

Abstract
Objective
The aim of this study was to investigate the impact of the modified ketogenic diet on DNA methylation in adults with epilepsy.
Methods
In this prospective study, we investigated the genome‐wide DNA methylation in whole blood in 58 adults with epilepsy treated with the modified ketogenic for 12 weeks. Patients were recruited from the National Center for Epilepsy, Norway, from March 1, 2011 to February 28, 2017. DNA methylation was analyzed using the Illumina Infinium MethylationEPIC BeadChip array. Analysis of variance and paired t‐test were used to identify differentially methylated loci after 4 and 12 weeks of dietary treatment. A false discovery rate approach with a significance threshold of <5% was used to adjust for multiple comparisons.
Results
We observed a genome‐wide decrease in DNA methylation, both globally and at specific sites, after 4 and 12 weeks of dietary treatment. A substantial share of the differentially methylated positions (CpGs) were annotated to genes associated with epilepsy (n = 7), lipid metabolism (n = 8), and transcriptional regulation (n = 10). Furthermore, five of the identified genes were related to inositol phosphate metabolism, which may represent a possible mechanism by which the ketogenic diet attenuates seizures.
Significance
A better understanding of the modified ketogenic diet's influence at the molecular level may be the key to unraveling the mechanisms by which the diet can ameliorate seizures and possibly to identifying novel therapeutic targets for epilepsy.
Keywords: epigenetics, high‐fat, Infinium MethylationEPIC BeadChip, low‐carbohydrate diet
Key Points.
In this prospective study, we investigated the impact of the modified ketogenic diet on DNA methylation in adults with epilepsy
Intraindividual comparisons of DNA methylation after the dietary treatment revealed a significant global decrease in DNA methylation
A substantial share of the differentially methylated CpGs were annotated to genes associated with epilepsy, lipid metabolism, and transcriptional regulation
Differentially methylated CpGs annotated to genes involved in inositol phosphate metabolism may represent a possible mechanism for the diet's antiseizure efficacy
Identifying the molecular consequences of the dietary treatment may reveal the diet's mechanisms by which it can ameliorate seizures
1. INTRODUCTION
Epilepsy is a heterogeneous neurological disorder characterized by unprovoked, recurrent seizures. Worldwide, >65 million people are affected and the disease itself, and associated comorbidities represent a huge burden of disease. 1 , 2 Antiseizure medications (ASMs) are the mainstay of epilepsy treatment. However, about one third of patients do not respond adequately to the currently available ASMs (drug‐resistant patients). 3 , 4 Moreover, treatment with ASMs offers only symptomatic relief by reducing the seizures without affecting the underlying epilepsy mechanisms. Thus, in most cases they are not able to prevent disease progression (epileptogenesis). 5
The ketogenic diet, a high‐fat, low‐carbohydrate diet, is an established treatment for patients with drug‐resistant epilepsy. The efficacy of the dietary treatment in children with epilepsy is well documented, 6 and recent years' research suggests that adults may also benefit from such diet therapy. 7 However, despite significant efforts to identify the underlying mechanisms behind the diet's seizure‐reducing effect, the mechanisms of action still remain elusive. Recent work suggests epigenetic mechanisms as an attractive candidate to explain the reduced neuronal excitability. 8 , 9 , 10
Epigenetic modification, including DNA methylation, is a dynamic process involved in regulation of gene expression and is essential for normal brain development and plasticity. Abnormal DNA methylation has been reported in a wide range of diseases, including epilepsy and other neurological disorders. 11 , 12 , 13 DNA methylation is the addition of a methyl group (CH3) at a cytosine base in the DNA. In mammals, this primarily occurs at cytosines followed by guanines, called cytosine–guanine dinucleotides (CpGs). Methylation of DNA is catalyzed by DNA methyltransferases (DNMTs), and methylation of promoters tends to induce gene silencing. Changes in DNA methylation can lead to altered expression of genes involved in neuronal excitability and inhibition, and thereby potentially promote epileptogenesis. “The methylation hypothesis” in epilepsy suggests that seizures themselves can induce DNA methylation changes that sustain, and even exacerbate, the epileptogenic process. 10 Studies both in animal models of epilepsy and in humans with temporal lobe epilepsy have shown a global increase in DNA methylation in epileptic brains compared to healthy controls. 8 , 9 , 11 , 14 Interestingly, inhibition of DNA methylation in animal models of epilepsy appears to prevent epileptogenesis. 8 Furthermore, the increase in DNA methylation has been shown to be counteracted by ketogenic dietary treatment, which also correlated with increased seizure threshold. 8 , 9
Because nutrition is a key environmental factor influencing DNA methylation, 15 we hypothesized that the drastic change in macronutrient composition that the ketogenic diet represents will have an impact on DNA methylation. Thus, we conducted a longitudinal genome‐wide DNA methylation study and investigated whether treatment with a modified ketogenic diet is associated with changes in DNA methylation in patients with drug‐resistant epilepsy.
2. MATERIALS AND METHODS
2.1. Study design and participants
Patients were recruited from the National Center for Epilepsy, Norway, between March 1, 2011, and February 28, 2017. The study cohort consisted of patients with focal epilepsy included in the randomized clinical trial by Kverneland et al. 16 and patients with generalized epilepsy included in an associated prospective, non‐randomized study by the same research group. 17 All participants followed the same diet intervention protocol. The baseline period was defined as the 12 weeks immediately preceding the 12‐week diet intervention period. In the baseline period, the participants ate their normal diet and recorded seizures systematically, and no changes in epilepsy treatment were allowed. In the intervention period, the participants ate a modified ketogenic diet and continued to keep a systematic record of seizures. All other epilepsy treatments were kept unchanged.
Inclusion criteria were generalized or focal epilepsy according to the International League Against Epilepsy classification, 18 ≥3 countable seizures per month, having tried ≥3 ASMs, age ≥ 16 years, body mass index > 18.5 kg/m2 (no upper limit), and the participants had to be motivated and capable of adhering to the diet for at least 12 weeks. Exclusion criteria were familial hypercholesterolemia, cardiovascular disease, kidney disease, treatment with a ketogenic diet for >1 week during the preceding year, status epilepticus in the past 6 months, epilepsy surgery (including vagus nerve stimulator implant in the past year), 4 continuous weeks free of seizures in the preceding 2 months, psychogenic nonepileptic seizures, known disease in which the dietary treatment is contraindicated, use of drugs or supplements that may interfere with the diet or ASMs, change of ASMs in the past 3 months before baseline, and pregnancy or planned pregnancy.
2.2. Procedures
2.2.1. Diet
The dietary intervention was previously described in detail. 16 Briefly, the diet contained a maximum of 16 g carbohydrate per day (excluding fibers), and the participants were encouraged to eat high‐fat foods to replace the carbohydrates in the diet. Proteins were eaten ad libitum, and the total energy content was not restricted. The diet was supplemented with one multivitamin and mineral tablet (Nycoplus Multi, Takeda) and 800 mg calcium (calcium carbonate, Takeda). A daily fluid intake of 2–3 L was recommended. To calculate the nutritional content of the meals, the participants used the Norwegian Food Composition Database. 19
2.2.2. Diet adherence
To assess adherence to the diet, the participants performed a 3‐day weighed food record prior to starting on the diet and before the hospital admissions at the 4‐ and 12‐week time points. In addition, the participants recorded urine ketones (acetoacetate) twice daily (morning and evening) at home during the diet intervention using urine dipsticks (Ketostix, Bayer Healthcare). Blood glucose and blood ketones (β‐hydroxybutyrate) were measured morning and evening during the hospital admissions (FreeStyle Precision Neo, Freestyle Precision Blood Glucose Test Strips, and FreeStyle Precision Xtra Blood β‐Ketone Test Strips, Abbott). The data have previously been reported by Kverneland et al. and indicate good compliance with the dietary treatment. 16 , 17 , 20
2.2.3. Biochemical analyses
Venous blood samples were collected after an overnight food and drug fast at baseline, and after 4 and 12 weeks of dietary treatment. All biochemical routine analyses were performed at Oslo University Hospital (Oslo, Norway). Folate, vitamin B12, and homocysteine serum (before June 6, 2012) or plasma concentrations were measured on a Roche Diagnostics platform using the Elecsys Folate III assay (Roche Diagnostics), the Elecsys Vitamin B12 II assay (Roche Diagnostics), and the Axis Homocysteine Enzyme Immunoassay (Axis‐Shield Diagnostics), respectively, according to the manufacturer's instructions.
DNA methylation analysis: Microarray preprocessing and quality control
Whole blood for DNA extraction was collected in VACUETTE K2EDTA blood collection tubes (Greiner Bio‐One International). DNA methylation was analyzed using the Infinium MethylationEPIC BeadChip, which quantitatively interrogates DNA methylation at >850 000 positions (CpGs) genome‐wide with single nucleotide resolution. The EPIC BeadChips were processed at the LIFE & BRAIN laboratory according to the manufacturer's instructions. All DNA methylation analyses were carried out using the R programming language (http://www.r‐project.org/). Preprocessing and quality assessment were performed using functions implemented in the minfi package. 21 First, normalization was performed with precrocessQuantile. Then, the data were preprocessed and filtered to remove probes with unreliable measurements (detection p‐values > .01, n = 20 283), probes located on the sex chromosomes (n = 18 654), probes with overlapping single nucleotide polymorphisms (n = 27 348), cross‐reactive probes (n = 39 112), and non‐CpG probes (n = 2458), 22 , 23 resulting in a final dataset consisting of 760 462 probes and 172 samples.
2.3. Statistical analysis
2.3.1. Statistical data analysis tools and presentation of data
DNA methylation analyses were carried out in R using packages specifically developed to analyze Illumina EPIC DNA methylation array data. Statistical analyses of other background variables were carried out in SPSS Statistics version 26 (IBM). Data are presented as mean (±SD) or median (quartiles), and minimum–maximum, or frequency (%), as appropriate. We tested for differences in blood biochemistry from baseline to 4 and 12 weeks of dietary treatment using paired t‐test.
2.3.2. Cell type composition
White blood cell differential counts consisting of relative proportions of lymphocytes, monocytes, and granulocytes were measured by standard methods at Oslo University Hospital. To explore potential differences in the lymphocytes, we also performed whole‐blood deconvolution and estimated proportions of CD4+ and CD8+ T cells, natural killer (NK) cells, B cells, monocytes, and granulocytes using the estimateCellCounts2 function in the FlowSorted.Blood.EPIC R package. 24 Deconvolution estimates were evaluated by calculating R2 and root mean square error comparing estimates to matched cell counts.
2.3.3. Differential DNA methylation analyses
A linear regression model implemented in limma 25 was fitted to M‐values (log2 of the β‐values) to identify intraindividual differentially methylated positions before and after treatment with the modified ketogenic diet. Intraindividual differences in global DNA methylation were tested using a paired t‐test between time points on mean DNA methylation across all CpGs in patients containing complete data from all time points. To adjust for multiple testing, a false discovery rate cutoff of <5% was used for genome‐wide significance by using the method of Benjamini and Hochberg. 26
2.4. Study outcomes
The primary outcome was changes in DNA methylation associated with 4 and 12 weeks treatment with the modified ketogenic diet. The secondary outcome was changes in DNA methylation associated with seizure response comparing responders with nonresponders after 12 weeks of dietary treatment. Because a threshold of 25% seizure reduction has been proposed as the lowest clinically relevant outcome of dietary treatment 27 and the seizure reduction in our study population was modest (Table S1), we chose to define participants who achieved ≥25% seizure reduction after 12 weeks of dietary treatment as responders. A nonresponder was defined as a participant with no seizure reduction (including participants with an increase in seizure frequency). Participants with .1%–24.9% seizure reduction at 12 weeks were included in neither the responder nor the nonresponder category, in an attempt to limit bias from participants with an uncertain seizure response to the dietary treatment.
3. RESULTS
3.1. Baseline demographics and clinical characteristics
Samples from 58 participants (age = 16–65 years) were included in the study (Figure 1). In general, the participants had a long history of epilepsy (mean = 25.0 ± SD 11.9 years), a high number of previously tried ASMs (mean = 8.7 ± SD 4.1), and multiple current ASMs (mean = 2.1 ± SD .9). Also, two thirds of the participants were occupationally disabled, indicating a high burden of disease. An overview of the main baseline demographics and clinical characteristics of the participants is presented in Table 1.
FIGURE 1.
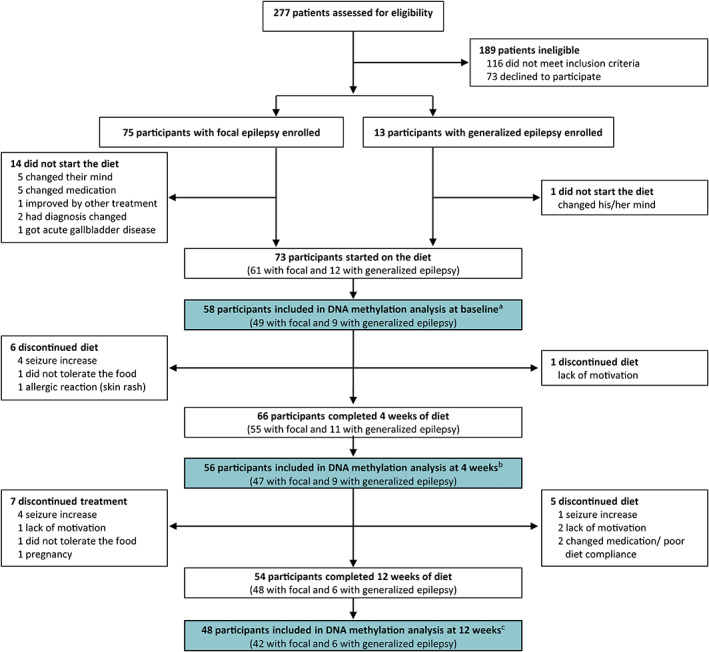
Study profile. aTwo participants did not give permission for analysis of the samples abroad, two participants were excluded from analysis due to poor diet compliance and change in medication, and 11 were missing blood samples or their blood samples were not analyzed because of lack of sample for DNA methylation analysis at 4 and 12 weeks of dietary treatment. bTwo participants did not give permission for analysis of the samples abroad, two participants were excluded from analysis due to poor diet compliance and change in medication, and six were missing blood samples. cTwo participants did not give permission for analysis of the samples abroad and four were missing blood samples.
TABLE 1.
Demographic and clinical characteristics of the participants at baseline, n = 58
| Characteristic | Mean (±SD) or median (quartiles) | Frequency (%) | Min–Max |
|---|---|---|---|
| Gender | |||
| Male | 24 (41%) | ||
| Female | 34 (59%) | ||
| Age, years | 36.5 (11.8) | 16–65 | |
| Epilepsy classification | |||
| Focal | 49 (85%) | ||
| Generalized | 9 (16%) | ||
| Age at first seizure, years | 7 (2–16) | 0–55 | |
| Epilepsy etiology | |||
| Structural | 16 (27.6%) | ||
| Genetic | 7 (12.1%) | ||
| Infectious | 4 (6.9%) | ||
| Unknown | 31 (53.4%) | ||
| Years with epilepsy | 25.0 (11.9) | 7–58 | |
| Seizure frequency per week | 3.5 (1.5–14.1) | .1–351.8 | |
| Intellectual disability | 21 (36%) | ||
| VNS, previous or current | 24 (41%) | ||
| Employment | |||
| Paid employment | 13 (22%) | ||
| Occupationally disabled | 37 (64%) | ||
| Other | 8 (14%) | ||
| Total number of ASMs tried | 8.7 (4.1) | 3–23 | |
| ASMs at diet initiation | 2.1 (.9) | 0–4 | |
Note: Data are presented as mean (±SD) or median (quartiles) and Min–Max for continuous variables, and frequency (percentage) for discrete variables.
Abbreviations: ASM, antiseizure medication; Min–Max, minimum–maximum; VNS, vagus nerve stimulator.
3.2. Energy and macronutrient intakes
Table 2 shows the estimated dietary intake of energy and macronutrients at baseline and 4 and 12 weeks after diet initiation based on the 3‐day weighed food records. On the ketogenic diet, the intake of fat was increased to about twice that of their baseline diet, whereas the intake of carbohydrate was greatly reduced to an average of only 13 g per day. The ketogenic ratio (grams fat to the sum of grams protein plus carbohydrate) was 1.7:1 at both 4 and 12 weeks after diet initiation as opposed to .3:1 at baseline. Thus, the macronutrient intake during the intervention period was in line with the study protocol.
TABLE 2.
Estimated intake of energy and macronutrients based on 3‐day weighed diet records at baseline, and after 4 and 12 weeks of treatment with modified ketogenic diet
| Baseline | 4 weeks on diet | 12 weeks on diet | ||||
|---|---|---|---|---|---|---|
| Mean (±SD) | n a | Mean (±SD) | n a | Mean (±SD) | n a | |
| Energy, kcal | 1856 (380) | 15 | 1987 (670) | 51 | 2007 (657) | 42 |
| Fat, g | 79 (19) | 15 | 170 (64) | 51 | 174 (61) | 42 |
| Fat, E% | 39 (7) | 15 | 76 (7) | 51 | 77 (6) | 42 |
| Protein, g | 81 (17) | 15 | 89 (31) | 51 | 92 (38) | 42 |
| Protein, E% | 18 (3) | 15 | 19 (5) | 51 | 18 (4) | 42 |
| Carbohydrates, g | 194 (58) | 15 | 13 (4) | 51 | 13 (3) | 42 |
| Carbohydrates, E% | 41 (8) | 15 | 3 (1) | 51 | 3 (1) | 42 |
| Ketogenic ratio b | .3:1 (.1) | 15 | 1.7:1 (.5) | 51 | 1.7:1 (.5) | 42 |
Abbreviation: E%, energy percentage.
Variation in n is due to missing values.
The ketogenic ratio defined as the ratio of grams fat to the sum of grams protein plus carbohydrate.
3.3. Folate, vitamin B12, and homocysteine status
Folate and vitamin B12 are essential micronutrients that together with homocysteine play an important role in the metabolism of methyl groups. The blood values for vitamin B12, folate, and homocysteine are given in Table 3. There was a significant increase in vitamin B12 and folate at both 4 and 12 weeks of dietary treatment compared to baseline (p < .001), whereas homocysteine was unchanged (p = .07 and p = .23 between baseline and 4 weeks and 12 weeks of dietary treatment, respectively). Hence, the folate and vitamin B12 status was improved after the diet intervention.
TABLE 3.
Blood biochemistry at baseline, and after 4 and 12 weeks of treatment with modified ketogenic diet
| Baseline | 4 weeks on diet | 12 weeks on diet | ||||
|---|---|---|---|---|---|---|
| Mean (±SD) | n a | Mean (±SD) | Mean (±SD) | n a | ||
| Folate, nmol·L−1 b | 19.3 (9.6) | 57 | 26.9 (8.0) c | 55 | 28.2 (9.4) c | 49 |
| Vitamin B12, pmol·L−1 b | 409.0 (172.6) | 58 | 527.0 (275.9) c | 55 | 473.0 (197.8) c | 49 |
| Homocysteine, μmol·L−1 b | 11.8 (6.7) | 58 | 10.6 (5.2) | 54 | 11.0 (5.8) | 49 |
Variation in n is due to missing values.
Paired t‐test was used as the statistical test.
Significantly different from baseline values (p < .001).
3.4. Cell type composition
As DNA methylation is highly tissue and cell type specific, changes in cell type composition can be a confounding factor. Therefore, we investigated potential alterations in cell type composition. There were no significant changes in the relative proportions of lymphocytes, monocytes, and granulocytes from baseline to 4 and 12 weeks of dietary treatment measured by routine cell counts (data not shown). To increase the precision, we also estimated the relative proportions of subpopulations of white blood cells. There were no significant changes in the estimated relative proportions of CD4+ and CD8+ T cells, B cells, NK cells, monocytes, and granulocytes, except a small increase in NK cells after 4 weeks of dietary treatment (.01 ± SD .02, p = .001). NK cells constitute only a small proportion of white blood cells, and we considered this minor change insufficient to be taken into account in the downstream analyses.
3.5. Epilepsy treatments
In accordance with the study protocol, none of the participants had any changes in epilepsy treatments, including type or doses of ASMs, neither during the baseline nor during 12 weeks of dietary intervention.
3.6. Global decrease in DNA methylation following treatment with the modified ketogenic diet
To investigate the influence of a modified ketogenic diet on global DNA methylation, we performed an intraindividual comparison of mean DNA methylation across all CpGs between baseline, and 4 and 12 weeks of dietary treatment, measuring the total content of DNA methylation at all CpGs included in this study (n = 760 462). This analysis revealed a significant decrease in global DNA methylation between baseline and 12 weeks, and between 4 and 12 weeks (paired t‐test p = .002 and .036, respectively; Figure 2).
FIGURE 2.
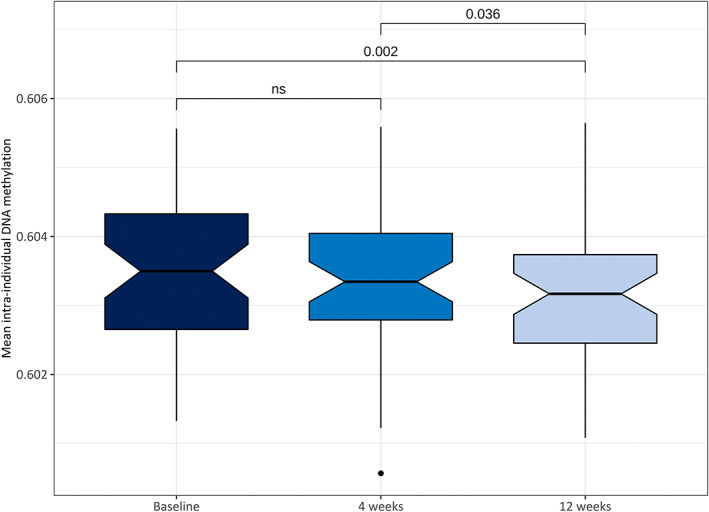
Intraindividual differences in mean global DNA methylation level across all time points (n = 47). Global DNA methylation levels were reduced after 4 and 12 weeks of dietary treatment; this was statistically significant between baseline and 12 weeks (p = .002), and between 4 and 12 weeks of dietary treatment (p = .036). Values are shown as boxplots (center lines, medians; notches, 95% confidence interval of medians; box limits, upper and lower quartiles; whiskers, 1.5 × interquartile range; points, outliers). ns, not significant.
3.7. Treatment with the modified ketogenic diet is associated with DNA methylation changes in genes related to epilepsy, metabolism, transcription, and various basic cell functions
Next, we performed analyses at a single nucleotide resolution to investigate whether the modified ketogenic diet was associated with changes in DNA methylation at specific CpGs. First, we performed an analysis of variance (ANOVA) in DNA methylation associated with the modified ketogenic diet across all time points. We identified 100 differentially methylated CpGs annotated to 75 genes (Table S1). Consistent with the observed reduction in global DNA methylation following dietary treatment, all CpGs displayed a decrease in DNA methylation compared to baseline. The genomic distribution of the differentially methylated CpGs in relation to genes shows that a large proportion are located in gene bodies (n = 54 sites, 54%) and gene promoters (n = 27 sites, 27%). With respect to CpG island context, the majority of differentially methylated CpGs were located outside CpG islands (i.e., open sea regions, n = 76 sites, 76%), whereas a smaller proportion were annotated to CpG islands (n = 22, 22%, in shores and shelfs, and n = 2, 2%, in the core islands).
To examine whether the changes in DNA methylation occur at specific times during the diet intervention, we performed pairwise comparisons between the different time points. These analyses identified 33 CpGs annotated to 21 genes from baseline to 4 weeks of dietary treatment (Figure 3A, Table S2), and 31 CpGs annotated to 24 genes from baseline to 12 weeks of dietary treatment (Figure 3B, Table S3). All differentially methylated CpGs showed a decrease in DNA methylation compared to baseline. There were no significant changes in DNA methylation between 4 and 12 weeks on diet (data not shown). The distribution of genomic locations of the differentially methylated CpGs at 4 and 12 weeks of dietary treatment was similar to the CpGs identified with the ANOVA analysis; the majority of the CpGs were located within or in close proximity to genes (52% in gene bodies, 21% in the promoters, and 2% in 3′ untranslated region). In the relation to CpG islands, most CpGs were located outside CpG islands (80% in open sea regions) and a smaller proportion within CpG islands (18% in shores and shelfs and 2% in core islands).
FIGURE 3.
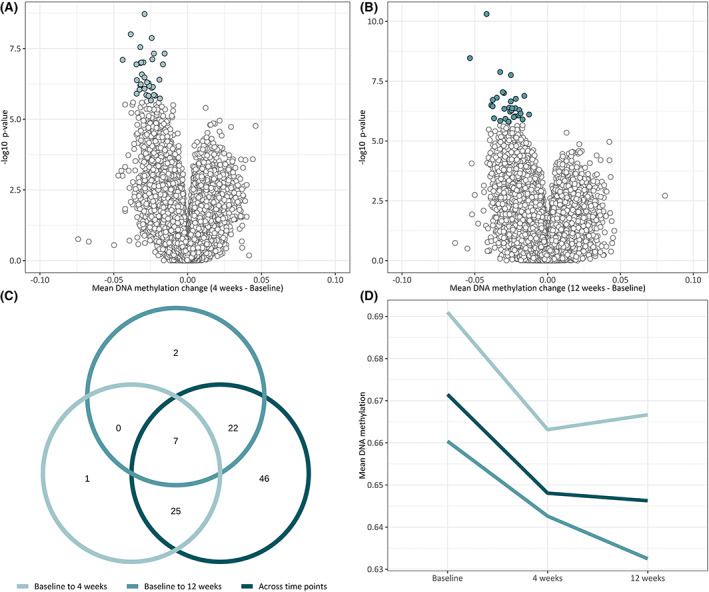
Differential DNA methylation from before to after treatment with a modified ketogenic diet. (A, B) volcano plot of log10 (p‐value) against mean delta–beta change, representing difference in DNA methylation from before to after treatment with the modified ketogenic diet. White or green circles indicate CpGs with significant differential DNA methylation. Thirty‐three CpGs were differentially methylated after 4 weeks of dietary treatment, and 31 CpGs were differentially methylated after 12 weeks of dietary treatment. (C) Venn diagram showing the overlap of CpGs differentially methylated in the three statistical analyses: analysis of variance, paired t‐test between baseline and after 4 weeks of dietary treatment, and paired t‐test between baseline and after 12 weeks of dietary treatment. (D) Mean linear trend of DNA methylation at differentially methylated CpGs during the intervention period. The decrease in DNA methylation at 4 weeks was slightly reversed after 12 weeks, whereas there was a linear decrease in DNA methylation of CpGs identified at 12 weeks of dietary treatment and across all time points.
An overview of all genes to which the differentially methylated CpGs are annotated is given in Table 4. Overall, the identified genes encode proteins involved in a broad range of biological functions, including epilepsy, lipid metabolism, transcriptional regulation, inositol phosphate metabolism, and regulation of cell growth and apoptosis.
TABLE 4.
Overview of the genes (sorted alphabetically by gene name) to which the differentially methylated CpGs are annotated
| Gene | Full gene name | Freq | Gene | Full gene name | Freq |
|---|---|---|---|---|---|
| ANXA11 | Annexin A11 | LEPREL1 | Prolyl 3‐hydroxylase 2 | ||
| APOB48R | Apolipoprotein B48 receptor | LTBP1 | Latent transforming growth factor beta binding protein 1 | ||
| ARHGEF28 | Rho guanine nucleotide exchange factor 28 | LY86 | Lymphocyte antigen 86 | ||
| B4GALT5 | Beta‐1.4‐galactosyltransferase 5 | MED13L | Mediator complex subunit 13 like | ||
| BCKDHB | Branched chain keto acid dehydrogenase E1 subunit beta | NAT8 | N‐acetyltransferase 8 | ||
| BLNK | B cell linker | NET1 | Neuroepithelial cell transforming 1 | ||
| CD93 | CD93 molecule | NMUR1 | Neuromedin U receptor 1 | ||
| CERS6 | Ceramide synthase 6 | NUCKS1 | Nuclear casein kinase and cyclin dependent kinase substrate 1 | ||
| CES1 | Carboxylesterase 1 | PDE4D | Phosphodiesterase 4D | 2 | |
| CIITA | Class II major histocompatibility complex transactivator | PDK4 | Pyruvate dehydrogenase kinase 4 | ||
| CLASP1 | Cytoplasmic linker associated protein 1 | PDZD8 | PDZ domain containing 8 | ||
| CPSF4L | Cleavage and polyadenylation specific factor 4 like | 2 | PLAGL1 | PLAG1 like zinc finger 1 | |
| CPT1A | Carnitine palmitoyltransferase 1A | PLCXD2 | Phosphatidylinositol specific phospholipase C X domain containing 2 | ||
| CSGALNACT1 | Chondroitin sulfate N‐acetylgalactosaminyltransferase 1 | PPAP2B | Phospholipid phosphatase 3 | ||
| DLGAP1 | DLG associated protein 1 | PRKCA | Protein kinase C alpha | ||
| DTD1 | D‐tyrosyl‐tRNA deacylase 1 | PSTPIP2 | Proline‐serine‐threonine phosphatase interacting protein 2 | ||
| DZIP1L | DAZ interacting zinc finger protein 1 like | PTH2R | Parathyroid hormone 2 receptor | ||
| EFNA5 | Ephrin A5 | RERE | Arginine‐glutamic acid dipeptide repeats | ||
| EHD1 | EH domain containing 1 | RNF166 | Ring finger protein 166 | ||
| EIF4E3 | Eukaryotic translation initiation factor 4E family member 3 | RNF19A | Ring finger protein 19A | ||
| ELMO1 | Engulfment and cell motility 1 | SLC22A23 | Solute carrier family 22 member 23 | ||
| FAM198B | Family with sequence similarity 198 member B | SNTB1 | Syntrophin beta 1 | ||
| FOXN3 | Forkhead box N3 | STARD9 | StAR related lipid transfer domain containing 9 | ||
| FTO | Fat mass and obesity‐associated protein | SUSD1 | Sushi domain containing 1 | ||
| GALNT2 | Polypeptide N‐acetylgalactosaminyltransferase 2 | 2 | SWT1 | SWT1, RNA endoribonuclease homolog | |
| GNAO1 | G protein subunit alpha o1 | TCF25 | Transcription factor 25 | ||
| HAL | Histidine ammonia‐lyase | TCFL5 | Transcription factor like 5 | ||
| HEPN1 | Hepatocellular carcinoma down‐regulated 1 | TEC | Tec protein tyrosine kinase | ||
| IMPA2 | Inositol monophosphatase 2 | TM4SF20 | Transmembrane 4 L six family member 20 | ||
| INPP1 | Inositol polyphosphate‐1‐phosphatase | TMEM45A | Transmembrane protein 45A | ||
| INPP4A | Inositol polyphosphate‐4‐phosphatase type I A | 2 | TPD52L1 | Tumor protein D52 like 1 | |
| INPP5A | Inositol polyphosphate‐5‐phosphatase A | 2 | TSPAN2 | Tetraspanin 2 | |
| KCNQ1 | Potassium voltage‐gated channel subfamily Q member 1 | 2 | TSSC1 | EARP complex and GARP complex interacting protein 1 | |
| KIAA1267 | KAT8 regulatory NSL complex subunit 1 | TULP4 | Tubby like protein 4 | ||
| LDB2 | LIM domain binding 2 | ZEB2 | Zinc finger E‐box binding homeobox 2 |
Note: Genes of uncertain function are not listed; these can be found in Tables S2–S4.
Abbreviations: Freq, frequency (the number of unique differentially methylated CpGs annotated to the gene concerned).
The majority of the differentially methylated CpGs identified between time points did not overlap (79% and 77% from baseline to 4 weeks and 12 weeks, respectively) and showed a time‐dependent change in DNA methylation during the diet intervention (Figure 3C). Only a small number of CpGs (n = 7), annotated to five genes (C5orf27, CD93, HEPN1, KCNQ1, and NAT8, two CpGs without gene annotation), were significant at both 4 and 12 weeks. Interestingly, there was a distinct difference in the mean linear trend of DNA methylation changes along the time course of the diet (Figure 3D). Whereas the decrease in DNA methylation identified at 4 weeks was slightly reversed after 12 weeks (light green line), the linear trend in decreased DNA methylation was consistent across the whole intervention period for the CpGs identified between baseline and 12 weeks or across time in general (medium green and dark green lines, respectively). These results may reflect rapid short‐term metabolic adaptations that are partly reversed after 4 weeks, whereas other metabolic adaptations occur more gradually and take more time.
3.8. No differences in DNA methylation between responders and nonresponders
As we suggest that DNA methylation may play a key role in exerting the seizure‐reducing effect in patients with epilepsy, we wanted to examine possible differences in DNA methylation between responders and nonresponders. However, intraindividual comparison of DNA methylation changes from baseline to 12 weeks of dietary treatment between responders (n = 20) and nonresponders (n = 21) did not reveal any significant differentially methylated CpGs between the two groups (data not shown).
4. DISCUSSION
This is the first report of the impact of the modified ketogenic diet on DNA methylation in humans with epilepsy. Adult epilepsy patients treated with the modified ketogenic diet had a decrease in DNA methylation, both globally and at specific genes associated with epilepsy, metabolism, transcriptional regulation, and various basic cell functions.
Despite the use of the ketogenic diet in epilepsy treatment for approximately 100 years, the mechanisms behind the diet's seizure‐reducing effect remain elusive. Evidence from preclinical studies suggests that epigenetic mechanisms, including DNA methylation, play a central role in disease development, as well as in successful dietary treatment of epilepsy. 8 , 9 , 28 , 29 Interestingly, a global increase in DNA methylation has been demonstrated in both animal models and in humans with epilepsy. 8 , 9 , 28 , 30 Moreover, studies in animal models of epilepsy have shown that treatment with the ketogenic diet counteracts the increased DNA methylation and attenuates seizures. 8 , 9 However, it is still unknown whether the observed reduction in DNA methylation after dietary treatment occurs at the same positions as those found to have increased DNA methylation, and whether these DNA methylation alterations are linked to the seizure‐reducing effect of the dietary treatment. Also, DNA methylation has been shown in experimental studies to be etiology‐dependent 31 ; thus, the baseline DNA methylation pattern of the participants may be influenced by etiology. However, even if baseline patterns differ between the participants, the diet‐induced DNA methylation alterations may be independent of epilepsy etiology, and the within‐subject design in our study may limit this potential bias.
Interestingly, we found that about 10% (n = 7) of the genes containing differentially methylated CpGs were associated or potentially associated with epilepsy (ELMO1, FTO, GNAO1, INPP4A, KCNQ1, MED13L, and ZEB2). 32 The identified genes are highly expressed in the central nervous system (CNS) and have essential roles in normal brain development and function. Of note, three of these genes play key roles in transcriptional regulation (MED13L, ZEB2) or posttranscriptional modifications (FTO). MED13L encodes a subunit of the Mediator complex, which is involved in transcriptional regulation of almost all genes transcribed by RNA polymerase II 33 ; the protein zinc finger E‐box homeobox, encoded by ZEB2, is an essential transcriptional repressor 34 ; and FTO was the first mRNA demethylase identified. 35 A large share of the identified genes encode proteins with transcriptional regulation as their main biological function.
Ten (12%) of the identified genes encode proteins involved in transcriptional regulation (CIITA, FOXN3, KIAA1267, LDB2, MED13L, PLAGL1, RERE, TCF25, TCFL5, and ZEB2). FOXN3, RERE, TCF25, and ZEB2 encode proteins that act as transcriptional repressors or corepressors, 34 , 36 , 37 , 38 whereas PLAGL1 and CIITA encode a transcriptional activator and coactivator, respectively. 39 , 40 Moreover, LIM domain binding 2, encoded by LDB2, is an adapter molecule that allows assembly of transcriptional regulatory complexes. 41 In addition, NUKCS1 is involved in chromatin remodeling and thereby may influence transcription. 42 Taken together, alterations in DNA methylation by these genes are likely to have a far‐reaching downstream effect on gene expression of a wide range of other genes.
Another group of genes with potential important effects in the CNS consists of five genes encoding for enzymes involved in inositol phosphate metabolism (IMPA2, INPP1, INPP4A, INPP5A, and PLCXD2). Inositol phosphate has important roles in signal transduction and Ca2+ homeostasis in the CNS, and imbalances in the inositol phosphate metabolism have been suggested to have a role in several neurological disorders, including epilepsy. 43 IMPA2 encodes inositol monophosphatase 2, an enzyme that catalyzes the conversion of myo‐inositol monophosphate to myo‐inositol. 44 Interestingly, Nakayama et al. reported IMPA2 to be a putative susceptibility gene for febrile seizures. 45 Furthermore, carbamazepine, a common ASM, has previous been shown to stimulate IMPA2 enzyme activity. 46 On the other hand, lithium, which is used in the treatment of bipolar disorders, inhibits IMPA2 46 and has a proconvulsive effect in rat lithium–pilocarpine‐induced seizures. 47 Interestingly, this effect can be reversed by administration of myo‐inositol. 47 Anticonvulsant effects of myo‐inositol have also been demonstrated in rats with pentylenetetrazol‐ or kainic acid‐induced seizures. 48 , 49 To our knowledge, an impact of the ketogenic diet on the inositol phosphate metabolism has not been described before, and may represent a plausible mechanism by which the ketogenic diet attenuates seizures.
As expected from the major shift in the whole‐body metabolism induced by the dietary treatment, a large proportion of the genes identified in our study are involved in lipid metabolism (APOB48R, B4GALT5, CERS6, CES1, CPT1A, GALNT2, PLCXD2, and PPAP2B) and regulation of carbohydrate metabolism (PDK4). Particularly interesting are CPT1A and PDK4, which play key roles in the regulation of fatty acid beta oxidation and glycolysis, respectively, as well as APOB48R, encoding the macrophage receptor apolipoprotein B48, which is decisive in postprandial uptake of lipids in macrophages. 50 Collectively, these findings demonstrate that our method captures important biological adaptations, thus underlining the validity of our results.
This study has some limitations. The dietary treatment was given as an adjunctive treatment, and all participants, except one, used ASMs. Although none of the participants changed type or dose of the ASMs during the 24‐week study period, the serum concentration of several ASMs was reduced during the diet intervention. 20 ASMs could potentially influence the DNA methylation profile either directly or indirectly through their influence on one‐carbon metabolism nutrients. 51 For instance, valproic acid, one of the most commonly used ASMs, 52 has been shown to inhibit DNMTs and induce a decrease in global DNA methylation. Thus, we cannot exclude that the unintentional drop in serum concentration could have influenced the DNA methylation.
Another limitation of the study is the lack of a control group, which means that we do not know whether continued epileptogenesis might have affected our results. However, our study population consists of patients with a very long history of epilepsy (on average 25 years). From this perspective, 12 weeks of continued epileptogenesis is a very short period of time, which we do not expect to constitute a relevant difference with regard to DNA methylation changes. In addition, we argue that the longitudinal study design with intraindividual comparisons of DNA methylation before and after the diet intervention also has the advantage of reducing the likelihood of potential confounding effects of genetic variations and interindividual differences in lifestyle and environmental exposures. Furthermore, the large proportion of differentially methylated sites annotated to genes associated with lipid metabolism supports that our study identifies meaningful biological changes that are genuine effects of the diet intervention.
We applied a pragmatic study design to investigate the impact of the dietary treatment on DNA methylation in a real‐life setting. Although the amount of carbohydrate was restricted to a maximum of 16 g per day, the ratio of fat to protein and the calorie intake were not specified. Hence, variations in the macronutrient compositions or the weight reduction 16 , 17 experienced by several of the participants may have influenced the results.
Our study used whole blood as a surrogate tissue for a disease that manifests in the brain. Currently, it is still unknown whether seizure‐associated DNA methylation changes occur in blood and how well diet‐induced DNA methylation alterations in blood correspond to DNA methylation changes in the brain. However, the ketogenic diet's antiseizure effect may be ascribed to a combination of mechanisms, involving both alterations at a systemic level and changes directly in the brain. 53 Finally, we were not able to detect any differences in DNA methylation between responders and nonresponders. However, our sample size was small, and the antiseizure effect of the dietary treatment in our study population was modest. 16
Importantly, we also find that our study has significant strengths. Compliance is a well‐known challenge in nutrition research, and in this study the intervention represents a significant change in the patients' diet and everyday life. However, we have robust objective measures of ketosis, regular follow‐up, and dietary assessments based on 3‐day weighed food records documenting compliance in our study.
In conclusion, we have identified a genome‐wide decrease in DNA methylation both globally and at specific loci in adult epilepsy patients treated with the modified ketogenic diet. Interestingly, a substantial share of the identified genes were associated with epilepsy and inositol phosphate metabolism. However, we were not able to identify any differences between responders and nonresponders; thus, the clinical implications of these findings remain to be elucidated. We believe that understanding the ketogenic diet's influence at the molecular level may be the key to unraveling the mechanisms by which the diet can ameliorate seizures and possibly to identifying novel therapeutic targets for epilepsy. Further studies, with larger sample size and with a control group of epilepsy patients eating their habitual diet while all epilepsy treatments are kept unchanged, are needed to elucidate the role of DNA methylation in successful dietary treatment of epilepsy.
AUTHOR CONTRIBUTIONS
All authors contributed to the study conception and design. Data collection was performed by Magnhild Kverneland and Karl Otto Nakken. Data analysis of DNA methylation was performed by Kristina Gervin; all other data analyses were performed by Sigrid Pedersen. The first draft of the manuscript was written by Sigrid Pedersen, and all authors commented on the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
M.K. has received two honoraria from Nutricia, which had no influence on data collection, analysis, or writing of the manuscript. The remaining authors have nothing to disclose.
ETHICAL APPROVAL
The study was approved by the Regional Committee for Medical and Health Research (2010/2326). All participants or parents/caregivers provided written informed consent before enrollment. All procedures in this study were in accordance with the Helsinki Declaration. The randomized clinical trial was registered at ClinicalTrials.gov (ID: NCT01311440). We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
This work was supported by the Dam Foundation, the Norwegian Epilepsy Association's Research Fund, the Novo Nordisk Foundation, and the National Advisory Unit on Rare Disorders, Norway. The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. All authors had full access to all the data in the study and had final responsibility for the decision to submit the manuscript for publication. We kindly thank the patients who, despite their load of disease, took part in the study. We thank Erik Taubøll for his continued support in planning and conducting the clinical trial. We would also like to thank the leadership and staff at the National Center for Epilepsy for all contributions.
Pedersen S, Kverneland M, Nakken KO, Rudi K, Iversen PO, Gervin K, Genome‐wide decrease in DNA methylation in adults with epilepsy treated with modified ketogenic diet: A prospective study. Epilepsia. 2022;63:2413–2426. 10.1111/epi.17351
DATA AVAILABILITY STATEMENT
Raw data from this project are not available due to privacy and ethical restrictions of the project approval and consent forms. Metadata generated in the study and code used in the analysis are available from the corresponding author upon reasonable request in accordance with the privacy policy of the informed consent by the participants.
REFERENCES
- 1. CJL M, Lopez AD. Global comparative assessments in the health sector: disease burden, expenditures and intervention packages. Geneva, Switzerland: World Health Organization; 1994. [Google Scholar]
- 2. Ngugi AK, Bottomley C, Kleinschmidt I, Sander JW, Newton CR. Estimation of the burden of active and life‐time epilepsy: a meta‐analytic approach. Epilepsia. 2010;51(5):883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51(6):1069–77. [DOI] [PubMed] [Google Scholar]
- 4. Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342(5):314–9. [DOI] [PubMed] [Google Scholar]
- 5. Löscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia. 2011;52(4):657–78. [DOI] [PubMed] [Google Scholar]
- 6. Martin K, Jackson CF, Levy RG, Cooper PN. Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst Rev. 2016;2:CD001903. [DOI] [PubMed] [Google Scholar]
- 7. Husari KS, Cervenka MC. The ketogenic diet all grown up‐ketogenic diet therapies for adults. Epilepsy Res. 2020;162:106319. [DOI] [PubMed] [Google Scholar]
- 8. Kobow K, Kaspi A, Harikrishnan KN, Kiese K, Ziemann M, Khurana I, et al. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol. 2013;126(5):741–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lusardi TA, Akula KK, Coffman SQ, Ruskin DN, Masino SA, Boison D. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology. 2015;99:500–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kobow K, Blumcke I. The methylation hypothesis: do epigenetic chromatin modifications play a role in epileptogenesis? Epilepsia. 2011;52(Suppl 4):15–9. [DOI] [PubMed] [Google Scholar]
- 11. Long HY, Feng L, Kang J, Luo ZH, Xiao WB, Long LL, et al. Blood DNA methylation pattern is altered in mesial temporal lobe epilepsy. Sci Rep. 2017;7:43810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Masliah E, Dumaop W, Galasko D, Desplats P. Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics. 2013;8(10):1030–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Semick SA, Bharadwaj RA, Collado‐Torres L, Tao R, Shin JH, Deep‐Soboslay A, et al. Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer's disease. Acta Neuropathol. 2019;137(4):557–69. [DOI] [PubMed] [Google Scholar]
- 14. Miller‐Delaney SF, Bryan K, Das S, McKiernan RC, Bray IM, Reynolds JP, et al. Differential DNA methylation profiles of coding and non‐coding genes define hippocampal sclerosis in human temporal lobe epilepsy. Brain. 2015;138(Pt 3):616–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tiffon C. The impact of nutrition and environmental epigenetics on human health and disease. Int J Mol Sci. 2018;19(11):3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kverneland M, Molteberg E, Iversen PO, Veierod MB, Tauboll E, Selmer KK, et al. Effect of modified Atkins diet in adults with drug‐resistant focal epilepsy: a randomized clinical trial. Epilepsia. 2018;59:1567–76. [DOI] [PubMed] [Google Scholar]
- 17. Kverneland M, Selmer KK, Nakken KO, Iversen PO, Tauboll E. A prospective study of the modified Atkins diet for adults with idiopathic generalized epilepsy. Epilepsy Behav. 2015;53:197–201. [DOI] [PubMed] [Google Scholar]
- 18. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Norwegian Food Safety Authority . Norwegian food composition database. Oslo, Norway: University of Oslo; 2018. [Google Scholar]
- 20. Kverneland M, Tauboll E, Molteberg E, Veierod MB, Selmer KK, Nakken KO, et al. Pharmacokinetic interaction between modified Atkins diet and antiepileptic drugs in adults with drug‐resistant epilepsy. Epilepsia. 2019;60:2235–44. [DOI] [PubMed] [Google Scholar]
- 21. Aryee MJ, Jaffe AE, Corrada‐Bravo H, Ladd‐Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole‐genome DNA methylation profiling. Genome Biol. 2016;17(1):208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCartney DL, Walker RM, Morris SW, McIntosh AM, Porteous DJ, Evans KL. Identification of polymorphic and off‐target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genom Data. 2016;9:22–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salas LA, Koestler DC, Butler RA, Hansen HM, Wiencke JK, Kelsey KT, et al. An optimized library for reference‐based deconvolution of whole‐blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol. 2018;19(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Ser B Stat Methodol. 1995;57(1):289–300. [Google Scholar]
- 27. Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 2008;7(6):500–6. [DOI] [PubMed] [Google Scholar]
- 28. Williams‐Karnesky RL, Sandau US, Lusardi TA, Lytle NK, Farrell JM, Pritchard EM, et al. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Invest. 2013;123(8):3552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Machnes ZM, Huang TC, Chang PK, Gill R, Reist N, Dezsi G, et al. DNA methylation mediates persistent epileptiform activity in vitro and in vivo. PLoS One. 2013;8(10):e76299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu Q, Wang L, Zhang Y, Zhao FH, Luo J, Xiao Z, et al. Increased expression of DNA methyltransferase 1 and 3a in human temporal lobe epilepsy. J Mol Neurosci. 2012;46(2):420–6. [DOI] [PubMed] [Google Scholar]
- 31. Debski KJ, Pitkanen A, Puhakka N, Bot AM, Khurana I, Harikrishnan KN, et al. Etiology matters—genomic DNA methylation patterns in three rat models of acquired epilepsy. Sci Rep. 2016;6:25668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Lin ZJ, Liu L, Xu HQ, Shi YW, Yi YH, et al. Epilepsy‐associated genes. Seizure. 2017;44:11–20. [DOI] [PubMed] [Google Scholar]
- 33. Allen BL, Taatjes DJ. The mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16(3):155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hegarty SV, Sullivan AM, O'Keeffe GW. Zeb2: a multifunctional regulator of nervous system development. Prog Neurobiol. 2015;132:81–95. [DOI] [PubMed] [Google Scholar]
- 35. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6‐methyladenosine in nuclear RNA is a major substrate of the obesity‐associated FTO. Nat Chem Biol. 2011;7(12):885–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kong X, Zhai J, Yan C, Song Y, Wang J, Bai X, et al. Recent advances in understanding FOXN3 in breast cancer, and other malignancies. Front Oncol. 2019;9:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang L, Tsai CC. Atrophin proteins: an overview of a new class of nuclear receptor corepressors. Nucl Recept Signal. 2008;6:e009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cai Z, Wang Y, Yu W, Xiao J, Li Y, Liu L, et al. hnulp1, a basic helix‐loop‐helix protein with a novel transcriptional repressive domain, inhibits transcriptional activity of serum response factor. Biochem Biophys Res Commun. 2006;343(3):973–81. [DOI] [PubMed] [Google Scholar]
- 39. Kas K, Voz ML, Hensen K, Meyen E, Van de Ven WJ. Transcriptional activation capacity of the novel PLAG family of zinc finger proteins. J Biol Chem. 1998;273(36):23026–32. [DOI] [PubMed] [Google Scholar]
- 40. Masternak K, Muhlethaler‐Mottet A, Villard J, Zufferey M, Steimle V, Reith W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000;14(9):1156–66. [PMC free article] [PubMed] [Google Scholar]
- 41. Bach I, Carrière C, Ostendorff HP, Andersen B, Rosenfeld MG. A family of LIM domain‐associated cofactors confer transcriptional synergism between LIM and Otx homeodomain proteins. Genes Dev. 1997;11(11):1370–80. [DOI] [PubMed] [Google Scholar]
- 42. Qiu B, Shi X, Wong ET, Lim J, Bezzi M, Low D, et al. NUCKS is a positive transcriptional regulator of insulin signaling. Cell Rep. 2014;7(6):1876–86. [DOI] [PubMed] [Google Scholar]
- 43. Frej AD, Otto GP, Williams RS. Tipping the scales: lessons from simple model systems on inositol imbalance in neurological disorders. Eur J Cell Biol. 2017;96(2):154–63. [DOI] [PubMed] [Google Scholar]
- 44. Ohnishi T, Ohba H, Seo KC, Im J, Sato Y, Iwayama Y, et al. Spatial expression patterns and biochemical properties distinguish a second myo‐inositol monophosphatase IMPA2 from IMPA1. J Biol Chem. 2007;282(1):637–46. [DOI] [PubMed] [Google Scholar]
- 45. Nakayama J, Yamamoto N, Hamano K, Iwasaki N, Ohta M, Nakahara S, et al. Linkage and association of febrile seizures to the IMPA2 gene on human chromosome 18. Neurology. 2004;63(10):1803–7. [DOI] [PubMed] [Google Scholar]
- 46. Vadnal R, Parthasarathy R. Myo‐inositol monophosphatase: diverse effects of lithium, carbamazepine, and valproate. Neuropsychopharmacology. 1995;12(4):277–85. [DOI] [PubMed] [Google Scholar]
- 47. Patishi Y, Belmaker RH, Bersudsky Y, Kofman O. A comparison of the ability of myo‐inositol and epi‐inositol to attenuate lithium‐pilocarpine seizures in rats. Biol Psychiatry. 1996;39(9):829–32. [DOI] [PubMed] [Google Scholar]
- 48. Nozadze M, Mikautadze E, Lepsveridze E, Mikeladze E, Kuchiashvili N, Kiguradze T, et al. Anticonvulsant activities of myo‐inositol and scyllo‐inositol on pentylenetetrazol induced seizures. Seizure. 2011;20(2):173–6. [DOI] [PubMed] [Google Scholar]
- 49. Tsverava L, Kandashvili M, Margvelani G, Lortkipanidze T, Gamkrelidze G, Lepsveridze E, et al. Long‐term effects of myoinositol on behavioural seizures and biochemical changes evoked by kainic acid induced epileptogenesis. Biomed Res Int. 2019;2019:4518160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brown ML, Ramprasad MP, Umeda PK, Tanaka A, Kobayashi Y, Watanabe T, et al. A macrophage receptor for apolipoprotein B48: cloning, expression, and atherosclerosis. Proc Natl Acad Sci U S A. 2000;97(13):7488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ni G, Qin J, Li H, Chen Z, Zhou Y, Fang Z, et al. Effects of antiepileptic drug monotherapy on one‐carbon metabolism and DNA methylation in patients with epilepsy. PLoS One. 2015;10(4):e0125656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tremolizzo L, Difrancesco JC, Rodriguez‐Menendez V, Riva C, Conti E, Galimberti G, et al. Valproate induces epigenetic modifications in lymphomonocytes from epileptic patients. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(1):47–51. [DOI] [PubMed] [Google Scholar]
- 53. Boison D. New insights into the mechanisms of the ketogenic diet. Curr Opin Neurol. 2017;30(2):187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
Raw data from this project are not available due to privacy and ethical restrictions of the project approval and consent forms. Metadata generated in the study and code used in the analysis are available from the corresponding author upon reasonable request in accordance with the privacy policy of the informed consent by the participants.