

To the Editor,
Airway inflammation in cystic fibrosis (CF) involves early and nonresolving activation of the innate immune system, characterized by neutrophil infiltration, production of serine proteases such as neutrophil elastase (NE), oxidative stress, and high levels of proinflammatory cytokines, even in the absence of infection. 1 Currently, there are no effective anti‐inflammatory therapies routinely used in clinical practice, however, significant resources are allocated to identifying such treatments. 1 Cystic fibrosis transmembrane conductance regulator (CFTR) modulator therapy is transforming the care of CF and is now used in infants as young as 4 months of age. The direct effect of CFTR modulator therapy on lung inflammation, and the potential role for synergistic anti‐inflammatory therapy, remains unclear.
Ivacaftor, the first CFTR modulator, has been shown to lower the levels of NE, interleukin (IL)‐8, and IL‐1β in the sputum of adults. 2 A study of sputum of adolescents and adults demonstrated treatment with lumacaftor/ivacaftor was associated with a significant reduction in IL‐1β but not IL‐6, IL‐8, tumor necrosis factor‐α (TNF‐α), or NE. 3 One study used peripheral blood monocyte‐derived macrophages from adults with CF to show that macrophage phagocytosis was restored to non‐CF levels in patients taking ivacaftor alone but not lumacaftor/ivacaftor. 4 Recently, whole blood transcriptomics revealed that people with CF show widespread overexpression of inflammation‐related genes that did not change following treatment with lumacaftor/ivacaftor. 5 In the only study of lower airway samples from early life, McNally et al. 6 analyzed bronchoalveolar lavage (BAL) obtained from children under 6 years of age, 1 year before commencing ivacaftor, immediately before treatment, and 1‐year posttreatment initiation. Inflammation was compared in children who had samples at similar time points but who were not eligible for ivacaftor treatment. This study showed ivacaftor did not reduce the levels of NE, IL‐8, or absolute neutrophil count.
Here, we profile airway inflammation in three clinical groups of children with CF (untreated [n = 40], lumacaftor/ivacaftor treated [n = 5], and ivacaftor treated [n = 9]) and in children without lung disease (children having bronchoscopy for investigation of stridor) (n = 8). Untreated children either have a genotype ineligible for modulator therapy, or their parents have elected not to start treatment. All children are enrolled in the AREST‐CF cohort, which involves collection of excess BAL, at the time of annual surveillance bronchoscopy, which is undertaken when a child is clinically well and is considered part of routine care at our CF center. Bronchoalveolar lavage was collected and processed as previously described. 7 Cytokines were measured in cell‐free BAL using the Human Soluble Protein Flex Set Cytometric Bead Array (BD Biosciences) according to the manufacturer's instructions. Data were acquired on an LSR II X‐20 Fortessa and analyzed using the BD FCAP Array software. The following nine inflammatory cytokines and chemokines were assessed: TNF‐α, chemokine ligand 5 (CCL5), chemokine ligand 3 (CCL3), chemokine (C‐X‐C motif) ligand 9 (CXCL9), chemokine ligand 2 (CCL2), IL‐8, IL‐1β, interferon‐α (IFN‐α), and IL‐6. Cytokine levels below the detection range, as supplied by the manufacturer, were arbitrarily reported as half the lower limit of detection for each cytokine and included in the analysis. Flow cytometry results for inflammatory cells in BAL were available in a sub‐cohort of participants (CF untreated [n = 9], lumacaftor/ivacaftor treated [n = 5], ivacaftor treated [n = 2], healthy controls [n = 4]) (see Table S1). For this study, BAL cells were processed fresh for flow cytometry as we have described previously. 8 Cell types captured in our panel include alveolar macrophages (CD45+CD206+), neutrophils (CD45+CD206−CD15+) and their subsets (CD66b and CD16 expression), classical monocytes (CD45+CD11c+CD14+CD16−), nonclassical monocytes (CD45+CD11c+CD14+CD16+), and dendritic cells (CD45+CD11c+HLADR+CD14−CD16−). Data were acquired on an LSR II X‐20 Fortessa and analyzed using FlowJo V10 software. The flow cytometry panel is shown in Table S2 and the manual gating strategy is depicted in Figure S1. For differential analysis of cytokine concentration (Figure 1A) and cell proportion data (Figure S2B), p values were determined by Kruskal–Wallis rank‐sum test and adjusted for multiple comparisons using the Benjamini and Hochberg approach to control the false discovery rate (FDR). FDR‐adjusted p < 0.05 were considered significant. For cytokines showing an FDR‐adjusted significant difference, Dunn's multiple comparison testing was used to find differences between clinical groups. Principal component analysis (Figure S2A) was conducted with age and sex as included variables. All analyses were performed in Prism version 9.1, with multiple comparison corrections performed in RStudio version 4.0.3.
Figure 1.
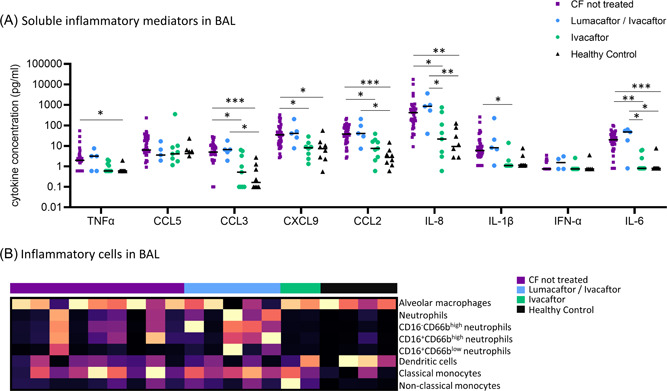
Early life inflammatory cytokine and cell profile in BAL of children with CF during treatment with CFTR modulators. (A) TNF‐α, CCL5, CCL3, CXCL9, CCL2, IL‐8, IL‐1β, IFNα, and IL‐6 were quantified in BAL using multiplex cytometric bead array for children with CF (untreated [purple, n = 40], lumacaftor/ivacaftor treated [blue, n = 5], and ivacaftor treated [green, n = 9]) and healthy controls (black, n = 8). FDR‐corrected p values are shown (p < 0.05, **p < 0.01, ***p < 0.001). Individual datapoints are shown. (B) Inflammatory cells (alveolar macrophages, neutrophils and their subsets, dendritic cells, classical monocytes, and nonclassical monocytes) were quantified in BAL using flow cytometry in a subset of children with CF (untreated [purple, n = 9], lumacaftor/ivacaftor treated [blue, n = 5], and ivacaftor treated [green, n = 2]) and healthy controls (black, n = 4). Individual datapoints are shown. BAL, bronchoalveolar lavage; CCL, chemokine ligand; CFTR, cystic fibrosis transmembrane conductance regulator; FDR, false discovery rate; IL, interleukin; IFNα, interferon‐α; TNF‐α, tumor necrosis factor‐α [Color figure can be viewed at wileyonlinelibrary.com]
Demographics of the study participants are described in Tables 1 and S1. Relative to untreated children, ivacaftor‐treated children had significantly lower median levels of CCL3 (0.52 vs. 4.99pg/ml, p = 0.013), CXCL9 (8.10 vs. 34.59 pg/ml, p = 0.01), CCL2 (7.61 vs. 37.96 pg/ml, p = 0.012), IL‐8 (21.69 vs. 422 pg/ml, p = 0.026), IL‐1β (1.10 vs. 5.93 pg/ml, p = 0.010) and IL‐6 (0.8 vs. 19.89 pg/ml, p = 0.001) (Figure 1A). There were no significant differences in BAL cytokines between lumacaftor/ivacaftor treated children and untreated children. Interestingly, median levels of IL‐8 and IL‐6 were significantly lower in ivacaftor treated children relative to lumacaftor/ivacaftor treated children (IL‐8: 21.69 vs. 851.48 pg/ml [p = 0.03], IL‐IL‐6: 0.8 vs. 47.38 pg/ml [p = 0.016]).
Table 1.
Demographics of study cohort
| CF untreated | CF lumacaftor/ivacaftor | CF ivacaftor | Healthy control | ||
|---|---|---|---|---|---|
| Number (n) | 40 | 5 | 9 | 8 | |
| Age (years), | 2.97 | 4.5 | 4.06 | 3.81 | |
| median (min–max) | (1–6) | (2–6) | (3–6) | (1–8) | |
| Sex (% female) | 60 | 60 | 22.2 | 25 | |
| Duration of treatment at sample collection (years), median (min–max) | 0.93 (0.17–1.39) | 1.88 (0.95–2.25) | |||
| Genotype (n) | Class I/Class I | 1 | |||
| Class I/Class VII | 3 | ||||
| Class II/Class I | 6 | ||||
| Class II/Class II | 23 | 5 | |||
| Class II/Class III | 1 | 8 | |||
| Class II/Class IV | 1 | ||||
| Class II/Class VII | 1 | ||||
| Class II/Unclassified | 4 | ||||
| Class III/Class IV | 0 | 1 | |||
| Pancreatic status (% insufficient) | 92.5 | 100 | 77.7 | ||
| Body Mass Index z‐score (median) | 0.45 | 0.53 | 0.50 | ||
| Infection status (% with bacterial or viral organism detected in BAL) | 80 | 100 | 55.5 | 62.5 | |
Note: Genotype was classified based on the system proposed by De Boeck and Amaral. 1
Abbreviations: BAL, bronchoalveolar lavage; CF, cystic fibrosis.
Relative to healthy controls, untreated children with CF showed significantly higher median levels of TNF‐α (1.97 vs. 0.6 pg/ml, p = 0.04), CCL3 (4.99 vs. 0.16 pg/ml, p = 0.0009), CXCL9 (34.59 vs. 7.41 pg/ml, p = 0.021), CCL2 (37.96 vs. 2.72 pg/ml, p = 0.0002), IL‐8 (422.41 vs. 9.38 pg/ml, p = 0.001), and IL‐6 (19.89 vs. 0.8 pg/ml, p = 0.001) (Figure 1A). Similarly, lumacaftor/ivacaftor‐treated children had significantly higher median levels of CCL3 (6.59 vs. 0.16 pg/ml, p = 0.036) CCL2 (40.76 vs. 2.72 pg/ml, p = 0.013), IL‐8 (851.48 vs. 9.38 pg/ml, p = 0.004), and IL‐6 (47.38 vs. 0.8 pg/ml, p = 0.01) relative to healthy controls.
The unsupervised principal component analysis depicted in Figure S2A confirms these findings, with ivacaftor treated children clustering together with the healthy controls, and lumacaftor/ivacaftor‐treated children clustering together with the untreated CF children.
We demonstrate that children treated with ivacaftor show lower pulmonary concentrations of a range of inflammatory mediators when compared to untreated children, as well as lumacaftor/ivacaftor treated children. In particular, ivacaftor treatment was associated with lower concentrations of two key cytokines: IL‐8 and IL‐1β which have previously been associated with the development of early life structural lung disease. Treatment with lumacaftor/ivacaftor, however, did not induce reductions of any inflammatory cytokine when compared to untreated children with CF.
To further assess the impact of CFTR modulator therapy on early life pulmonary inflammation, we defined the inflammatory cell profile of BAL in a sub‐cohort of participants (Figures 1B and S2B). Whilst more limited in numbers, this exploratory analysis showed a similar response to that observed for soluble inflammatory mediators, where ivacaftor treated children are more similar to healthy controls, and lumacaftor/ivacaftor treated children are more similar to untreated CF children. This effect is most prominent in the neutrophil populations (Figures 1B and S2B).
There are several limitations of this study that should be highlighted. There are relatively small numbers of patients in the CF treatment groups and participants treated with ivacaftor had been treated for a longer duration than those treated with lumacaftor/ivacaftor (1.88years vs. 0.93 years). In addition, the median age of the untreated CF group was lower than those in the treated group. These limitations reflect the difficulty in obtaining lower airway samples from preschool children. We are only able to obtain samples at the time of clinically indicated procedures which means limits the ability to control the duration of therapy and age at sampling. Highly effective modulator therapy has been shown to improve lung function and sweat chloride after 2 weeks of therapy and this effect is sustained for 52 weeks. The impact of lumacaftor/ivacaftor on lung function follows a similar trajectory. 9 Based on this we think it is it is unlikely that a longer duration of therapy of lumacaftor/ivacaftor would result in an altered inflammatory profile, however, this is a possibility and hence a limitation of this study. The small sample size also limits the potential significance of the finding of lower infection rates in BAL of ivacaftor‐treated children relative to lumacaftor/ivacaftor‐treated children (55.5% vs. 100%, p = 0.07) (Table 1). Previously ivacaftor was not associated with reduced lower airway infection in preschool children with CF. 6 However, data from international registries have shown a trend to reduced infections with organisms including Pseudomonas aeruginosa, Staphylococcus aureus, and Aspergillus species in people treated with ivacaftor. As the concentration of proinflammatory cytokines increase with age, the younger age of the CF untreated group likely leads to underestimation of the effect of modulator treatment. The absence of pretreatment measurements in those treated with modulators is also a limitation. Despite these limitations, these data still give important insights into the lower airway environment in preschool children with CF and in particular the anti‐inflammatory effect of ivacaftor.
The findings of this study are in contrast to those of McNally et al. 6 who found there was no change in IL‐8 concentration posttreatment with ivacaftor. Potential explanations for the differences in findings include a larger CF control group in our study allowing more accurate estimation of untreated inflammatory profile, different cytokine measurement platforms (cytokine bead array vs. enzyme‐linked immunosorbent assay), and different analysis approaches (reporting concentration as opposed to presence/absence). Regardless, further studies are needed to confirm the findings of our work.
While the difference in response to the two treatments is not surprising given prior data showing that ivacaftor is more effective at improving CFTR function and lung disease outcomes, this is the first work to illustrate a difference in airway inflammation in early life. The altered inflammatory environment may be secondary to an altered infection, or a direct effect of modulator therapy on CFTR function given the previous evidence that aberrant inflammation can occur in the absence of infection. 10 Of note, some children treated with ivacaftor still exhibited a proinflammatory cytokine profile suggesting the anti‐inflammatory effect of modulator therapy may vary between individuals. These results, which may not be generalizable to older patients with established lung disease, suggest that a reduction in early life pulmonary inflammation may be one of the mechanisms by which ivacaftor improves lung disease outcomes. As trials in early life using this drug were predicated on safety and tolerability rather than efficacy, our findings highlight the need for larger, longitudinal studies to determine the real‐world effectiveness of ivacaftor in optimizing lung function (including that determined by more sensitive techniques such as multiple breath washout) and lung structure (preventing the early onset of bronchiectasis). If this was demonstrated with ivacaftor, as well as other novel CFTR modulators such as elexacaftor/tezacaftor/ivacaftor, it would suggest that synergistic anti‐inflammatory therapy may not be needed by all patients and that BAL inflammatory profile may represent a biomarker of therapeutic response.
AUTHOR CONTRIBUTIONS
Shivanthan Shanthikumar and Melanie R. Neeland conducted the experiments and co‐wrote the first draft of the manuscript. Shivanthan Shanthikumar and Sarath C. Ranganathan recruited the patients and collected the biospecimens. All authors contributed to and approved the final version of the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
SS is supported by a Vertex Mentored Research Innovation Award. We thank the children and parents who participated in the AREST‐CF study. Open access publishing facilitated by The University of Melbourne, as part of the Wiley ‐ The University of Melbourne agreement via the Council of Australian University Librarians.
Shanthikumar S, Ranganathan S, Neeland MR. Ivacaftor, not ivacaftor/lumacaftor, associated with lower pulmonary inflammation in preschool cystic fibrosis. Pediatric Pulmonology. 2022;57:2549‐2552. 10.1002/ppul.26063
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016;4(8):662‐674. [DOI] [PubMed] [Google Scholar]
- 2. Hisert KB, Heltshe SL, Pope C, et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am J Respir Crit Care Med. 2017;195(12):1617‐1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Graeber SY, Boutin S, Wielpütz MO, et al. Effects of lumacaftor‐ivacaftor on lung clearance index, magnetic resonance imaging, and airway microbiome in Phe508del homozygous patients with cystic fibrosis. Ann Am Thorac Soc. 2021;18(6):971‐980. [DOI] [PubMed] [Google Scholar]
- 4. Zhang S, Shrestha CL, Kopp BT. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci Rep. 2018;8(1):17066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kopp BT, Fitch J, Jaramillo L, et al. Whole‐blood transcriptomic responses to lumacaftor/ivacaftor therapy in cystic fibrosis. J Cyst Fibros. 2020;19(2):245‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McNally P, Butler D, Karpievitch YV, et al. Ivacaftor and airway inflammation in preschool children with cystic fibrosis. Am J Respir Crit Care Med. 2021;204:605‐608. [DOI] [PubMed] [Google Scholar]
- 7. Sly PD, Gangell CL, Chen L, et al. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. 2013;368(21):1963‐1970. [DOI] [PubMed] [Google Scholar]
- 8. Shanthikumar S, Ranganathan SC, Saffery R, Neeland MR. Mapping pulmonary and systemic inflammation in preschool aged children with cystic fibrosis. Front Immunol. 2021;12:733217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor‐ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tirouvanziam R, de Bentzmann S, Hubeau C, et al. Inflammation and infection in naive human cystic fibrosis airway grafts. Am J Respir Cell Mol Biol. 2000;23(2):121‐127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.