

Abstract
Focal segmental glomerulosclerosis (FSGS) is not a disease, rather a pattern of histological injury occurring from a variety of causes. The exact pathogenesis has yet to be fully elucidated but is likely varied based on the type of injury and the primary target of that injury. However, the approach to treatment is often based on the degree of podocyte foot process effacement and clinical presentation without sufficient attention paid to etiology. In this regard, there are many monogenic causes of FSGS with variable presentation from nephrotic syndrome with histological features of primary podocytopathy to more modest degrees of proteinuria with limited evidence of podocyte foot process injury. It is likely that genetic causes are largely underdiagnosed, as the role and the timing of genetic testing in FSGS is not established and genetic counseling, testing options, and interpretation of genotype in the context of phenotype may be outside the scope of practice for both nephrologists and geneticists. Yet most clinicians believe that a genetic diagnosis can lead to targeted therapy, limit the use of high‐dose corticosteroids as a therapeutic trial, and allow the prediction of the natural history and risk for recurrence in the transplanted kidney. In this manuscript, we emphasize that genetic FSGS is not monolithic in its presentation, opine on the importance of genetic testing and provide an algorithmic approach to deployment of genetic testing in a timely fashion when faced with a patient with FSGS.
Keywords: albuminuria, foot process effacement, genetic variant, Mendelian disease, nephrotic syndrome, podocytopathy, proteinuria
1. INTRODUCTION
Focal segmental glomerulosclerosis (FSGS) is a descriptive term for a histological pattern of disease seen on kidney biopsy that is the final common pathway of a variety of injuries sustained by the kidneys. It was first described in 1925 in adults with nephritic proteinuria (Cameron, 2003; Rich, 1957). FSGS may be idiopathic, genetic, autoimmune, secondary to drugs, infections, or a (mal)adaptive response to many forms of chronic kidney disease with reduced nephron mass. Several recent studies have highlighted the burden of undiagnosed monogenic causes of FSGS and steroid‐resistant nephrotic syndrome (SRNS).
The incidence of FSGS appears to have increased over the last few decades (Kitiyakara, Eggers, & Kopp, 2004). However, the true incidence of FSGS is not known, as many probably remain undiagnosed, either because they present late with advanced renal failure or because the presentation is non‐specific, and a glomerular disease is not suspected or because the cause of end stage kidney disease (ESKD), even if known, is not reported. As per the 2019 annual report of US Renal Data System (USRDS), approximately 134,608 individuals were newly diagnosed with ESKD, a 16% increase since 2009 (USRDS, 2021). In the same report, it was noted that 14% patients in the United States who start dialysis do not have a clear diagnosis for their kidney failure. The European Renal Association‐European Dialysis and Transplant Association (ERA‐EDTA) registry data report the number of ESKD cases that remain unsolved to be as high as 27% (Kramer et al., 2021).
The current classifications of FSGS into primary, secondary, and genetic forms is overly simplistic as it considers genetic causes as monolithic with similar presentations and approaches to therapy (De Vriese, Sethi, Nath, Glassock, & Fervenza, 2018; Rosenberg & Kopp, 2017). To complicate matters, current KDIGO guidelines for patients with FSGS do not support the routine use of genetic testing and therefore the incidence of genetic disease in various forms of FSGS cannot be ascertained (Kidney Disease: Improving Global Outcomes Glomerular Diseases Work Group, 2021).
In this manuscript, we provide background information on FSGS, describe the heterogeneity of genetic FSGS, and provide a new conceptual framework and an algorithmic approach to evaluation of FSGS with integration of genetic testing into the evaluation and management of FSGS. We believe that, where appropriate, there should be a greater emphasis on early genetic testing to direct management.
2. CURRENT DEFINITION OF FSGS
FSGS is a pattern of histological injury where there is a segmental increase in glomerular matrix leading to sclerosis in “some” part (segmental) of at least one glomerulus (focal) seen on light microscopy (LM), with electron microscopy (EM) showing varying degrees of podocyte foot process effacement (Figure 1). Immunofluorescence examination may show trace IgM and C3 in sclerosed areas, thought to be from non‐specific trapping. FSGS may be very focal and may arise first in juxtamedullary glomeruli and the titular lesion may be missed with an inadequate or superficial sample. With disease progression, segmental sclerosis progresses to global sclerosis. In late stages of disease, all sampled glomeruli may show global sclerosis and without a single segmental lesion it cannot be identified as FSGS which has progressed beyond recognition, as it is not distinguishable from other forms of advanced chronic kidney disease where non‐specific global glomerulosclerosis, interstitial fibrosis, and tubular atrophy predominate.
FIGURE 1.
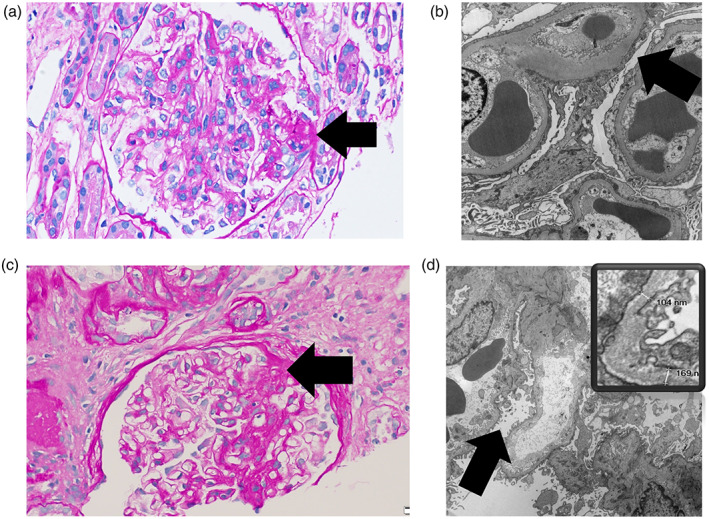
Histological features in two genetic forms of focal segmental glomerulosclerosis (FSGS) manifesting as primary and secondary FSGS. Panels (a) and (b): Patient with autosomal dominant FSGS from a heterozygous TRPC6 variant. Light microscopy (PAS stain, 40× objective) in panel (a) shows a glomerulus with an area of segmental sclerosis (arrow). Electron microscopy (10,000× direct magnification) in panel (b) shows a glomerulus with diffuse podocyte epithelial foot process effacement (arrow). Panels (c) and (d): Patient with X‐linked FSGS from a hemizygous COL4A5 variant. Light microscopy (PAS stain, 40× objective) in panel (c) shows a glomerulus with an area of segmental sclerosis (arrow). Electron microscopy (10,000× direct magnification) in panel (d) shows a glomerulus with relatively limited, focal podocyte epithelial foot process effacement (arrow). Inset shows thin basement membranes. The glomerular basement membranes ranged from 59.8 to 203 nm (average 120.2 nm)
3. CLINICAL PRESENTATION AND PROGRESSION
Clinically, untreated FSGS is characterized by varying degrees of proteinuria with or without a decline in kidney function. Some patients can present with nephrotic syndrome, which is defined as proteinuria >3.5 g in 24 hr, with hypoalbuminemia and edema, whereas others may be completely asymptomatic and incidentally discovered to have proteinuria with or without a change in kidney function. It is important to note that most patients with FSGS have proteinuria without edema or hypoalbuminemia. Furthermore, patients, including those that present with nephrotic syndrome can undergo spontaneous, or treatment‐induced remission that can be long lasting (Troyanov et al., 2005). Infants and children presenting with nephrotic syndrome with relatively preserved glomerular filtration rate (GFR) are often not biopsied unless a therapeutic trial of corticosteroids fails to induce remission. In contrast, adults presenting with varying degrees of proteinuria or the nephrotic syndrome are almost always biopsied before treatment is contemplated. Thus, some children with steroid‐sensitive nephrotic syndrome may have unrecognized FSGS, although most clinicians believe that patients who have durable remission with corticosteroids likely have minimal change disease. A significant number of children with frequently relapsing or SRNS, if biopsied, will demonstrate FSGS and in the literature, SRNS and FSGS are often used interchangeably (Lee, Kronbichler, Shin, & Oh, 2021; Reidy & Kaskel, 2007).
4. PATHOGENESIS OF FSGS
The glomerular filtration barrier has three core constituents, the podocyte foot processes with their slit diaphragms, the glomerular basement membrane, and capillary endothelial cells. Any injury to this filtration barrier can cause proteinuria and some of these injuries progress to FSGS. FSGS is often a consequence of direct podocyte injury as it is seen in patients with some genetic variants that affect the podocyte and can be recapitulated in experimental animals with deliberate podocyte damage or depletion (Lepori, Zand, Sethi, Fernandez‐Juarez, & Fervenza, 2018; Lim, Yang, Do, & Fogo, 2016; Wharram et al., 2005). The precise cause of podocyte injury in primary FSGS, if not known, is termed idiopathic. After the podocyte injury, some podocytes drop out and the remaining podocytes hypertrophy to cover the glomerular capillary surface with flattening or effacement of the foot processes. FSGS may be caused by pathogenic variants in any of several genes that affect podocyte structure or function or the slit diaphragm as well as those that affect the integrity of the glomerular basement membrane. In other cases, FSGS can be secondary to viral infections, drugs or autoantibodies that injure the podocyte or follows a reduction in nephron mass (maladaptive) from genetic or non‐genetic causes.
Minimal change disease is also considered to be secondary to podocyte injury, although it is generally reversible. As the name implies, in minimal change disease, the glomeruli are normal by LM but show diffuse foot process effacement on EM. The etiology of minimal change disease is largely unknown although emerging evidence indicates that some of these are secondary to autoantibodies against podocyte proteins like nephrin and Annexin A2 (Watts et al., 2022; Ye, Zhou, et al., 2021; Ye, Zhang, et al., 2021). FSGS and minimal change disease are collectively called “podocytopathies” and these two conditions may be on the spectrum of podocyte injury with mild reversible disease usually presenting as minimal change disease with a good long‐term prognosis and more severe, often progressive disease presenting as FSGS. One of the explanations for the proposition that these are on the same spectrum is that the sclerotic lesions take time to develop, so the timing of the biopsy plays an important role in diagnosis since podocyte foot process effacement is common to both minimal change disease and FSGS and in the earliest stage maybe the only abnormality identified. The other explanation is that sclerotic FSGS lesions maybe missed on initial biopsy and a diagnosis of minimal change disease may be erroneously made (Howie, Pankhurst, Sarioglu, Turhan, & Adu, 2005).
5. LIMITATION OF CURRENT FSGS CLASSIFICATIONS
Over the years, there have been many attempts to classify FSGS and these schemes fall into two main taxonomies: the histological and etiopathogenic classifications. The histological classification (“Columbia Classification”) proposed in 2003 has five different subtypes: perihilar, tip, collapsing, cellular, and not otherwise specified (D'Agati, Fogo, Bruijn, & Jennette, 2004). The Columbia classification is however not widely accepted; it does not help with diagnosis or treatment and has no correlation with causes of FSGS except in some cases of the collapsing form. There are a few proposed variations on an etiopathogenic classification of FSGS, primarily combining clinical and electron microscopic features and the presumed etiology to identify subgroups for further investigation, to prognosticate on clinical course, or for a trial of corticosteroid or immunosuppressive therapy (De Vriese et al., 2018; Rosenberf & Kopp, 2017). In this schema, primary FSGS is recognized as presenting with nephrotic syndrome and demonstrating the segmental sclerosing lesions on LM together with diffuse foot process effacement on EM (Figure 1). Secondary FSGS is everything else and is caused by genetic diseases or by certain drugs or viral infections or as a maladaptive response to reduction in nephron number. These secondary causes of FSGS are thought to have focal or limited foot process effacement and to not present with nephrotic syndrome, although the data to support that premise is not robust. In fact, some genetic causes of FSGS, especially those that disrupt the slit diaphragm (e.g., NPHS2, TRPC6) do present with diffuse FPE and the nephrotic syndrome (Lepori et al., 2018). Conversely, whether FSGS with diffuse foot process effacement without nephrotic syndrome should be called primary FSGS is not a settled matter (Howie, 2020). Adding to the nosologic uncertainty is the notion that primary FSGS, as defined by pathology is homogenous and synonymous with idiopathic FSGS.
There is debate on what constitutes primary FSGS and on the accepted “secondary” causes of FSGS that should be excluded first but if primary FSGS is a diagnosis of exclusion, then a thorough evaluation for an etiological cause should always be the goal. The consensus opinion developed by KDIGO in 2021, states that when considering a diagnosis of primary FSGS there should be no other identifiable cause of FSGS yet concluded that the role of genetic testing for FSGS is uncertain, and therefore should not be routinely used to determine a cause for FSGS (Kidney Disease: Improving Global Outcomes Glomerular Diseases Work Group, 2021). Despite the absence of any randomized placebo‐controlled trial of corticosteroids in FSGS, these guidelines recommend a trial of high‐dose steroids in those with primary FSGS (Hodson, Sinha, & Cooper, 2022).
6. GENETIC FSGS DOES NOT HAVE A COMMON HISTOLOGICAL PATTERN
It is generally accepted that knowing the etiology of FSGS is important as it has implications for predicting natural history, for determining the likelihood of responsiveness to immunosuppressive therapy, for optimal management, for predicting disease recurrence post‐transplant. However, the etiology of FSGS cannot be reliably ascertained by histopathology alone although some have suggested there is a correlation between genetic causes of FSGS and the degree of foot process effacement.
Some authors have attempted to address this question by measuring the degree of podocyte foot process effacement and foot process width (FPW) in primary compared to genetic causes of FSGS (Ishizuka et al., 2021). In one study of nine primary and seven genetic FSGS patients, the percentage of foot process effacement was significantly higher in those with primary FSGS as compared to genetic FSGS (>88% vs. <38%). Moreover, FPW was significantly larger in primary FSGS than in genetic FSGS (>3,000 nm vs. <2,000 nm). While the pattern of foot process effacement was segmental in genetic disease, it was diffuse in primary. However, the study was limited to a small sample with the monogenic cases secondary to variants in NUP107, WT1, LAMB2, INF, and NUP93B, and most of these presented without nephrotic syndrome (Ishizuka et al., 2021). The authors also performed a literature search and identified 38 patients where the genetic mutation and the degree of foot process effacement were available for review. Patients with variants in genes that encode slit diaphragm proteins such as NPHS2, CD2AP, and TRPC6 showed diffuse foot process effacement (Ishizuka et al., 2021). In another study of a cohort of sporadic FSGS, the percentage of patients with >50% foot process effacement were not different between genetic and non‐genetic causes of FSGS (Yao et al., 2019). These studies indicate that there is significant heterogeneity in presentation and degree of foot process effacement between different genetic forms of FSGS, which is not surprising because genetic variants that affect the podocyte slit diaphragm directly are likely to behave differently from those that affect the GBM and those that reduce nephron mass and secondarily affect the podocyte.
7. PREVALENCE OF GENETIC CAUSES OF FSGS
As proposed in Table 1, genetic FSGS is a broad term that includes all monogenic causes of FSGS including those which have a phenotype of primary FSGS with nephrotic proteinuria and other forms where FSGS is secondary and the renal phenotype may include congenital anomalies of development, nephronophthisis, chronic tubulointerstitial disease or a proximal tubulopathy. Furthermore, monogenic FSGS may be renal limited or involve kidneys as a part of a syndrome with other extrarenal manifestations such as Pierson syndrome or nail‐patella syndrome (NPS). FSGS may also occur in the setting of genetic susceptibility where the presence of sequence variants is not sufficient for disease: APOL1 is one such risk gene for FSGS. Thus, FSGS can occur in association with the inheritance of two copies of the APOL1 FSGS susceptibility alleles, G1 and G2, where a second or additional hits are needed for the development of renal disease (Kruzel‐Davila, Wasser, & Skorecki, 2017).
TABLE 1.
Proposed etiopathogenic classification of FSGS
| 1. Primary (direct podocyte effect—“podocytopathic”) |
|
| 2. Secondary |
|
Abbreviations: ADTKD, autosomal dominant tubulointerstitial kidney disease; FSGS, focal segmental glomerulosclerosis.
The prevalence of monogenic FSGS appears to be higher with a lower age of disease onset with 20–30% of children with SRNS or FSGS having a genetic cause (Bierzynska et al., 2017; Nagano et al., 2020; Park et al., 2020; Sadowski et al., 2015; Trautmann et al., 2015; Wang et al., 2017; Warejko et al., 2018). The prevalence of monogenic FSGS in adult FSGS is not well known since testing is generally limited to those with early onset disease, resistance to immunosuppression, positive family history, or syndromic features. In a series of adults from Britain, 22% of those with a family history and 10% of those without a family history had pathogenic variants that explained their FSGS (Gast et al., 2016). In another series, 22% of adults presenting with SRNS had an identifiable genetic cause and in patients with FSGS in the Toronto GN Registry, the genetic diagnosis rate was 11% (Sen et al., 2017; Yao et al., 2019). In a French cohort of patients with sporadic SRNS or FSGS and lack of response to glucocorticoids and cyclosporin, 11% had pathogenic variants in monogenic SRNS genes while another 10% had two high‐risk APOL1 risk alleles (Gribouval et al., 2018). In another recent study from Europe of sporadic FSGS where other secondary causes had been excluded, comprehensive genetic testing yielded a genetic cause of non‐syndromic FSGS in 12% patients (Braunisch et al., 2021). The most common genes responsible for adult onset FSGS are the Alport genes, COL4A3, COL4A4 and COL4A5 with rates of 38–55% in those with genetic FSGS (Gast et al., 2016; Yao et al., 2019) followed by INF2 (~17%), TRPC6 (~12%) and ACTN4 (3.5%) (Rood, Deegens, & Wetzels, 2012). With the slow adoption of genetic testing in the evaluation and management of FSGS, there can be a long delay (~9 years) from the time of disease onset to establish a genetic diagnosis (Braunisch et al., 2021).
8. RELEVANCE OF FAMILY HISTORY IN GENETIC FSGS
The presence of a family history in a patient with FSGS substantially increases the likelihood of a monogenic cause. As is true for most Mendelian diseases, childhood onset FSGS generally arises from loss of function of both copies of a gene in an autosomal recessive pattern of disease while a majority of those with adult onset FSGS have monoallelic gene variants inherited in an autosomal dominant fashion (Trautmann et al., 2015). However, the absence of a family history does not exclude a monogenic cause. For example, in a recent study of apparently sporadic non‐syndromic FSGS in adults, 12% of patients had pathogenic variants in FSGS genes, 44% of whom had COL4 variants (Gribouval et al., 2018). In another study, 10% of adults with FSGS and no family history had a genetic cause (Santin et al., 2011).
There are several reasons for lack of family history with monogenic disorders. With autosomal recessive diseases, affected individuals are seen in just one generation unless there is extensive consanguinity. With small family sizes, and the expected likelihood of biallelic variants in just 1 in 4 offspring from two heterozygous carrier parents, the proband may be the only affected individual. With autosomal dominant conditions, there may be limited penetrance or variable expressivity, or the phenotype may not be apparent in other affected family members without formal testing (e.g., microscopic hematuria or low‐level proteinuria) or there may be lack of knowledge of family history because of adoption, estrangement, or misattributed paternity. In other situations, heterozygous dominant variants may arise de novo and thus affect just one individual and if the condition causes early onset ESKD or otherwise affects fertility, the disease may not be transmitted to the next generation. In X‐linked recessive disorders like Alport syndrome, heterozygous carriers may be minimally symptomatic and there may be no first‐degree relatives that are also affected to the same degree.
9. BENEFITS OF GENETIC TESTING
There are many potential benefits to genetic testing in patients with SRNS and/or FSGS. One is the ability to identify a genetic cause and thus make a genetic diagnosis and this can be done at any stage of the illness. This can be complementary to a renal biopsy and in some situations, especially in children with SRNS or in those with classic manifestations of a syndromic disease, can take the place of a kidney biopsy. Further, identification of a genetic cause of FSGS at any age significantly correlates with corticosteroid resistance, and avoiding ineffective and potentially toxic corticosteroid therapies is another benefit. However, it is worth noting that FSGS from causal variants in EMP2 or PLCG2 may be steroid responsive (Dorval et al., 2018; Parker et al., 2019). Generally, genetic forms of FSGS are also resistant to therapy with cyclosporin and/or MMF although it has not been systematically studied (Kemper & Lemke, 2018).
Knowledge of the specific cause of SRNS or FSGS can also guide specific therapy. For example, CoQ10 deficiency with renal disease from variants in COQ6 appears to respond to CoQ10 supplementation, a simple, well tolerated, and inexpensive therapy (Ashraf et al., 2013; Heeringa et al., 2011). In general, genetic forms of FSGS do not recur post‐transplant and would not be expected to benefit from prophylactic or therapeutic plasmapheresis following a kidney transplant. Another benefit of identifying a genetic cause is the ability to evaluate for relevant extrarenal features where associated conditions may benefit from earlier diagnosis and/or intervention such as immunodeficiency with variants in SMARCAL1 or karyotyping for sex chromosome determination in Frasier syndrome (Milunsky, 1993; Morimoto, Lewis, Lucke, & Boerkoel, 1993). In other conditions, future serious complications can be anticipated and preemptively addressed such as with prophylactic gonadectomy or bilateral nephrectomy to eliminate the risk of gonadoblastoma or nephroblastoma with WT1 variants in Frasier syndrome and Denys–Drash syndrome respectively (Lipska‐Zietkiewicz, 1993).
A genetic diagnosis can also help with evaluating risk of disease in asymptomatic family members and can also assist with pre‐pregnancy counseling and in preimplantation diagnosis for affected patients who may be considering starting a family. Finally, identifying the genetic cause in an affected individual will allow the targeted evaluation for the familial variant (cascade screening) in a biologically related individual who volunteers to be a kidney donor (Thomas et al., 2021).
10. WHO SHOULD GET GENETIC TESTING?
Children and young adults have a genetic cause in as many as 20–30% of SRNS and FSGS. Patients with extrarenal features or with a family history of FSGS or even uncharacterized nephropathy are also more likely to have a monogenic cause for their renal disease. It is reasonable to consider genetic testing in those under 30 years with SRNS or as soon as the diagnosis of FSGS is made. Genetic testing should also be considered for those with other system involvement that might be part of the spectrum of a renal syndromic disease or those who have a positive family history (Figure 2). Children that present with nephrotic syndrome are usually first treated with steroids without a biopsy as the vast majority have steroid‐sensitive minimal change disease. Those with SRNS should undergo genetic testing which can be performed in lieu of or prior to a biopsy. As discussed above, histological and ultrastructural kidney biopsy findings are of limited value, as some genetic diseases cause primary FSGS with diffuse foot process effacement and nephrotic syndrome whereas others cause secondary FSGS with more modest proteinuria and focal or limited foot process effacement. For example, pathogenic variants in podocyte expressed genes such as NPHS2 and TRPC6 affect podocyte structure or the integrity of the glomerular slit diaphragm causing diffuse glomerular foot process effacement while pathogenic variants in genes that affect the glomerular basement membrane such as the Alport spectrum nephropathy genes COL4A3, COL4A4, and COL4A5 cause more focal foot process effacement and a secondary FSGS (Figure 1).
FIGURE 2.
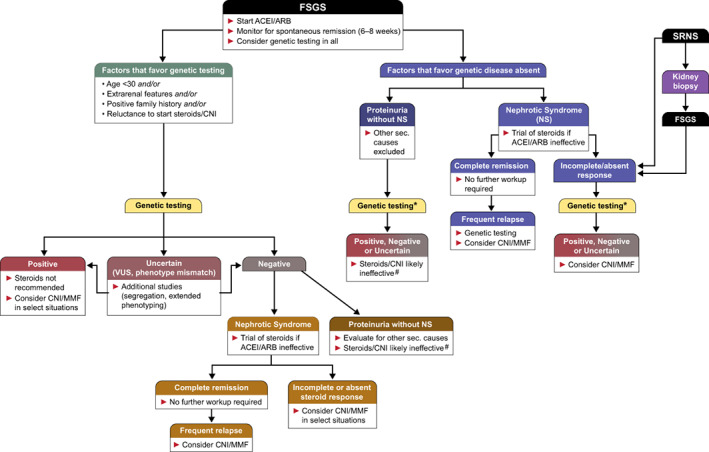
Flowsheet summarizing an algorithmic approach to genetic testing and management of FSGS and SRNS. Patients who have FSGS by biopsy (typically adults) enter the flowsheet at the top (left) and either get genetic testing first to guide corticosteroid therapy or get a trial of corticosteroids first if they have nephrotic syndrome and a lower suspicion for genetic disease. Genetic testing after a failed steroid trial is generally for information purposes only*. In all situations, patients without nephrotic syndrome are not considered as likely to respond to corticosteroids or other immunosuppressive therapy#. ACEI, angiotensin converting enzyme inhibitor; ARB, angiotensin receptor blocker; CNI, calcineurin inhibitors; FSGS, focal segmental glomerulosclerosis; MMF, mycophenolate mofetil; NS, nephrotic syndrome; SRNS, steroid‐resistant nephrotic syndrome; VUS, variants of uncertain significance
Therefore, genetic testing should also be considered at any age where there is no clear etiology of FSGS or if they present with nephrotic syndrome and fail a trial of corticosteroids.
11. GENETIC TESTING APPROACH
The traditional method of genetic testing utilizes PCR amplification of a single gene, or a limited number of genes followed by Sanger sequencing. With a heterogenous condition like FSGS where there are over 40 genes that cause FSGS, Sanger sequencing is of limited value unless there are distinct features that limit the differential diagnosis to a very small subset of genes. A patient with FSGS who has characteristic skeletal manifestations, or macrothrombocytopenia or microcoria could be tested in isolation for just LMX1B, MYH9, or LAMB2 respectively. In the last few years, advances in sequencing and in bioinformatic processing have led to the ability to rapidly sequence and analyze many, many genes or the exome or genome with next generation sequencing and with falling costs this has become the preferred approach to evaluate genetic sequences even for a limited number of genes. Many clinical testing companies offer exome sequencing with analysis restricted to a preselected phenotype‐specific subset of genes, for example, an FSGS panel, which may be at a lower cost than a more comprehensive panel or the entire exome or genome. Limiting analysis to a subset of genes, would restrict the identified genetic variants to those relevant to the phenotype and avoids the problem of variants of uncertain significance (VUS) or likely pathogenic or pathogenic variants in genes unrelated to the phenotype. However, the overall success of this approach will be determined by the completeness of the disease or phenotype‐specific panel, and one could miss less common genetic causes and non‐glomerular causes of FSGS such as TTC21B if they are not included on the panel (Riedhammer et al., 2020). A better approach may be the use of a comprehensive gene panel that includes all genes relevant to any renal phenotype or an exome analysis with analysis limited to kidney disease genes, which will increase the sensitivity but may reduce the specificity (Jayasinghe et al., 2021; Mansilla et al., 2021).
It is important to be aware that population prevalence of certain genetic variants is much higher than the known clinical prevalence of disease and that not all predicted pathogenic or likely pathogenic variants identified in a given individual may be relevant to their kidney disease (Barua & Paterson, 2021; Gibson et al., 2021; Wang et al., 2019). The appropriate interpretation of some genetic variants may require knowledge of ancestry‐specific allele frequencies in the population and the ability to confirm that the given genotype segregates with disease within the affected family.
Every patient who is being considered for genetic testing should have pretest and post‐test counseling about the impact of making a genetic diagnosis so that they can properly assess the pros and cons of testing. These include a discussion of the limitations to testing including the possibility of confounding VUSs, the implications of a positive diagnosis for the predicted course of disease, the impact on the ability to obtain, health, life or disability insurance, and possible consequences for asymptomatic family members including children. Since most nephrologists are not trained in genetic counseling or in the interpretation of sequence variants, access to a genetic counselor and/or a clinical geneticist is desirable.
12. OVERVIEW OF GENETIC CAUSES OF FSGS/SRNS
Nephrotic syndrome that manifests in utero or presents within 3 months of birth is called congenital nephrotic syndrome (CNS). Eighty percent or more are genetic with the majority of these caused by autosomal recessive variants in NPHS1 with the rest accounted for by recessive variants in NPHS2 or LAMB2 or heterozygous variants in WT1 and PLCE1. While most nephrotic syndrome that occurs later in childhood represent minimal change disease and are steroid responsive and not biopsied, steroid resistance can prompt a biopsy revealing FSGS. Monogenic causes can be identified in a significant fraction of childhood onset SRNS or FSGS and a smaller fraction of adults. There are now over 40 different genetic causes of FSGS (Table 2; Table S1) and the more common or distinctive ones are discussed below.
TABLE 2.
Genetic causes of FSGS*
| Gene | Protein and its function | Gene locus | Inheritance | Phenotype |
|---|---|---|---|---|
| 1. Glomerular causes | ||||
| A. Slit diaphragm protein | ||||
| NPHS1 | Nephrin | 19q13.1 | AR | CNS, SRNS, childhood FSGS |
| NPHS2 | Podocin | 1q25 | AR | CNS, SRNS, FSGS |
| TRPC6 | TRPC6 | 11q22. | AD | Adult onset FSGS |
| CD2AP | CD2 associated protein | 6p12 | AD/AR | Childhood and adult FSGS |
| CRB2 | Crumbs family member 2 | 9q33 | AR | SRNS, childhood FSGS, Ventriculomegaly with cystic kidney disease |
| MAGI2 | Membrane‐associated guanylate kinase, WW, PDZ domain containing 2 | 7q21 | AR | CNS |
| B. Actin cytoskeleton and cytosol protein | ||||
| ACTN4 | Alpha‐actinin 4 | 19q13.1 | AD | Adult onset FSGS |
| INF2 | Inverted formin 2 | 14q32.33 | AD | Adult onset FSGS, Charcot–Marie–Tooth disease E |
| MYO1E | Myosin 1E | 15q22.2 | AR | Early onset AR FSGS |
| ARHGDIA | Arhgdia | 17q25.3 | AR | CNS, SRNS, DMS, |
| ANLN | Anillin | 7p14 | AD | Childhood and adult FSGS |
| ARHGAP24 | Rho GTPase‐activating protein 24 | 4q21 | AD a | Adolescent onset FSGS |
| PLCE1 | Phospholipase C epsilon 1 | 10q23.33 | AR | SRNS, DMS, childhood FSGS |
| KANK2 | Kidney ankyrin repeat containing protein 2 | 19p13.2 | AR | SRNS |
| PTPRO | Receptor‐type tyrosine‐protein phosphatase‐O | 12p12.3 | AR | SRNS, FSGS |
| COQ8B /ADCK4 | aarF domain containing kinase 4 | 19q13 | AR | SRNS, FSGS, collapsing FSGS |
| TBC1D8B | TBC1 domain family member 8B | Xq22 | XL | SRNS, FSGS |
| C. Nuclear protein | ||||
| WT1 | Wilms tumor protein 1 | 11p13 | AD | Isolated FSGS |
| NUP107 | Nucleoporin, 107‐KD | 12q15 | AR | SRNS, DMS, FSGS |
| NUP93 | Nucleoporin, 93‐KD | 16q13 | AR | SRNS, DMS, FSGS |
| NUP205 | Nucleoporin, 205‐KD | 7q33 | AR | SRNS, FSGS |
| NUP85 | Nucleoporin, 85‐KD | 17q25 | AR | SRNS |
| NUP133 | Nucleoporin, 133‐KD | 1q42 | AR | SRNS |
| PAX2 | Paired box 2 nuclear transcription factor | 10q24 | AD | FSGS |
| D. Glomerular basement membrane | ||||
| LAMB2 | Laminin subunit beta 2 | 3p21 | AR | CNS |
| COL4A3 | α3 type 4 collagen | 2q36‐37 | AR, AD | Alport syndrome, isolated FSGS |
| COL4A4 | α4 type 4 collagen | 2q35‐37 | AR, AD | Alport syndrome, isolated FSGS |
| COL4A5 | α5 type 4 collagen | Xq22 | X‐linked | Alport syndrome, isolated FSGS |
| LMX1B | LIM homeobox nuclear transcription factor 1B | 9q31.1 | AD | Isolated FSGS |
| 2. Syndromic causes of FSGS | ||||
| WT1 b | Wilms' tumor 1 | 11p13 | AD | Frasier syndrome, DDS, Meacham syndrome, isolated diffuse mesangial sclerosis, isolated FSGS, Wilms tumor, WAGR |
| LMX1B b | LIM homeobox nuclear transcription factor 1B | 9q31.1 | AD | Nail‐patella syndrome, isolated FSGS |
| PAX2 b | Paired box 2 nuclear transcription factor | 10q24 | AD | Papillorenal syndrome, isolated FSGS |
| LAMB2 b | Laminin subunit beta 2 | 3p21 | AR | Pierson syndrome |
| COQ2 | Polyprenyltransferase, coenzyme Q2 | 4q21 | AR | Encephalomyopathy, SRNS |
| COQ6 | Ubiquinine biosynthesis monooxygenase COQ6 | 14q24.3 | AR | Childhood FSGS, DMS, deafness |
| PDSS2 | Decaprenyl diphosphate synthase subunit 2 | 6q21 | AR | Encephalomyopathy, SRNS |
| NUP107 b | Nucleoporin, 107‐KD | 12q15 | AR | Galloway‐Mowat syndrome, ovarian dysgenesis, SRNS |
| WDR73 | WD‐repeat containing protein 73 | 15q25 | AR | Galloway‐Mowat syndrome, SRNS |
| OSGEP | O‐sialoglycoprotein endopeptidase | 14q11 | AR | Galloway‐Mowat syndrome, SRNS, FSGS |
| WDR4 | WD‐repeat containing protein 5 | 21q22 | AR | Galloway‐Mowat syndrome, microcephaly, seizures, brain malformations, FSGS |
| LAGE3 | L antigen family member 3 | Xq28 | XL | Galloway‐Mowat syndrome, SRNS, FSGS |
| SGPL1 | Sphingosine‐1‐phosphate lyase‐1 | 10q22 | AR | SRNS with icthyosis and adrenal insufficiency and immunodeficiency |
| MYH9 | Nonmuscle myosin heavy chain IIA | 22q12 | AD | Macrothrombocytopenia, deafness, SRNS, FSGS |
| ITGA3 | Alpha‐3 integrin | 17q21 | AR | Epidermolysis bullosa, interstitial lung disease, congenital nephrotic syndrome, FSGS |
| SMARCAL1 | SWI/SNF related matrix associated, Actin‐dependent regular of chromatin, subfamily A‐like 1 | 2q35 | AR | Schimke immunoosseous dysplasia, SRNS, FSGS |
| 3. High risk for FSGS | ||||
| APOL1 | Apolipoprotein L1 | 22q12.3 | AR a | FSGS, collapsing FSGS |
| 4. Non‐glomerular causes | ||||
| CLCN5 | Chloride channel 5 | Xp11 | XL | Dent disease, FSGS |
| OCRL | Inositol polyphosphate 5‐phosphatase | Xq26 | XL | Dent disease, Lowe syndrome, FSGS |
| UMOD | Uromodulin | 16p12 | AD | ADTKD, FSGS |
| TTC21B | Tetratricopeptide repeat domain‐containing protein 21B | 2q24 | AR | Nephronophthisis, FSGS |
| NPHP4 | Nephrocystin 4 | 1p36 | AR | Nephronophthisis, FSGS |
| CC2D2A | Coiled‐coil and C2 domains‐containing protein 2A | 4p15 | AR | Nephronophthisis, FSGS |
| MTTL1 | Mitochondrial tRNA‐leucine | MtDNA | Mito | MELAS syndrome, FSGS |
Abbreviations: AD, autosomal dominant; ADTKD, autosomal dominant tubulointerstitial kidney disease; AR, autosomal recessive; CNS, congenital nephrotic syndrome; DMS, diffuse mesangial sclerosis; FSGS, focal segmental glomerulosclerosis; SRNS, steroid‐resistant nephrotic syndrome; XL, X‐linked.
Reported in just one pedigree with FSGS. Unclear if this is an FSGS susceptibility gene or a bona fide monogenic cause of FSGS.
Can present with isolated non‐syndromic FSGS.
12.1. Selected genes
12.1.1. NPHS1
NPHS1 (OMIM 602716) encodes nephrin, a transmembrane protein expressed in the podocyte slit diaphragm where it interacts with another nephrin molecule and with podocin and CD2AP. Biallelic null variants in NPHS1 is the most common cause of CNS. A variety of homozygous and compound heterozygous variants in NPHS1 have been reported in CNS. Two founder mutations in NPHS1 labeled Finnmajor and Finnminor cause the Finnish type of CNS. Finnmajor is a frameshifting 2 bp deletion in exon 2 with loss of protein expression while Finnminor is a premature stop gain variant within exon 26 predicting a truncated protein although it is not clear if this variant protein is expressed and functional (Kestila et al., 1998). More recently, NPHS1 biallelic variants were identified in a cohort of sporadic and familial FSGS with at least one presenting as a 27‐year‐old suggesting that the inheritance of at least one hypomorphic NPHS1 allele can lead to childhood or later onset FSGS (Santin, Garcia‐Maset, et al., 2009). Some patients with CNS or FSGS from NPHS1 variants who receive a kidney transplant develop recurrent proteinuria from the development of anti‐nephrin antibodies (Holmberg & Jalanko, 2014).
12.1.2. Podocin ( NPHS2 )
NPHS2 (OMIM 604766) encodes podocin, a transmembrane protein localized to the intercellular junction of podocyte foot processes where it interacts with nephrin (Li & He, 2015). Podocin is necessary for nephrin recruitment to lipid raft domains and podocin variants likely cause disease by disrupting this association (Huber et al., 2003). Biallelic truncating and missense variants were described in a series of patients with familial SRNS but has since been rep in sporadic SRNS as well (Boute, 2000). The most common pathogenic variant described is an R138Q substitution. One variant, the R229Q substitution is found in 3.5% of non‐Finnish Europeans and is pathogenic only when inherited in trans with certain missense variants affecting exons 7 and 8 (Tory et al., 2014). The incidence of sporadic FSGS from biallelic variants in NPHS2 has been reported to be ~13% in various studies. Causal variants in NPHS2 have been detected in adults with FSGS as well (Machuca et al., 2009; Tsukaguchi et al., 2002). One study reported that no patient with homozygous or compound heterozygous NPHS2 mutations had post‐transplant recurrence (Jungraithmayr et al., 2011).
12.1.3. INF2
INF2 (OMIM 610982) encodes Inverted Formin 2, one of the formin family of proteins which is involved in remodeling of actin and microtubule cytoskeleton. INF2 expression happens in various organs including kidney, liver, heart, and the peripheral nervous system. In kidneys, INF2 is expressed predominantly in podocytes. Heterozygous variants in INF2 are usually missense or in‐frame indels and can lead to either renal limited FSGS or Charcot–Marie–Tooth (CMT) disease‐related FSGS (Boyer, Benoit, et al., 2011; Boyer, Nevo, et al., 2011). The CMT phenotype is characterized by hand/foot deformities, absent deep tendon reflexes, progressive distal muscle weakness, and atrophy leading to inability to walk or use hand muscles. White matter changes along with mental retardation and hearing loss are also found in some patients. All variants causing disease appear to localize within the diaphanous inhibitory domain in exons 2–4 (Labat‐de‐Hoz & Alonso, 2020). Renal disease is more severe and occurs earlier in life when associated with CMT disease (Mademan et al., 2013). INF2 is the cause of autosomal dominant FSGS in 12–17% of cases, but, in the presence of CMT, in as many as 75% of the cases of FSGS (Boyer, Nevo, et al., 2011). In this study, the mean age of proteinuria was 18 years with 5 of 12 having classic nephrotic syndrome and ESKD developed in nearly all patients with a mean age of 21 years.
12.1.4. TRPC6
TRPC6 (OMIM 603652) encodes transient receptor potential cationic channel, subfamily C, member 6 which belongs to a family of TRP cationic channels. TRPC6 is a non‐selective cell surface expressed cation channel, activated by diacylglycerol which mediates calcium entry (Hofmann et al., 1999). In the kidney, TRPC6 is primarily expressed in podocytes and is a component of the slit diaphragm (Reiser et al., 2005). Heterozygous missense variants are identified in familial as well as in sporadic FSGS with most affected individuals presenting as young adults (Santin, Ars, et al., 2009). Most of the disease‐causing TRPC6 mutations are activating/gain of function variants causing upregulation of channel activity, with the remaining being loss of function variants with dominant negative activity (Riehle et al., 2016). Overexpression of wild type or mutant trpc6 in mice causes proteinuria with diffuse foot process effacement (Figure 1a,b) with an FSGS phenotype (Krall et al., 2010).
12.1.5. ACTN4
ACTN4 (OMIM 604368) encodes α‐actinin‐4, a widely expressed actin‐binding protein. In the kidney, α‐actinin‐4 is expressed in the podocyte and may play a role in foot process adhesion to the GBM (Smoyer, Mundel, Gupta, & Welsh, 1997). Heterozygous ACTN4 missense variants cause FSGS in approximately 3.5% cases of familial FSGS and <1% of sporadic FSGS (Henderson, Alexander, & Pollak, 2009; Weins et al., 2005). Disease‐causing variants appear to demonstrate increased affinity for F‐actin suggestive of a gain of function mechanism (Kaplan et al., 2000; Weins et al., 2007). Interestingly, ACTN4 knockout mice developed glomerular disease with altered podocyte morphology and foot process effacement suggesting that both gain of function and loss of function can cause a similar phenotype. In humans, typical disease onset is in adulthood with variable degrees of proteinuria and slow progression to ESKD. Kidney biopsies show LM findings typical of FSGS and variable effacement of foot processes on EM (Henderson et al., 2009). Thickening and irregularity of the lamina densa of the GBM and irregularly aggregated electron‐dense material within the podocyte cytoplasm may also be seen.
12.1.6. CD2AP
CD2AP (OMIM 60421) encodes CD2‐associated protein, an adaptor protein that regulates the actin cytoskeleton and plays a role in cytoskeletal remodeling, cell motility, and cell survival (Kirsch, Georgescu, Ishimaru, & Hanafusa, 1999; Shih et al., 1999). CD2AP binds to nephrin and podocin forming a component of the slit diaphragm (Schwarz et al., 2001; Shih et al., 2001). CD2AP‐associated FSGS was first reported in two unrelated adults where a heterozygous variant predicted to disrupt splicing was identified (Kim et al., 2003). Three additional patients with heterozygous missense or in‐frame indel variants in CD2AP were identified in an Italian cohort with sporadic FSGS (Gigante et al., 2009). A young child of consanguineous parents who developed FSGS carried a homozygous, predicted null CD2AP stop gain variant in the penultimate coding exon (Lowik et al., 2007). A second report of three offspring of consanguineous parents all of whom inherited homozygous frame shift mutations clearly established that recessive loss of function variants in CD2AP causes FSGS (Takano et al., 2019). Mice lacking CD2AP develop CNS or FSGS while haploinsufficiency increased susceptibility to glomerular injury from nephrotoxins (Kim et al., 2003; Shih et al., 1999; Takano et al., 2019). It is likely that haploinsufficiency for CD2AP is merely a risk factor for FSGS and that two loss of function variants are required to develop monogenic disease.
12.1.7. MYO1E
MYO1E (OMIM 601479) encodes for a non‐muscle class 1 myosin, myosin 1E (Myo1E) that is expressed in the podocyte plasma membrane and appears to bind the tips of F‐actin and regulate the foot process cytoskeleton (Mele et al., 2011). Homozygous loss of function mutations in MYO1E in two different pedigrees were first reported in 2011 (Mele et al., 2011). All reported cases are inherited in an autosomal recessive pattern, with most presenting as childhood onset SRNS, the youngest reported case being in a 6‐month‐old (Al‐Hamed et al., 2013; Guaragna et al., 2020; Lennon et al., 2015). The light microscopic biopsy findings are consistent with FSGS, although some rare cases showed a minimal change disease pattern (Al‐Hamed et al., 2013). EM may reveal a thickened and disorganized GBM and effacement of foot processes. While most patients progress to ESKD within 3–10 years of diagnosis, some remain stable until early adulthood. MYO1E knockout mice also exhibit proteinuria with glomerulosclerosis and foot process effacement (Krendel et al., 2009).
12.1.8. ANLN
ANLN (OMIM 616027) encodes an F‐actin‐binding cytoskeletal protein anillin, which is involved in the cell cycle. Anillin is ubiquitously expressed in all organs but is very sparsely expressed in normal podocytes on immunohistochemistry (Gbadegesin et al., 2014). In the podocyte, anillin associates with CD2AP. Heterozygous mutations in ANLN causing autosomal dominant FSGS were first reported in 2014 in two kindred and expression studies suggested the mutant protein showed reduced binding to CD2AP and reduced podocyte migration. Zebrafish with ANLN knockdown developed complete effacement of podocyte foot processes and edema (Gbadegesin et al., 2014).
12.1.9. MYO9A
MYO9A (OMIM 604875) encodes myosin 9A, a non‐muscle myosin with a Rho‐GAP domain that binds F‐actin. MYO9A mRNA is expressed in multiple tissues including kidneys and brain (Gorman et al., 1999). In kidneys MYO9A is expressed in podocytes and colocalizes with F‐actin and podocin and reduces active RhoA. A heterozygous nonsense variant, R701X, in MYO9A was recently identified in a sibling pair as a cause of monogenic FSGS (Li et al., 2021). Kidney biopsy of the index case showed FSGS with progressive decline in renal function while the sibling's proteinuria and kidney function remained stable. Exome sequencing identified heterozygous MYO9A VUS in a cohort of childhood and young adult FSGS. Mice carrying a single copy of the R701X variant were normal at birth but developed proteinuria with FSGS and foot process effacement recapitulating the human phenotype (Li et al., 2021).
12.1.10. TRIM8
TRIM8 (OMIM 606125) encodes tripartite motif containing 8 (TRIM8), an E3 ubiquitin ligase that is a member of the TRIM family of proteins. TRIM8 is highly expressed in kidneys, CNS, and lens of the eye (Reymond et al., 2001). TRIM8 promotes degradation of the suppressor of cytokine signaling 1 protein (SOCS1) and regulates interferon‐gamma and NF‐kB signaling pathways (Toniato et al., 2002). While heterozygous truncating TRIM8 variants were first reported in a patient with early onset developmental delay and seizures, the clinical spectrum was expanded to include childhood onset FSGS (Assoum et al., 2018; Sakai et al., 2016). Subsequently, heterozygous variants in TRIM8 were identified in eight patients from a large cohort of childhood FSGS/SRNS and in three patients from a cohort of patients with epilepsy indicating that the same TRIM8 variants cause a renal and neuronal phenotype (Weng et al., 2021). Thus far, just one patient with a TRIM8 variant has been reported with nephrotic syndrome without any CNS manifestation (Shirai et al., 2021). All reported patients have de novo truncating variants in the last exon suggesting that variant proteins are expressed and have a dominant negative effect. Expression of causal variants in human podocytes showed mislocalization to the nucleoplasm. Patients present with SRNS or FSGS on biopsy, rapidly progressing to ESKD with no reported recurrence post‐transplant (Weng et al., 2021).
12.1.11. COL4A3 , COL4A4 , COL4A5 (Alport genes)
COL4A3, COL4A4, and COL4A5 encode the alpha 3 (α3[IV]), alpha 4 (α4[IV]), and alpha 5 (α5[IV]) chains of type IV collagen that together form a heterotrimer and are present in the glomerular basement membrane. These collagen chains have a short N‐terminal 7S domain, a long triple helical collagenous domain consisting of Gly‐X‐Y repeats and a C‐terminal globular non‐collagenous domain (Hudson, Tryggvason, Sundaramoorthy, & Neilson, 2003). All three collagen chains must interact at the NC1 domain and assemble as a triple helix along the collagenous domain to form the collagen network of the GBM. COL4A5 is on the X‐chromosome and COL4A3 and COL4A4 are expressed head‐to‐head on chromosome 2 and these have long been known to cause Alport syndrome inherited in an X linked or autosomal dominant or autosomal recessive manner. In its classic form Alport syndrome typically presents with the renal manifestations of hematuria, proteinuria, and progressive decline in kidney function and the extrarenal manifestations of sensorineural deafness and ocular abnormalities such as anterior lenticonus and dot and fleck retinopathy (Kashtan, 1993). Not all patients have extrarenal or syndromic manifestations and it may be more accurate to call the renal disease Alport nephropathy. Microscopic hematuria is universal in males with X‐linked Alport nephropathy and is very common in other forms of Alport nephropathy and is often the first sign and observed in early childhood. Proteinuria gradually worsens as disease progresses. The majority of men with X‐linked Alport nephropathy have ESKD by their 40s while women present as heterozygous carriers or with milder disease with lower and slower rates of progression. The variants seen in X‐linked Alport nephropathy include missense variants, large deletions, and null variants including small indels. Males with hemizygous null variants were more likely to develop early onset of ESKD and manifest deafness and ocular abnormalities compared to those with missense variants (Bekheirnia et al., 2010).
A smaller number of patients have autosomal recessive disease and present with early and progressive disease. With increased genetic testing, it has become apparent that a significant number of patients with monoallelic variants in COL4A3, COL4A4 and a few with COL4A5 present with non‐syndromic renal disease ranging from isolated microscopic hematuria to varying degrees of proteinuria to more advanced CKD. The EM findings on renal biopsy are abnormalities of the GBM ranging from diffuse thinning to variable splitting and or thickening of the GBM and variable degrees of foot process effacement (Figure 1c,d). The majority of patients with heterozygous variants in COL4A3 or COL4A4 Alport may only have isolated persistent microscopic hematuria with diffuse thinning of the GBM which was previously called thin basement membrane disease and assumed to be benign although it is now clear that some patients develop progressive disease (Matthaiou, Poulli, & Deltas, 2020; Savige et al., 2021). Some patients with Alport nephropathy develop FSGS on biopsy, a form of secondary FSGS, and COL4 variants are now known to be the commonest cause of genetic FSGS in adults (Gast et al., 2016; Papazachariou et al., 2017).
Genotype–phenotype correlations were attempted in a recent systematic review of over 700 published cases of patients with heterozygous COL4A3 and COL4A4 variants, almost all of whom had microscopic hematuria (Matthaiou et al., 2020). Contrary to widely held beliefs, 46% of patients with single variants had proteinuria in addition to hematuria, 29% had a decline in eGFR and 15% had ESKD, although publication bias may have led to preferential reporting of more severe cases. Many patients presented with missense variants, typically substituting glycine (Gly) in one of the Gly‐X‐Y repeats, compared to null variants. Missense variants causing glycine substitution in one subunit appear to reduce the structural stability of the heterotrimer and reduce the binding of collagen to integrin indicative of a dominant negative effect while null variants indicate that haploinsufficiency is also a mechanism for autosomal dominant Alport nephropathy (Yeo et al., 2020).
12.1.12. The special case of APOL1
Advanced CKD has long been known to occur at a disproportionately higher rate in those with sub‐Saharan African ancestry compared to that of other ancestries. In 2010, the high‐risk alleles (termed G1 and G2) of the apolipoprotein L1 (APOL1) gene on chromosome 22 were found to account for a significant portion of this risk (Genovese et al., 2010; Tzur et al., 2010). Inheritance of two copies of the high‐risk alleles (G1/G1, G1/G2, or G2/G2) substantially increases the risk of FSGS and global glomerulosclerosis in people with hypertension and with certain infections like HIV, CoVID 19 and malaria (Amoura et al., 2020; Kruzel‐Davila et al., 2017; Shetty et al., 2021). The high‐risk APOL1 genotype (G1/G1, G1/G2, or G2/G2) is present in as many as 75% of those with African ancestry who have FSGS, with a significant fraction having an aggressive, treatment‐resistant form of collapsing FSGS (Genovese et al., 2010; Kopp et al., 2011). The exact mechanism by which APOL1 causes or facilitates kidney disease in humans remains unknown although animal models suggest recessive toxic gain of function in the expressed podocyte (Beckerman et al., 2017; McCarthy et al., 2021).
Population‐based studies such as REGARDS (REasons for Geographic and Racial Differences in Stroke) demonstrate that the high‐risk genotype increases the risk of ESKD only about twofold (Naik et al., 2017). Clearly most with two copies of the high‐risk APOL1 alleles do not develop FSGS, or other kidney disease and it is likely that HTN or certain infections or other environmental factors are the required “second hit” for the development and/or progression of disease. Many patients of African ancestry are presumed to have hypertensive nephrosclerosis by clinicians, when in fact they could have undiagnosed hypertension‐related APOL1‐associated FSGS. Healthcare disparities including delay in referral to a nephrologist, and lack of biopsy and genetic testing can contribute to a missed or delayed diagnosis in these individuals.
12.2. Syndromic causes of FSGS
12.2.1. WT1
WT1 (OMIM 607102) encodes a zinc‐finger tumor protein that functions as a transcriptional activator or repressor and plays a crucial role in urogenital development. The male sex determining gene SRY is one of its important transcriptional targets (Hossain & Saunders, 2001). One of the two principal WT1 transcripts is an alternately spliced isoform that has an additional three amino acids (+KTS) between exons 8 and 9. Heterozygous variants in WT1 have been identified in patients with isolated Wilms tumor, with isolated nephrotic syndrome, and with Denys–Drash syndrome (DDS) and Frasier syndromes. WT1 kidney disease may be diffuse mesangial sclerosis (DMS) in early childhood and FSGS in those that present later. DDS is a syndrome characterized by early onset SRNS, progressing to ESKD together with ambiguous genitalia and risk of early onset Wilms tumor (Lipska‐Zietkiewicz, 1993). Generally, de novo, heterozygous, missense, or premature truncation variants of WT1 is identified in DDS. Patients with DDS may need bilateral nephrectomies to prevent Wilms tumor formation or be monitored frequently until age 8 to detect and treat early tumors.
Frasier syndrome is characterized by steroid‐resistant FSGS in early childhood with disorders of sexual development in males and high risk of gonadoblastoma in adolescence. It has a relatively slower progressive course of renal failure as compared to DDS and frequently is diagnosed when a phenotypic female is evaluated for primary amenorrhea and found to have a 46XY karyotype. Patients with Frasier syndrome require gonadectomy due to high risk of developing gonadoblastoma in the streak gonad. Frasier syndrome usually arises from a variant in intron 9 that alters the ratio of alternate transcripts that include or exclude KTS (+KTS and −KTS). DDS can be recapitulated in mice that express a single copy of a zinc finger truncated DDS mutant in mice (Patek et al., 1999). Likewise, heterozygous mice with reduced expression of the +KTS isoform develop glomerulosclerosis while homozygous mice lacking +KTS have complete XY sex reversal (Hammes et al., 2001). However, the distinction between DDS and Frasier may not be absolute as DDS type variants have been found in some patients with Frasier syndrome. Identifying a WT1 variant during the assessment of SRNS or FSGS in children should prompt an evaluation of sexual development and screening for or preemptive management to prevent the development of nephroblastoma or gonadoblastoma (Lipska‐Zietkiewicz, 1993).
12.2.2. LMX1B
LMX1B (OMIM 602575) encodes LIM homeobox transcription factor 1 beta, a member of the LIM‐homeodomain family of transcription factors with two zinc finger motifs. LMX1B gene is located on chromosome 9q34 and is strongly expressed in human fetal and adult kidneys and in dorsal mesenchymal tissues and anterior limb structures during mouse embryonic development (Dreyer et al., 1998; Dreyer et al., 2000). Nail patella syndrome (NPS) or onycho‐osteodysplasia is characterized by dysplastic nails, absent patellae, iliac horns, and nephropathy (Beals & Eckhardt, 1969). Skeletal involvement in NPS is varied with absent, hypoplastic, or dystrophic, pitted nails; and absent, small, or irregularly shaped patella with recurrent subluxation. Heterozygous de novo missense and null variants were first identified in three patients with NPS (Dreyer et al., 1998). Subsequently, heterozygous LMX1B variants were identified in 37 of 41 families with NPS and most mutations were null mutations suggesting that haploinsufficiency was a mechanism of disease (McIntosh et al., 1998). LMX1B null mice have absent patella, hypoplastic nails, and a form of renal dysplasia thus recapitulating the human phenotype (Chen et al., 1998). LMX1B binds cis‐acting elements in intron 1 of COL4A4 and can stimulate expression of reporter constructs containing this putative enhancer sequence (Morello et al., 2001). LMX1B null mice also show marked reduction in expression of α3(IV) and α4(IV) collagen in GBM, suggesting that LMX1B may be required for normal GBM development and when dysregulated may contribute to the renal pathology in NPS (Chen et al., 1998). Recently, missense variants in LMX1B were identified in three families with isolated autosomal dominant FSGS without nail or skeletal manifestations suggesting that variants in LMX1B can cause non‐syndromic FSGS (Boyer et al., 2013).
12.2.3. LAMB2
LAMB2 (OMIM 150325) encodes the β2 subunit of laminin, a constituent of the GBM. Laminins are heterotrimeric matrix proteins composed of α, β, and γ subunits and of these Laminin‐521, a heterotrimer of laminins (α5, β2, and γ1) is expressed in the glomerular basement membrane along with collagen type IV isoforms α3, α4, and α5, nidogen, and sulfated proteoglycan. Laminin 521 is also found in ocular structures and the neuromuscular system. Homozygous or compound heterozygous null variants in LAMB2 cause Pierson syndrome, clinical features being CNS often rapidly progressing to ESKD, and severe ocular, and neuromuscular defects (Matejas et al., 2010). The ocular manifestations include microcoria, hypoplasia of the ciliary body, and blindness and the neurological abnormalities include hypotonia, areflexia, and psychomotor retardation. Renal limited disease or those with an added mild ocular phenotype are seen with less severe missense variants that are predicted to diminish but not eliminate protein function (Hasselbacher et al., 2006; Mohney et al., 2011). Kidney biopsy shows DMS with severe podocyte foot process effacement and irregular thickening of the GBM on EM and absent beta2 laminin staining on immunofluorescence (VanDeVoorde, Witte, Kogan, & Goebel, 2006).
12.2.4. Coenzyme Q‐related nephropathies
Coenzyme Q10 (CoQ10, ubiquinone) plays an essential role in oxidative phosphorylation in mitochondria by shuttling electrons from Complex 1 and II to complex III but it also functions as an antioxidant and co‐factor for mitochondrial dehydrogenases and in nucleoside biosynthesis (Ozaltin, 2014). The CoQ10 synthesis pathway involves at least 16 enzymes including COQ2‐COQ7, COQ8A, COA8B, COQ9, COQ10A, COQ10B, PDSS1, PDSS2, FDX1L, FDXR, ALDH3A1 (Tan & Airik, 2021). Primary CoQ10 deficiency is seen with genetic variants in several genes including COQ2, COQ4, COQ6, COQ7, COQ8A/ADCK3, COQ8B/ADCK4, COQ9, PDSS1, and PDSS2. Of these biallelic variants in COQ2, COQ6, ADCK4, and PDSS2 are associated with glomerular involvement with proteinuria and progressive renal disease and are known as the CoQ10 nephropathies (Schijvens et al., 2020). The CoQ10 nephropathies can cause isolated SRNS or be associated with other phenotypic features such as encephalomyopathy, sensorineural hearing loss, seizures, and developmental abnormalities. Most affected patients with PDSS2, COQ2, COQ6 are infants or very young children while the median age for ADCK4 is 12 years. In case reports, most affected patients were children or adolescents (Schijvens et al., 2020). Between 40 and 60% of these patients showed some improvement in clinical condition with high‐dose COQ10 supplementation.
12.2.5. SMARCAL1
SMARCAL1 (OMIM 606622) encodes for a protein involved in chromatin remodeling, SWI/SNF related matrix associated, actin‐dependent regular of chromatin, subfamily A‐like 1 protein. Biallelic variants in SMARCAL1 were identified as the cause in 16 of 23 cases of Schimke immune‐osseous dysplasia (SIOD) (Boerkoel et al., 2002). SIOD is an autosomal recessive disorder with multisystem involvement characterized by spondyloepiphyseal dysplasia, growth failure, T cell immunodeficiency, and pre‐pubertal onset SRNS/FSGS causing progressive CKD (Boerkoel et al., 2000; Saraiva et al., 1999; Spranger et al., 1991). The disease severity appears to inversely correlate with the degree of residual SMARCAL1 activity (Elizondo et al., 2009). In one report prior to the identification of the genetic basis of disease, a majority (27 of 39 patients) had FSGS on biopsy which was resistant to steroids/cyclosporine/cyclophosphamide (Boerkoel et al., 2000). Twenty‐two of 39 progressed to ESKD within 9 months to 11 years of diagnosis. Half the patients with ESKD received a transplant in that report and none had recurrent FSGS in the allograft.
12.3. Non‐glomerular causes of genetic FSGS
12.3.1. Dent disease ( CLCN5 , OCRL )
Dent disease is an X‐linked inherited proximal tubulopathy typically manifesting in childhood or early adulthood with low molecular weight proteinuria, nephrocalcinosis, hypercalciuria, and nephrolithiasis causing progressive renal failure and ESKD in the third–fifth decade of life. CLCN5 (OMIM 300008) encodes a voltage‐gated ion channel of the CLC family and accounts for 60–65% of cases (Dent disease type 1), while OCRL (OMIM 300535) which encodes a phosphatidylinositol 4,5‐bisphosphate‐5‐phosphatase account for 10–15% cases (Dent disease type 2) and another 20–25% do not have identified variants in either gene and are classified as Dent disease type 3 (Anglani et al., 2015; Hoopes et al., 2004; Wang et al., 2016). CLCN5 is expressed in proximal tubule, collecting duct and epithelial cells of thick ascending limb (TAL) and is involved in acidification of early endosomes (Devuyst, Christie, Courtoy, Beauwens, & Thakker, 1999; Gunther, Luchow, Cluzeaud, Vandewalle, & Jentsch, 1998; Lloyd et al., 1996). OCRL localizes mainly in endoplasmic reticulum, is widely distributed in human tissues, and its exact role in causing disease is not yet clear. Inactivating variants in either gene is responsible for Dent disease in hemizygous males. Dent disease type 2 is allelic with a more severe phenotype called Lowe syndrome which is characterized by ocular abnormalities (prenatal dense congenital cataracts, glaucoma, microphthalmia, decreased visual acuity), neonatal hypotonia, areflexia, and intellectual impairment (Bezdicka, Langer, Hacek, & Zieg, 2020; Hichri et al., 2011). Dent disease type 2 arises from truncating variants in exon 1 through 7 while the Lowe phenotype arises from variants in exons 8 through 24.
The proteinuria in Dent disease is primarily tubular but can reach nephrotic range and is sometimes mistaken for a glomerular phenotype. However, FSGS was reported in 7% and focal global glomerulosclerosis in 83% of patients with a diagnosis of Dent disease 1 (Wang et al., 2016). In the same study, foot process effacement was reported in 57% of patients, being mild and segmental in most of them. No clear explanation exists for the podocyte injury.
12.3.2. UMOD
UMOD (OMIM 191845) encodes for tubular protein uromodulin, previously known as Tamm‐Horsfall glycoprotein. Heterozygous variants in UMOD are the most frequent cause of autosomal dominant tubulointerstitial kidney disease (ADTKD). Previously presumed to have a low prevalence, one study found UMOD to be the most common cause of inherited kidney disease after ADPKD in those with Stages 3–5 CKD (Gast et al., 2018). In another study, UMOD variants were found to be responsible for 3% of cases amongst a cohort of 3,315 patients with CKD (Groopman, Goldstein, & Gharavi, 2019). While the classic clinical features of UMOD‐ADTKD are bland urine, minimal to absent proteinuria, normal‐sized kidneys sometimes with a few corticomedullary cysts, and hyperuricemia with or without a history of juvenile gout, biopsies of individuals with confirmed UMOD identified FSGS in 21% of them, none of whom had nephrotic range proteinuria (Chun et al., 2020).
12.3.3. Nephronophthisis genes ( TTC21B , NPHP4 , CC2D2A )
Nephronophthisis is an autosomal recessive inherited tubulointerstitial kidney disease classically presenting in early childhood with polyuria, polydipsia, poor growth, bland urine, and progressive decline in kidney function and ESKD in the second–third decade of life. This disease is classified as a ciliopathy as variants that cause nephronophthisis come from genes that encode ciliary proteins. Patients tend to have normal to slightly smaller kidneys with occasional renal cysts. About 20% of patients have extra‐renal features including oculomotor involvement (Cogan's oculomotor apraxia) and pigmentary retinal dystrophy (Senior Loken syndrome) and others have more extensive CNS involvement (Joubert syndrome, Meckel‐Gruber syndrome) or hepatic fibrosis and cerebral involvement (COACH syndrome) (Braun & Hildebrandt, 2017; Mistry, Ireland, Ng, Henderson, & Pollak, 2007; Srivastava, Molinari, Raman, & Sayer, 2017). Biallelic variants in at least 20 different genes are now known to cause nephronophthisis with or without extrarenal features (Stokman, Lilien, & Knoers, 1993). Of these NPHP1 is the commonest cause of nephronophthisis and the most prevalent genetic variant of NPHP1 is a homozygous deletion of the whole gene (Konig et al., 2017; Zhang et al., 2018).
Of the many reported genetic causes of nephronophthisis, at least three have been reported to also present with FSGS. TTC21B encodes for ciliary protein IFT139, a component of intra‐flagellar transport (IFT) complex required for retrograde ciliary transport (Tran et al., 2008). The same homozygous missense variant in TTC21B, p.P209L, was found in seven French families with FSGS and in two additional families from Spain (Bullich et al., 2017; Huynh Cong et al., 2014). NPHP4 presenting with FSGS was reported in just one consanguineous family where multiple siblings presented with advanced renal disease and proteinuria and carried the same homozygous NPHP4 missense variants although only one patient was biopsied to confirm FSGS (Mistry et al., 2007). CC2D2A is the third ciliary gene that has been reported to cause FSGS and this was in a single patient with heavy proteinuria and hypoalbuminemia where compound heterozygous missense variants were identified (Awazu et al., 2022). Renal biopsy revealed FSGS with focal foot process effacement. The mechanism of FSGS with nephronophthisis genes is unknown but may simply represent a secondary adaptive response to nephron loss and any of the nephronophthisis genes may cause this type of injury. Interestingly IFT139 is expressed at the base of the primary cilium of developing podocytes and knockdown of IFT139 leads to abnormal podocyte migration and cytoskeletal alterations in culture and may explain why TTC21B may be associated with FSGS (Huynh Cong et al., 2014).
12.3.4. PAX2
PAX2 (OMIM 167409) encodes a transcription factor Paired Box gene 2 which plays a role in embryonic kidney, ocular and brain development. A heterozygous frameshift deletion in PAX2 was first reported in a family with optic nerve coloboma, vesicoureteric reflux, and renal hypoplasia (Sanyanusin et al., 1995). Subsequently, a large number of reports have established that PAX2 variants cause papillorenal syndrome (previously called renal‐coloboma syndrome) which consists of any one of a variety of CAKUT (congenital abnormalities of the kidney and urinary tract) ranging from vesicoureteric reflux to renal hypoplasia to multicystic dysplastic kidneys combined with ocular abnormalities (Bower et al., 2012). Some patients can present with renal abnormalities without any ocular findings (Barua et al., 2014). PAX2 was first reported to associate with FSGS with the report of an index family with FSGS, where PAX2 variant segregated with disease and it was subsequently identified in 4% of a familial FSGS cohort (Barua et al., 2014). Most PAX2 variants that cause FSGS appear to be missense and outside the transactivation domain and likely function as dominant negative whereas PAX2 variants that cause papillorenal syndrome are generally truncating variants establishing haploinsufficiency as a mechanism of disease although there is intrafamilial variability in presentation. How PAX2 causes FSGS is unclear but PAX2 is known to regulate WT1 which is important for podocyte development and may therefore contribute to FSGS via WT1. Alternatively, FSGS may be maladaptive and secondary to reduction in nephron mass from CAKUT.
13. OVERVIEW OF NON‐GENETIC CAUSES OF FSGS
13.1. Idiopathic/autoimmune FSGS
Idiopathic FSGS has been hypothesized to be secondary to a circulating permeability factor. This hypothesis is tenable given the numerous reports of FSGS recurrence occurring hours to days following unrelated kidney transplants (Chang et al., 2012). Furthermore, in some situations, plasmapheresis appears to induce remission once post‐transplant recurrence occurs. Anecdotally, a transplanted kidney with early recurrence was successfully retransplanted into a different patient without the development of proteinuria in the new host (Gallon, Leventhal, Skaro, Kanwar, & Alvarado, 2012). A number of circulating factors believed to be responsible have been proposed including soluble urine‐type plasminogen activator (suPAR), and apoA1b. Recently, autoantibodies to Annexin A2 were identified in 18% of a large cohort of children with primary nephrotic syndrome which included patients with minimal change disease and FSGS (Ye, Zhang, et al., 2021). Reduction of antibodies with immunosuppression correlated with an improvement in albuminuria and consistent with a causal role, these antibodies induced proteinuria when injected into BALB/c mice. Although there are no rigorous randomized controlled trials, plasmapheresis with or without rituximab has become standard of care for post‐transplant recurrence of FSGS. Yet plasmapheresis is rarely employed to manage primary FSGS with nephrotic syndrome when it occurs in the native kidneys.
13.2. Infection‐induced FSGS
Some infections cause a type of FSGS called collapsing FSGS (cFSGS). The most common infections associated with FSGS are HIV, malaria, and CoVID‐19 (Greze et al., 2018; Laboux, Gibier, Pottier, Glowacki, & Hamroun, 2021; Shetty et al., 2021). The mechanism is believed to be direct podocyte injury, best studied with HIV‐associated nephropathy (HIVAN) where HIV gene expression in podocytes leads to development of HIVAN (Zhong et al., 2005). More recently several case reports have described HIVAN‐like presentation with cFSGS on biopsy, rapidly declining renal function, nephrotic range proteinuria in native and transplant patients with SARS‐CoV‐2 infection, referred to as COVAN (Lazareth et al., 2020). Most if not all patients who develop infection‐related cFSGS carry two risk alleles for APOL1 (Muehlig, Gies, Huber, & Braun, 2021).
13.3. Drug‐induced FSGS
Various medications are known to cause both collapsing and non‐collapsing type FSGS from a variety of mechanisms, but most common postulated reason being direct podocyte injury from drugs like IFN and bisphosphonates. Sirolimus can present with nephrotic range proteinuria and de novo FSGS lesions have been seen on biopsy (Letavernier et al., 2007). Similarly, androgenic anabolic steroids can cause FSGS presenting as AKI with nephrotic proteinuria (Herlitz et al., 2010). The tumor necrosis factor‐alpha (TNF‐α) inhibitor infliximab has also been reported to cause FSGS in a patient who was found to have asymptomatic nephrotic range proteinuria and hematuria on routine follow up (Yarkan Tugsal et al., 2019).
13.4. Maladaptive/hyperfiltration injury
Maladaptive FSGS is typically a result of mismatch between the glomerular filtration load and the number of functioning nephrons, occurring in those with genetic and non‐genetic etiologies and occurring secondary to glomerular hyperfiltration/hypertrophy. Podocytes have limited tensile strength and when exposed to high filtration rate, may be susceptible to podocyte detachment and foot process effacement. This foot process effacement may be limited and uneven but predisposes to the development of segmental sclerosis. Some have posited that FPE may be a protective phenomenon to prevent podocyte detachment by re‐distributing the mechanical forces (De Vriese et al., 2018). Glomerulomegaly is a common feature, with clinical presentation being sub‐nephrotic range proteinuria and sometimes, gradually declining renal function.
14. SUMMARY
FSGS is a nonspecific term that refers to a pattern of glomerular histological and ultrastructural changes that may be a direct result of podocyte injury or the indirect result of a deleterious process elsewhere in the kidney and may occur from idiopathic, genetic, or other secondary causes. The approach to an etiological evaluation and treatment is often based on the degree of podocyte foot process effacement and the clinical presentation, although there is limited evidence to justify this approach. In this regard, there are many monogenic causes of FSGS with variable presentation from nephrotic syndrome with histological features of primary podocytopathy to more modest degrees of proteinuria with limited evidence of podocyte foot process injury. Yet genetic causes are largely underdiagnosed, as the role and the timing of genetic testing in FSGS is not established and genetic counseling, testing options, and interpretation of genotype in the context of phenotype may be outside the scope of practice for both nephrologists and geneticists. In this review, we have provided an overview of the current definition and understanding of FSGS, propose a new etiopathogenic classification that recognizes the heterogeneity of genetic FSGS and offer an algorithmic framework for approaching the management of FSGS that is informed by genetic testing.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Table S1 Genetic causes of FSGS (sorted by mode of inheritance).
ACKNOWLEDGMENT
The authors thank Sonali Gupta for her assistance with the manuscript.
Sambharia, M. , Rastogi, P. , & Thomas, C. P. (2022). Monogenic focal segmental glomerulosclerosis: A conceptual framework for identification and management of a heterogeneous disease. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 190C:377–398. 10.1002/ajmg.c.31990
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Al‐Hamed, M. H. , Al‐Sabban, E. , Al‐Mojalli, H. , Al‐Harbi, N. , Faqeih, E. , Al Shaya, H. , … Meyer, B. F. (2013). A molecular genetic analysis of childhood nephrotic syndrome in a cohort of Saudi Arabian families. Journal of Human Genetics, 58(7), 480–489. 10.1038/jhg.2013.27 [DOI] [PubMed] [Google Scholar]
- Amoura, A. , Moktefi, A. , Halfon, M. , Karras, A. , Rafat, C. , Gibier, J. B. , … Audard, V. (2020). Malaria, collapsing glomerulopathy, and focal and segmental glomerulosclerosis. Clinical Journal of the American Society of Nephrology, 15(7), 964–972. 10.2215/CJN.00590120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglani, F. , D'Angelo, A. , Bertizzolo, L. M. , Tosetto, E. , Ceol, M. , Cremasco, D. , … Network, D. D. I. (2015). Nephrolithiasis, kidney failure and bone disorders in Dent disease patients with and without CLCN5 mutations. Springerplus, 4, 492. 10.1186/s40064-015-1294-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf, S. , Gee, H. Y. , Woerner, S. , Xie, L. X. , Vega‐Warner, V. , Lovric, S. , … Hildebrandt, F. (2013). ADCK4 mutations promote steroid‐resistant nephrotic syndrome through CoQ10 biosynthesis disruption. The Journal of Clinical Investigation, 123(12), 5179–5189. 10.1172/JCI69000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assoum, M. , Lines, M. A. , Elpeleg, O. , Darmency, V. , Whiting, S. , Edvardson, S. , … Faivre, L. (2018). Further delineation of the clinical spectrum of de novo TRIM8 truncating mutations. American Journal of Medical Genetics Part A, 176(11), 2470–2478. 10.1002/ajmg.a.40357 [DOI] [PubMed] [Google Scholar]
- Awazu, M. , Yamada, M. , Asada, N. , Hashiguchi, A. , Kosaki, K. , & Matsumura, K. (2022). A girl with a mutation of the ciliary gene CC2D2A presenting with FSGS and nephronophthisis. CEN Case Reports, 11(1), 116–119. 10.1007/s13730-021-00640-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barua, M. , & Paterson, A. D. (2021). Population‐based studies reveal an additive role of type IV collagen variants in hematuria and albuminuria. Pediatric Nephrology, 37(2), 253–262. 10.1007/s00467-021-04934-y [DOI] [PubMed] [Google Scholar]
- Barua, M. , Stellacci, E. , Stella, L. , Weins, A. , Genovese, G. , Muto, V. , … Pollak, M. R. (2014). Mutations in PAX2 associate with adult‐onset FSGS. Journal of the American Society of Nephrology, 25(9), 1942–1953. 10.1681/ASN.2013070686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beals, R. K. , & Eckhardt, A. L. (1969). Hereditary onycho‐osteodysplasia (nail‐patella syndrome). A report of nine kindreds. The Journal of Bone and Joint Surgery. American Volume, 51(3), 505–516. [PubMed] [Google Scholar]
- Beckerman, P. , Bi‐Karchin, J. , Park, A. S. , Qiu, C. , Dummer, P. D. , Soomro, I. , … Susztak, K. (2017). Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nature Medicine, 23(4), 429–438. 10.1038/nm.4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekheirnia, M. R. , Reed, B. , Gregory, M. C. , McFann, K. , Shamshirsaz, A. A. , Masoumi, A. , & Schrier, R. W. (2010). Genotype‐phenotype correlation in X‐linked Alport syndrome. Journal of the American Society of Nephrology, 21(5), 876–883. 10.1681/ASN.2009070784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezdicka, M. , Langer, J. , Hacek, J. , & Zieg, J. (2020). Dent disease type 2 as a cause of focal segmental glomerulosclerosis in a 6‐year‐old boy: A case report. Frontiers in Pediatrics, 8, 583230. 10.3389/fped.2020.583230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierzynska, A. , McCarthy, H. J. , Soderquest, K. , Sen, E. S. , Colby, E. , Ding, W. Y. , … Saleem, M. A. (2017). Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney International, 91(4), 937–947. 10.1016/j.kint.2016.10.013 [DOI] [PubMed] [Google Scholar]
- Boerkoel, C. F. , O'Neill, S. , Andre, J. L. , Benke, P. J. , Bogdanovic, R. , Bulla, M. , … Weksberg, R. (2000). Manifestations and treatment of Schimke immuno‐osseous dysplasia: 14 new cases and a review of the literature. European Journal of Pediatrics, 159(1–2), 1–7. 10.1007/s004310050001 [DOI] [PubMed] [Google Scholar]
- Boerkoel, C. F. , Takashima, H. , John, J. , Yan, J. , Stankiewicz, P. , Rosenbarker, L. , … Stockton, D. W. (2002). Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno‐osseous dysplasia. Nature Genetics, 30(2), 215–220. 10.1038/ng821 [DOI] [PubMed] [Google Scholar]
- Boute, N. (2000). NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid‐resistant nephrotic syndrome (vol 24, pg 349, 2000). Nature Genetics, 25(1), 125. [DOI] [PubMed] [Google Scholar]
- Bower, M. , Salomon, R. , Allanson, J. , Antignac, C. , Benedicenti, F. , Benetti, E. , … Heidet, L. (2012). Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus‐specific database. Human Mutation, 33(3), 457–466. 10.1002/humu.22020 [DOI] [PubMed] [Google Scholar]
- Boyer, O. , Benoit, G. , Gribouval, O. , Nevo, F. , Tete, M. J. , Dantal, J. , … Antignac, C. (2011). Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. Journal of the American Society of Nephrology, 22(2), 239–245. 10.1681/ASN.2010050518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer, O. , Nevo, F. , Plaisier, E. , Funalot, B. , Gribouval, O. , Benoit, G. , … Mollet, G. (2011). INF2 mutations in Charcot‐Marie‐Tooth disease with glomerulopathy. The New England Journal of Medicine, 365(25), 2377–2388. 10.1056/NEJMoa1109122 [DOI] [PubMed] [Google Scholar]
- Boyer, O. , Woerner, S. , Yang, F. , Oakeley, E. J. , Linghu, B. , Gribouval, O. , … Antignac, C. (2013). LMX1B mutations cause hereditary FSGS without extrarenal involvement. Journal of the American Society of Nephrology, 24(8), 1216–1222. 10.1681/Asn.2013020171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, D. A. , & Hildebrandt, F. (2017). Ciliopathies. Cold Spring Harbor Perspectives in Biology, 9(3), a028191. 10.1101/cshperspect.a028191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunisch, M. C. , Riedhammer, K. M. , Herr, P. M. , Draut, S. , Gunthner, R. , Wagner, M. , … Hoefele, J. (2021). Identification of disease‐causing variants by comprehensive genetic testing with exome sequencing in adults with suspicion of hereditary FSGS. European Journal of Human Genetics, 29(2), 262–270. 10.1038/s41431-020-00719-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullich, G. , Vargas, I. , Trujillano, D. , Mendizabal, S. , Pinero‐Fernandez, J. A. , Fraga, G. , … Ars, E. (2017). Contribution of the TTC21B gene to glomerular and cystic kidney diseases. Nephrology, Dialysis, Transplantation, 32(1), 151–156. 10.1093/ndt/gfv453 [DOI] [PubMed] [Google Scholar]
- Cameron, J. S. (2003). Focal segmental glomerulosclerosis in adults. Nephrology, Dialysis, Transplantation, 18(Suppl 6), vi45–vi51. 10.1093/ndt/gfg1058 [DOI] [PubMed] [Google Scholar]
- Chang, J. W. , Pardo, V. , Sageshima, J. , Chen, L. , Tsai, H. L. , Reiser, J. , … Fornoni, A. (2012). Podocyte foot process effacement in postreperfusion allograft biopsies correlates with early recurrence of proteinuria in focal segmental glomerulosclerosis. Transplantation, 93(12), 1238–1244. 10.1097/TP.0b013e318250234a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. , Lun, Y. , Ovchinnikov, D. , Kokubo, H. , Oberg, K. C. , Pepicelli, C. V. , … Johnson, R. L. (1998). Limb and kidney defects in Lmx1b mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nature Genetics, 19(1), 51–55. 10.1038/ng0598-51 [DOI] [PubMed] [Google Scholar]
- Chun, J. , Wang, M. X. , Wilkins, M. S. , Knob, A. U. , Benjamin, A. , Bu, L. H. , & Pollak, M. R. (2020). Autosomal dominant tubulointerstitial kidney disease‐uromodulin misclassified as focal segmental glomerulosclerosis or hereditary glomerular disease. Kidney International Reports, 5(4), 519–529. 10.1016/j.ekir.2019.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Agati, V. D. , Fogo, A. B. , Bruijn, J. A. , & Jennette, J. C. (2004). Pathologic classification of focal segmental glomerulosclerosis: A working proposal. American Journal of Kidney Diseases, 43(2), 368–382. 10.1053/j.ajkd.2003.10.024 [DOI] [PubMed] [Google Scholar]
- De Vriese, A. S. , Sethi, S. , Nath, K. A. , Glassock, R. J. , & Fervenza, F. C. (2018). Differentiating primary, genetic, and secondary FSGS in adults: A clinicopathologic approach. Journal of the American Society of Nephrology, 29(3), 759–774. 10.1681/ASN.2017090958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devuyst, O. , Christie, P. T. , Courtoy, P. J. , Beauwens, R. , & Thakker, R. V. (1999). Intra‐renal and subcellular distribution of the human chloride channel, CLC‐5, reveals a pathophysiological basis for Dent's disease. Human Molecular Genetics, 8(2), 247–257. 10.1093/hmg/8.2.247 [DOI] [PubMed] [Google Scholar]
- Dorval, G. , Gribouval, O. , Martinez‐Barquero, V. , Machuca, E. , Tete, M. J. , Baudouin, V. , … Boyer, O. (2018). Clinical and genetic heterogeneity in familial steroid‐sensitive nephrotic syndrome. Pediatric Nephrology, 33(3), 473–483. 10.1007/s00467-017-3819-9 [DOI] [PubMed] [Google Scholar]
- Dreyer, S. D. , Morello, R. , German, M. S. , Zabel, B. , Winterpacht, A. , Lunstrum, G. P. , … Lee, B. (2000). LMX1B transactivation and expression in nail‐patella syndrome. Human Molecular Genetics, 9(7), 1067–1074. 10.1093/hmg/9.7.1067 [DOI] [PubMed] [Google Scholar]
- Dreyer, S. D. , Zhou, G. , Baldini, A. , Winterpacht, A. , Zabel, B. , Cole, W. , … Lee, B. (1998). Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nature Genetics, 19(1), 47–50. 10.1038/ng0598-47 [DOI] [PubMed] [Google Scholar]
- Elizondo, L. I. , Cho, K. S. , Zhang, W. , Yan, J. , Huang, C. , Huang, Y. , … Boerkoel, C. F. (2009). Schimke immuno‐osseous dysplasia: SMARCAL1 loss‐of‐function and phenotypic correlation. Journal of Medical Genetics, 46(1), 49–59. 10.1136/jmg.2008.060095 [DOI] [PubMed] [Google Scholar]
- Gallon, L. , Leventhal, J. , Skaro, A. , Kanwar, Y. , & Alvarado, A. (2012). Resolution of recurrent focal segmental glomerulosclerosis after retransplantation. The New England Journal of Medicine, 366(17), 1648–1649. 10.1056/NEJMc1202500 [DOI] [PubMed] [Google Scholar]
- Gast, C. , Marinaki, A. , Arenas‐Hernandez, M. , Campbell, S. , Seaby, E. G. , Pengelly, R. J. , … Venkat‐Raman, G. (2018). Autosomal dominant tubulointerstitial kidney disease – UMOD is the most frequent non polycystic genetic kidney disease. BMC Nephrology, 19, 301. 10.1186/s12882-018-1107-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gast, C. , Pengelly, R. J. , Lyon, M. , Bunyan, D. J. , Seaby, E. G. , Graham, N. , … Ennis, S. (2016). Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrology Dialysis Transplantation, 31(6), 961–970. 10.1093/ndt/gfv325 [DOI] [PubMed] [Google Scholar]
- Gbadegesin, R. A. , Hall, G. , Adeyemo, A. , Hanke, N. , Tossidou, I. , Burchette, J. , … Winn, M. P. (2014). Mutations in the gene that encodes the F‐actin binding protein anillin cause FSGS. Journal of the American Society of Nephrology, 25(9), 1991–2002. 10.1681/Asn.2013090976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese, G. , Friedman, D. J. , Ross, M. D. , Lecordier, L. , Uzureau, P. , Freedman, B. I. , … Pollak, M. R. (2010). Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science, 329(5993), 841–845. 10.1126/science.1193032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, J. , Fieldhouse, R. , Chan, M. M. Y. , Sadeghi‐Alavijeh, O. , Burnett, L. , Izzi, V. , … Consortium, G. E. R . (2021). Prevalence estimates of predicted pathogenic COL4A3‐COL4A5 variants in a population sequencing database and their implications for Alport syndrome. Journal of the American Society of Nephrology, 32(9), 2273–2290. 10.1681/Asn.2020071065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigante, M. , Pontrelli, P. , Montemurno, E. , Roca, L. , Aucella, F. , Penza, R. , … Gesualdo, L. (2009). CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS). Nephrology, Dialysis, Transplantation, 24(6), 1858–1864. 10.1093/ndt/gfn712 [DOI] [PubMed] [Google Scholar]
- Gorman, S. W. , Haider, N. B. , Grieshammer, U. , Swiderski, R. E. , Kim, E. , Welch, J. W. , … Duhl, D. M. (1999). The cloning and developmental expression of unconventional myosin IXA (MYO9A) a gene in the Bardet‐Biedl syndrome (BBS4) region at chromosome 15q22‐q23. Genomics, 59(2), 150–160. 10.1006/geno.1999.5867 [DOI] [PubMed] [Google Scholar]
- Greze, C. , Garrouste, C. , Kemeny, J. L. , Philipponnet, C. , Aniort, J. , & Heng, A. E. (2018). Collapsing focal segmental glomerulosclerosis induced by cytomegalovirus: A case report (Hyalinose segmentaire et focale collapsante secondaire au cytomegalovirus: a propos d'un cas). Néphrologie & Thérapeutique, 14(1), 50–53. 10.1016/j.nephro.2017.06.002 [DOI] [PubMed] [Google Scholar]
- Gribouval, O. , Boyer, O. , Hummel, A. , Dantal, J. , Martinez, F. , Sberro‐Soussan, R. , … Servais, A. (2018). Identification of genetic causes for sporadic steroid‐resistant nephrotic syndrome in adults. Kidney International, 94(5), 1013–1022. 10.1016/j.kint.2018.07.024 [DOI] [PubMed] [Google Scholar]
- Groopman, E. , Goldstein, D. , & Gharavi, A. (2019). Diagnostic utility of exome sequencing for kidney disease. Reply. The New England Journal of Medicine, 380(21), 2080–2081. 10.1056/NEJMc1903250 [DOI] [PubMed] [Google Scholar]
- Guaragna, M. S. , de Brito Lutaif, A. C. G. , de Souza, M. L. , Maciel‐Guerra, A. T. , Belangero, V. M. S. , Guerra‐Junior, G. , & de Mello, M. P. (2020). Promises and pitfalls of whole‐exome sequencing exemplified by a nephrotic syndrome family. Molecular Genetics and Genomics, 295(1), 135–142. 10.1007/s00438-019-01609-0 [DOI] [PubMed] [Google Scholar]
- Gunther, W. , Luchow, A. , Cluzeaud, F. , Vandewalle, A. , & Jentsch, T. J. (1998). ClC‐5, the chloride channel mutated in Dent's disease, colocalizes with the proton pump in endocytotically active kidney cells. Proceedings of the National Academy of Sciences of the United States of America, 95(14), 8075–8080. 10.1073/pnas.95.14.8075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes, A. , Guo, J. K. , Lutsch, G. , Leheste, J. R. , Landrock, D. , Ziegler, U. , … Schedl, A. (2001). Two splice variants of the Wilms' tumor 1 gene have distinct functions during sex determination and nephron formation. Cell, 106(3), 319–329. 10.1016/S0092-8674(01)00453-6 [DOI] [PubMed] [Google Scholar]
- Hasselbacher, K. , Wiggins, R. C. , Matejas, V. , Hinkes, B. G. , Mucha, B. , Hoskins, B. E. , … Hildebrandt, F. (2006). Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2‐associated disorders. Kidney International, 70(6), 1008–1012. 10.1038/sj.ki.5001679 [DOI] [PubMed] [Google Scholar]
- Heeringa, S. F. , Chernin, G. , Chaki, M. , Zhou, W. , Sloan, A. J. , Ji, Z. , … Hildebrandt, F. (2011). COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. The Journal of Clinical Investigation, 121(5), 2013–2024. 10.1172/JCI45693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, J. M. , Alexander, M. P. , & Pollak, M. R. (2009). Patients with ACTN4 mutations demonstrate distinctive features of glomerular injury. Journal of the American Society of Nephrology, 20(5), 961–968. 10.1681/Asn.2008060613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitz, L. C. , Markowitz, G. S. , Farris, A. B. , Schwimmer, J. A. , Stokes, M. B. , Kunis, C. , … D'Agati, V. D. (2010). Development of focal segmental glomerulosclerosis after anabolic steroid abuse. Journal of the American Society of Nephrology, 21(1), 163–172. 10.1681/ASN.2009040450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hichri, H. , Rendu, J. , Monnier, N. , Coutton, C. , Dorseuil, O. , Poussou, R. V. , … Lunardi, J. (2011). From Lowe syndrome to Dent disease: Correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Human Mutation, 32(4), 379–388. 10.1002/humu.21391 [DOI] [PubMed] [Google Scholar]
- Hodson, E. M. , Sinha, A. , & Cooper, T. E. (2022). Interventions for focal segmental glomerulosclerosis in adults. Cochrane Database of Systematic Reviews, 2, CD003233. 10.1002/14651858.CD003233.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, T. , Obukhov, A. G. , Schaefer, M. , Harteneck, C. , Gudermann, T. , & Schultz, G. (1999). Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature, 397(6716), 259–263. 10.1038/16711 [DOI] [PubMed] [Google Scholar]
- Holmberg, C. , & Jalanko, H. (2014). Congenital nephrotic syndrome and recurrence of proteinuria after renal transplantation. Pediatric Nephrology, 29(12), 2309–2317. 10.1007/s00467-014-2781-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopes, R. R. , Raja, K. M. , Koich, A. , Hueber, P. , Reid, R. , Knohl, S. J. , & Scheinman, S. J. (2004). Evidence for genetic heterogeneity in Dent's disease. Kidney International, 65(5), 1615–1620. 10.1111/j.1523-1755.2004.00571.x [DOI] [PubMed] [Google Scholar]
- Hossain, A. , & Saunders, G. F. (2001). The human sex‐determining gene SRY is a direct target of WT1. Journal of Biological Chemistry, 276(20), 16817–16823. 10.1074/jbc.M009056200 [DOI] [PubMed] [Google Scholar]
- Howie, A. J. (2020). Genetic studies of focal segmental glomerulosclerosis: A waste of scientific time? Pediatric Nephrology, 35(1), 9–16. 10.1007/s00467-018-4161-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie, A. J. , Pankhurst, T. , Sarioglu, S. , Turhan, N. , & Adu, D. (2005). Evolution of nephrotic‐associated focal segmental glomerulosclerosis and relation to the glomerular tip lesion. Kidney International, 67(3), 987–1001. 10.1111/j.1523-1755.2005.00162.x [DOI] [PubMed] [Google Scholar]
- Huber, T. B. , Simons, M. , Hartleben, B. , Sernetz, L. , Schmidts, M. , Gundlach, E. , … Benzing, T. (2003). Molecular basis of the functional podocin‐nephrin complex: Mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Human Molecular Genetics, 12(24), 3397–3405. 10.1093/hmg/ddg360 [DOI] [PubMed] [Google Scholar]
- Hudson, B. G. , Tryggvason, K. , Sundaramoorthy, M. , & Neilson, E. G. (2003). Alport's syndrome, Goodpasture's syndrome, and type IV collagen. The New England Journal of Medicine, 348(25), 2543–2556. 10.1056/NEJMra022296 [DOI] [PubMed] [Google Scholar]
- Huynh Cong, E. , Bizet, A. A. , Boyer, O. , Woerner, S. , Gribouval, O. , Filhol, E. , … Antignac, C. (2014). A homozygous missense mutation in the ciliary gene TTC21B causes familial FSGS. Journal of the American Society of Nephrology, 25(11), 2435–2443. 10.1681/ASN.2013101126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka, K. , Miura, K. , Hashimoto, T. , Kaneko, N. , Harita, Y. , Yabuuchi, T. , … Hattori, M. (2021). Degree of foot process effacement in patients with genetic focal segmental glomerulosclerosis: A single‐center analysis and review of the literature. Scientific Reports, 11(1), 12008. 10.1038/s41598-021-91520-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasinghe, K. , Stark, Z. , Kerr, P. G. , Gaff, C. , Martyn, M. , Whitlam, J. , … Quinlan, C. (2021). Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genetics in Medicine, 23(1), 183–191. 10.1038/s41436-020-00963-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungraithmayr, T. C. , Hofer, K. , Cochatt, P. , Chemin, G. , Cortina, G. , Fargue, S. , … Zimmerhackl, L. B. (2011). Screening for NPHS2 mutations may help predict FSGS recurrence after transplantation. Journal of the American Society of Nephrology, 22(3), 579–585. 10.1681/Asn.2010010029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, J. M. , Kim, S. H. , North, K. N. , Rennke, H. , Correia, L. A. , Tong, H. Q. , … Pollak, M. R. (2000). Mutations in ACTN4, encoding alpha‐actinin‐4, cause familial focal segmental glomerulosclerosis. Nature Genetics, 24(3), 251–256. 10.1038/73456 [DOI] [PubMed] [Google Scholar]
- Kashtan, C. E. (1993). Alport syndrome. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Gripp K. W., et al. (Eds.), GeneReviews®. Seattle, WA: University of Washington. Retrieved from. https://www.ncbi.nlm.nih.gov/pubmed/20301386 [Google Scholar]
- Kemper, M. J. , & Lemke, A. (2018). Treatment of genetic forms of nephrotic syndrome. Frontiers in Pediatrics, 6, 72. 10.3389/fped.2018.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kestila, M. , Lenkkeri, U. , Mannikko, M. , Lamerdin, J. , McCready, P. , Putaala, H. , … Tryggvason, K. (1998). Positionally cloned gene for a novel glomerular protein – nephrin – is mutated in congenital nephrotic syndrome. Molecular Cell, 1(4), 575–582. 10.1016/S1097-2765(00)80057-X [DOI] [PubMed] [Google Scholar]
- Kidney Disease: Improving Global Outcomes Glomerular Diseases Work Group . (2021). KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney International, 100(4S), S1–S276. 10.1016/j.kint.2021.05.021 [DOI] [PubMed] [Google Scholar]
- Kim, J. M. , Wu, H. , Green, G. , Winkler, C. A. , Kopp, J. B. , Miner, J. H. , … Shaw, A. S. (2003). CD2‐associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science, 300(5623), 1298–1300. 10.1126/science.1081068 [DOI] [PubMed] [Google Scholar]
- Kirsch, K. H. , Georgescu, M. M. , Ishimaru, S. , & Hanafusa, H. (1999). CMS: An adapter molecule involved in cytoskeletal rearrangements. Proceedings of the National Academy of Sciences of the United States of America, 96(11), 6211–6216. 10.1073/pnas.96.11.6211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitiyakara, C. , Eggers, P. , & Kopp, J. B. (2004). Twenty‐one‐year trend in ESRD due to focal segmental glomerulosclerosis in the United States. American Journal of Kidney Diseases, 44(5), 815–825. 10.1053/j.ajkd.2004.07.008 [DOI] [PubMed] [Google Scholar]
- Konig, J. , Kranz, B. , Konig, S. , Schlingmann, K. P. , Titieni, A. , Tonshoff, B. , … Gesellschaft fur Padiatrische Nephrologie . (2017). Phenotypic spectrum of children with nephronophthisis and related ciliopathies. Clinical Journal of the American Society of Nephrology, 12(12), 1974–1983. 10.2215/CJN.01280217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp, J. B. , Nelson, G. W. , Sampath, K. , Johnson, R. C. , Genovese, G. , An, P. , … Winkler, C. A. (2011). APOL1 genetic variants in focal segmental glomerulosclerosis and HIV‐associated nephropathy. Journal of the American Society of Nephrology, 22(11), 2129–2137. 10.1681/ASN.2011040388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall, P. , Canales, C. P. , Kairath, P. , Carmona‐Mora, P. , Molina, J. , Carpio, J. D. , … Walz, K. (2010). Podocyte‐specific overexpression of wild type or mutant Trpc6 in mice is sufficient to cause glomerular disease. PLoS One, 5(9), e12859. 10.1371/journal.pone.0012859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, A. , Boenink, R. , Stel, V. S. , Santiuste de Pablos, C. , Tomovic, F. , Golan, E. , … Jager, K. J. (2021). The ERA‐EDTA registry annual report 2018: A summary. Clinical Kidney Journal, 14(1), 107–123. 10.1093/ckj/sfaa271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krendel, M. , Kim, S. V. , Willinger, T. , Wang, T. , Kashgarian, M. , Flavell, R. A. , & Mooseker, M. S. (2009). Disruption of myosin 1e promotes podocyte injury. Journal of the American Society of Nephrology, 20(1), 86–94. 10.1681/ASN.2007111172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruzel‐Davila, E. , Wasser, W. G. , & Skorecki, K. (2017). APOL1 nephropathy: A population genetics and evolutionary medicine detective story. Seminars in Nephrology, 37(6), 490–507. 10.1016/j.semnephrol.2017.07.002 [DOI] [PubMed] [Google Scholar]
- Labat‐de‐Hoz, L. , & Alonso, M. A. (2020). The formin INF2 in disease: Progress from 10 years of research. Cellular and Molecular Life Sciences, 77(22), 4581–4600. 10.1007/s00018-020-03550-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laboux, T. , Gibier, J. B. , Pottier, N. , Glowacki, F. , & Hamroun, A. (2021). COVID‐19‐related collapsing glomerulopathy revealing a rare risk variant of APOL1: Lessons for the clinical nephrologist (Feb, 10.1007/s40620‐020‐00935‐6, 2021). Journal of Nephrology, 34(2), 379. 10.1007/s40620-021-01037-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazareth, H. , Pere, H. , Binois, Y. , Chabannes, M. , Schurder, J. , Bruneau, T. , … Pallet, N. (2020). COVID‐19‐related collapsing glomerulopathy in a kidney transplant recipient. American Journal of Kidney Diseases, 76(4), 590–594. 10.1053/j.ajkd.2020.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. M. , Kronbichler, A. , Shin, J. I. , & Oh, J. (2021). Current understandings in treating children with steroid‐resistant nephrotic syndrome. Pediatric Nephrology, 36(4), 747–761. 10.1007/s00467-020-04476-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon, R. , Stuart, H. M. , Bierzynska, A. , Randles, M. J. , Kerr, B. , Hillman, K. A. , … Woolf, A. S. (2015). Coinheritance of COL4A5 and MYO1E mutations accentuate the severity of kidney disease. Pediatric Nephrology, 30(9), 1459–1465. 10.1007/s00467-015-3067-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepori, N. , Zand, L. , Sethi, S. , Fernandez‐Juarez, G. , & Fervenza, F. C. (2018). Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clinical Kidney Journal, 11(2), 179–190. 10.1093/ckj/sfx143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letavernier, E. , Bruneval, P. , Mandet, C. , Van Huyen, J. P. D. , Peraldi, M. N. , Helal, I. , … Legendre, C. (2007). High sirolimus levels may induce focal segmental glomerulosclerosis de novo. Clinical Journal of the American Society of Nephrology, 2(2), 326–333. 10.2215/Cjn.03751106 [DOI] [PubMed] [Google Scholar]
- Li, Q. , Gulati, A. , Lemaire, M. , Nottoli, T. , Bale, A. , & Tufro, A. (2021). Rho‐GTPase activating protein myosin MYO9A identified as a novel candidate gene for monogenic focal segmental glomerulosclerosis. Kidney International, 99(5), 1102–1117. 10.1016/j.kint.2020.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , & He, J. C. (2015). An update: The role of nephrin inside and outside the kidney. Science China. Life Sciences, 58(7), 649–657. 10.1007/s11427-015-4844-1 [DOI] [PubMed] [Google Scholar]
- Lim, B. J. , Yang, J. W. , Do, W. S. , & Fogo, A. B. (2016). Pathogenesis of focal segmental glomerulosclerosis. Journal of Pathology and Translational Medicine, 50(6), 405–410. 10.4132/jptm.2016.09.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipska‐Zietkiewicz, B. S. (1993). WT1 disorder. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Gripp K. W., et al. (Eds.), GeneReviews®. Seattle, WA: University of Washington. Retrieved from. https://www.ncbi.nlm.nih.gov/pubmed/32352694 [Google Scholar]
- Lloyd, S. E. , Pearce, S. H. , Fisher, S. E. , Steinmeyer, K. , Schwappach, B. , Scheinman, S. J. , … Thakker, R. V. (1996). A common molecular basis for three inherited kidney stone diseases. Nature, 379(6564), 445–449. 10.1038/379445a0 [DOI] [PubMed] [Google Scholar]
- Lowik, M. M. , Groenen, P. J. , Pronk, I. , Lilien, M. R. , Goldschmeding, R. , Dijkman, H. B. , … van den Heuvel, L. P. (2007). Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney International, 72(10), 1198–1203. 10.1038/sj.ki.5002469 [DOI] [PubMed] [Google Scholar]
- Machuca, E. , Hummel, A. , Nevo, F. , Dantal, J. , Martinez, F. , Al‐Sabban, E. , Baudouin, V. , Abel, L. , Grünfeld, J.‐P. , & Antignac, C. (2009). Clinical and epidemiological assessment of steroid‐resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney International, 75(7), 727–735. 10.1038/ki.2008.650 [DOI] [PubMed] [Google Scholar]
- Mademan, I. , Deconinck, T. , Dinopoulos, A. , Voit, T. , Schara, U. , Devriendt, K. , … Baets, J. (2013). De novo INF2 mutations expand the genetic spectrum of hereditary neuropathy with glomerulopathy. Neurology, 81(22), 1953–1958. 10.1212/01.wnl.0000436615.58705.c9 [DOI] [PubMed] [Google Scholar]
- Mansilla, M. A. , Sompallae, R. R. , Nishimura, C. J. , Kwitek, A. E. , Kimble, M. J. , Freese, M. E. , … Thomas, C. P. (2021). Targeted broad‐based genetic testing by next‐generation sequencing informs diagnosis and facilitates management in patients with kidney diseases. Nephrology, Dialysis, Transplantation, 36(2), 295–305. 10.1093/ndt/gfz173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matejas, V. , Hinkes, B. , Alkandari, F. , Al‐Gazali, L. , Annexstad, E. , Aytac, M. B. , … Zenker, M. (2010). Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Human Mutation, 31(9), 992–1002. 10.1002/humu.21304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthaiou, A. , Poulli, T. , & Deltas, C. (2020). Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: A systematic review. Clinical Kidney Journal, 13(6), 1025–1036. 10.1093/ckj/sfz176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, G. M. , Blasio, A. , Donovan, O. G. , Schaller, L. B. , Bock‐Hughes, A. , Magraner, J. M. , … Pollak, M. R. (2021). Recessive, gain‐of‐function toxicity in an APOL1 BAC transgenic mouse model mirrors human APOL1 kidney disease. Disease Models & Mechanisms, 14(8), dmm048952. 10.1242/dmm.048952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh, I. , Dreyer, S. D. , Clough, M. V. , Dunston, J. A. , Eyaid, W. , Roig, C. M. , … Lee, B. (1998). Mutation analysis of LMX1B gene in nail‐patella syndrome patients. American Journal of Human Genetics, 63(6), 1651–1658. 10.1086/302165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele, C. , Iatropoulos, P. , Donadelli, R. , Calabria, A. , Maranta, R. , Cassis, P. , … PodoNet, C. (2011). MYO1E mutations and childhood familial focal segmental glomerulosclerosis. The New England Journal of Medicine, 365(4), 295–306. 10.1056/NEJMoa1101273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milunsky, J. M. (1993). Waardenburg syndrome type I. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Gripp K. W., et al. (Eds.), GeneReviews®. Seattle, WA: University of Washington. Retrieved from. https://www.ncbi.nlm.nih.gov/pubmed/20301703 [Google Scholar]
- Mistry, K. , Ireland, J. H. , Ng, R. C. , Henderson, J. M. , & Pollak, M. R. (2007). Novel mutations in NPHP4 in a consanguineous family with histological findings of focal segmental glomerulosclerosis. American Journal of Kidney Diseases, 50(5), 855–864. 10.1053/j.ajkd.2007.08.009 [DOI] [PubMed] [Google Scholar]
- Mohney, B. G. , Pulido, J. S. , Lindor, N. M. , Hogan, M. C. , Consugar, M. B. , Peters, J. , … Harris, P. C. (2011). A novel mutation of LAMB2 in a multigenerational mennonite family reveals a new phenotypic variant of Pierson syndrome. Ophthalmology, 118(6), 1137–1144. 10.1016/j.ophtha.2010.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello, R. , Zhou, G. , Dreyer, S. D. , Harvey, S. J. , Ninomiya, Y. , Thorner, P. S. , … Lee, B. (2001). Regulation of glomerular basement membrane collagen expression by LMX1B contributes to renal disease in nail patella syndrome. Nature Genetics, 27(2), 205–208. 10.1038/84853 [DOI] [PubMed] [Google Scholar]
- Morimoto, M. , Lewis, D. B. , Lucke, T. , & Boerkoel, C. F. (1993). Schimke immunoosseous dysplasia. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Gripp K. W., et al. (Eds.), GeneReviews®. Seattle, WA: University of Washington. Retrieved from. https://www.ncbi.nlm.nih.gov/pubmed/20301550 [Google Scholar]
- Muehlig, A. K. , Gies, S. , Huber, T. B. , & Braun, F. (2021). Collapsing focal segmental glomerulosclerosis in viral infections. Frontiers in Immunology, 12, 800074. 10.3389/fimmu.2021.800074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano, C. , Yamamura, T. , Horinouchi, T. , Aoto, Y. , Ishiko, S. , Sakakibara, N. , … Nozu, K. (2020). Comprehensive genetic diagnosis of Japanese patients with severe proteinuria. Scientific Reports, 10(1), 270. 10.1038/s41598-019-57149-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik, R. P. , Irvin, M. R. , Judd, S. , Gutierrez, O. M. , Zakai, N. A. , Derebail, V. K. , … Cushman, M. (2017). Sickle cell trait and the risk of ESRD in blacks. Journal of the American Society of Nephrology, 28(7), 2180–2187. 10.1681/ASN.2016101086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaltin, F. (2014). Primary coenzyme Q10 (CoQ 10) deficiencies and related nephropathies. Pediatric Nephrology, 29(6), 961–969. 10.1007/s00467-013-2482-z [DOI] [PubMed] [Google Scholar]
- Papazachariou, L. , Papagregoriou, G. , Hadjipanagi, D. , Demosthenous, P. , Voskarides, K. , Koutsofti, C. , … Deltas, C. (2017). Frequent COL4 mutations in familial microhematuria accompanied by later‐onset Alport nephropathy due to focal segmental glomerulosclerosis. Clinical Genetics, 92(5), 517–527. 10.1111/cge.13077 [DOI] [PubMed] [Google Scholar]
- Park, E. , Lee, C. , Kim, N. K. D. , Ahn, Y. H. , Park, Y. S. , Lee, J. H. , … Cheong, H. I. (2020). Genetic study in Korean pediatric patients with steroid‐resistant nephrotic syndrome or focal segmental glomerulosclerosis. Journal of Clinical Medicine, 9(6), 2013. 10.3390/jcm9062013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, L. , Bahat, H. , Appel, M. Y. , Baum, D. V. , Forer, R. , Pillar, N. , … Goldman, M. (2019). Phospholipase C‐gamma 2 activity in familial steroid‐sensitive nephrotic syndrome. Pediatric Research, 85(5), 719–723. 10.1038/s41390-018-0259-6 [DOI] [PubMed] [Google Scholar]
- Patek, C. E. , Little, M. H. , Fleming, S. , Miles, C. , Charlieu, J. P. , Clarke, A. R. , … Hastie, N. D. (1999). A zinc finger truncation of murine WT1 results in the characteristic urogenital abnormalities of Denys‐Drash syndrome. Proceedings of the National Academy of Sciences of the United States of America, 96(6), 2931–2936. 10.1073/pnas.96.6.2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy, K. , & Kaskel, F. J. (2007). Pathophysiology of focal segmental glomerulosclerosis. Pediatric Nephrology, 22(3), 350–354. 10.1007/s00467-006-0357-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser, J. , Polu, K. R. , Moller, C. C. , Kenlan, P. , Altintas, M. M. , Wei, C. L. , … Pollak, M. R. (2005). TRPC6 is a glomerular slit diaphragm‐associated channel required for normal renal function. Nature Genetics, 37(7), 739–744. 10.1038/ng1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond, A. , Meroni, G. , Fantozzi, A. , Merla, G. , Cairo, S. , Luzi, L. , … Ballabio, A. (2001). The tripartite motif family identifies cell compartments. The EMBO Journal, 20(9), 2140–2151. 10.1093/emboj/20.9.2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich, A. R. (1957). A hitherto undescribed vulnerability of the juxtamedullary glomeruli in lipoid nephrosis. Bulletin of the Johns Hopkins Hospital, 100(4), 173–186. Retrieved from. https://www.ncbi.nlm.nih.gov/pubmed/13426687 [PubMed] [Google Scholar]
- Riedhammer, K. M. , Braunisch, M. C. , Gunthner, R. , Wagner, M. , Hemmer, C. , Strom, T. M. , … Hoefele, J. (2020). Exome sequencing and identification of phenocopies in patients with clinically presumed hereditary nephropathies. American Journal of Kidney Diseases, 76(4), 460–470. 10.1053/j.ajkd.2019.12.008 [DOI] [PubMed] [Google Scholar]
- Riehle, M. , Buscher, A. K. , Gohlke, B. O. , Kassmann, M. , Kolatsi‐Joannou, M. , Brasen, J. H. , … Harteneck, C. (2016). TRPC6 G757D loss‐of‐function mutation associates with FSGS. Journal of the American Society of Nephrology, 27(9), 2771–2783. 10.1681/Asn.2015030318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rood, I. M. , Deegens, J. K. , & Wetzels, J. F. (2012). Genetic causes of focal segmental glomerulosclerosis: Implications for clinical practice. Nephrology, Dialysis, Transplantation, 27(3), 882–890. 10.1093/ndt/gfr771 [DOI] [PubMed] [Google Scholar]
- Rosenberg, A. Z. , & Kopp, J. B. (2017). Focal segmental glomerulosclerosis. Clinical Journal of the American Society of Nephrology, 12(3), 502–517. 10.2215/CJN.05960616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski, C. E. , Lovric, S. , Ashraf, S. , Pabst, W. L. , Gee, H. Y. , Kohl, S. , … Hildebrandt, F. (2015). A single‐gene cause in 29.5% of cases of steroid‐resistant nephrotic syndrome. Journal of the American Society of Nephrology, 26(6), 1279–1289. 10.1681/ASN.2014050489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, Y. , Fukai, R. , Matsushita, Y. , Miyake, N. , Saitsu, H. , Akamine, S. , … Hara, T. (2016). De novo truncating mutation of TRIM8 causes early‐onset epileptic encephalopathy. Annals of Human Genetics, 80(4), 235–240. 10.1111/ahg.12157 [DOI] [PubMed] [Google Scholar]
- Santin, S. , Ars, E. , Rossetti, S. , Salido, E. , Silva, I. , Garcia‐Maset, R. , … de los Rios, F. J. (2009). TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrology, Dialysis, Transplantation, 24(10), 3089–3096. 10.1093/ndt/gfp229 [DOI] [PubMed] [Google Scholar]
- Santin, S. , Bullich, G. , Tazon‐Vega, B. , Garcia‐Maset, R. , Gimenez, I. , Silva, I. , … Ars, E. (2011). Clinical utility of genetic testing in children and adults with steroid‐resistant nephrotic syndrome. Clinical Journal of the American Society of Nephrology, 6(5), 1139–1148. 10.2215/CJN.05260610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santin, S. , Garcia‐Maset, R. , Ruiz, P. , Gimenez, I. , Zamora, I. , Pena, A. , … FSGS Spanish Study Group . (2009). Nephrin mutations cause childhood‐ and adult‐onset focal segmental glomerulosclerosis. Kidney International, 76(12), 1268–1276. 10.1038/ki.2009.381 [DOI] [PubMed] [Google Scholar]
- Sanyanusin, P. , Schimmenti, L. A. , McNoe, L. A. , Ward, T. A. , Pierpont, M. E. , Sullivan, M. J. , … Eccles, M. R. (1995). Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nature Genetics, 9(4), 358–364. 10.1038/ng0495-358 [DOI] [PubMed] [Google Scholar]
- Saraiva, J. M. , Dinis, A. , Resende, C. , Faria, E. , Gomes, C. , Correia, A. J. , … da Fonseca, N. (1999). Schimke immuno‐osseous dysplasia: Case report and review of 25 patients. Journal of Medical Genetics, 36(10), 786–789. 10.1136/jmg.36.10.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savige, J. , Lipska‐Zietkiewicz, B. S. , Watson, E. , Hertz, J. M. , Deltas, C. , Mari, F. , … Flinter, F. (2021). Guidelines for genetic testing and management of Alport syndrome. Clinical Journal of the American Society of Nephrology, 17, 143–154. 10.2215/Cjn.04230321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schijvens, A. M. , van de Kar, N. C. , Bootsma‐Robroeks, C. M. , Cornelissen, E. A. , van den Heuvel, L. P. , & Schreuder, M. F. (2020). Mitochondrial disease and the kidney with a special focus on CoQ10 deficiency. Kidney International Reports, 5(12), 2146–2159. 10.1016/j.ekir.2020.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, K. , Simons, M. , Reiser, J. , Saleem, M. A. , Faul, C. , Kriz, W. , … Mundel, P. (2001). Podocin, a raft‐associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. The Journal of Clinical Investigation, 108(11), 1621–1629. 10.1172/JCI12849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen, E. S. , Dean, P. , Yarram‐Smith, L. , Bierzynska, A. , Woodward, G. , Buxton, C. , … Saleem, M. A. (2017). Clinical genetic testing using a custom‐designed steroid‐resistant nephrotic syndrome gene panel: Analysis and recommendations. Journal of Medical Genetics, 54(12), 795–804. 10.1136/jmedgenet-2017-104811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty, A. A. , Tawhari, I. , Safar‐Boueri, L. , Seif, N. , Alahmadi, A. , Gargiulo, R. , … Quaggin, S. E. (2021). COVID‐19‐associated glomerular disease. Journal of the American Society of Nephrology, 32(1), 33–40. 10.1681/ASN.2020060804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih, N. Y. , Li, J. , Cotran, R. , Mundel, P. , Miner, J. H. , & Shaw, A. S. (2001). CD2AP localizes to the slit diaphragm and binds to nephrin via a novel C‐terminal domain. The American Journal of Pathology, 159(6), 2303–2308. 10.1016/S0002-9440(10)63080-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih, N. Y. , Li, J. , Karpitskii, V. , Nguyen, A. , Dustin, M. L. , Kanagawa, O. , … Shaw, A. S. (1999). Congenital nephrotic syndrome in mice lacking CD2‐associated protein. Science, 286(5438), 312–315. 10.1126/science.286.5438.312 [DOI] [PubMed] [Google Scholar]
- Shirai, Y. , Miura, K. , Kaneko, N. , Ishizuka, K. , Endo, A. , Hashimoto, T. , … Hattori, M. (2021). A novel de novo truncating TRIM8 variant associated with childhood‐onset focal segmental glomerulosclerosis without epileptic encephalopathy: A case report. BMC Nephrology, 22(1), 417. 10.1186/s12882-021-02626-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoyer, W. E. , Mundel, P. , Gupta, A. , & Welsh, M. J. (1997). Podocyte alpha‐actinin induction precedes foot process effacement in experimental nephrotic syndrome. The American Journal of Physiology, 273(1 Pt 2), F150–F157. 10.1152/ajprenal.1997.273.1.F150 [DOI] [PubMed] [Google Scholar]
- Spranger, J. , Hinkel, G. K. , Stoss, H. , Thoenes, W. , Wargowski, D. , & Zepp, F. (1991). Schimke immuno‐osseous dysplasia – A newly recognized multisystem disease. Journal of Pediatrics, 119(1), 64–72. 10.1016/S0022-3476(05)81040-6 [DOI] [PubMed] [Google Scholar]
- Srivastava, S. , Molinari, E. , Raman, S. , & Sayer, J. A. (2017). Many genes‐one disease? Genetics of nephronophthisis (NPHP) and NPHP‐associated disorders. Frontiers in Pediatrics, 5, 287. 10.3389/fped.2017.00287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokman, M. , Lilien, M. , & Knoers, N. (1993). Nephronophthisis. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Gripp K. W., et al. (Eds.), GeneReviews®. Seattle, WA: University of Washington. Retrieved from. https://www.ncbi.nlm.nih.gov/pubmed/27336129 [PubMed] [Google Scholar]
- Takano, T. , Bareke, E. , Takeda, N. , Aoudjit, L. , Baldwin, C. , Pisano, P. , … Gupta, I. R. (2019). Recessive mutation in CD2AP causes focal segmental glomerulosclerosis in humans and mice. Kidney International, 95(1), 57–61. 10.1016/j.kint.2018.08.014 [DOI] [PubMed] [Google Scholar]
- Tan, W. , & Airik, R. (2021). Primary coenzyme Q10 nephropathy, a potentially treatable form of steroid‐resistant nephrotic syndrome. Pediatric Nephrology, 36(11), 3515–3527. 10.1007/s00467-020-04914-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, C. P. , Gupta, S. , Freese, M. E. , Chouhan, K. K. , Dantuma, M. I. , Holanda, D. G. , … Smith, R. J. (2021). Sequential genetic testing of living‐related donors for inherited renal disease to promote informed choice and enhance safety of living donation. Transplant International, 34(12), 2696–2705. 10.1111/tri.14133 [DOI] [PubMed] [Google Scholar]
- Toniato, E. , Chen, X. P. , Losman, J. , Flati, V. , Donahue, L. , & Rothman, P. (2002). TRIM8/GERP RING finger protein interacts with SOCS‐1. The Journal of Biological Chemistry, 277(40), 37315–37322. 10.1074/jbc.M205900200 [DOI] [PubMed] [Google Scholar]
- Tory, K. , Menyhard, D. K. , Woerner, S. , Nevo, F. , Gribouval, O. , Kerti, A. , … Antignac, C. (2014). Mutation‐dependent recessive inheritance of NPHS2‐associated steroid‐resistant nephrotic syndrome. Nature Genetics, 46(3), 299–304. 10.1038/ng.2898 [DOI] [PubMed] [Google Scholar]
- Tran, P. V. , Haycraft, C. J. , Besschetnova, T. Y. , Turbe‐Doan, A. , Stottmann, R. W. , Herron, B. J. , … Beier, D. R. (2008). THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nature Genetics, 40(4), 403–410. 10.1038/ng.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautmann, A. , Bodria, M. , Ozaltin, F. , Gheisari, A. , Melk, A. , Azocar, M. , … PodoNet, C. (2015). Spectrum of steroid‐resistant and congenital nephrotic syndrome in children: The PodoNet registry cohort. Clinical Journal of the American Society of Nephrology, 10(4), 592–600. 10.2215/CJN.06260614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanov, S. , Wall, C. A. , Miller, J. A. , Scholey, J. W. , Cattran, D. C. , & Toronto Glomerulonephritis Registry Group . (2005). Focal and segmental glomerulosclerosis: Definition and relevance of a partial remission. Journal of the American Society of Nephrology, 16(4), 1061–1068. 10.1681/ASN.2004070593 [DOI] [PubMed] [Google Scholar]
- Tsukaguchi, H. , Sudhakar, A. , Le, T. C. , Nguyen, T. , Yao, J. , Schwimmer, J. A. , … Pollak, M. R. (2002). NPHS2 mutations in late‐onset focal segmental glomerulosclerosis: R229Q is a common disease‐associated allele. Journal of Clinical Investigation, 110(11), 1659–1666. 10.1172/Jci200216242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzur, S. , Rosset, S. , Shemer, R. , Yudkovsky, G. , Selig, S. , Tarekegn, A. , … Skorecki, K. (2010). Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Human Genetics, 128(3), 345–350. 10.1007/s00439-010-0861-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- USRDS . (2021). 2021 USRDS annual data report: Epidemiology of kidney disease in the United States . Retrieved from https://adr.usrds.org/2021
- VanDeVoorde, R. , Witte, D. , Kogan, J. , & Goebel, J. (2006). Pierson syndrome: A novel cause of congenital nephrotic syndrome. Pediatrics, 118(2), e501–e505. 10.1542/peds.2005-3154 [DOI] [PubMed] [Google Scholar]
- Wang, F. , Zhang, Y. , Mao, J. , Yu, Z. , Yi, Z. , Yu, L. , … Hildebrandt, F. (2017). Spectrum of mutations in Chinese children with steroid‐resistant nephrotic syndrome. Pediatric Nephrology, 32(7), 1181–1192. 10.1007/s00467-017-3590-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Chun, J. , Genovese, G. , Knob, A. U. , Benjamin, A. , Wilkins, M. S. , … Pollak, M. R. (2019). Contributions of rare gene variants to familial and sporadic FSGS. Journal of the American Society of Nephrology, 30(9), 1625–1640. 10.1681/ASN.2019020152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Anglani, F. , Beara‐Lasic, L. , Mehta, A. J. , Vaughan, L. E. , Herrera Hernandez, L. , … Investigators of the Rare Kidney Stone, C . (2016). Glomerular pathology in dent disease and its association with kidney function. Clinical Journal of the American Society of Nephrology, 11(12), 2168–2176. 10.2215/CJN.03710416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warejko, J. K. , Tan, W. , Daga, A. , Schapiro, D. , Lawson, J. A. , Shril, S. , … Hildebrandt, F. (2018). Whole exome sequencing of patients with steroid‐resistant nephrotic syndrome. Clinical Journal of the American Society of Nephrology, 13(1), 53–62. 10.2215/CJN.04120417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts, A. J. B. , Keller, K. H. , Lerner, G. , Rosales, I. , Collins, A. B. , Sekulic, M. , … Weins, A. (2022). Discovery of autoantibodies targeting Nephrin in minimal change disease supports a novel autoimmune etiology. Journal of the American Society of Nephrology, 33(1), 238–252. 10.1681/ASN.2021060794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weins, A. , Kenlan, P. , Herbert, S. , Le, T. C. , Villegas, I. , Kaplan, B. S. , … Pollak, M. R. (2005). Mutational and biological analysis of alpha‐actinin‐4 in focal segmental glomerulosclerosis. Journal of the American Society of Nephrology, 16(12), 3694–3701. 10.1681/ASN.2005070706 [DOI] [PubMed] [Google Scholar]
- Weins, A. , Schlondorff, J. S. , Nakamura, F. , Denker, B. M. , Hartwig, J. H. , Stossel, T. P. , & Pollak, M. R. (2007). Disease‐associated mutant alpha‐actinin‐4 reveals a mechanism for regulating its F‐actin‐binding affinity. Proceedings of the National Academy of Sciences of the United States of America, 104(41), 16080–16085. 10.1073/pnas.0702451104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng, P. L. , Majmundar, A. J. , Khan, K. , Lim, T. Y. , Shril, S. , Jin, G. , … Sanna‐Cherchi, S. (2021). De novo TRIM8 variants impair its protein localization to nuclear bodies and cause developmental delay, epilepsy, and focal segmental glomerulosclerosis. American Journal of Human Genetics, 108(2), 357–367. 10.1016/j.ajhg.2021.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharram, B. L. , Goyal, M. , Wiggins, J. E. , Sanden, S. K. , Hussain, S. , Filipiak, W. E. , … Wiggins, R. C. (2005). Podocyte depletion causes glomerulosclerosis: Diphtheria toxin‐induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. Journal of the American Society of Nephrology, 16(10), 2941–2952. 10.1681/ASN.2005010055 [DOI] [PubMed] [Google Scholar]
- Yao, T. , Udwan, K. , John, R. , Rana, A. , Haghighi, A. , Xu, L. , … Barua, M. (2019). Integration of genetic testing and pathology for the diagnosis of adults with FSGS. Clinical Journal of the American Society of Nephrology, 14(2), 213–223. 10.2215/CJN.08750718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarkan Tugsal, H. , Zengin, B. , Kenar, G. , Can, G. , Unlu, M. , Onen, F. , & Birlik, M. (2019). Infliximab‐associated focal segmental glomerulosclerosis in a patient with ankylosing spondylitis. Rheumatology International, 39(3), 561–567. 10.1007/s00296-019-04241-8 [DOI] [PubMed] [Google Scholar]
- Ye, Q. , Zhang, Y. , Zhuang, J. , Bi, Y. , Xu, H. , Shen, Q. , … Mao, J. (2021). The important roles and molecular mechanisms of annexin A2 autoantibody in children with nephrotic syndrome. Annals of Translational Medicine, 9(18), 1452. 10.21037/atm-21-3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, Q. , Zhou, C. , Wang, D. J. , Fu, H. D. , Wang, J. J. , & Mao, J. H. (2021). Seven novel podocyte autoantibodies were identified to diagnosis a new disease subgroup‐autoimmune Podocytopathies. Clinical Immunology, 232, 108869. 10.1016/j.clim.2021.108869 [DOI] [PubMed] [Google Scholar]
- Yeo, J. , Qiu, Y. , Jung, G. S. , Zhang, Y. W. , Buehler, M. J. , & Kaplan, D. L. (2020). Adverse effects of Alport syndrome‐related Gly missense mutations on collagen type IV: Insights from molecular simulations and experiments. Biomaterials, 240, 119857. 10.1016/j.biomaterials.2020.119857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Su, B. , Liu, X. , Xiao, H. , Ding, J. , & Yao, Y. (2018). Mutations in TTC21B cause different phenotypes in two childhood cases in China. Nephrology (Carlton), 23(4), 371–376. 10.1111/nep.13008 [DOI] [PubMed] [Google Scholar]
- Zhong, J. Y. , Zuo, Y. Q. , Ma, J. , Fogo, A. B. , Jolicoeur, P. , Ichikawa, I. , & Matsusaka, T. (2005). Expression of HIV‐1 genes in podocytes alone can lead to the full spectrum of HIV‐1‐associated nephropathy. Kidney International, 68(3), 1048–1060. 10.1111/j.1523-1755.2005.00497.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Genetic causes of FSGS (sorted by mode of inheritance).
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.