

Abstract
Aim
Using Niemann–Pick type C disease (NPC) as a paradigm, we aimed to improve biomarker discovery in patients with neurometabolic disorders.
Method
Using a multiplexed liquid chromatography tandem mass spectrometry dried bloodspot assay, we developed a selective intelligent biomarker panel to monitor known biomarkers N‐palmitoyl‐O‐phosphocholineserine and 3β,5α,6β‐trihydroxy‐cholanoyl‐glycine as well as compounds predicted to be affected in NPC pathology. We applied this panel to a clinically relevant paediatric patient cohort (n = 75; 35 males, 40 females; mean age 7 years 6 months, range 4 days–19 years 8 months) presenting with neurodevelopmental and/or neurodegenerative pathology, similar to that observed in NPC.
Results
The panel had a far superior performance compared with individual biomarkers. Namely, NPC‐related established biomarkers used individually had 91% to 97% specificity but the combined panel had 100% specificity. Moreover, multivariate analysis revealed long‐chain isoforms of glucosylceramide were elevated and very specific for patients with NPC.
Interpretation
Despite advancements in next‐generation sequencing and precision medicine, neurological non‐enzymatic disorders remain difficult to diagnose and lack robust biomarkers or routine functional testing for genetic variants of unknown significance. Biomarker panels may have better diagnostic accuracy than individual biomarkers in neurometabolic disorders, hence they can facilitate more prompt disease identification and implementation of emerging targeted, disease‐specific therapies.
What this paper adds
Intelligent biomarker panel design can help expedite diagnosis in neurometabolic disorders.
In Niemann–Pick type C disease, such a panel performed better than individual biomarkers.
Biomarker panels are easy to implement and widely applicable to neurometabolic conditions.
What this paper adds
Intelligent biomarker panel design can help expedite diagnosis in neurometabolic disorders.
In Niemann–Pick type C disease, such a panel performed better than individual biomarkers.
Biomarker panels are easy to implement and widely applicable to neurometabolic conditions.

Improved time to diagnosis for neurometabolic disorders, with Niemann Pick C (NPC) as an example, using a multiplex assay panel. Top panel displays the current scenario with diagnostic confirmation taking months and often requiring lengthy biochemical testing, especially in cases of genetic variants of unknown significance. Improved diagnosis using a bloodspot assay can achieve biochemical confirmation in 2 to 6 weeks and reduce need for subsequent biochemical confirmation after genetic diagnosis. Abbreviations: CE, clinical exome; CDH, ceramide dihexoside; DBS, dried blood spot; GlcCer, glucosylceramide; NPC, Niemann‐Pick type C; PPCS, N‐palmitoyl‐O‐phosphocholineserine; SM, sphingomyelin; WES, whole exome sequencing; WGS, whole genome sequencing.
This original article is commented on by Pearl on pages 1441–1442 of this issue.
Abbreviations
- CDH
ceramide dihexoside
- DBS
dried blood spot
- GBA
glucocerebrosidase
- GlcCer
glucosylceramide
- NPC
Niemann–Pick type C disease
- PPCS
N‐palmitoyl‐O‐phosphocholineserine
Despite advances made in next‐generation sequencing, 1 , 2 timely diagnosis and subsequent initiation of appropriate treatments remain problematic in many neurological conditions. This is particularly true in the field of rare disorders, where clinical presentations are often insidious, and expertise is only available in a few specialist centres. Genomic DNA sequencing can detect most disease‐causing variants, but functional assays and relevant biomarkers are often essential to confirm pathogenicity of genetic changes. Additionally, early detection is paramount for patients with disorders affecting the developing brain or resulting in neurodegeneration in order to achieve the best clinical outcomes. 3 This is more pertinent in the current era of personalized medicine, where an ever‐increasing number of novel, targeted, and disease‐specific therapeutic approaches are becoming available for neurological diseases that in the past were considered untreatable. 4 , 5 , 6
Disease‐relevant biomarkers can greatly facilitate timely diagnosis and disease monitoring. However, in cases lacking a single ultra‐sensitive and ultra‐specific marker (which is the norm for most disorders), intelligent design of a biochemical marker panel could be considered as a way to increase diagnostic specificity and accuracy. In this project we used Niemann–Pick type C disease (NPC) as such an example of a clinically heterogeneous neurodegenerative disorder.
NPC is caused by biallelic mutations in the NPC1 gene (in most cases) or, rarely, NPC2. 7 , 8 , 9 Its prevalence is reportedly 0.35 to 2.2 per 100 000 live births, depending on the country, but this might be an underestimate in view of failure to reach diagnosis in many cases. 9 , 10 At a cellular level, NPC results in accumulation of various lipids including cholesterol and sphingolipids in the endo‐lysosomal system. 11 , 12 , 13 Biomarkers recently established to improve NPC detection include cholesterol oxidation products (oxysterols), 14 , 15 the 3β,5α,6β‐trihydroxy‐cholanoyl‐glycine bile acid, 16 , 17 lyso‐sphingomyelin 509 (now identified as N‐palmitoyl‐O‐phosphocholineserine [PPCS] 18 ), and lyso‐sphingomyelin. 19 , 20 , 21 So far, the sensitivity and specificity for most of these biomarkers have been assessed in patients with NPC against typically developing comparison individuals, rather than against a relevant age‐matched symptomatic patient cohort with similar manifestations. Hence, for our study we recruited a cohort of paediatric patients with neurodevelopmental and neurodegenerative symptoms and signs, in which NPC was part of the differential diagnosis.
To test the hypothesis that a panel of intelligently selected biomarkers could improve detection of NPC, we combined currently available biomarkers and additional glycosphingolipids, sphingomyelins, and bile acids (predicted to be abnormal on the basis of disease pathology) 22 , 23 into one multiplexed dried blood spot (DBS) assay.
METHOD
Ethics
This study was approved by the National Research Ethics Service in the UK (National Research Ethics Service Committee: London – Bloomsbury, REC reference 13/LO/0168, IRAS project 95005) and Great Ormond Street Hospital Research and Development Audit Department (reference 12CM29). Written informed consent from patients or guardians and family members was obtained in all cases.
Patient ascertainment
Undiagnosed paediatric patients from a single UK paediatric hospital's neurology and metabolic disease clinics, presenting between March 2015 and September 2016, with a possible neurometabolic disorder, were prospectively recruited. Inclusion criteria firstly included disease of childhood onset (0–18 years). Moreover, patients with the presence of one or more neurological feature (such as developmental delay, neurological regression, movement disorder, gaze palsy, epilepsy, autistic features), with or without other pointers towards an underlying inborn error of metabolism (such as suggestive biochemical markers, neuroimaging abnormalities, or organomegaly), as identified through routine clinical care, were eligible to participate. Characterization of the clinical phenotype was undertaken by direct clinical examination and case note review. For comparison, a cohort of population norm adults was also prospectively established.
Sample collection
All patients underwent blood sampling for research purposes after informed consent. Blood samples in ethylenediaminetetraacetic acid‐containing vials were collected for genomic DNA analysis and sequencing. DBS samples from patients and comparison individuals were also collected for biomarker analysis and stored at −80°C.
Genetic sample analysis
In cases of very suggestive clinical, biochemical, and/or radiological presentations, targeted gene testing was performed in the first instance. If this was negative, or in the absence of features pointing towards a specific underlying genetic diagnosis, gene panel testing, whole‐exome, clinical exome, and/or whole‐genome sequencing analysis was done, either as part of NHS clinical care or on a research basis. Research next‐generation sequencing largely occurred through sample contribution to larger studies such as Deciphering Developmental Disorders or the Genomics England 100,000 Genomes Project. Copy number variants (deletions, duplications) were also investigated by comparative genomic hybridization microarray analysis.
Multiplex bloodspot panel
A complete method description is given in [Link], [Link], Tables [Link], [Link], and Figure S1). In summary, a 6 mm punch out of each DBS sample was added to 100 μL of extraction solution containing internal standards. Samples were briefly incubated and centrifuged at 16 000 g to fully submerge. Samples were extracted for 10 minutes in a sonication bath at room temperature and centrifuged for 5 minutes at 16 000 g to remove any particulate. Each sample was diluted in a 1:1 ratio with methanol and subjected to liquid chromatography with tandem mass spectrometry analysis. All compounds were standardized to an internal standard (Tables S1 and S5). Kuchar et al. have demonstrated the use of both PPCS and lyso‐sphingomyelin help discriminate NPC; 21 therefore, we have shown our data as a ratio of PPCS to lyso‐sphingomyelin.
Data analysis
Raw liquid chromatography with tandem mass spectrometry data were analysed using TargetLynx version 4.0 (Waters, Milford, MA, USA). Standardized data were exported. GraphPad Prism version 6 (GraphPad Software, CA, USA) was used for comparative analysis. Kruskal–Wallis non‐parametric analysis of variance was used to determine statistical significance. Multivariate analysis was done using SIMCA version 15 (Sartorius, Goettingen, Germany) (Table S6).
RESULTS
Patient cohort characteristics
Seventy‐five patients were recruited, presenting with a heterogeneous range of symptoms and signs suggestive of neurogenetic, neurometabolic, or neurodegenerative conditions. Additionally, 10 population norm adults (age range 23–48 years) were recruited as a comparison group. Mean and median ages were 7 years 6 months and 5 years 2 months respectively (age range 4 days–19 years 8 months). Both sexes were almost equally represented (35 males, 40 females, ratio 0.88:1). Clinical manifestations are summarized in Figure 1. The most common presenting features were neurodevelopmental delay (61 out of 75), non‐epileptic movement disorders such as ataxia or dystonia (38 out of 75), epilepsy (24 out of 75), and neurological regression (14 out of 75).
FIGURE 1.

Clinical characteristics of the patient cohort (n = 75). Left: developmental delay (n = 61), non‐epileptic movement disorders (n = 38), and epilepsy (n = 24) were the most encountered presenting features. Other less frequent clinical manifestations included neurological regression, muscle weakness, liver involvement (including organomegaly and/or deranged function), and gaze palsy. Right: Venn diagram depicting the co‐manifestation of the three most common presenting clinical features encountered in the cohort.
All patients underwent extensive genetic, biochemical, and/or neuroimaging studies as part of their clinical care. Genetic investigations included whole‐exome, whole‐genome, or gene panel analysis in all cases (75 out of 75). Eventually a definitive genetic diagnosis was established in 43 cases. Of those, five were diagnosed with NPC (NPC1 mutations), while NPC was sufficiently excluded in the 32 out of 75 undiagnosed patients. Other diagnoses established are detailed in Table S7.
Design and multivariate analysis of the DBS biomarker panel
Compounds suspected of being relevant to NPC disease pathology (see Appendix S2 and Figure S1) were included in the multiplex assay (Table S5). Because of known issues with artefactual conversion, 24 particularly for DBS, oxysterols were not included. Using multivariate analysis, compounds that best allowed discrimination of patients with NPC, and new biomarkers, were identified. Principal component analysis of all the samples from the patient cohort, including the five genetically confirmed patients with NPC, showed that all NPC‐positive patients clustered away from other patients, and the comparison group (Figure 2a). This indicated that NPC profiles are unique, and that there were no unidentified cases of NPC in the proportion of patients who remained undiagnosed. Moreover, to establish which compounds contribute to the NPC profile and potentially identify novel disease‐specific markers, orthogonal projections to latent structures discriminant analysis (Figure 2b, Figure S2, Table S6) was subsequently performed between the diagnosed, non‐NPC, and NPC subgroups of patients. Several compounds, which had either elevated or decreased concentrations in NPC, were identified (Figure 2c). Elevated compounds included the ratio of PPCS to lyso‐sphingomyelin, 3β,5α,6β‐trihydroxy‐cholanoyl‐glycine, and C24 isoforms of glucosylceramide (GlcCer). Decreased compounds consisted mainly of ceramide dihexoside (CDH), in particular the long‐chain C24 and C26 isoforms.
FIGURE 2.
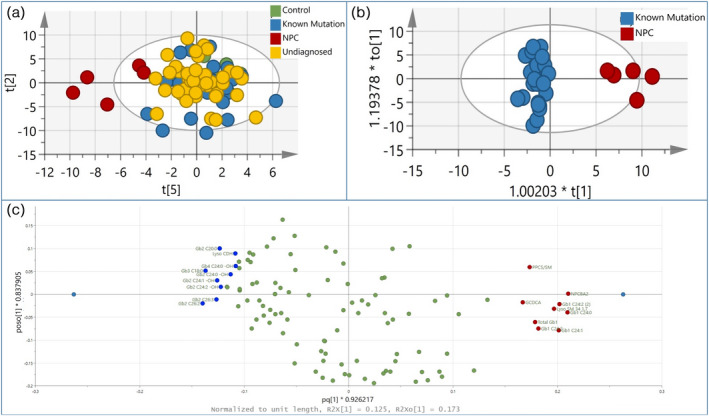
Multivariate analysis of multiplex bloodspot assay. (a) Principal component analysis of neurodevelopmental cohort and comparison group with 116 variables (compounds). Score plot for the seven‐component principal component analysis model shows t[2] and t[5] account for 13.6% and 5.9% of the variation respectively. (b) Orthogonal projections to latent structures discriminant analysis of comparison between Niemann–Pick type C disease (NPC) and known mutation samples. (c) Corresponding loading plot of (b) showing compounds elevated (right) and decreased (left) in NPC.
Univariate analysis of multiplex assay biomarkers
Compounds identified in the multivariate analysis were subsequently also subjected to univariate analysis (Figure 3). Known NPC biomarkers PPCS/lyso‐sphingomyelin and 3β,5α,6β‐trihydroxy‐cholanoyl‐glycine, when analysed individually, had a specificity of 91% and 96.7% respectively (values between the 2.5th and 97.5th centiles were used as the reference range for each group; Table S8). The PPCS:lyso‐sphingomyelin ratio, previously identified as a very specific marker, 20 showed a significant increase in the NPC group compared with population norm adults and paediatric patients, both diagnosed and undiagnosed (Figure 3a). However, three patients from the known mutation group (harbouring mutations in SCN1A, KMT2B, and ABA‐T) also demonstrated a PPCS:lyso‐sphingomyelin ratio in the NPC range. The 3β,5α,6β‐trihydroxy‐cholanoyl‐glycine bile acid was also elevated in the NPC cohort, by 30‐ to 50‐fold compared with the other groups (Figure 3a). However, one patient (patient 509, with a chromosome 7 paternal uniparental disomy, presenting with cholestatic disease in infancy and developmental delay) also had a level in the NPC range. New biomarkers, used to increase the specificity of NPC diagnosis, included long‐chain C24 isoforms of GlcCer. These were increased in NPC‐positive cases (Figure 3b); however, some patients from the known mutation group also demonstrated levels within the NPC range. Finally, the multivariate analysis had indicated reductions in isoforms of CDH. As these were likely to represent the natural precursor of the elevated GlcCer isoforms (Figure S1), the ratio of the corresponding isoform product (GlcCer) to substrates (CDH) was examined. Ratios for hydroxylated C24 isoforms (GlcCer‐C24‐OH:CDH C24‐OH) allowed a significant separation of the NPC group compared with the others (Figure 3c), with no other patient samples overlapping.
FIGURE 3.

Univariate analysis of Niemann–Pick type C disease (NPC) biomarkers from multivariate analysis comparing unaffected comparison individuals (n = 10), confirmed NPC (n = 5), neurodevelopmental patients with known mutations (n = 30), and undiagnosed patients with unknown mutations (n = 40). Significance determined by Kruskal–Wallis analysis of variance with Dunn's post hoc test; **p < 0.01, ***p < 0.001, ****p < 0.0001. Patients with known mutations with biomarker values in the NPC range are indicated on the graphs. (a) Known NPC biomarkers N‐palmitoyl‐O‐phosphocholineserine (PPCS) ratioed to lyso‐sphingomyelin and the bile acid 3β,5α,6β‐trihydroxy‐cholanoyl‐glycine. (b) C24 isoforms of hexosylceramides implicated in orthogonal projections to latent structures discriminant analysis. (c) Ratio of hexosylceramide isoforms to corresponding dihexosylceramide precursor isoforms improves specificity. Abbreviation: Lyso‐SM, lyso‐sphingomyelin.
DISCUSSION
NPC is clinically heterogeneous, with patients often manifesting non‐specific signs early in the disease course. 9 , 25 Diagnostic delays often ensue, especially because of the condition's rarity and limited suspicion among non‐specialists, the lack of ultra‐specific diagnostic markers, and because many rare neurogenetic and neurometabolic disorders have similar presentations. Our patient cohort reflected this, with recruited children manifesting a wide array of often non‐specific neurodevelopmental issues and an insidious disease onset. Even with access to next‐generation sequencing, difficulties in establishing a diagnosis of NPC often arise, for example when trying to decipher identified variants of unknown significance. Hence, the development of robust disease‐specific markers for diagnosis, accurate disease monitoring, and assessment of efficacy of emerging novel, targeted therapies 5 is crucial.
Until recently, filipin staining of unesterified cholesterol in cultured skin fibroblasts was used as the confirmatory test for NPC. However, this test has many limitations as it is invasive, time consuming, and requires a high level of expertise in the analysis. 26 More recently, plasma oxysterols, PPCS with lyso‐sphingomyelin, and the 3β,5α,6β‐trihydroxy‐cholanoyl‐glycine bile acid have shown promise as NPC‐specific biomarkers; 14 , 16 , 17 , 18 , 19 , 21 however, apart from their occasional non‐specificity and lack of data about efficacy in accurately monitoring disease progression or response to therapeutic interventions, their levels have not been sufficiently examined in populations of similarly affected patients without NPC with other neurological disorders.
To address the continuous need for improved biomarkers in NPC, we developed a panel of markers that included both established markers and others with levels predicted to be affected by disease pathophysiology. Our final panel included measurements of PPCS, lyso‐sphingomyelin, 3β,5α,6β‐trihydroxy‐cholanoyl‐glycine, GlcCer C24‐OH, and CDH C24‐OH. Other studies have assumed similar approaches of combining biomarker analysis for NPC diagnosis, but using both DBS and serum. 27 We selected performing the assay only on DBS, which has advantages of simplified sample collection, transport, and preservation. Another advantage of using DBS was that most compounds of interest were quickly, easily, and reliably extractable, hence our assay would be associated with low costs and rapid turnaround times. Moreover, this approach also removed the need for multiple laboratories to test single analytes. Importantly, multivariate analysis of the biomarker panel in the multiplex assay significantly increased the specificity in discriminating NPC from non‐NPC cases.
Furthermore, we examined whether any other compounds were altered in NPC. Multivariate analysis revealed that, as well as the known biomarkers, an elevated ratio of hydroxylated C24 species of GlcCer to CDH (GlcCer‐C24‐OH:CDH C24‐OH) gave good specificity for NPC. GlcCer has previously been observed to be increased in NPC 28 , 29 but its role in the disease is not well studied. It is hydrolysed to ceramide by glucocerebrosidase (GBA) for which there are two forms: GBA1 (lysosomal, usually implicated in Gaucher disease) and GBA2 (membrane‐associated). 30 , 31 GBA activity is influenced by other lipids such as cholesterol and sphingomyelin, 32 which are affected in NPC, while GBA2 has been shown to regulate endo‐lysosomal function in NPC. 33 Hydroxylated isoforms of glycosphingolipids are particularly enriched in biological membranes, 34 and are therefore more likely to be substrates of GBA2 than GBA1. More studies are warranted to determine this new biomarker's clinical utility and to identify links to NPC pathophysiology.
Univariate analysis of other identified biomarkers distinguished patients with NPC, but with occasional overlap with other patient groups, which suggests that using just one marker can be of limited use. We observed that PPCS/lyso‐sphingomyelin was increased in three patients without NPC who had gene defects in SCN1A, ABA‐T, and KMT2B (Figure 3a). However, all three of these patients had normal levels of 3β,5α,6β‐trihydroxy‐cholanoyl‐glycine, which eliminated suspicions of NPC (Figure 3a). Moreover, only one patient without NPC (chromosome 7 paternal uniparental disomy presenting with cholestasis) had 3β,5α,6β‐trihydroxy‐cholanoyl‐glycine levels in the NPC range; conversely, PPCS/lyso‐sphingomyelin levels were normal; therefore, the use of more than one biomarker in a multiplex assay can greatly reduce false‐positive results and improve the specificity of NPC diagnosis.
There were limitations in our work. Our cohort was fairly large, but only five patients with NPC were included and analysed. Our population norm comparison cohort was not age‐matched, hence more studies are warranted to ensure that paediatric metabolite values from typically developing children do not overlap with NPC. Patients with Niemann–Pick disease types A and B, as well as NPC1 carriers, should also be included in future studies, as they can exhibit biomarker (e.g. PPCS) values that overlap with those of patients with NPC. 19 Moreover, oxysterols were not included in the analysis owing to expected artefacts in extraction from DBS samples. 24 Nevertheless, our results clearly separate patients with NPC from age‐matched patients with similar clinical presentations and provide a paradigm for similar approaches in other monogenic conditions.
In summary, we describe an approach to aiding diagnosis in NPC by designing a biochemically relevant liquid chromatography with tandem mass spectrometry‐based biomarker panel, and subsequent multivariate analysis. This tool accurately distinguished patients with NPC within a clinically and genetically heterogeneous cohort of age‐matched children presenting with symptoms and signs of neurometabolic disease. This selection of biomarkers could be applicable in the interpretation of NPC1/2 genetic variants, thus expediting diagnosis and subsequent implementation of appropriate treatments. Overall, our results provide proof‐of‐concept that such methodologies can be useful in many other neurological and metabolic disorders that, like NPC, are not linked to specific enzyme deficiencies and lack reliable functional assays or single clinical, radiological, or biochemical diagnostic markers.
Supporting information
Appendix S1: Sample preparation protocol.
Appendix S2: Multiplex compound inclusion criteria.
Figure S1: Glycosphingolipid degradation pathway.
Figure S2: Orthogonal projections to latent structures discriminant analysis biplot of Figure 2 data.
Table S1: Internal standard compound stock solutions.
Table S2: Table of mass spectral analysis parameters.
Table S3: Gradient parameters 1.
Table S4: Gradient parameters 2.
Table S5: Mass transitions and conditions for all compounds and internal standards.
Table S6: Orthogonal projections to latent structures discriminant analysis parameters.
Table S7: Genetic diagnoses established in our patient cohort.
Table S8: Individual biomarker statistics.
ACKNOWLEDGMENTS
Members of the clinical cohort recruitment and characterization group are as follows: Matthew P. Wilson, Youssef Khalil, Emma Footitt, Spyros Batzios, James Davison, Lucinda J. Carr, Marios Kaliakatsos, Robert Robinson, Hywel Williams, and Peter T. Clayton.
All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health was made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Papandreou A, Doykov I, Spiewak J, Komarov N, Habermann S, Kurian MA, Clinical cohort recruitment and characterization group . Niemann–Pick type C disease as proof‐of‐concept for intelligent biomarker panel selection in neurometabolic disorders. Dev Med Child Neurol. 2022;64(12):1539–1546. 10.1111/dmcn.15334
Funding informationThis research was supported, and funded, by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. A.P. and P.G. received funding from Actelion for the study of undiagnosed neurodegenerative disorders. This work was also supported by a Clinical Research Training Fellowship (Action Medical Research and the British Paediatric Neurology Association, GN2465) to A.P. The authors have stated that they had no interests which might be perceived as posing a conflict or bias.
Members of the clinical cohort recruitment and characterization group are listed in the Acknowledgements.
Apostolos Papandreou and Ivan Doykov contributed equally to this work; Kevin Mills, Paul Gissen and Wendy Heywood contributed equally to this work.
DATA AVAILABILITY STATEMENT
Most data that supports the findings of this study are available in the supplementary material of this article. Other data that support the findings of this study are available on request from the corresponding author, but are currently not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Lanza G, Calì F, Vinci M, Cosentino FII, Tripodi M, Spada RS, et al. A Customized Next‐Generation Sequencing‐Based Panel to Identify Novel Genetic Variants in Dementing Disorders: A Pilot Study. Neural Plast. 2020;2020:8078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lunke S, Eggers S, Wilson M, Patel C, Barnett CP, Pinner J, et al. Feasibility of Ultra‐Rapid Exome Sequencing in Critically Ill Infants and Children With Suspected Monogenic Conditions in the Australian Public Health Care System. Jama. 2020;323(24):2503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Piroth T, Boelmans K, Amtage F, Rijntjes M, Wierciochin A, Musacchio T, et al. Adult‐Onset Niemann‐Pick Disease Type C: Rapid Treatment Initiation Advised but Early Diagnosis Remains Difficult. Frontiers in neurology. 2017;8:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. The New England journal of medicine. 2018;378(20):1898–907. [DOI] [PubMed] [Google Scholar]
- 5. Mengel E, Patterson MC, Da Riol RM, Del Toro M, Deodato F, Gautschi M, et al. Efficacy and safety of arimoclomol in Niemann‐Pick disease type C: Results from a double‐blind, randomised, placebo‐controlled, multinational phase 2/3 trial of a novel treatment. Journal of inherited metabolic disease. 2021;44(6):1463–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vinciguerra L, Lanza G, Puglisi V, Fisicaro F, Pennisi M, Bella R, et al. Update on the Neurobiology of Vascular Cognitive Impairment: From Lab to Clinic. International journal of molecular sciences. 2020;21(8), 2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, et al. Niemann‐Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277(5323):228–31. [DOI] [PubMed] [Google Scholar]
- 8. Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, et al. Identification of HE1 as the second gene of Niemann‐Pick C disease. Science. 2000;290(5500):2298–301. [DOI] [PubMed] [Google Scholar]
- 9. Papandreou A, Gissen P. Diagnostic workup and management of patients with suspected Niemann‐Pick type C disease. Therapeutic advances in neurological disorders. 2016;9(3):216–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Labrecque M, Touma L, Bhérer C, Duquette A, Tétreault M. Estimated prevalence of Niemann‐Pick type C disease in Quebec. Scientific reports. 2021;11(1):22621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pentchev PG, Comly ME, Kruth HS, Tokoro T, Butler J, Sokol J, et al. Group C Niemann‐Pick disease: faulty regulation of low‐density lipoprotein uptake and cholesterol storage in cultured fibroblasts. FASEB J. 1987;1(1):40–5. [DOI] [PubMed] [Google Scholar]
- 12. Pentchev PG, Comly ME, Kruth HS, Vanier MT, Wenger DA, Patel S, et al. A defect in cholesterol esterification in Niemann‐Pick disease (type C) patients. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(23):8247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liscum L, Ruggiero RM, Faust JR. The intracellular transport of low density lipoprotein‐derived cholesterol is defective in Niemann‐Pick type C fibroblasts. J Cell Biol. 1989;108(5):1625–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Porter FD, Scherrer DE, Lanier MH, Langmade SJ, Molugu V, Gale SE, et al. Cholesterol oxidation products are sensitive and specific blood‐based biomarkers for Niemann‐Pick C1 disease. Science translational medicine. 2010;2(56):56ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schroepfer GJ, Jr. Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev. 2000;80(1):361–554. [DOI] [PubMed] [Google Scholar]
- 16. Mazzacuva F, Mills P, Mills K, Camuzeaux S, Gissen P, Nicoli ER, et al. Identification of novel bile acids as biomarkers for the early diagnosis of Niemann‐Pick C disease. FEBS Lett. 2016;590(11):1651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang X, Sidhu R, Mydock‐McGrane L, Hsu FF, Covey DF, Scherrer DE, et al. Development of a bile acid‐based newborn screen for Niemann‐Pick disease type C. Science translational medicine. 2016;8(337):337ra63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sidhu R, Mondjinou Y, Qian M, Song H, Kumar AB, Hong X, et al. N‐acyl‐O‐phosphocholineserines: structures of a novel class of lipids that are biomarkers for Niemann‐Pick C1 disease. Journal of lipid research. 2019;60(8):1410–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giese AK, Mascher H, Grittner U, Eichler S, Kramp G, Lukas J, et al. A novel, highly sensitive and specific biomarker for Niemann‐Pick type C1 disease. Orphanet journal of rare diseases. 2015;10:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geberhiwot T, Moro A, Dardis A, Ramaswami U, Sirrs S, Marfa MP, et al. Consensus clinical management guidelines for Niemann‐Pick disease type C. Orphanet journal of rare diseases. 2018;13(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuchar L, Sikora J, Gulinello ME, Poupetova H, Lugowska A, Malinova V, et al. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin‐509 for differential screening of Niemann‐Pick A/B and C diseases. Analytical biochemistry. 2017;525:73–7. [DOI] [PubMed] [Google Scholar]
- 22. Vanier MT. Niemann‐Pick disease type C. Orphanet journal of rare diseases. 2010;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sevin M, Lesca G, Baumann N, Millat G, Lyon‐Caen O, Vanier MT, et al. The adult form of Niemann‐Pick disease type C. Brain. 2007;130(Pt 1):120–33. [DOI] [PubMed] [Google Scholar]
- 24. van Reyk DM, Brown AJ, Hult'en LM, Dean RT, Jessup W. Oxysterols in biological systems: sources, metabolism and pathophysiological relevance. Redox Rep. 2006;11(6):255–62. [DOI] [PubMed] [Google Scholar]
- 25. Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F. Recommendations for the diagnosis and management of Niemann‐Pick disease type C: an update. Molecular genetics and metabolism. 2012;106(3):330–44. [DOI] [PubMed] [Google Scholar]
- 26. Vanier MT, Latour P. Laboratory diagnosis of Niemann‐Pick disease type C: the filipin staining test. Methods in cell biology. 2015;126:357–75. [DOI] [PubMed] [Google Scholar]
- 27. Wu C, Iwamoto T, Hossain MA, Akiyama K, Igarashi J, Miyajima T, et al. A combination of 7‐ketocholesterol, lysosphingomyelin and bile acid‐408 to diagnose Niemann‐Pick disease type C using LC‐MS/MS. PloS one. 2020;15(9):e0238624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vanier MT. Complex lipid trafficking in Niemann‐Pick disease type C. Journal of inherited metabolic disease. 2015;38(1):187–99. [DOI] [PubMed] [Google Scholar]
- 29. Vanier MT. Biochemical studies in Niemann‐Pick disease. I. Major sphingolipids of liver and spleen. Biochimica et biophysica acta. 1983;750(1):178–84. [DOI] [PubMed] [Google Scholar]
- 30. Korschen HG, Yildiz Y, Raju DN, Schonauer S, Bonigk W, Jansen V, et al. The non‐lysosomal beta‐glucosidase GBA2 is a non‐integral membrane‐associated protein at the endoplasmic reticulum (ER) and Golgi. The Journal of biological chemistry. 2013;288(5):3381–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aureli M, Masilamani AP, Illuzzi G, Loberto N, Scandroglio F, Prinetti A, et al. Activity of plasma membrane beta‐galactosidase and beta‐glucosidase. FEBS Lett. 2009;583(15):2469–73. [DOI] [PubMed] [Google Scholar]
- 32. Abdul‐Hammed M, Breiden B, Schwarzmann G, Sandhoff K. Lipids regulate the hydrolysis of membrane bound glucosylceramide by lysosomal beta‐glucocerebrosidase. Journal of lipid research. 2017;58(3):563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wheeler S, Haberkant P, Bhardwaj M, Tongue P, Ferraz MJ, Halter D, et al. Cytosolic glucosylceramide regulates endolysosomal function in Niemann‐Pick type C disease. Neurobiology of disease. 2019;127:242–52. [DOI] [PubMed] [Google Scholar]
- 34. Westerlund B, Slotte JP. How the molecular features of glycosphingolipids affect domain formation in fluid membranes. Biochimica et biophysica acta. 2009;1788(1):194–201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Sample preparation protocol.
Appendix S2: Multiplex compound inclusion criteria.
Figure S1: Glycosphingolipid degradation pathway.
Figure S2: Orthogonal projections to latent structures discriminant analysis biplot of Figure 2 data.
Table S1: Internal standard compound stock solutions.
Table S2: Table of mass spectral analysis parameters.
Table S3: Gradient parameters 1.
Table S4: Gradient parameters 2.
Table S5: Mass transitions and conditions for all compounds and internal standards.
Table S6: Orthogonal projections to latent structures discriminant analysis parameters.
Table S7: Genetic diagnoses established in our patient cohort.
Table S8: Individual biomarker statistics.
Data Availability Statement
Most data that supports the findings of this study are available in the supplementary material of this article. Other data that support the findings of this study are available on request from the corresponding author, but are currently not publicly available due to privacy or ethical restrictions.