results from the Vienna cancer and thrombosis study. Res Pract Thromb Haemost. 2019;3(3):503‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Desch A, Strozyk EA, Bauer AT, et al. Highly invasive melanoma cells activate the vascular endothelium via an MMP‐2/integrin αvβ5‐induced secretion of VEGF‐A. Am J Pathol. 2012;181(2):693‐705. [DOI] [PubMed] [Google Scholar]
- 15. Goerge T, Barg A, Schnaeker E‐M, et al. Tumor‐derived matrix metalloproteinase‐1 targets endothelial proteinase‐activated receptor 1 promoting endothelial cell activation. Cancer Res. 2006;66(15):7766‐7774. [DOI] [PubMed] [Google Scholar]
- 16. Goertz L, Schneider SW, Desch A, et al. Heparins that block VEGF‐A‐mediated von Willebrand factor fiber generation are potent inhibitors of hematogenous but not lymphatic metastasis. Oncotarget. 2016;7(42):68527‐68545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. John A, Robador JR, Vidal‐Y‐Sy S, et al. Urothelial carcinoma of the bladder induces endothelial cell activation and hypercoagulation. Mol Cancer Res. 2020;18(7):1099‐1109. [DOI] [PubMed] [Google Scholar]
- 18. Zhang P, Goodrich C, Fu C, Dong C. Melanoma upregulates ICAM‐1 expression on endothelial cells through engagement of tumor CD44 with endothelial E‐selectin and activation of a PKCα‐p38‐SP‐1 pathway. FASEB J. 2014;28(11):4591‐4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bauer AT, Suckau J, Frank K, et al. von Willebrand factor fibers promote cancer‐associated platelet aggregation in malignant melanoma of mice and humans. Blood. 2015;125(20):3153‐3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goerge T, Kleinerüschkamp F, Barg A, et al. Microfluidic reveals generation of platelet‐strings on tumor‐activated endothelium. Thromb Haemost. 2007;98(2):283‐286. [PubMed] [Google Scholar]
- 21. McCarty OJ, Mousa SA, Bray PF, Konstantopoulos K. Immobilized platelets support human colon carcinoma cell tethering, rolling, and firm adhesion under dynamic flow conditions. Blood. 2000;96(5):1789‐1797. [PubMed] [Google Scholar]
- 22. Morganti M, Carpi A, Amo‐Takyi B, et al. Von Willebrand's factor mediates the adherence of human tumoral cells to human endothelial cells and ticlopidine interferes with this effect. Biomed Pharmacother. 2000;54(8–9):431‐436. [DOI] [PubMed] [Google Scholar]
- 23. Sudha T, Phillips P, Kanaan C, Linhardt RJ, Borsig L, Mousa SA. Inhibitory effect of non‐anticoagulant heparin (S‐NACH) on pancreatic cancer cell adhesion and metastasis in human umbilical cord vessel segment and in mouse model. Clin Exp Metastasis. 2012;29(5):431‐439. [DOI] [PubMed] [Google Scholar]
- 24. Djaafar S, Dunand‐Sautier I, Gonelle‐Gispert C, et al. Enoxaparin attenuates mouse colon cancer liver metastases by inhibiting Heparanase and interferon‐γ‐inducible chemokines. Anticancer Res. 2016;36(8):4019‐4032. [PubMed] [Google Scholar]
- 25. Harvey JR, Mellor P, Eldaly H, Lennard TWJ, Kirby JA, Ali S. Inhibition of CXCR4‐mediated breast cancer metastasis: a potential role for heparinoids? Clin Cancer Res. 2007;13(5):1562‐1570. [DOI] [PubMed] [Google Scholar]
- 26. Kirwan CC, Descamps T, Castle J. Circulating tumour cells and hypercoagulability: a lethal relationship in metastatic breast cancer. Clin Transl Oncol. 2020;22(6):870‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shaker H, Bundred NJ, Landberg G, et al. Breast cancer stromal clotting activation (tissue factor and thrombin): a pre‐invasive phenomena that is prognostic in invasion. Cancer Med. 2020;9(5):1768‐1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ward SE, Fogarty H, Karampini E, et al. ADAMTS13 regulation of VWF multimer distribution in severe COVID‐19. J Thromb Haemost. 2021; 19(8):1914‐1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fazavana J, Brophy TM, Chion A, et al. Investigating the clearance of VWF A‐domains using site‐directed PEGylation and novel N‐linked glycosylation. J Thromb Haemost. 2020;18(6):1278‐1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ward SE, Curley GF, Lavin M, et al. Von Willebrand factor propeptide in severe coronavirus disease 2019 (COVID‐19): evidence of acute and sustained endothelial cell activation. Br J Haematol. 2021;192(4):714‐719. [DOI] [PubMed] [Google Scholar]
- 31. Carpentier G, Berndt S, Ferratge S, et al. Angiogenesis analyzer for ImageJ – a comparative morphometric analysis of “Endothelial Tube Formation Assay ” and “Fibrin Bead Assay”. Sci Rep. 2020;10: 11568. 10.1038/s41598-020-67289-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nossent AY, Van Marion V, Van Tilburg NH, et al. von Willebrand factor and its propeptide: the influence of secretion and clearance on protein levels and the risk of venous thrombosis. J Thromb Haemost. 2006;4:2556‐2562. [DOI] [PubMed] [Google Scholar]
- 33. Mangan SH, Van Campenhout A, Rush C, Golledge J. Osteoprotegerin upregulates endothelial cell adhesion molecule response to tumor necrosis factor‐alpha associated with induction of angiopoietin‐2. Cardiovasc Res. 2007;76(3):494‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wohner N, Sebastian S, Muczynski V, et al. Osteoprotegerin modulates platelet adhesion to von Willebrand factor during release from endothelial cells. J Thromb Haemost. 2022;20(3):755‐766. [DOI] [PubMed] [Google Scholar]
- 35. Takagi S, Tsukamoto S, Kawano Y, et al. Platelets/megakaryocytes are critical regulators of tumor progression in multiple myeloma. Blood. 2015;126(23):1793‐1793. [Google Scholar]
- 36. Battinelli EM, Markens BA, Kulenthirarajan RA, Machlus KR, Flaumenhaft R, Italiano JE Jr. Anticoagulation inhibits tumor cell‐mediated release of platelet angiogenic proteins and diminishes platelet angiogenic response. Blood. 2014;123(1):101‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang L, Luan Y, Miao X, et al. Platelet releasate promotes breast cancer growth and angiogenesis via VEGF–integrin cooperative signalling. Br J Cancer. 2017;117(5):695‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23(5):1011‐1027. [DOI] [PubMed] [Google Scholar]
- 39. Sopo M, Anttila M, Hämäläinen K, et al. Expression profiles of VEGF‐A, VEGF‐D and VEGFR1 are higher in distant metastases than in matched primary high grade epithelial ovarian cancer. BMC Cancer. 2019;19(1):584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Salven P, Perhoniemi V, Tykkä H, Mäenpää H, Joensuu H. Serum VEGF levels in women with a benign breast tumor or breast cancer. Breast Cancer Res Treat. 1999;53(2):161‐166. [DOI] [PubMed] [Google Scholar]
- 41. Zhao W, McCallum SA, Xiao Z, Zhang F, Linhardt RJ. Binding affinities of vascular endothelial growth factor (VEGF) for heparin‐derived oligosaccharides. Biosci Rep. 2012;32(1):71‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zuo X‐X, Yang Y, Zhang Y, Zhang ZG, Wang XF, Shi YG. Platelets promote breast cancer cell MCF‐7 metastasis by direct interaction: surface integrin α2β1‐contacting‐mediated activation of Wnt‐β‐catenin pathway. Cell Commun Signal. 2019;17(1):142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2(10):795‐803. [DOI] [PubMed] [Google Scholar]
- 44. Randi AM, Smith KE, Castaman G. von Willebrand factor regulation of blood vessel formation. Blood. 2018;132(2):132‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gisterek I, Matkowski R, Lacko A, et al. Serum vascular endothelial growth factors a, C and d in human breast tumors. Pathol Oncol Res. 2010;16(3):337‐344. [DOI] [PubMed] [Google Scholar]
- 46. Norrby K. 2.5 kDa and 5.0 kDa heparin fragments specifically inhibit microvessel sprouting and network formation in VEGF165‐mediated mammalian angiogenesis. Int J Exp Pathol. 2000;81(3):191‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guerrero PA, McCarty JH. TGF‐β activation and signaling in angiogenesis. In: Simionescu D, Simionescu A eds. Physiol Pathol Angiogenes Signal Mech Target Ther. IntechOpen; 2017. 10.5772/66405 [DOI] [Google Scholar]
- 48. Feinauer MJ, Schneider SW, Berghoff AS, et al. Local blood coagulation drives cancer cell arrest and brain metastasis in a mouse model. Blood. 2021;137(9):1219‐1232. [DOI] [PubMed] [Google Scholar]
- 49. Sobel M, Soler DF, Kermode JC, Harris RB. Localization and characterization of a heparin binding domain peptide of human von Willebrand factor. J Biol Chem. 1992;267(13):8857‐8862. [PubMed] [Google Scholar]
- 50. Kevane B, Egan K, Allen S, et al. Endothelial barrier protective properties of low molecular weight heparin: a novel potential tool in the prevention of cancer metastasis? Res Pract Thromb Haemost. 2017;1(1):23‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lal I, Dittus K, Holmes CE. Platelets, coagulation and fibrinolysis in breast cancer progression. Breast Cancer Res. 2013;15(4):207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hjortoe GM, Petersen LC, Albrektsen T, et al. Tissue factor‐factor VIIa‐specific up‐regulation of IL‐8 expression in MDA‐MB‐231 cells is mediated by PAR‐2 and results in increased cell migration. Blood. 2004;103(8):3029‐3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bourcy M, Suarez‐Carmona M, Lambe J, et al. Tissue factor induced by epithelial‐mesenchymal transition triggers a procoagulant state that drives metastasis of circulating tumor cells. Cancer Res. 2016;76(14):4270‐4282. [DOI] [PubMed] [Google Scholar]
- 54. Dent R, Trudeau M, Pritchard KI, et al. Triple‐negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15):4429‐4434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1
Figure S1
Figure S2
Figure S3
Figure S4
Table S1
Appendix S1