

Summary
Despite the success of BCR‐ABL‐specific tyrosine kinase inhibitors (TKIs) such as imatinib in chronic phase (CP) chronic myeloid leukaemia (CML), patients with blast phase (BP)‐CML continue to have a dismal outcome with median survival of less than one year from diagnosis. Thus BP‐CML remains a critical unmet clinical need in the management of CML. Our understanding of the biology of BP‐CML continues to grow; genomic instability leads to acquisition of mutations which drive leukaemic progenitor cells to develop self‐renewal properties, resulting in differentiation block and a poor‐prognosis acute leukaemia which may be myeloid, lymphoid or bi‐phenotypic. Similar advances in therapy are urgently needed to improve patient outcomes; however, this is challenging given the rarity and heterogeneity of BP‐CML, leading to difficulty in designing and recruiting to prospective clinical trials. This review will explore the treatment of BP‐CML, evaluating the data for TKI therapy alone, combinations with intensive chemotherapy, the role of allogeneic haemopoietic stem cell transplantation, the use of novel agents and clinical trials, as well as discussing the most appropriate methods for diagnosing BP and assessing response to therapy, and factors predicting outcome.
Keywords: allogeneic stem cell transplantation, blast phase chronic myeloid leukaemia, clinical trials, combination therapy, tyrosine kinase inhibitors
Abbreviations
- ACA
additional cytogenetic abnormality
- alloHSCT
allogeneic haemopoietic stem cell transplantation
- AML
acute myeloid leukaemia
- AP
accelerated phase
- BP
blast phase
- CCyR
complete cytogenetic response
- cGVHD
chronic graft‐versus‐host disease
- CHR
complete haematological response
- CML
chronic myeloid leukaemia
- CNA
copy number abnormalities
- CNS
central nervous system
- CP
chronic phase
- DLI
donor lymphocyte infusion
- DMR
deep molecular response
- ELN
European LeukemiaNet
- ELTS
EUTOS Long Term Survival
- MMR
major molecular remission
- MUL
molecularly undetectable leukaemia
- NGS
next‐generation sequencing
- OS
overall survival
- PFS
progression‐free survival
- PRC
polycomb repressive complex
- RT‐qPCR
reverse transcription quantitative real‐time polymerase chain reaction
- TKI
tyrosine kinase inhibitor
- WHO
World Health Organisation
INTRODUCTION
Chronic myeloid leukaemia (CML) is a relatively rare form of myeloproliferative neoplasm, with an incidence of 0.7–1.0/100 000. 1 CML has the hallmark genetic lesion the Philadelphia (Ph) chromosome, a reciprocal translocation between the long arms of chromosomes 9 and 22, with the resultant shortened chromosome 22 being the afore‐mentioned Ph chromosome, producing the oncogenic fusion protein BCR‐ABL1, a constitutively active tyrosine kinase. 2
CML is a triphasic disorder. The majority of patients present in chronic phase (CP), associated with a raised white cell count, splenomegaly and, occasionally, symptoms of hyperviscosity. However, according to the EUTOS population‐based registry, 4%–5% of patients present in accelerated phase (AP) 3 ; a transitional stage of disease associated with further genetic mutations, an increase in blasts and primitive cells, cytopenias and, often, treatment resistance. A further 1%–2% present in blast phase (BP; sometimes referred to as ‘blast crisis’) CML, a poor‐prognosis acute leukaemia, which may be myeloid, lymphoid or mixed phenotype. 4 In addition to presenting with de novo BP‐CML, patients may progress on therapy. With the introduction of BCR‐ABL1‐specific tyrosine kinase inhibitors (TKIs), the rate of progression to AP or BP has fallen from 5%–20% to 1%–5% annually, with the greatest risk of progression during the first year following diagnosis. 5 While TKIs provide excellent outcomes for patients with CP‐CML, with the majority of CP patients expected to have a normal life expectancy, 6 responses to TKIs in BP are infrequent and of short duration, with most patients dying from refractory disease within a year of developing BP‐CML. 5 Therefore BP‐CML continues to be a critical area of clinical need where new approaches to improve patient outcomes are urgently required.
DIAGNOSIS OF BLAST PHASE CHRONIC MYELOID LEUKAEMIA
The World Health Organisation (WHO) and European LeukemiaNet (ELN) differ on the diagnostic criteria for BP‐CML, with the WHO citing 20% or more blasts and the ELN 30% or more blasts in the peripheral blood or bone marrow. 7 , 8 A study by Cortes et al. 9 evaluated outcomes for BP‐CML patients according to both WHO and ELN criteria. Overall survival (OS) was significantly better in patients with 20%–29% blasts as compared to those with blasts at 30% or more with three‐year OS of 42% versus 10%, respectively. Within the recent MATCHPOINT clinical trial for UK patients with BP‐CML, the ELN definition of 30% or more blasts was used as an entry criterion. 10 Extramedullary blast proliferation is also diagnostic of BP‐CML, regardless of disease phase in the bone marrow. 7 , 8
Where a diagnosis of BP‐CML is suspected, a bone marrow aspirate should be performed to evaluate morphology, immunophenotyping to confirm if myeloid, lymphoid or mixed phenotype, and full karyotype to identify any additional cytogenetic abnormalities (ACAs). 11 The ACAs which may be diagnostic of AP‐CML, e.g. extra Ph chromosome, isochromosome 17q, trisomy 8 or 19, may also be present at diagnosis of de novo BP‐CML or following progression from CP‐ or AP‐CML. 12 Where the aspirate is a dry tap, a trephine should be performed, and where possible a portion of this sent for karyotyping. A BCR‐ABL1 kinase domain mutation should be performed at diagnosis of de novo BP or on progression to AP‐ or BP‐CML. 11 , 13 Next‐generation sequencing (NGS) panel assessment to identify commonly mutated lymphoid or myeloid genes may also be considered, but remains an experimental approach in CML.
MECHANISMS UNDERLYING PROGRESSION TO BP‐CML
The molecular mechanisms underlying progression to BP‐CML are not fully understood, and are likely multifactorial (Figure 1). While CP‐CML results from acquisition of BCR‐ABL1 in a primitive haemopoietic stem cell, in BP, progenitor cells acquire self‐renewal potential and undergo differentiation arrest. 14 Progenitors in BP‐CML have more stem cell‐like properties, 15 with upregulation of β‐catenin and C‐MYC activity, 16 , 17 and vulnerability to oxidative DNA damage leading to genomic instability and a high mutation burden, 18 , 19 including acquisition of ACAs and molecular lesions, e.g. mutation of TP53 which occurs in up to 20% of BP‐CML cases. 20 A detailed review of the biology and mechanisms of progression to BP‐CML is beyond the scope of this review. This topic has been reviewed elsewhere. 21 , 22 Very recently, epigenetic reprogramming via the polycomb repressive complex (PRC) complex has been identified as important for progression to BP‐CML. 23
FIGURE 1.
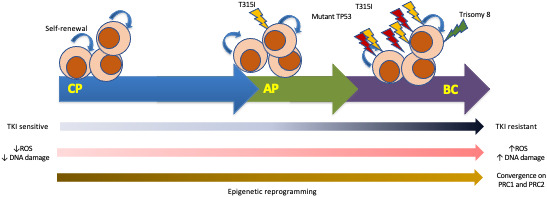
Clonal evolution in CML. The schematic diagram proposes a model for clonal evolution in CML. Acquisition of BCR‐ABL1 within a stem/progenitor cell results in an increase in ROS, genomic instability and epigenetic reprogramming with convergence on PRC1 and PRC2 complexes. 21 , 22 , 23 AP‐CML may develop when additional mutations, e.g. T315I, occur and treatment resistance ensues. Further progression to BP occurs when leukaemic progenitor cells acquire self‐renewal potential, which is combined with worsening genomic instability leading to acquisition of further genetic abnormalities, e.g. TP53 mutation, trisomy 8 and differentiation block. AP, accelerated phase; BP, blast phase; CML, chronic myeloid leukaemia; CP, chronic phase; ROS, reactive oxygen species; TKI, tyrosine kinase inhibitor.
PROGNOSTIC RISK FACTORS IN BP‐CML
Several clinical, cytogenetic and molecular features have been identified as having prognostic importance in BP‐CML. Recent studies by Jain et al., and Lauseker et al., undertaken in the TKI era, have identified clinical features associated with either poorer OS or increased risk of treatment failure. 24 , 25 These include older age (>58 years), thrombocytopenia, anaemia, high LDH, prior TKI therapy, progression from CP or AP, and myeloid immunophenotype. Interestingly, in the study by Lauseker et al. 25 the EUTOS Long Term Survival (ELTS) score, developed for CP‐CML, defined two prognostic groups in BP‐CML. The probability of two‐year OS for patients with a high‐risk ELTS score was 0.4 (95% confidence interval 0.29–0.56) versus 0.69 (95% confidence interval 0.49–0.96) for non‐high‐risk patients.
The German CML IV study demonstrated that the presence of major route ACAs, defined as a second Ph chromosome, trisomy 8 or 19 and isochromosome (17q), were associated with progression to advanced‐phase CML. 26 More recently, Chen et al. 27 also identified 3q26.2 rearrangement as a high‐risk genetic lesion in TKI‐treated patients which correlated with the presence of ABL1 mutations (associated with BCR‐ABL1 kinase domain mutations). This is confirmed by Gong et al. 28 who demonstrated that TKI therapy substantially reduces the risk of progression to BP in low‐risk, but not high‐risk ACAs, namely 3q26.2 rearrangement, 7(del7q), isochromosome (17q) and high‐risk complex karyotype. The German CML Study Group has also recently shown that the presence of high‐risk ACAs in patients with low blast percentages are an early marker of CML progression and predict poor OS. 29
Using whole‐exome and RNA sequencing, Branford et al. 30 showed that all patients in their cohort (n = 39) had mutated cancer genes at progression to BP‐CML with patients having up to five genetic lesions at diagnosis of BP. A very recent comprehensive genetic analysis has shown that specific genetic lesions are better predictors of survival in BP, compared to clinical parameters. 31 They demonstrate in a large patient cohort, with 52 matched CP and BP samples, that the number of mutations acquired during progression correlated with the time to progression, and inversely correlated with exposure to TKI therapy during CP. This suggests that accumulation of mutations is suppressed by TKI therapy, which may also prevent progression to BP in some patients. Driver mutations implicated in other myeloid malignancies, e.g. RUNX1, ABL1, TP53, ASXL1, BCOR/BCORL1 and WT1, were found in BP‐CML. Interestingly, ASXL1 was already present in CP, and, in 14 of 15 patients with an ASXL1 mutation, other mutations, e.g. TP53 or RUNX1 developed during progression to BP. They also demonstrated that specific copy number abnormalities (CNAs) and genetic lesions were enriched in myeloid BP; these were trisomies 21, 8 and 9, ASXL1 and TP53 mutations. CDKN2A/B, IKZF1 deletion, −7/del(7p) and −9/del(9p) were enriched in lymphoid BP. RUNX1 and extra Ph chromosome were seen in both lymphoid and myeloid BP.
ABL1 mutations occurred almost exclusively in patients who had previously received TKIs, and were the most frequent mutation type. 30 , 31 In the study by Ochi et al., 31 all BP‐CML patients with an ABL1 mutation, had additional cytogenetic or molecular lesions. The study by Branford et al. 30 demonstrated that only 2/19 patients had an ABL1 mutation as the only genetic lesion, and that ABL1 mutations were more common with progression to lymphoid BP (16/20 patients) versus myeloid BP (3/19 patients), and in lymphoid BP, were frequently associated with IKZF1 mutation. Mutations of TP53, BCOR and RUNX1 were less frequent in TKI‐treated patients. Independent risk factors predicting OS in patients treated with TKI‐based therapy were ASXL1 mutations, complex CNAs, isochomosome (17q), and trisomy 21. 31
TREATMENT OF BP‐CML
When deciding on appropriate therapy for BP‐CML, many factors need to be considered which will affect the choice of therapy. Principal among these are (1): is BP a de novo presentation or a progression on TKI or other therapy? (2) Is BP myeloid, lymphoid or mixed phenotype? (3) What prior TKIs (if any) has the patient been exposed to? (4) Is the patient known to have a BCR‐ABL1 kinase domain mutation? (5) Is the patient fit for intensive chemotherapy and is consideration of allogeneic haemopoietic stem cell transplantation (alloHSCT) for post‐remission consolidation therapy appropriate?
In the pre‐TKI era, patients were treated with chemotherapy protocols designed for acute leukaemias, with acute myeloid leukaemia (AML)‐type intensive induction approaches for myeloid BP, 32 and vincristine and prednisolone‐based multidrug approaches used for lymphoid BP. 33 The outcomes for intensive chemotherapy approaches alone in BP‐CML were inferior to responses in de novo acute leukaemias. Responses tended to be of short duration, with patients relapsing within a few months in the absence of alloHSCT consolidation. The introduction of TKIs changed the treatment landscape for patients with BP‐CML, and enabled some older, frailer patients to obtain remissions (albeit of short duration) when used as single agents. However, all patients considered appropriate and with a suitable donor, should proceed to alloHSCT on achievement of a second CP (CP2), as this remains the therapeutic option with the highest likelihood of long‐term remission or cure. 11 , 34
MONITORING RESPONSE TO THERAPY AND APPROPRIATE TREATMENT END‐POINTS IN BP‐CML
Appropriate investigations for monitoring response to therapy in BP‐CML are shown in Table 1. Historically, the initial therapeutic goal in BP‐CML was to return patients to a CP2, as the outcome of alloHSCT is better if patients are transplanted in CP2 compared to active BP disease. 4
TABLE 1.
Suggested investigations at diagnosis of BP‐CML and to monitor response to therapy
| Diagnosis | On therapy | |
|---|---|---|
| Full blood count, blood film and 200‐cell differential | X | X |
| Immunophenotyping by multi‐parameter flow cytometry | X | X a |
| Cytogenetics for full karyotype | X | X a |
| BCR‐ABL qRT‐PCRIS | X | X b |
| BCR‐ABL kinase domain mutation testing by NGS | X | X c |
| NGS myeloid/lymphoid panel assessment | X | |
| Tissue typing of patient, siblings and VUD search if transplant being considered | X | |
| Lumbar puncture and CSF cytology in lymphoid and mixed phenotype BP | X | X d |
Abbreviations: CSF, cerebrospinal fluid; IS, International Scale; MMR, major molecular remission; NGS, next generation sequencing; VUD, volunteer unrelated donor.
Bone marrow assessment three‐monthly until achievement of MMR and then as clinically indicated.
Three‐monthly indefinitely. More frequent monitoring may be required if concerned about relapse of losing molecular response.
Three‐monthly until achievement of MMR and then if loss of response.
To correspond with intrathecal therapy and as clinically indicated.
Studies in CP‐CML have shown that responses deeper than complete cytogenetic response (CCyR; defined as the absence of the Ph chromosome evaluated in at least 20 cells in metaphase in a bone marrow aspirate) confer no additional survival benefit. 35 The situation in BP‐CML is very different. Five‐year OS for BP‐CML patients achieving CCyR is an abysmal 12.2%. 36 This is only modestly improved in BP patients achieving major molecular remission (MMR; defined as a BCR‐ABL1:control gene ratio of ≤0.1% on the International Scale) at 34.4%. In BP‐CML, optimal responses are seen in patients with molecularly undetectable leukaemia (MUL; with specification of the number of control gene transcripts), 11 with a five‐year OS of 71.8%. 36 Of note, in this study, 80/92 (87%) of patients achieving MUL had received an alloHSCT. The remaining 12 patients achieved MUL following a combination of TKI and chemotherapy. Median OS for patients achieving MUL was 132.2 months, and this should be considered the treatment goal for optimal long‐term survival in BP‐CML.
SINGLE‐AGENT TKI THERAPY
In BP‐CML, responses to single‐agent TKIs are seen, but are not maintained. The choice of TKI is dependent on whether or not the presentation is of de novo BP, or progression on a TKI; if progression, then knowledge of prior TKI use, responses and presence of BCR‐ABL1 mutations is critical. There are no randomised controlled trials of TKI therapy in BP‐CML. However, there are multiple single‐arm studies with imatinib in de novo BP‐CML, 37 , 38 , 39 , 40 , 41 , 42 dasatinib and nilotinib in imatinib failure, 43 , 44 , 45 , 46 , 47 and bosutinib and ponatinib following resistance or intolerance to two or more TKIs. 48 , 49 Nilotinib is not licensed for BP‐CML. There are limited data for front‐line use of second‐generation TKIs in de novo BP‐CML. There are no data for asciminib in BP‐CML. Response rates are shown in Table 2, and the available data for all approved TKIs are considered below. However, these responses are not durable, with a median response duration of less than 12 months for all TKIs.
TABLE 2.
Outcomes to single agent TKIs in BP CML
| TKI | Prior TKI (yes/no) | Reference(s) | No. of patients | CHR (%) | CCyR (%) | alloHSCT (%) | Median OS (mo) |
|---|---|---|---|---|---|---|---|
| Imatinib | No | 37 |
mBP = 38 lBP = 20 |
10.5 20 |
7.9 10 |
4.5 N/A |
N/A N/A |
| No | 40, 41 | 229 (mBP) | 15.3 | 7.4 | 5 | 6.9 | |
| No | 38 | 75 | 21.3 | 18.7 | 9.3 | 6.5 | |
| No | 42 | 30 | 33 | 0 | N/A | OS 36% at 1 year | |
| No | 39 | 92 | 26.1 | 9.8 | 9.8 | 7.0 | |
| Dasatinib | Yes | 47 |
mBP = 23 lBP/Ph+ALL = 10 |
34.8 70 |
26.1 30 |
N/A 10 |
N/A N/A |
| Yes | 43, 46 |
mBP = 149 lBP = 61 |
17.4 18.0 |
17.5 36.8 |
6.0 9.8 |
7.7–7.9 9.0–11.4 |
|
| Nilotinib | Yes | 45 | 33 | 6.1 | 6.1 | N/A | N/A |
| Yes | 44 |
mBP = 105 lBP = 31 |
24 21 |
30 32 |
11 6 |
10.1 7.9 |
|
| Yes | 51 |
mBP = 133 lBP = 50 |
6.8 14.0 |
8.3 26.0 |
N/A N/A |
OS 63% at 18 months for mBP/lBP | |
| Bosutinib | Yes | 49 |
Second line = 36 ≥Third line = 28 |
27 4 |
37 17 |
N/A N/A |
11.2 8.9 |
| Ponatinib | Yes | 48, 52 | 62 (incl. 24 with T315I) | 21 | 17.4 | 9.7 | OS 29% at 1 year & 9% at 3 years |
Abbreviations: alloHSCT, allogeneic haemopoietic stem cell transplantation; BP, blast phase; CCyR, complete cytogenetic remission; CHR, complete haematologic remission; CML, chronic myeloid leukaemia; lBP, lymphoid blast phase; mBP, myeloid blast phase; N/A, data not available; OS, overall survival; Ph+ALL, Philadelphia chromosome‐positive acute lymphoid leukaemia; TKI, tyrosine kinase inhibitor.
Imatinib
The recommended dose of imatinib in BP‐CML is 800 mg once daily; however many patients will be intolerant of this dose, and 400 or 600 mg once daily may be more achievable. Clinical trials in BP‐CML, with no prior exposure to TKIs, have investigated doses ranging from 300 to 1000 mg daily. Whilst the majority of patients achieved some form of haematological response, complete haematological response (CHR; defined as normal full blood count and blood film as well as no splenomegaly on examination) was much less common, occurring in 10.5%–33% of patients in early trials of imatinib (Table 2). CCyR was also rare, occurring in 0%–18.7% of patients. Survival was short, with median OS ranging from 6.5–10 months. Not all patients had de novo CML in these early imatinib trials, with some having previously received alloHSCT in an earlier phase of disease. Only a minority of patients subsequently proceeded to alloHSCT. Molecular responses were not reported in these early imatinib trials.
Dasatinib
The recommended dose of dasatinib in BP‐CML is 140 mg once daily. This dosing schedule has a better safety profile than 70 mg twice daily. 46 Clinical trials of dasatinib in BP‐CML have been performed in patients resistant or intolerant to imatinib. 43 , 46 , 47 There are no prospective clinical trials, and limited data on the front‐line use of dasatinib in de novo BP‐CML. The START‐B (myeloid BP) and START‐L (lymphoid BP) trials reported high rates of BCR‐ABL1 kinase domain mutations, with 42% and 65% of patients, respectively, having these mutations detected by Sanger sequencing. 43 CHR rates varied widely between the studies from 17.4% to 70.0% (Table 2). 43 , 46 , 47 A minority of patients achieved CCyR (17.5%–36.8%). There was a trend towards higher CCyR rates in lymphoid compared to myeloid BP (30.0%–36.8% vs. 17.5%–26.1%, respectively). Median OS was short, ranging from 7.7–11.4 months. As with imatinib, only a minority of patients proceeded to alloHSCT, and previous alloHSCT was associated with a poorer outcome. 43 Cytopenias were almost universal in BP‐CML patients treated with dasatinib, and there was an increased risk of pleural effusion compared with CP‐CML, 46 in part related to the higher dose (140 mg in BP vs. 100 mg in CP). There is some evidence that dasatinib crosses the blood–brain barrier which makes it a more attractive option for patients with lymphoid BP or evidence of central nervous system (CNS) disease. 50
Nilotinib
Nilotinib is not licensed in BP‐CML. Clinical trials of nilotinib in BP‐CML were conducted in patients resistant or intolerant to imatinib, 44 , 45 , 51 with the most commonly assessed dose being 400 mg twice daily. There are no prospective clinical trials on the front‐line use of nilotinib in de novo BP‐CML. As with dasatinib, a high rate of BCR‐ABL1 kinase domain mutations was reported, with 44% of resistant patients and 10% of intolerant patients having these mutations detected by Sanger sequencing. 44 CHR rates were low, ranging from 6.1%–24%, with no clear difference between lymphoid and myeloid phenotype (Table 2). 44 , 45 , 51 CCyR was achieved in 6.1%–32% of patients, with a trend towards higher CCyR rates in lymphoid compared to myeloid BP (26%–32% vs. 8.3%–30%, respectively), 44 , 51 however, where OS was evaluated, this did not translate into a survival advantage in lymphoid BP, with OS being 10.1 months compared with 7.9 months for myeloid and lymphoid BP, respectively. 44 Only a minority of patients proceeded to alloSCT. Grade 3/4 cytopenias were very common in BP‐CML patients treated with nilotinib, and the most common non‐haematological adverse events were hyperbilirubinaemia, elevation of transaminases, lipase elevation, rash and nausea. 44 , 51
Bosutinib
The recommended dose of bosutinib in BP‐CML is 500 mg once daily. Available data for bosutinib in BP‐CML is from the Phase 1/2 Study 200 in patients resistant or intolerant to imatinib (second line; n = 36) and dasatinib and/or nilotinib (third or higher line; n = 28). 49 Of the 64 patients recruited, 23 had myeloid, 10 lymphoid and 31 unspecified BP‐CML. 53% of recruited patients had at least one, and 11% has two or more BCR‐ABL1 kinase domain mutations. Baseline T315I mutations were present in 16% of patients. The CHR rates for bosutinib were 27% and 4% and CCyR rates 37% and 17% in second line and third or higher line, respectively. Median OS was 11.2 months in second line and 8.9 months in third or higher line. As with other second‐generation TKIs, cytopenias were common. Grade 1/2 biochemical abnormalities were also common, but grade 3/4 adverse events much less frequent.
Ponatinib
In the UK, ponatinib is approved for use in patients with BP‐CML who have failed a second ‐generation TKI or have a T315I mutation (www.nice.org.uk/guidance/ta451). The PACE (Ponatinib Ph+ ALL and CML Evaluation) clinical trial assessed the efficacy of 45 mg ponatinib in patients with all phases of CML and Ph+ALL, including 62 patients with BP‐CML. 48 , 52 Ponatinib was administered as second, third or fourth line therapy, and 24 BP‐CML patients had a T315I mutation. CHR, CCyR and MMR were achieved in 21%, 17.4% and 12.9% of patients, respectively. Responses were short‐lived, with a median progression‐free survival (PFS) of 3.7 months. 9.4% of patients proceeded to alloHSCT. Overall survival was 29% at one year and 9% at three years.
No data are available for the second‐generation TKI radotinib or the STAMP (specifically targeting the myristoyl pocket) inhibitor asciminib in BP‐CML. In conclusion, outcomes are unsatisfactory for all single‐agent TKIs, with only a minority of patients achieving a CCyR (6.1%–37%) which was short‐lived. Median OS ranged from 6.5 to 11.4 months with few patients proceeding to potentially curative alloHSCT consolidation.
COMBINATION THERAPIES
With the dismal outcomes seen with TKI alone in BP‐CML, attention has focussed on TKIs in combination with chemotherapy or immunological approaches. Available data are from either short case series or early‐phase single‐arm clinical trials. Due to the rarity of BP‐CML in the TKI era, large randomised clinical trials comparing different treatment approaches are not feasible, and often different therapies have been adopted for myeloid versus lymphoid BP, based on treatments for AML and ALL, respectively. TKI combination clinical trials and outcomes for myeloid and lymphoid BP‐CML are summarised in Tables 3 and 4, respectively.
TABLE 3.
Outcomes to TKI in combination with AML chemotherapy in myeloid BP‐CML
| Combination | Reference(s) | No. of patients | CHR (%) | CCyR (%) | alloHSCT (%) | Median OS (mo) |
|---|---|---|---|---|---|---|
| Ponatinib + FLAG‐IDA | 10 | 17 (incl. 4 lBP) | 19 | 50 | 70.6 | 12 |
| Dasatinib + FLAG‐IDA | 98 | 4 (incl 1 lBP) | 100 | 75 | 100 | N/A |
| Imatinib + ‘7+3’ daunorubicin/cytarabine | 99 | 36 | 56 | 30.6 | 30.6 | 16 |
| Imatinib + idarubicin/idarubicin + cytarabine | 100 | 19 | 47 | 15.8 | 26.3 | 5 |
| Imatinib + mitoxantrone/etoposide | 101 | 16 | 81 | N/A | 37.5 | 6.4 |
| Imatinib + decitabine | 53 | 10 | 20 | 10 | N/A | 3.5 |
| Dasatinib + decitabine | 54 | 30 (incl AP n = 7 and Ph+AML n = 4) | 48 | 33.3 | 26.7 | 13.8 |
| Dasatinib or nilotinib or ponatinib + azacytidine | 55 | 7 | 71 | 43 | N/A | 27.4 |
| Imatinib + omacetaxine | 102 | 12 | 58 | 27.3 | 72.7 | N/A; 75% OS at 12 months |
Abbreviations: alloHSCT, allogeneic haemopoietic stem cell transplantation; AP, accelerated phase; CCyR, complete cytogenetic remission; CHR, complete haematologic remission; FLAG‐IDA, fludarabine, cytarabine, granulocyte‐colony stimulating factor, idarubicin; lBP, lymphoid blast phase; N/A, data not available; OS, overall survival; Ph+AML, Philadelphia chromosome‐positive acute myeloid leukaemia.
TABLE 4.
Outcomes to TKI in combination with ALL chemotherapy in lymphoid BP‐CML
| Combination | Reference(s) | No. of patients | CHR (%) | CCyR (%) | alloHSCT (%) | Median OS (mo) |
|---|---|---|---|---|---|---|
| Imatinib + vincristine + dexamethasone | 58 | 12 | 91.7 | 33.3 | 58.3 | 13 |
| Imatinib or dasatinib + HyperCVAD | 56 | 42 | 90.4 | 58 | 42.9 | 17 |
Abbreviations: alloHSCT, allogeneic haemopoietic stem cell transplantation; CCyR, complete cytogenetic remission; CHR, complete haematologic remission; HyperCVAD, hyperfractionated cyclophosphamide, vincristine, adriamycin and dexamethasone; OS, overall survival.
Myeloid blast phase
Within the UK, we have recently published the Phase 1/2 MATCHPOINT clinical trial, using an Eff‐Tox design, combining ponatinib with the chemotherapy regimen FLAG‐IDA (fludarabine, cytarabine, idarubicin and granulocyte‐colony stimulating factor [G‐CSF]). 10 Patients received either one or two cycles of FLAG‐IDA with ponatinib 30 mg daily, and then proceeded to alloHSCT. Where feasible, ponatinib was re‐started as maintenance post alloHSCT. Of the 17 patients recruited, 16 were evaluable for the primary end‐point; 11 (69%) achieved a return to CP2, with five achieving MMR after one cycle of ponatinib–FLAG‐IDA. Interestingly, 8/16 (50%) patients achieved CCyR after the first cycle of therapy, with no additional patients achieving CCyR after a second cycle of FLAG‐IDA. Median OS was 12 months and 41% of patients were alive at three years. Long‐term survival was critically dependent on successful transplantation, and not a second cycle of FLAG‐IDA chemotherapy. Post‐transplant maintenance ponatinib, at a dose of 15 mg for those patients in MMR, was well tolerated, with no excess toxicity observed when ponatinib was used both pre and post alloHSCT. The study identified no new safety signals for ponatinib when combined with FLAG‐IDA chemotherapy.
Studies combining a TKI with a hypomethylating agent have been published (Table 3). This approach may be more suitable for older or less fit patients. In the study by Oki et al., 53 results with imatinib in combination with decitabine were poor with a CCyR rate of 10% and median OS of 3.5 months. However, the majority of these patients had previously received imatinib therapy. More recent small, single‐arm studies have combined a second‐generation TKI or ponatinib with decitabine or azacytidine, and these look to hold more promise with CCyR rates of 33%–43% and a median OS of 13.8–27.4 months. 54 , 55 An ongoing Phase 2 study (PONAZA; www.clinicaltrials.gov NCT03895671; accessed 8th April 2022) aims to recruit up to 40 patients with AP‐ or BP‐CML and assess the combination of ponatinib at a starting dose of 45 mg daily with azacytidine. It will be interesting to compare the results of this study with MATCHPOINT to better understand the optimal approach to treating myeloid BP‐CML. 10
Lymphoid blast phase
For patients with lymphoid BP, considering an ALL‐type approach to combination therapy is also an option. In the largest study to date, Strati et al. 56 evaluated imatinib (400–800 mg daily) or dasatinib (50–140 mg daily) with hyperfractionated cyclophosphamide, vincristine, adriamycin and dexamethasone (hyperCVAD) in 42 patients with lymphoid BP. CHR and CCyR rates were 90% and 58%, respectively. The commonest grade 3/4 adverse events were haematological toxicity and infection, occurring in 100% and 59% of patients, respectively. Despite this, the treatment discontinuation rate was less than 10%. 53% of patients proceeded to alloHSCT and median OS was 17 months. Remission duration was longer for patients receiving dasatinib (not reached vs. 14 months; p = 0.15), achieving CCyR (not reached vs. 8 months; p < 0.015), undergoing alloHSCT (not reached vs. 7 months; p < 0.01). Interestingly, a recent Phase 2 study combining ponatinib with hyperCVAD in Ph+ALL has reported a five‐year OS of 71%. 57 The ponatinib was reduced to 30 mg daily after cycle 1 in the latter part of the study due to concerns about cardiovascular toxicity.
In a less intensive approach, Rea et al. 58 evaluated IM 800 mg together with vincristine and dexamethasone in 13 lymphoid BP patients. Of the 12 evaluable patients, 11 (91.7%) achieved CHR and 4 (33.3%) achieved CCyR. Seven patients proceeded to alloHSCT and median OS was 13 months. Results were inferior in those patients that had received prior imatinib with all four patients relapsing within nine months.
More recently short case series of TKI in combination with immunotherapy, either inotuzumab or blinatumomab, have been published. Of two reported patients with lymphoid BP, treated with bosutinib 400 mg in combination with the anti‐CD22 drug–antibody conjugate inotuzumab ozogamicin, one responded. 59 Assi et al. 60 reported three patients with lymphoid BP treated with the bi‐specific anti‐CD3/CD19 monoclonal antibody blinatumomab in combination with TKI (ponatinib n = 2 and dasatinib n = 1). All three patients achieved CCyR and even deep molecular response (DMR), defined as the absence of a detectable BCR‐ABL1 transcript by RT‐qPCR with a sensitivity of 0.01%. One patient proceeded to alloHSCT and all three were alive at the time of the report, eight months after treatment initiation. Further to this, Patel et al. 61 report a single case of multiply relapsed CD19+ lymphoid BP achieving DMR and proceeding to alloHSCT after ponatinib 45 mg in combination with blinatumomab.
ALLOGENEIC HAEMOPOIETIC STEM CELL TRANSPLANTATION
Early referral to a transplant centre should be considered when there is evidence of molecular progression (loss of MMR, acquisition of BCR‐ABL kinase domain mutations or acquisition of major route or high‐risk ACAs) 5 after the failure of two or more TKIs as progression to frank BP‐CML is associated with poor transplant outcomes, despite TKI therapy (Table 5). As evidenced above, outcomes for patients with BP‐CML are universally poor after TKI or TKI in combination with chemotherapy. Therefore, patients that are biologically fit enough and with a suitable donor should be offered alloHSCT. However, limited data are available reporting on transplant outcomes in the TKI era; available studies are presented in Table 5. Peripheral‐blood stem cells are preferred over bone marrow as the stem cell source, with lower non‐relapse mortality. 62 Patients who achieve a CP2 or better have a superior outcome compared to those transplanted in frank BP. 63 , 64 Khoury et al. and Radujkovic et al. reported a three‐year OS of 35%–40% and 51.1%, respectively, for BP‐CML in remission compared to 8%–11% and 23.3%, respectively, for patients transplanted with active BP‐CML, likely due to a higher rate of relapse in the active BP‐CML cohorts. Other adverse prognostic factors include a high EBMT risk score (≥5), 63 , 65 reduced performance status, 64 low CD34+ cell count in graft, and donor age. 62 Radujkovic et al. 64 also reported transplant prior to 2010 was associated with increased non‐relapse mortality. There was no difference in outcomes between related versus matched unrelated donor or reduced‐intensity versus fully myeloablative conditioning regimens. The impact of chronic graft‐versus‐host disease (cGVHD) is less clear. Niederweiser et al. 62 reported improved OS for patients that develop cGVHD, but Radujkovic et al. 64 did not. In a subanalysis, in patients with active disease, use of a matched unrelated donor was associated with improved OS, and in patients in remission at the time of transplant, a high EBMT score was associated with worse OS. Niederweiser et al. 62 reported improved outcomes for BP‐CML patients receiving post‐transplant TKI and donor lymphocyte infusions (DLI).
TABLE 5.
Allogeneic stem cell transplantation outcomes for patients with blast phase CML 1990–2018
| References | Dates of study | No. of patients | 3‐year OS (%) | 3‐year PFS (%) | Relapse incidence (%) | Non‐relapse mortality (%) |
|---|---|---|---|---|---|---|
| 65 , a | 2000–2009 | 63 | 36 | 23 | 40 | 33 |
| 63 | 1999–2004 | 80 (active BP‐CML) | 14 | 10 | 36 | 54 |
| 64 | 2004–2016 | 170 | 39 | 26 | 51 | 23 |
| 62 , b | 1990–2018 | 147 (incl. 51 AP‐CML) | 34 | 26 | 43 | 28 |
Abbreviations: OS, overall survival; PFS, progression‐free survival.
Median follow‐up since transplant 25 months.
Median follow‐up since transplant 15 years.
In summary, while outcomes following alloHSCT for BP‐CML remain poor compared to alloHSCT for CP‐CML, 63 they are superior to non‐transplant outcomes. Novel strategies are required to reduce relapse risk post alloHSCT, including reducing disease burden pre transplant and, where possible, returning patients to CP2. Further strategies to improve outcome include post‐transplant TKI and optimising the graft‐versus‐leukaemia effect through early reduction of immunosuppression and consideration of prophylactic DLI. 66 However, it is unlikely that frank BP relapse post alloHSCT will respond to DLI or TKI. 67
THE ROLE OF TKI MAINTENANCE THERAPY POST ALLOHSCT
Data on the use of TKI post transplant are limited. 68 Where BP‐CML patients have demonstrated TKI sensitivity pre alloSCT or have not exhausted all TKI options, then post‐transplant TKI is recommended, 10 , 13 , 39 , 69 especially following reduced‐intensity alloHSCT. 70 The recent study by Neiderweiser et al. 62 confirms this with a significant improvement in OS with post‐transplant TKI in BP‐CML. However, the optimal duration of therapy is unclear. As discussed earlier, achievement of deep molecular remission (MUL) is important for long‐term survival. 36 Post‐transplant BCR‐ABL1 monitoring in BP‐CML should be performed three‐monthly for first two years and then 3–6‐monthly indefinitely. 13
SPECIAL SITUATIONS
Extramedullary disease
BP‐CML may rarely present as an extramedullary blast cell proliferation of either myeloid, lymphoid or bi‐phenotypic origin. 71 It is reported in approximately 8%–16% of cases of BP‐CML, and may involve multiple sites. 72 , 73 This is usually associated with BP‐CML in the bone marrow, although may be the first indication of BP. The most common sites of involvement are lymph nodes, skin (leukaemia cutis), bone and central nervous system (CNS). 8 There is a high rate of delayed or mis‐diagnosis which can compromise therapy. While data on extramedullary BP‐CML is scant, and is predominantly from case reports, patients should be managed as other patients with BP‐CML. 74 , 75 , 76 Where possible, intensive chemotherapy combined with TKI is indicated, and should be followed with alloHSCT and post‐transplant TKI maintenance. There is insufficient data to determine if TKIs have altered the incidence or disease course of patients with extramedullary BP‐CML. However, a recent case series of 42 patients with myeloid sarcoma identified that they are more common in men (M:F ratio = 4.3:1), and in the TKI era, the outcome is better as compared with myeloid BP‐CML with median OS of 18.4 months compared to 8 months for myeloid BP‐CML (p = 0.01). 77 Furthermore, the study reported superior OS for de novo myeloid sarcoma as compared to development of a myeloid sarcoma while on TKI therapy (36.0 vs. 8.3 months; p = 0.002). There was no difference in OS for progression to myeloid sarcoma or myeloid BP on TKI therapy (8.3 vs. 7.0 months, respectively; p = 0.55).
Central nervous system disease
Extramedullary BP‐CML may present as CNS leukaemia. Although more common with lymphoid and bi‐phenotypic BP‐CML, it can also occur with myeloid BP. 78 It is more common with a high white cell count at presentation. 79 , 80 CNS relapse has been identified in approximately 20% of BP‐CML or Ph+ ALL previously treated with imatinib. 81 Often patients will show a complete response in the bone marrow with isolated CNS relapse, supporting the role of the CNS as a sanctuary site for leukaemia stem cells. 82
Imatinib has relatively poor CNS penetration, and there are multiple case reports of CNS leukaemia/relapse on imatinib. 81 There is some evidence that dasatinib crosses the blood–brain barrier 50 ; it has demonstrated improved survival in a murine model of BCR‐ABL1‐positive CNS leukaemia, and clinical responses in patients with BCR‐ABL1‐positive leukaemia and CNS disease. There are isolated case reports for the use of nilotinib and bosutinib in BCR‐ABL1‐positive CNS leukaemia. 80 , 83 He et al. 84 report a series of nine cases of Ph+ leukaemia with CNS disease and T315I mutation (five Ph+ALL and four BP‐CML), post alloHSCT who were treated with ponatinib. There were five isolated CNS relapses and four CNS and marrow relapses. Eight of nine patients, including all four BP‐CML patients were alive a median of 18 months after first CNS relapse, suggesting that ponatinib may have efficacy in recurrent Ph+ CNS leukaemia. Regardless of which TKI is preferred, prevention and treatment of CNS leukaemia should also include systemic and/or intrathecal therapy. CNS prophylaxis should be administered to patients with lymphoid and bi‐phenotypic BP‐CML. 13 , 34
Blast phase CML and COVID‐19
While patients with CP‐CML in MMR on TKIs or in treatment‐free remission do not have an increased risk of severe COVID‐19 disease and death, this is not the case for advanced‐phase CML. Patients with AP‐ or BP‐CML are 8.2‐fold more likely to die from COVID‐19 compared to patients in MMR. 85 Thus, patients with advanced‐phase CML should be considered for antibody (sotrovimab) or antiviral therapy (nirmatrelvir, remdesivir or molnupiravir) to reduce the risk of hospitalisation and death. 86 , 87
Blast phase CML in children
A detailed consideration of BP‐CML in children is beyond the scope of this review and readers are referred to the recent excellent article by Hijuya and Suttorp. 88 Due to the rarity of CML, and BP‐CML, specifically, in children, paediatric guidelines are lacking, and it is recommended that, where possible, adult guidelines are followed, with patients proceeding to alloHSCT wherever possible.
NOVEL THERAPEUTIC APPROACHES
Outcomes for BP‐CML remain poor, and new therapeutic approaches are urgently needed. Phase 1 clinical trials of asciminib and oliverembatinib (HQP1351) did include patients with BP‐CML, although these data remain unpublished. 89 , 90 Recently, the novel TKI K0706 has demonstrated an acceptable toxicity profile and preliminary evidence of response in advanced‐phase CML in a Phase 1 clinical trial. 91 Despite the success of the anti‐CD33 drug–antibody conjugate gemtuzumab ozogamicin in AML, 92 this has not been extensively assessed in myeloid BP‐CML. One early trial showed responses in 2/9 patients with BP‐CML when combined with fludarabine and cytarabine, 93 but subsequent to this, only isolated cases have been reported. 71 , 94 A recent single‐centre, retrospective study of venetoclax with TKI‐based regimens has shown some promising results in BP‐CML (n = 9), 95 with a median OS of 10.9 months, and further studies are warranted. The aurora kinase inhibitor danusertib has demonstrated responses in a Phase 1 trial in patients with the T315I mutation, 96 including BP‐CML, but further clinical development was halted due to practicalities of the drug infusion frequency. Immunotherapies, for example CAR‐T cell therapy, are also a rapidly expanding field, and with the immune dysfunction associated with BP‐CML, may offer hope for the future. 97 Table 6 summarises ongoing clinical trials including BP‐CML patients.
TABLE 6.
Currently recruiting BP‐CML clinical trials in adult patients using novel therapeutic approaches (excluding alloHSCT) on the www.clinicaltrials.gov website (accessed on April 26, 2022)
| Phase and trial title | Drug | Patient population | Primary end‐point | NCT number |
|---|---|---|---|---|
| Phase 1. ABL001 + dasatinib + prednisone for BCR‐ABL+ B‐ALL or CML |
Asciminib Dasatinib Prednisone |
Ph+ALL; lBP‐CML | MTD of asciminib | NCT03595917 |
| Phase 1. Study of HQP1351 in refractory CML and Ph+ALL |
Olverembatinib (HQP1351) Blinatumomab |
CP‐, AP‐ and BP‐CML, Ph+ALL | Cmax and AUC for HQP1351 | NCT04260022 |
| Phase 1. Fludarabine, cytarabine and pegcrisantaspase for the treatment of relapsed or refractory leukaemia |
Fludarabine Cytarabine Pegcrisantaspase |
AML, ALL, T‐PLL, biphenotypic AL, BP‐CML | Safety and tolerability of fludarabine, cytarabine and pegcrisantaspase | NCT04526785 |
| Phase 1. Hu8F4 in treating patients with advanced haematologic malignancies | Hu8F4 (anti‐PR1/HLA‐A2 monoclonal antibody) | HR‐MDS, CMML, AML, BP‐CML, HR‐MF | DLT and minimum safe and biologically‐effective dose | NCT02530034 |
| Phase 1/2. Venetoclax, ponatinib, and dexamethasone in participants with PH+ or BCR‐ABL positive relapsed or refractory ALL or chronic myelogenous leukemia |
Dexamethasone Ponatinib Venetoclax Rituximab |
Ph+ALL; lBP‐CML | MTD of venetoclax in combination with dexamethasone and ponatinib | NCT03576457 |
| Phase 2. PONAZA: A combination of ponatinib and 5‐azacitidine in chronic myelogenous leukaemia in AP or in myeloid blast crisis |
Ponatinib 5‐azacitidine |
AP‐CML and mBP‐CML | 2‐year OS | NCT03895671 |
| Phase 2. Cladribine, idarubicin, cytarabine and venetoclax in treating patients with AML, high‐risk myelodysplastic syndrome or BP‐CML |
Cladribine Idarubicin Cytarabine Venetoclax |
AML, HR‐MDS, BP‐CML | CR rate up to 12 months | NCT 02115295 |
| Phase 2. Decitabine, venetoclax and ponatinib for the treatment of Ph+ALL or mBP‐ or AP‐CML |
Decitabine Venetoclax Ponatinib |
Ph+AML, mBP‐CML, AP‐CML | CR/CRi rate | NCT04188405 |
| Phase 2. Low‐dose chemotherapy, ponatinib and blinatumomab in treating patients with Philadelphia chromosome‐positive and/or BCR‐ABL‐positive ALL |
Blinatumomab Cyclophosphamide Cytarabine Filgrastim Methotrexate PEGFilgrastim Ponatinib Rituximab Vincristine |
Newly diagnosed Ph+ALL and lBP‐CML | CMR in newly diagnosed Ph+ and/or BCR‐ABL+ recipients | NCT03147612 |
| Phase 2. Blinatumomab, cytarabine, methotrexate and ponatinib in treating patients with Philadelphia chromosome positive, or BCR‐ABL‐positive, or relapsed/refractory AL |
Blinatumomab Cytarabine Methotrexate Ponatinib |
Newly diagnosed and R/R Ph+ALL and lBP‐CML | CMR at 18 weeks | NCT03263572 |
Abbreviations: AL, acute leukaemia; A ALL, acute lymphoblastic leukaemia; alloHSCT, allogeneic haemopoietic stem cell transplantation; AML, acute myeloid leukaemia; AP, accelerated phase; AUC, area under curve; B‐ALL, B‐cell acute lymphoblastic leukaemia; BP, blast phase; Cmax, maximum plasma concentration; CMML, chronic myelomonocytic leukaemia; CMR, complete molecular response; CML, chronic myeloid leukaemia; CP, chronic phase; CR, complete response; CRi complete response with incomplete count recovery; DLT, dose‐limiting toxicity; HR‐MDS, high‐risk myelodysplastic syndrome; HR‐MF, high‐risk myelofibrosis; lBP, lymphoid blast phase; mBP, myeloid blast phase; MTD, maximum tolerated dose; OS, overall survival; Ph+ALL, Philadelphia chromosome‐positive ALL; T‐PLL, T‐cell prolymphocytic leukaemia.
CONCLUSIONS
While TKIs have transformed outcomes for patients with CP‐CML, outcomes remain poor for BP. Thus, a major focus needs to be prevention of progression to BP‐CML, with early intervention for those patients failing to achieve milestone responses. 11 Due to the rarity of BP‐CML, clinical trial data are limited, and guidelines are often based on expert opinion rather than Phase 2 and Phase 3 trial outcomes. Figure 2 summarises the broad approach to managing BP‐CML in the UK. 34
FIGURE 2.
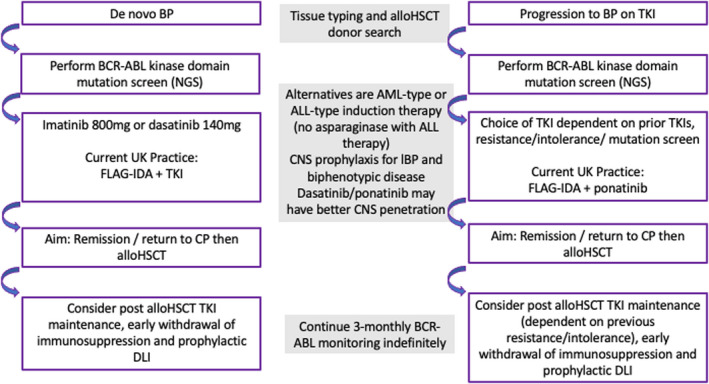
Current management of BP‐CML. The flow chart summarises the current approach(es) for management of BP‐CML. Although there are some similarities in management, de novo BP‐CML and progression to BP‐CML on therapy are considered separately. ALL, acute lymphoblastic leukaemia; alloHSCT, allogeneic haemopoietic stem cell transplantation; AML, acute myeloid leukaemia; BP, blast phase; CML, chronic myeloid leukaemia; CNS, central nervous system; CP, chronic phase; DLI, donor lymphocyte infusion; FLAG‐IDA, fludarabine, cytarabine, granulocyte‐colony stimulating factor, idarubicin chemotherapy; lBP, lymphoid blast phase; NGS, next‐generation sequencing; TKI, tyrosine kinase inhibitor.
To improve the therapeutic approach for BP‐CML, we need to work collaboratively across countries and continents, with industry, charities and academic institutions to clinically develop more effective therapies. Available data shows that MUL is required for optimal survival outcomes in BP‐CML, 36 and this should be our goal.
FUNDING INFORMATION
Mhairi Copland is an employee of the University of Glasgow. No additional funding was required for preparation of this review article.
CONFLICT OF INTEREST
Mhairi Copland has received research funding from Cyclacel and Incyte, is/has been an advisory board member for Novartis, Incyte, Jazz Pharmaceuticals, Pfizer and Servier, and has received honoraria from Astellas, Novartis, Incyte, Pfizer and Jazz Pharmaceuticals.
Copland M. Treatment of blast phase chronic myeloid leukaemia: A rare and challenging entity. Br J Haematol. 2022;199:665–678. 10.1111/bjh.18370
DATA AVAILABILITY STATEMENT
Not applicable as review article and no datasets included.
REFERENCES
- 1. Höglund M, Sandin F, Simonsson B. Epidemiology of chronic myeloid leukaemia: an update. Ann Hematol. 2015;94(Suppl 2):S241–S247. [DOI] [PubMed] [Google Scholar]
- 2. Heisterkamp N, Stam K, Groffen J, de Klein A, Grosveld G. Structural organization of the bcr gene and its role in the Ph' translocation. Nature. 1985;315(6022):758–61. [DOI] [PubMed] [Google Scholar]
- 3. Hoffmann VS, Baccarani M, Hasford J, Castagnetti F, Di Raimondo F, Casado LF, et al. Treatment and outcome of 2904 CML patients from the EUTOS population‐based registry. Leukemia. 2017;31(3):593–601. [DOI] [PubMed] [Google Scholar]
- 4. DeFilipp Z, Khoury HJ. Management of advanced‐phase chronic myeloid leukemia. Curr Hematol Malig Rep. 2015;10(2):173–81. [DOI] [PubMed] [Google Scholar]
- 5. Hehlmann R. How I treat CML blast crisis. Blood. 2012;120(4):737–47. [DOI] [PubMed] [Google Scholar]
- 6. Bower H, Björkholm M, Dickman PW, Höglund M, Lambert PC, Andersson TM. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J Clin Oncol. 2016;34(24):2851–7. [DOI] [PubMed] [Google Scholar]
- 7. Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2006;108(6):1809–20. [DOI] [PubMed] [Google Scholar]
- 8. Vardiman JW, Melo JV, Baccarani M, Thiele J. Chronic myelogenous leukemia, BCR‐ABL1 positive. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: International Agency for Research on Cancer; 2008. [Google Scholar]
- 9. Cortes JE, Talpaz M, O'Brien S, Faderl S, Garcia‐Manero G, Ferrajoli A, et al. Staging of chronic myeloid leukemia in the imatinib era: an evaluation of the World Health Organization proposal. Cancer. 2006;106(6):1306–15. [DOI] [PubMed] [Google Scholar]
- 10. Copland M, Slade D, McIlroy G, Horne G, Byrne JL, Rothwell K, et al. Ponatinib with fludarabine, cytarabine, idarubicin, and granulocyte colony‐stimulating factor chemotherapy for patients with blast‐phase chronic myeloid leukaemia (MATCHPOINT): a single‐arm, multicentre, phase 1/2 trial. Lancet Haematol. 2022;9(2):E121–E132. [DOI] [PubMed] [Google Scholar]
- 11. Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34(4):966–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fabarius A, Kalmanti L, Dietz CT, Lauseker M, Rinaldetti S, Haferlach C, et al. Impact of unbalanced minor route versus major route karyotypes at diagnosis on prognosis of CML. Ann Hematol. 2015;94(12):2015–24. [DOI] [PubMed] [Google Scholar]
- 13. Deininger MW, Shah NP, Altman JK, Berman E, Bhatia R, Bhatnagar B, et al. Chronic myeloid leukemia, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2020;18(10):1385–415. [DOI] [PubMed] [Google Scholar]
- 14. Kinstrie R, Karamitros D, Goardon N, Morrison H, Hamblin M, Robinson L, et al. Heterogeneous leukemia stem cells in myeloid blast phase chronic myeloid leukemia. Blood Adv. 2016;1(3):160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vetrie D, Helgason GV, Copland M. The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML. Nat Rev Cancer. 2020;20(3):158–73. [DOI] [PubMed] [Google Scholar]
- 16. Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granulocyte‐macrophage progenitors as candidate leukemic stem cells in blast‐crisis CML. N Engl J Med. 2004;351(7):657–67. [DOI] [PubMed] [Google Scholar]
- 17. Abraham SA, Hopcroft LE, Carrick E, Drotar ME, Dunn K, Williamson AJ, et al. Dual targeting of p53 and c‐MYC selectively eliminates leukaemic stem cells. Nature. 2016;534(7607):341–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cramer‐Morales K, Nieborowska‐Skorska M, Scheibner K, Padget M, Irvine DA, Sliwinski T, et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. 2013;122(7):1293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grossmann V, Kohlmann A, Zenger M, Schindela S, Eder C, Weissmann S, et al. A deep‐sequencing study of chronic myeloid leukemia patients in blast crisis (BC‐CML) detects mutations in 76.9% of cases. Leukemia. 2011;25(3):557–60. [DOI] [PubMed] [Google Scholar]
- 20. Stuppia L, Calabrese G, Peila R, Guanciali‐Franchi P, Morizio E, Spadano A, et al. p53 loss and point mutations are associated with suppression of apoptosis and progression of CML into myeloid blastic crisis. Cancer Genet Cytogenet. 1997;98(1):28–35. [DOI] [PubMed] [Google Scholar]
- 21. Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120(7):2254–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bavaro L, Martelli M, Cavo M, Soverini S. Mechanisms of disease progression and resistance to tyrosine kinase inhibitor therapy in chronic myeloid leukemia: an update. Int J Mol Sci. 2019;20(24):6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ko TK, Javed A, Lee KL, Pathiraja TN, Liu X, Malik S, et al. An integrative model of pathway convergence in genetically heterogeneous blast crisis chronic myeloid leukemia. Blood. 2020;135(26):2337–53. [DOI] [PubMed] [Google Scholar]
- 24. Jain P, Kantarjian HM, Ghorab A, Sasaki K, Jabbour EJ, Nogueras Gonzalez G, et al. Prognostic factors and survival outcomes in patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era: cohort study of 477 patients. Cancer. 2017;123(22):4391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lauseker M, Bachl K, Turkina A, Faber E, Prejzner W, Olsson‐Strömberg U, et al. Prognosis of patients with chronic myeloid leukemia presenting in advanced phase is defined mainly by blast count, but also by age, chromosomal aberrations and hemoglobin. Am J Hematol. 2019;94(11):1236–43. [DOI] [PubMed] [Google Scholar]
- 26. Fabarius A, Leitner A, Hochhaus A, Müller MC, Hanfstein B, Haferlach C, et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long‐term observation of 1151 patients from the randomized CML Study IV. Blood. 2011;118(26):6760–8. [DOI] [PubMed] [Google Scholar]
- 27. Chen Z, Shao C, Wang W, Zuo Z, Mou X, Hu SJ, et al. Cytogenetic landscape and impact in blast phase of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Leukemia. 2017;31(3):585–92. [DOI] [PubMed] [Google Scholar]
- 28. Gong Z, Medeiros LJ, Cortes JE, Chen Z, Zheng L, Li Y, et al. Cytogenetics‐based risk prediction of blastic transformation of chronic myeloid leukemia in the era of TKI therapy. Blood Adv. 2017;1(26):2541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hehlmann R, Voskanyan A, Lauseker M, Pfirrmann M, Kalmanti L, Rinaldetti S, et al. High‐risk additional chromosomal abnormalities at low blast counts herald death by CML. Leukemia. 2020;34(8):2074–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Branford S, Wang P, Yeung DT, Thomson D, Purins A, Wadham C, et al. Integrative genomic analysis reveals cancer‐associated mutations at diagnosis of CML in patients with high‐risk disease. Blood. 2018;132(9):948–61. [DOI] [PubMed] [Google Scholar]
- 31. Ochi Y, Yoshida K, Huang YJ, Kuo MC, Nannya Y, Sasaki K, et al. Clonal evolution and clinical implications of genetic abnormalities in blastic transformation of chronic myeloid leukaemia. Nat Commun. 2021;12(1):2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iacoboni SJ, Plunkett W, Kantarjian HM, Estey E, Keating MJ, McCredie KB, et al. High‐dose cytosine arabinoside: treatment and cellular pharmacology of chronic myelogenous leukemia blast crisis. J Clin Oncol. 1986;4(7):1079–88. [DOI] [PubMed] [Google Scholar]
- 33. Canellos GP, DeVita VT, Whang‐Peng J, Carbone PP. Hematologic and cytogenetic remission of blastic transformation in chronic granulocytic leukemia. Blood. 1971;38(6):671–9. [PubMed] [Google Scholar]
- 34. Smith G, Apperley J, Milojkovic D, Cross NCP, Foroni L, Byrne J, et al. A British Society for Haematology Guideline on the diagnosis and management of chronic myeloid leukaemia. Br J Haematol. 2020;191(2):171–93. [DOI] [PubMed] [Google Scholar]
- 35. Kantarjian H, Cortes JE. Complete cytogenetic response, not deep molecular response, is associated with survival in chronic myeloid leukemia. J Clin Oncol. 2014;32(27):3077. [DOI] [PubMed] [Google Scholar]
- 36. Chen Z, Medeiros LJ, Kantajian HM, Zheng L, Gong Z, Patel KP, et al. Differential depth of treatment response required for optimal outcome in patients with blast phase versus chronic phase of chronic myeloid leukemia. Blood Cancer J. 2017;7(2):e521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR‐ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. [Erratum appears in N Engl J Med 2001 Jul 19;345(3):232]. N Engl J Med. 2001;344(14):1038–42. [DOI] [PubMed] [Google Scholar]
- 38. Kantarjian HM, Cortes J, O'Brien S, Giles FJ, Albitar M, Rios MB, et al. Imatinib mesylate (STI571) therapy for Philadelphia chromosome‐positive chronic myelogenous leukemia in blast phase. Blood. 2002;99(10):3547–53. [DOI] [PubMed] [Google Scholar]
- 39. Palandri F, Castagnetti F, Testoni N, Luatti S, Marzocchi G, Bassi S, et al. Chronic myeloid leukemia in blast crisis treated with imatinib 600 mg: outcome of the patients alive after a 6‐year follow‐up. Haematologica. 2008;93(12):1792–6. [DOI] [PubMed] [Google Scholar]
- 40. Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99(10):3530–9. [DOI] [PubMed] [Google Scholar]
- 41. Silver RT, Cortes J, Waltzman R, Mone M, Kantarjian H. Sustained durability of responses and improved progression‐free and overall survival with imatinib treatment for accelerated phase and blast crisis chronic myeloid leukemia: long‐term follow‐up of the STI571 0102 and 0109 trials. Haematologica. 2009;94(5):743–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sureda A, Carrasco M, de Miguel M, Martinez JA, Conde E, Sanz MA, et al. Imatinib mesylate as treatment for blastic transformation of Philadelphia chromosome positive chronic myelogenous leukemia. Haematologica. 2003;88(11):1213–20. [PubMed] [Google Scholar]
- 43. Cortes J, Kim DW, Raffoux E, Martinelli G, Ritchie E, Roy L, et al. Efficacy and safety of dasatinib in imatinib‐resistant or ‐intolerant patients with chronic myeloid leukemia in blast phase. Leukemia. 2008;22(12):2176–83. [DOI] [PubMed] [Google Scholar]
- 44. Giles FJ, Kantarjian HM, le Coutre PD, Baccarani M, Mahon FX, Blakesley RE, et al. Nilotinib is effective in imatinib‐resistant or ‐intolerant patients with chronic myeloid leukemia in blastic phase. Leukemia. 2012;26(5):959–62. [DOI] [PubMed] [Google Scholar]
- 45. Kantarjian H, Giles F, Wunderle L, Bhalla K, O'Brien S, Wassmann B, et al. Nilotinib in imatinib‐resistant CML and Philadelphia chromosome‐positive ALL. N Engl J Med. 2006;354(24):2542–51. [DOI] [PubMed] [Google Scholar]
- 46. Saglio G, Hochhaus A, Goh YT, Masszi T, Pasquini R, Maloisel F, et al. Dasatinib in imatinib‐resistant or imatinib‐intolerant chronic myeloid leukemia in blast phase after 2 years of follow‐up in a phase 3 study: efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer. 2010;116(16):3852–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib‐resistant Philadelphia chromosome‐positive leukemias. N Engl J Med. 2006;354(24):2531–41. [DOI] [PubMed] [Google Scholar]
- 48. Cortes JE, Kim DW, Pinilla‐Ibarz J, le Coutre P, Paquette R, Chuah C, et al. A phase 2 trial of ponatinib in Philadelphia chromosome‐positive leukemias. N Engl J Med. 2013;369(19):1783–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gambacorti‐Passerini C, Kantarjian HM, Kim D‐W, Khoury HJ, Turkina AG, Brummendorf TH, et al. Long‐term efficacy and safety of bosutinib in patients with advanced leukemia following resistance/intolerance to imatinib and other tyrosine kinase inhibitors. Am J Hematol. 2015;90(9):755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Porkka K, Koskenvesa P, Lundán T, Rimpiläinen J, Mustjoki S, Smykla R, et al. Dasatinib crosses the blood‐brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome‐positive leukemia. Blood. 2008;112(4):1005–12. [DOI] [PubMed] [Google Scholar]
- 51. Nicolini FE, Masszi T, Shen Z, Gallagher NJ, Jootar S, Powell BL, et al. Expanding Nilotinib Access in Clinical Trials (ENACT), an open‐label multicenter study of oral nilotinib in adult patients with imatinib‐resistant or ‐intolerant chronic myeloid leukemia in accelerated phase or blast crisis. Leuk Lymphoma. 2012;53(5):907–14. [DOI] [PubMed] [Google Scholar]
- 52. Cortes JE, Kim DW, Pinilla‐Ibarz J, le Coutre PD, Paquette R, Chuah C, et al. Ponatinib efficacy and safety in Philadelphia chromosome‐positive leukemia: final 5‐year results of the phase 2 PACE trial. Blood. 2018;132(4):393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Oki Y, Kantarjian HM, Gharibyan V, Jones D, O'brien S, Verstovsek S, et al. Phase II study of low‐dose decitabine in combination with imatinib mesylate in patients with accelerated or myeloid blastic phase of chronic myelogenous leukemia. Cancer. 2007;109(5):899–906. [DOI] [PubMed] [Google Scholar]
- 54. Abaza Y, Kantarjian H, Alwash Y, Borthakur G, Champlin R, Kadia T, et al. Phase I/II study of dasatinib in combination with decitabine in patients with accelerated or blast phase chronic myeloid leukemia. Am J Hematol. 2020;95(11):1288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ruggiu M, Oberkampf F, Ghez D, Cony‐Makhoul P, Beckeriche F, Cano I, et al. Azacytidine in combination with tyrosine kinase inhibitors induced durable responses in patients with advanced phase chronic myelogenous leukemia. Leuk Lymphoma. 2018;59(7):1659–65. [DOI] [PubMed] [Google Scholar]
- 56. Strati P, Kantarjian H, Thomas D, O'Brien S, Konoplev S, Jorgensen JL, et al. HCVAD plus imatinib or dasatinib in lymphoid blastic phase chronic myeloid leukemia. Cancer. 2014;120(3):373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jabbour E, Short NJ, Ravandi F, Huang X, Daver N, DiNardo CD, et al. Combination of hyper‐CVAD with ponatinib as first‐line therapy for patients with Philadelphia chromosome‐positive acute lymphoblastic leukaemia: long‐term follow‐up of a single‐centre, phase 2 study. Lancet Haematol. 2018;5(12):e618–e627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rea D, Legros L, Raffoux E, Thomas X, Turlure P, Maury S, et al. High‐dose imatinib mesylate combined with vincristine and dexamethasone (DIV regimen) as induction therapy in patients with resistant Philadelphia‐positive acute lymphoblastic leukemia and lymphoid blast crisis of chronic myeloid leukemia. Leukemia. 2006;20(3):400–3. [DOI] [PubMed] [Google Scholar]
- 59. Jain N, Maiti A, Ravandi F, Konopleva M, Daver N, Kadia T, et al. Inotuzumab ozogamicin with bosutinib for relapsed or refractory Philadelphia chromosome positive acute lymphoblastic leukemia or lymphoid blast phase of chronic myeloid leukemia. Am J Hematol. 2021;96(8):1000–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Assi R, Kantarjian H, Short NJ, Daver N, Takahashi K, Garcia‐Manero G, et al. Safety and efficacy of blinatumomab in combination with a tyrosine kinase inhibitor for the treatment of relapsed Philadelphia chromosome‐positive leukemia. Clin Lymphoma Myeloma Leuk. 2017;17(12):897–901. [DOI] [PubMed] [Google Scholar]
- 61. Patel SA, Bledsoe JR, Higgins AW, Hutchinson L, Gerber JM. Rapid and deep remission induced by blinatumomab for CD19‐positive chronic myeloid leukemia in lymphoid blast phase. JCO Precis Oncol. 2021;5:PO.21.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Niederwieser C, Morozova E, Zubarovskaya L, Zabelina T, Klyuchnikov E, Janson D, et al. Risk factors for outcome after allogeneic stem cell transplantation in patients with advanced phase CML. Bone Marrow Transplant. 2021;56(11):2834–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Khoury HJ, Kukreja M, Goldman JM, Wang T, Halter J, Arora M, et al. Prognostic factors for outcomes in allogeneic transplantation for CML in the imatinib era: a CIBMTR analysis. Bone Marrow Transplant. 2012;47(6):810–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Radujkovic A, Dietrich S, Blok HJ, Nagler A, Ayuk F, Finke J, et al. Allogeneic stem cell transplantation for blast crisis chronic myeloid leukemia in the era of tyrosine kinase inhibitors: a retrospective study by the EBMT Chronic Malignancies Working Party. Biol Blood Marrow Transplant. 2019;25(10):2008–16. [DOI] [PubMed] [Google Scholar]
- 65. Nicolini F, Modolo L, Raus N, Milpied N, Socie G, Yakoub‐Agha I, et al. Allogeneic stem cell transplantation for blast crisis (BC) chronic myelogenous leukemia (CML) in the tyrosine kinase inhibitors (TKIs) era. Analysis of pre‐transplant variables on transplant outcome. On behalf of the Society Francaise de Greffe de Moelle et de Therapie Cellulaire and the French Group of CML. Blood. 2010;116(21):2266.20574047 [Google Scholar]
- 66. Craddock CF. We do still transplant CML, don't we? Hematology Am Soc Hematol Educ Program. 2018;2018(1):177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chalandon Y, Passweg JR, Schmid C, Olavarria E, Dazzi F, Simula MP, et al. Outcome of patients developing GVHD after DLI given to treat CML relapse: a study by the Chronic Leukemia Working Party of the EBMT. Bone Marrow Transplant. 2010;45(3):558–64. [DOI] [PubMed] [Google Scholar]
- 68. Lübking A, Dreimane A, Sandin F, Isaksson C, Märkevärn B, Brune M, et al. Allogeneic stem cell transplantation for chronic myeloid leukemia in the TKI era: population‐based data from the Swedish CML registry. Bone Marrow Transplant. 2019;54(11):1764–74. [DOI] [PubMed] [Google Scholar]
- 69. Oyekunle A, Zander AR, Binder M, Ayuk F, Zabelina T, Christopeit M, et al. Outcome of allogeneic SCT in patients with chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Ann Hematol. 2013;92(4):487–96. [DOI] [PubMed] [Google Scholar]
- 70. Olavarria E, Siddique S, Griffiths MJ, Avery S, Byrne JL, Piper KP, et al. Posttransplantation imatinib as a strategy to postpone the requirement for immunotherapy in patients undergoing reduced‐intensity allografts for chronic myeloid leukemia. Blood. 2007;110(13):4614–7. [DOI] [PubMed] [Google Scholar]
- 71. Ovilla‐Martinez R, Weber Sánchez LA, Cota‐Rangel X, Baez‐Islas PE. Gemtuzumab‐ozogamicin and blinatumomab as treatment for refractory mixed‐phenotype blast crisis in chronic myeloid leukaemia. BMJ Case Rep. 2021;14(11):e243745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Terjanian T, Kantarjian H, Keating M, Talpaz M, McCredie K, Freireich EJ. Clinical and prognostic features of patients with Philadelphia chromosome‐positive chronic myelogenous leukemia and extramedullary disease. Cancer. 1987;59(2):297–300. [DOI] [PubMed] [Google Scholar]
- 73. Specchia G, Palumbo G, Pastore D, Mininni D, Mestice A, Liso V. Extramedullary blast crisis in chronic myeloid leukemia. Leuk Res. 1996;20(11‐12):905–8. [DOI] [PubMed] [Google Scholar]
- 74. Levy RA, Mardones MA, Burch MM, Krause JR. Myeloid sarcoma as the presenting symptom of chronic myelogenous leukemia blast crisis. Proc (Baylor Univ Med Cent). 2014;27(3):246–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tsukamoto S, Ota S, Ohwada C, Takeda Y, Takeuchi M, Sakaida E, et al. Extramedullary blast crisis of chronic myelogenous leukemia as an initial presentation. Leuk Res Rep. 2013;2(2):67–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Qi J, Zhang F, Liu Y, Yao J, Xu Y, He H. Extramedullary blast crisis of chronic myelogenous leukemia with a skin lesion: a case report and literature review. Am J Dermatopathol. 2021;43(6):450–3. [DOI] [PubMed] [Google Scholar]
- 77. Chen Z, Wang W, Cortes JE, Liu E, Miranda RN, Zhao C, et al. Differential clinical and prognostic impact of myeloid sarcoma vs medullary myeloid blast phase of chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood Cancer J. 2016;6:e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rytting ME, Wierda WG. Central nervous system relapse in two patients with chronic myelogenous leukemia in myeloid blastic phase on imatinib mesylate therapy. Leuk Lymphoma. 2004;45(8):1623–6. [DOI] [PubMed] [Google Scholar]
- 79. Altintas A, Cil T, Kilinc I, Kaplan MA, Ayyildiz O. Central nervous system blastic crisis in chronic myeloid leukemia on imatinib mesylate therapy: a case report. J Neuro‐Oncol. 2007;84(1):103–5. [DOI] [PubMed] [Google Scholar]
- 80. Mansoori H, Faraz M, Qadir H, Rashid A, Ali M. Precursor lymphoblastic lymphoma in the extramedullary tissue: a rare manifestation of chronic myeloid leukemia in blast crisis. Cureus. 2020;12(10):e11009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Aftimos P, Nasr F. Isolated CNS lymphoid blast crisis in a patient with imatinib‐resistant chronic myelogenous leukemia: case report and review of the literature. Leuk Res. 2009;33(11):e178–e180. [DOI] [PubMed] [Google Scholar]
- 82. Bujassoum S, Rifkind J, Lipton JH. Isolated central nervous system relapse in lymphoid blast crisis chronic myeloid leukemia and acute lymphoblastic leukemia in patients on imatinib therapy. Leuk Lymphoma. 2004;45(2):401–3. [DOI] [PubMed] [Google Scholar]
- 83. Martinez‐Cordero H, Patiño‐Escobar B, Enciso LJ, Otero DM, Spirko P. Myelomastocytic blast cell crisis in resistant tyrosine kinase inhibitor chronic myelogenous leukemia: case report and review of literature. Cureus. 2019;11(5):e4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. He JB, Zhang X, Guo ZW, Liu MM, Xu N, Huang F, et al. Ponatinib therapy in recurrent Philadelphia chromosome‐positive central nervous system leukemia with T315I mutation after Allo‐HSCT. Int J Cancer. 2020;147(4):1071–7. [DOI] [PubMed] [Google Scholar]
- 85. Pagnano KB, Hoow Kok C, Mauro MJ, Cortes JE, Evans N, Jiang Q, et al. COVID‐19 in patients with chronic myeloid leukemia: poor outcomes for patients with comorbidities, older age, advanced phase disease and those from low‐income countries: an update of the Candid study. Blood. 2021;138:634. [Google Scholar]
- 86. Kreuzberger N, Hirsch C, Chai KL, Tomlinson E, Khosravi Z, Popp M, et al. SARS‐CoV‐2‐neutralising monoclonal antibodies for treatment of COVID‐19. Cochrane Database Syst Rev. 2021;9:CD013825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. García‐Lledó A, Gómez‐Pavón J, González Del Castillo J, Hernández‐Sampelayo T, Martín‐Delgado MC, Martín Sánchez FJ, et al. Pharmacological treatment of COVID‐19: an opinion paper. Rev Esp Quimioter. 2022;35(2):115–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hijiya N, Suttorp M. How I treat chronic myeloid leukemia in children and adolescents. Blood. 2019;133(22):2374–84. [DOI] [PubMed] [Google Scholar]
- 89. Hughes TP, Mauro MJ, Cortes JE, Minami H, Rea D, DeAngelo DJ, et al. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. N Engl J Med. 2019;381(24):2315–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Qian J, Shi D, Li Z, Qin Y, Zhao T, Liu B, et al. Updated safety and efficacy results of Phase 1 study of olverembatinib (HQP1351), a novel third‐generation BCR‐ABL tyrosine kinase inhibitor (TKI), in patients with TKI‐resistant chronic myeloid leukemia (CML). Blood. 2021;138:311. [Google Scholar]
- 91. Cortes JE, Kim D‐W, Nicolini FE, Saikia T, Charbonnier A, Apperley JF, et al. Phase 1 trial of K0706, a novel oral BCR‐ABL1 tyrosine kinase inhibitor (TKI): in patients with chronic myelogenous leukemia (CML) and Philadelphia positive Acute Lymphoblastic Leukemia (Ph+ ALL) failing ≥3 prior TKI therapies: initial safety and efficacy. Blood. 2019;134:4158. [Google Scholar]
- 92. Burnett A, Stone R. AML: new drugs but new challenges. Clin Lymphoma Myeloma Leuk. 2020;20(6):341–50. [DOI] [PubMed] [Google Scholar]
- 93. Jabbour E, Garcia‐Manero G, Cortes J, Ravandi F, Plunkett W, Gandhi V, et al. Twice‐daily fludarabine and cytarabine combination with or without gentuzumab ozogamicin is effective in patients with relapsed/refractory acute myeloid leukemia, high‐risk myelodysplastic syndrome, and blast‐ phase chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2012;12(4):244–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Parsi M, Budak‐Alpdogan T. Promyelocytic blast crisis of chronic myeloid leukemia in a patient undergoing therapy with a tyrosine kinase inhibitor. Cureus. 2020;12(3):e7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Maiti A, Franquiz MJ, Ravandi F, Cortes JE, Jabbour EJ, Sasaki K, et al. Venetoclax and BCR‐ABL tyrosine kinase inhibitor combinations: outcome in patients with philadelphia chromosome‐positive advanced myeloid leukemias. Acta Haematol. 2020;143(6):567–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Borthakur G, Dombret H, Schafhausen P, Brummendorf TH, Boissel N, Jabbour E, et al. A phase I study of danusertib (PHA‐739358) in adult patients with accelerated or blastic phase chronic myeloid leukemia and Philadelphia chromosome‐positive acute lymphoblastic leukemia resistant or intolerant to imatinib and/or other second generation c‐ABL therapy. Haematologica. 2015;100(7):898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hsieh YC, Kirschner K, Copland M. Improving outcomes in chronic myeloid leukemia through harnessing the immunological landscape. Leukemia. 2021;35(5):1229–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Milojkovic D, Ibrahim A, Reid A, Foroni L, Apperley J, Marin D. Efficacy of combining dasatinib and FLAG‐IDA for patients with chronic myeloid leukemia in blastic transformation. Haematologica. 2012;97(3):473–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Deau B, Nicolini FE, Guilhot J, Huguet F, Guerci A, Legros L, et al. The addition of daunorubicin to imatinib mesylate in combination with cytarabine improves the response rate and the survival of patients with myeloid blast crisis chronic myelogenous leukemia (AFR01 study). Leuk Res. 2011;35(6):777–82. [DOI] [PubMed] [Google Scholar]
- 100. Quintás‐Cardama A, Kantarjian H, Garcia‐Manero G, O'Brien S, Faderl S, Ravandi F, et al. A pilot study of imatinib, low‐dose cytarabine and idarubicin for patients with chronic myeloid leukemia in myeloid blast phase. Leuk Lymphoma. 2007;48(2):283–9. [DOI] [PubMed] [Google Scholar]
- 101. Fruehauf S, Topaly J, Buss EC, Fischer T, Ottmann OG, Emmerich B, et al. Imatinib combined with mitoxantrone/etoposide and cytarabine is an effective induction therapy for patients with chronic myeloid leukemia in myeloid blast crisis. Cancer. 2007;109(8):1543–9. [DOI] [PubMed] [Google Scholar]
- 102. Fang B, Li N, Song Y, Han Q, Zhao RC. Standard‐dose imatinib plus low‐dose homoharringtonine and granulocyte colony‐stimulating factor is an effective induction therapy for patients with chronic myeloid leukemia in myeloid blast crisis who have failed prior single‐agent therapy with imatinib. Ann Hematol. 2010;89(11):1099–105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable as review article and no datasets included.