

Summary
Myeloproliferative neoplasms can be associated with bleeding manifestations which can cause significant morbidities. Although haematologists are aware of the likelihood of this complication in the setting of myeloproliferative neoplasms, it may often be overlooked especially in patients with no extreme elevation of blood counts and those with myelofibrosis. Acquired von Willebrand syndrome and platelet dysfunction are the two common diagnoses to be considered in this regard. In this review article, we discuss the mechanisms for the development of these rare bleeding disorders, their diagnosis and practical management.
Keywords: acquired von Willebrand, ET, MF, myeloproliferative neoplasm, platelets, PV
INTRODUCTION
Bleeding in myeloproliferative neoplasms (MPNs) was first reported in 1934, the initial case being one of spontaneous bleeding in a patient with underlying thrombocythaemia. 1 Frequency of reported bleeding in MPNs varies within the literature, but is consistently elevated when compared with the general population. A meta‐analysis of 13 436 patients with primary myelofibrosis (PMF), essential thrombocytosis (ET) or polycythaemia vera (PV) reported a pooled prevalence of bleeding complications of 6.2%. 2 Another recent meta‐analysis by Nicol et al. included 12 papers concerning ET (10 370 patients) and seven concerning PV (5683 patients). The reported incidence of major bleeding in ET was 0.43–5.3/100 patient years, and was 0.3–5.3/100 patient years for PV. Given the reported rate of gastrointestinal bleeding secondary to aspirin use in the general population has been calculated at 0.48–3.64/1000 patients years, 3 , 4 risk of bleeding is clearly increased in the MPN cohort. Smaller studies have reported even higher bleeding rates. 5 For example, Kander et al. observed at least a single bleeding episode in 15.6% of 351 MPN patients. Of the 55 patients in this study who experienced bleeding, more than 50% required hospitalisation, blood transfusion or a corrective procedure. 6 Use of anticoagulants and antiplatelet agents may further increase this risk. 7
Incidence and prevalence of bleeding varies according to the type of MPN, with accurate histopathological diagnosis of paramount importance for these patients. For example, one study showed that bleeding occurred twice as frequently in patients with PMF than ET, and the reported prevalences for PV, ET and PMF in Nicol et al.'s meta‐analysis were 6.9%, 7.3% and 8.9% respectively. 2 , 8 Further scrutiny of this trend noted bleeding in the setting of thrombocytosis to be more specific to prefibrotic PMF than to ET. Prefibrotic PMF may mimic ET and requires bone marrow examination for the presence of increased reticulin fibres to distinguish between these conditions. 9 Not only does prefibrotic MF have a higher risk of progression to PMF, leukaemic transformation, and death, but it also confers a greater risk of haemorrhagic complications than ET. 10 Of 1104 patients, 180 had a revised diagnosis of prefibrotic PMF after histological review. The incidence of bleeding in this cohort was double that of those with ET. 8 Barbui et al. also found the incidence of bleeding to be higher in patients with PMF receiving anticoagulant agents compared to patients with ET or PV. The cumulative incidence of bleeding in patients with PMF receiving a direct oral anticoagulant (DOAC) was found to be 5.6% of patients per year compared with 2.1% and 1.6% for patients with PV and ET respectively. 11
The natural history of disease can also impact bleeding. A study by Kaifie et al. found that bleeding events in MPNs peak after diagnosis rather than at time of diagnosis, thereby suggesting that bleeding may be related to progression of the underlying disease or to its treatment. This study described a major bleeding event in 8% of patients with a relatively equal distribution between PV, ET and PMF. 12
The aetiology of bleeding in MPNs is multifactorial, 6 , 13 , 14 with up to 39 different potential risk factors suggested. 5 In particular, the role of Acquired von Willebrand Syndrome (AVWS) and acquired platelet dysfunction have been studied in some detail with the complex pathogenesis of these disorders remaining a matter of debate. The effect of underlying treatments on extrinsic platelet function, most notably anti‐platelet drugs and cytoreductive agents, have also been a focus of study. 15 , 16
ACQUIRED VON WILLEBRAND SYNDROME
Incidence
Acquired von Willebrand Syndrome (AVWS) is a rare, acquired bleeding disorder caused by non‐hereditary, structural, functional or qualitative defects in von Willebrand factor (VWF). 13 , 17 The first report of AVWS in MPN described a case of bleeding in a patient with myelofibrosis that had transformed from PV. 18 The exact incidence of AVWS in MPNs is difficult to quantify and challenges to diagnosis mean that the true incidence is likely underreported. 19 , 20 It is widely accepted that AVWS is more common in lymphoproliferative than myeloproliferative diseases; however MPNs have been shown to account for approximately 15% of all reported cases of the disorder. 21 One study, encompassing 116 patients with ET and 57 patients with PV, revealed an incidence of AVWS in 55% and 49% of ET and PV patients respectively. 20 Of note, this study showed the incidence of AVWS to be highest in those with a platelet count of higher than 1000 ×109/l (68.3%) compared with lower platelet counts. Although selection bias may have contributed to the high frequency identified in this study, it is an indication of how common AVWS may be when tested for in patients with an underlying MPN.
Mechanisms
Several different hypotheses for the development of AVWS in MPN are outlined in the literature. In short, these can be due to:
sequestration of high molecular weight VWF multimers (HMWMs) by excessive numbers of platelets, most likely through GP1b; 17 , 22
proteolysis in setting of high platelets likely mediated through ADAMTS13 and enhanced sheer stress — this theory may be more relevant to AVWS caused by high sheer stress than in MPNs, where ADAMST13 levels are not significantly altered; 23
the presence of antibodies which either interfere with VWF activity or enhance its clearance; 22 , 24 , 25 , 26 , 27
High platelet counts and AVWS
High platelet counts in disorders such as ET can result in alterations to VWF multimer composition. 29 Qualitative defects in VWF through cleavage of HMWMs in the presence of thrombocytosis can develop, while VWF antigen levels are preserved, similar to the pattern seen in patients with Type 2A Von Willebrand Disease. 29 , 30 , 31 An inverse relationship between platelet count and VWF activity (demonstrated using the VWF ristocetin cofactor assay; VWF:RCo) has been outlined, upon which a basis of understanding for the pathogenesis of AVWS began to be formulated. The abnormality in VWF:RCo was attributed to increased proteolysis of VWF multimers by circulating platelets, demonstrated by increased VWF degradation products in gel electrophoresis. 24 , 32 In addition, normalisation of the platelet count has been shown to restore VWF multimeric composition. 33 In keeping with this hypothesis, the general consensus had been to advise testing for AVWS in a patient with an underlying MPN such as PV or ET with a platelet count higher than 1000 × 109/l. 34 , 35 , 36
Non‐elevated platelet counts and AVWS
In contrast to the findings described above, the median platelet count in patients with MPN and AVWS in a prospective study of 68 patients by Mohri et al. was only 638 × 109/l (697 × 109/l for PV and 1249 × 109/l for ET). 37 When comparing AVWS in patients with ET and PV, another study reported similar median platelet counts of 920 × 109/l for ET patients and 679 × 109/l for PV patients respectively. 20 This suggests that increased VWF proteolysis due to thrombocytosis may not be the only mechanism underpinning development of AVWS, and that other factors such as platelet function defects may play a role in haemorrhagic risk, particularly for non‐ET patients with lower platelet counts. 20 , 38 , 39 Patients with platelet counts over 1000–1500 × 109/l are usually considered high‐risk for developing AVWS; however as demonstrated above, extreme thrombocytosis is not always required. Indeed, AVWS has been reported in PV with an essentially normal platelet count at the time of diagnosis. 39 In this case, the patient had a near absence of HMWMs, which resolved after venesection and cytoreductive therapy was employed to reduce the elevated haematocrit. A high haematocrit has been reported to increase AVWS risk in both ET and PV, whereas leucocytosis appears to be more relevant to patients with PV only. 40 , 41 Increased blood viscosity related to an elevated haematocrit and white cell count may increase sheer stress and proteolysis, leading to destruction of HMWMs in the absence of marked thrombocytosis. Leucocytes also contain proteases with the ability to cleave VWF, which may bear relevance in patients with leucocytosis. 42 Of note, in a small series of patients with PMF, no difference in haematocrit or platelet counts was identified between cases with or without AVWS. 41
JAK‐2 mutations linked to AVWS?
The JAK‐2 V617F mutation is critically important to the pathogenesis of MPNs, most notably PV in which it is present in 95% of cases. 43 , 44 One study of bleeding in MPNs found the presence of the JAK‐2 V617F mutation to be an independent risk for development of AVWS in patients with ET. 20 Interestingly, JAK‐2 mutated ET patients were more likely to develop AVWS than calreticulin (CALR) mutated patients, despite a lower average platelet count in this group. Another has attributed high JAK‐2 V617F allele burden to an increased incidence of bleeding in these disorders. 28 Finally, the role of shear stress in contributing toward VWF proteolysis has been explored with studies suggestive of a conformational change in the VWF multimer, predisposing it to interaction and subsequent proteolysis by ADAMTS13 in response to shear stress. However, no defined link between this mechanism and MPNs has yet been proven. 45 , 46
Sites of bleeding with AVWS
The gastrointestinal tract (GIT) and central nervous system (CNS) and are frequent sites of major bleeding in MPN accounting for up to half of reported cases each (GIT bleeding in 48% and 54.7% of PV and ET patients respectively, and CNS in 44% of PV patients). 5 Spontaneous bleeding including epistaxis, other mucocutaneous bleeding, and easy bruising as well as drug‐associated or procedure‐related bleeding are also commonly observed. 5
A recent meta‐analysis of 38 studies reported the GI tract to be the most common bleeding site in patients with ET and PV (40.8% and 35.7% of cases respectively). In ET, the second most common site was the ear, nose and throat (12.2%), whereas it was mucocutaneous bleeding in PV patients (17.2%). This review found 37 lethal bleeding events described in the literature. The site of bleeding was stated in only eight cases, of which seven were secondary to intracranial bleeding. Overall, the incidence of all bleeding events analysed within this meta‐analysis was 1.39–6.6/100 patients years in ET and 0.3–5.3/100 patients years in PV. 5
An International society of thrombosis and haemostasis (ISTH) AVWS registry of 186 cases, of whom 15% were secondary to MPN, reported mucocutaneous bleeding in 68%. 34 A German MPN registry study also found the GI tract to be the most common location of bleeding in a registry of patients with ET, PV, PMF and unclassified MPN (MPN‐U). 12 Procedural and post‐operative bleeding is another important factor to consider. Procedural bleeding was reported in only six of the 38 studies considered in Nicol et al.'s meta‐analysis, encompassing a total of 21 bleeding episodes. 5 The most representative data of surgical interventions in ET and PV patients was published in 2008, with data analysed from 255 patients with a diagnosis of either PV or ET with a total of 311 surgical interventions. A total of 30 cases of bleeding were described with 23 of these cases deemed to be a major bleeding event. 47
Diagnosis
AVWS is suspected in individuals with no personal or family history of bleeding. In such patients, reduced von Willebrand (VW) activity/antigen ratio (VWF:Ac/Ag or VWF:CB/Ag) can be an indicator for structural or functional VW disorders even in the presence of a normal overall activity. If the activity is normal, multimer analysis is the most helpful tool to detect structural abnormalities of von Willebrand factor (VWF). In particular, decreased high molecular weight (HMW) multimers in the presence of otherwise normal VWF parameters indicate the presence of AWVS. 48
Since VW multimer analysis is difficult to perform outside highly specialised laboratories, an alternate diagnostic approach is to use half‐life studies. In most patients with inherited von Willebrand disease (VWD) (in the absence of the very rare situation of inhibitors to VWF), administration of VWF concentrates would be expected to raise VW activity levels to a predetermined level, and for levels to remain elevated for at least 12 h. If activity levels drop to baseline in less than 4–6 h, this may suggest a diagnosis of AVWS. 48 The exogenously administered VW factor is rapidly cleared by one of the several mechanisms described above. This practical test can also inform the management of AVWS, providing information regarding how frequently VWF concentrates may need to be administered.
Management
Studies of AVWS with thrombocytosis have shown a role for cytoreduction in improving VWF:RCo and associated normalisation of bleeding symptoms. 20 , 24 One such study has shown cytoreductive treatment to result in remission of 12 of 14 patients with MPN‐associated AWVS. 37
Management of acute bleeding in these patients can be challenging. Desmopressin (DDAVP) is a common treatment for inherited VWD, stimulating the release of HMWMs from endothelial Weibel–Palade bodies. However, it has been shown to have poor efficacy in the treatment of bleeding in AVWS, producing haemostasis in only around a fifth of MPN patients in an ISTH registry study. 34 It is thought that this may be due to prior exhaustion of endothelial VWF stores, or adsorption or proteolytic cleavage of the released VWF. 26 , 49 There is also a theoretical concern regarding the use of DDAVP in patients who may have underlying atherosclerotic heart disease, which may involve a significant proportion of elderly MPN patients. There are little data on the use of VWF concentrates in this setting, but some studies have suggested better outcomes than achieved using DDAVP. 19 High doses (50–100 iu/kg) and more frequent dosing based on VWF levels may be required to achieve adequate responses. 26 , 34 , 50 Anti‐fibrinolytic agents such as tranexamic acid may be used as adjuncts with DDAVP or VWF concentrates, or as single agents for minor bleeding episodes. 49 High‐dose intravenous immunoglobulin (IVIg) may be an option in patients failing to respond to other measures. This treatment is effective in cases of AVWS caused by pathogenic antibodies, particularly IgG monoclonal gammopathy of uncertain significance (MGUS). 51 Such antibodies have been reported in MPN patients who may therefore benefit from a trial of IVIg. 52 Finally, recombinant factor VIIa may be considered in patients with severe refractory bleeding. 26 , 53 , 54
Based on the available evidence, in Figure 1 we provide a proposed management algorithm for patients with AVWS presenting with or without bleeding symptoms.
FIGURE 1.
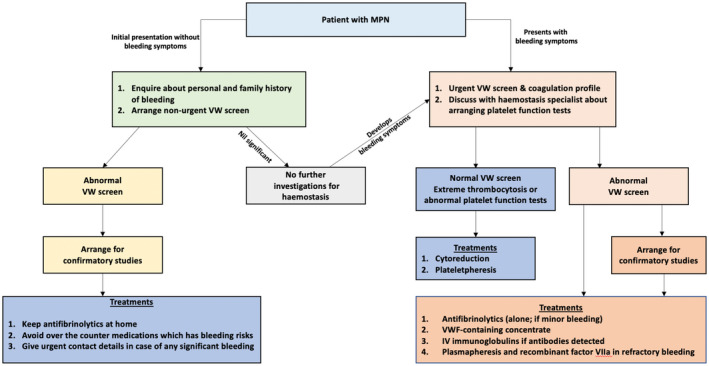
This proposal for bleed management in myeloproliferative neoplasms (MPN) is not derived from evidence‐based trials which are yet to be performed. Since abnormal Von Willebrand levels can be seen in those patients with non‐extremely elevated platelet counts, non‐urgent testing may be undertaken in all patients. This can assist in the clinical management if found to be abnormal including consideration of haemostatic treatment for surgery and interventional procedures. It will also help in early intervention if bleeding were to develop in the future. In addition, in those with abnormal Von Willebrand (VW) levels but who are ‘asymptomatic’ from a bleeding perspective, discussion about bleeding risks when starting aspirin becomes highly relevant. Management approaches in minor bleeding are with antifibrinolytics while in major bleeding situations, different options in addition to antifibrinolytics should be considered.
Antiplatelets and anticoagulants in those with AVWS
Incidence of thrombosis is high in MPN, with one meta‐analysis of 13 436 patients estimating an incidence of 20% at presentation. 12 Thus, use of anticoagulants is common in this patient cohort for treatment of thrombotic complications and prevention of thrombosis recurrence. A role also exists for antiplatelet agents, particularly aspirin, in thrombosis prevention and prophylaxis. The ECLAP study in 2004 illustrated the anti‐thrombotic value of prophylactic aspirin use in PV patients, with a reduction in the incidence of thrombotic events by 50%–60%. 55 However, use of these agents can increase risk of bleeding and precipitate bleeding episodes, especially in the setting of AVWS. Such episodes of bleeding can lead to the premature discontinuation of anticoagulant agents which in turn is associated with a significantly increased risk of recurrent thrombosis. 56 , 57 , 58 In the general population, rates of gastrointestinal bleeding attributed to long‐term aspirin use have been reported as 0.48–3.64/1000 patients years, 3 , 4 which is approximately 10 times less frequent than bleeding episodes in MPN overall. 5 Considering DOACs, an analysis of data from Danish registries of over 30 000 non‐MPN patients reported one‐year bleeding rates of 2.8%, 1.8% and 2.2% for rivaroxaban, dabigatran and apixaban, respectively. 59
In a study of bleeding complications in BCR‐ABL‐negative MPN, Wille et al. reported a bleeding event in 143 of 829 (17.2%) patients with an underlying diagnosis of BCR‐ABL‐negative MPN. 7 Of those who experienced a bleeding episode, 60.1% were prescribed an antiplatelet agent and 20.3% were prescribed an anticoagulant. Of the 829 patients studied, 6.2% were found to have a diagnosis of AVWS, which was not significantly increased, at 7.7%, in those who experienced a bleeding event. Therefore, although this study showed increased rates of bleeding associated with anticoagulant and antiplatelet use, it did not directly implicate AVWS as an independent risk factor for bleeding in MPN patients receiving these classes of drugs. There is also interest in whether more frequent aspirin administration might provide superior anti‐thrombotic activity compared with once‐daily use in ET. A randomised study comparing 100 mg of aspirin once, twice or thrice daily, showed improved biochemical evidence of platelet inhibition, without increased bleeding or objective evidence of clinical benefit. 60 Longer follow‐up is required to ascertain whether this is an effective or safe option in these patients.
Another study focused on bleeding rates in MPN patients receiving DOACs, with a reported one‐year cumulative incidence of bleeding of 12.3% (6.4%–18.2%). 61 Presence of AVWS was not assessed in this study; however leucocytosis was found to be associated with a higher risk of bleeding on a DOAC. In another study of MPN patients treated with antiplatelets or anticoagulation after a transient ischaemic attack (TIA) or ischaemic stroke, only 4% of the 597 patients analysed experienced a major bleeding event. Given the relatively low rate of severe bleeding in this study, a favourable risk profile for the use of antiplatelet/anticoagulant therapy in the setting of ischaemic stroke and TIA was outlined, with benefit proven by a reduction in recurrence of cardiovascular events. 62
ACQUIRED PLATELET DYSFUNCTION
Incidence
The understanding of platelet dysfunction in MPN is less clear. There is significant evidence of platelet hyperreactivity and increased risk of thrombosis. 63 , 64 Conversely, extreme thrombocytosis, meaning a platelet count of greater than 1500 × 109/l, confers a higher risk of bleeding, which may be independent of AVWS. 65 A study of 776 ET patients with platelet count higher than 450 × 109/l confirmed the risk of bleeding to increase with higher platelet counts. 66 Use of non‐steroidal anti‐inflammatory drugs to treat constitutional symptoms, alongside routine administration of anti‐platelet agents may further exacerbate the bleeding risk. 13 , 67
Mechanisms
Reduced platelet responsiveness to platelet agonists has been described in some studies, but poorly correlates with bleeding risk. 68 Platelets from patients with ET were observed to have impaired binding to fibrinogen, and reduced activation of phosphoinositol‐3 (PI3) kinase‐mediated pathways in response to several platelet agonists, resulting in impaired thrombin generation. 69 Intrinsic platelet dysfunction in MPN patients has also been described as a consequence of the JAK‐2 V617F mutation itself. JAK‐2 V617F transgenic mice demonstrate defective primary haemostasis including hyporeactive platelets with reduced activation in response to collagen, and reduced thrombus formation and size. 70 , 71 Mice lacking JAK2 develop megakaryocyte hyperplasia, extreme thrombocytosis and bleeding, 72 suggesting that normal JAK2 functionality rather than absence of overactivity is required for megakaryocyte homeostasis.
Furthermore, it has been proposed that the reduced response to platelet agonists seen in platelet aggregation studies reflects a secondary storage pool defect that occurs following spontaneous platelet aggregation. 73 Patients with ET and PV with microvascular ischaemic or thrombotic features have evidence of reduced platelet survival with increased levels of platelet activation markers detectable. These findings are reversed by aspirin treatment. 74 Similarly, another study showed increased in vivo platelet activation in ET and PV patients, demonstrated by elevated P selectin expression. Platelet activation was associated with an increase in plasma vascular cell adhesion molecule‐1, indicating the presence of endothelial activation and VWF‐mediated spontaneous platelet aggregation. 75 It has therefore been proposed that hyperreactive platelets spontaneously activate, particularly in areas of high sheer stress, leading to symptoms of erythromelalgia, then de‐aggregate and recirculate as exhausted platelets with a secondary storage pool defect. 73 , 76 , 77
The role of CALR mutations in platelet function in patients with ET is less certain. Approximately 60% of ET patients have a pathogenic JAK2 mutation and around 30% have a CALR mutation. 78 The phenotype of CALR‐mutated ET differs to its JAK2‐associated counterpart, with higher platelet counts, lower haematocrits and white cell counts, and less frequent thrombosis seen. 79 CALR mutations have been shown to affect both platelet number and function. One group reported reduced response to ADP stimulation in CALR‐mutated platelets compared with normal or JAK2‐mutated platelets, with reduced ability to attach to fibrinogen. 80 The aetiology of bleeding in CALR‐mutated MPNs may therefore differ from those with an underlying JAK2 mutation. Even less is known about patients with thrombopoietin receptor gene (MPL)‐driven MPNs. 81 Similar to CALR‐associated ET, baseline platelet counts are often higher and haemoglobin may be lower, however whether there is an increased haemorrhagic or thrombotic risk is unknown. 81
Management
As with AVWS, one of the main strategies for the treatment of non‐therapy‐associated platelet dysfunction is that of cytoreduction, as normalisation of thrombocytosis may correct the haemostatic defects predisposing to bleeding. 8 Therapeutic plateletpheresis is another potential strategy for treatment of acquired platelet dysfunction in cases of extreme thrombocytosis. 82 Plateletpheresis can selectively remove larger, dysfunctional platelets from circulation, and has also been shown to normalise morphological and functional platelet properties. 83 , 84 Although use of apheresis in this setting has become more limited with the advent of effective pharmacological cytoreduction, plateletpheresis can still play a role in effective reduction of platelet numbers in an emergency setting. 82 Importantly, although plateletpheresis can offer a rapid reduction in platelet numbers, this will not target the aetiology of the thrombocytosis, and can even induce a transient increase in haematopoietic stem cell factors, leading to a compensatory rise in platelet count. Thus, it is recommenced that cytoreductive therapy is commenced concomitantly. 85 , 86
Drug‐associated platelet dysfunction is another relevant factor to consider. As has been mentioned previously, platelet dysfunction due to underlying use of antiplatelet agents and non‐steroidal anti‐inflammatories in MPNs can lead to significant bleeding. 48 Antiplatelet agents play a role in thrombosis prevention in MPNs and are recommended especially in high‐risk disease. 3 However, given the bleeding risk associated with extreme thrombocytosis, it has been suggested that omission of aspirin when the platelet count is higher than 1000 × 109/l should be considered, regardless of thrombotic risk, to offset the additional effect of therapy‐associated platelet dysfunction. 87 , 88 There are little robust data to specify a safe threshold for anti‐platelet use. One group analysed bleeding rates in patients with ET and early PMF. While they showed an increased risk of bleeding in ET patients with a platelet count higher than 1000 × 109/l, this was not statistically significant. 8 A study of low‐dose aspirin in 433 patients with low‐risk ET, 162 of whom had an underlying JAK2 mutation and 271 a CALR mutation, reported increased rates of bleeding in CALR‐mutated patients receiving aspirin compared with observation alone (12.9 vs 1.8 episodes/1000 patient years), but no reduction in thrombosis. Conversely, those with JAK2‐mutated disease treated with aspirin suffered fewer thrombotic events without any increase in bleeding observed. 89 Of note, 35% of the CALR group had a platelet count of over 1000 × 109/l compared with 14% of the JAK2 group. Differences in platelet biology between JAK2‐ and CALR‐mutated ET, might account for some of the increased bleeding risk noted in patients with extreme thrombocytosis.
Targeting JAK2
JAK pathway inhibitors are playing a growing role in the treatment of MPNs. This has stemmed in part from knowledge of the importance of the JAK‐2 V617F mutation in terms of thrombosis risk. 90 However, as this pathway may also impact the haemorrhagic phenotype seen, these therapies could potentially offer additional haemostatic benefits. Studies of these drugs have not been designed with this end‐point in mind, and few have specifically reported bleeding events.
Ruxolitinib is a JAK1/2 inhibitor used in patients with PMF and PV. The pivotal COMFORT‐1 study was a randomised placebo‐controlled trial of ruxolitinib in PMF. Ecchymoses were reported in 17.4% of the investigator arm versus 9.3% of the control arm, all of which were grade 1–2. This may have reflected increased rates of thrombocytopenia associated with ruxolitinib treatment (69.7% vs 30.5%). No other bleeding outcomes were reported. 91 The COMFORT‐2 study compared ruxolitinib with best available therapy (BAT). Rates of epistaxis and haematomas were not significantly different between the treatment arms. 92 The RESPONSE study investigated ruxolitinib in PV; however, no bleeding events were reported. 93
Fedratinib is a specific JAK2 inhibitor. In the placebo‐controlled randomised JAKARTA study in intermediate‐ to high‐risk PMF patients, thrombocytopenia was more common in patients treated with fedratinib (47% vs. 26%); however, bleeding was not reported. 94 Results of the FREEDOM 2 study of fedratinib compared with BAT are awaited (NCT03952039).
Momelotinib is a JAK1/2 inhibitor. The phase 3 SIMPLIFY 1 and 2 studies have compared momelotinib with ruxolitinib in treatment‐naïve PMF, and momelotinib with BAT in PMF patients previously treated with ruxolitinib respectively. Momelotinib may cause less thrombocytopenia than ruxolitinib, with epistaxis reported in 8% of the momelotinib arm of SIMPLIFY‐2 and 12% of the BAT arm. 95 , 96
Pacritinib is an inhibitor of JAK2/IRAK1 (interleukin‐1 receptor associated kinase 1). The PERSIST‐1 study compared pacritinib with BAT excluding JAK2 inhibitors in higher‐risk MF, 97 whereas the PERSIST‐2 study allowed ruxolitinib use in the BAT arm. 98 PERSIST‐1 reported similar rates of thrombocytopenia (pacritinib 17%, BAT 14%). PERSIST‐2 reported grade 3–4 bleeding in 7%, 14% and 7% of patients receiving once‐daily pacritinib, twice daily pacritinib and BAT, respectively. Thrombocytopenia is a common side effect of all these therapies, and is seen more frequently in PMF than the other MPNs, leading to treatment discontinuation in nearly 4% of patients in COMFORT‐1, and dose reduction in 48%. 99 At the current time, it is not possible to say whether JAK inhibition significantly impacts risk of haemorrhage, which may be partly confounded by concomitant thrombocytopenia.
In addition to JAK2, as understanding of the role of the PI3 kinase pathway in defective haemostasis progresses, this may provide a further therapeutic target for management of acquired platelet dysfunction in MPNs. 69
CONCLUSION
Risk of bleeding in patients with MPNs is poorly understood. AVWS may be present in up to 50% of patients when sought for, although does not always correlate with bleeding phenotype. Additional platelet function defects and intrinsic changes in platelet physiology caused by the underlying driver mutation, alongside the impact of aspirin, may worsen bleeding tendency. To compound the situation, these patients have an increased thrombotic potential, with up to 20% presenting with a thrombotic event, caused by a combination of endothelial activation and platelet hyperreactivity. 2 , 63 , 64 Such patients require anticoagulation, although predicting which patients will bleed and which will clot is currently not possible, nor is it possible to predict which patients will tolerate anticoagulation and which will become unacceptably haemorrhagic. In this review, we have discussed the available evidence regarding the aetiology of AVWS and platelet defects, alongside bleeding phenotype and potential management strategies. We have provided a practical algorithmic approach to peri‐operative management in patients with AVWS; however, further study is required to determine the best approach to these complex patients.
AUTHOR CONTRIBUTIONS
Edward Jones and Bryan Dillon wrote the manuscript. Dawn Swan critically reviewed and edited the manuscript. Jecko Thachil conceived and reviewed the manuscript.
CONFLICT OF INTEREST
No author has any conflict of interest.
Acknowledgements
Open access funding provided by IReL. [Correction added on 15 July 2022, after first online publication: IReL funding statement has been added.]
Jones E, Dillon B, Swan D, Thachil J. Practical management of the haemorrhagic complications of myeloproliferative neoplasms. Br J Haematol. 2022;199(3):313–321. 10.1111/bjh.18322
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analysed in this review.
REFERENCES
- 1. Epstein E, Goedel A. Hemorrhagic thrombocythemia with a cascular, sclerotic spleen. Virchows Arch. 1934;293:233–48. [Google Scholar]
- 2. Rungjirajittranon T, Owattanapanich W, Ungprasert P, Siritanaratkul N, Ruchutrakool T. A systematic review and meta‐analysis of the prevalence of thrombosis and bleeding at diagnosis of Philadelphia‐negative myeloproliferative neoplasms. BMC Cancer. 2019;19(1):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Whitlock EP, Burda BU, Williams SB, Guirguis‐Blake JM, Evans CV. Bleeding risks with aspirin use for primary prevention in adults: a systematic review for the U.S. preventive services task force. Ann Intern Med. 2016;164(12):826–35. [DOI] [PubMed] [Google Scholar]
- 4. García Rodríguez LA, Martín‐Pérez M, Hennekens CH, Rothwell PM, Lanas A. Bleeding risk with long‐term low‐dose aspirin: a systematic review of observational studies. PLoS One. 2016;11(8):e0160046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nicol C, Lacut K, Pan‐Petesch B, Lippert E, Ianotto JC. Hemorrhage in essential thrombocythemia or polycythemia vera: epidemiology, location, risk factors, and lessons learned from the literature. Thromb Haemost. 2021;121(5):553–64. [DOI] [PubMed] [Google Scholar]
- 6. Kander EM, Raza S, Zhou Z, Gao J, Zakarija A, McMahon BJ, et al. Bleeding complications in BCR‐ABL negative myeloproliferative neoplasms: prevalence, type, and risk factors in a single‐center cohort. Int J Hematol. 2015;102(5):587–93. [DOI] [PubMed] [Google Scholar]
- 7. Wille K, Huenerbein K, Jagenberg E, Sadjadian P, Becker T, Kolatzki V, et al. Bleeding complications in bcr‐abl‐negative myeloproliferative neoplasms (MPN): a retrospective single‐center study of 829 MPN patients. Eur J Haematol. 2022;108(2):154–62. [DOI] [PubMed] [Google Scholar]
- 8. Finazzi G, Carobbio A, Thiele J, Passamonti F, Rumi E, Ruggeri M, et al. Incidence and risk factors for bleeding in 1104 patients with essential thrombocythemia or prefibrotic myelofibrosis diagnosed according to the 2008 WHO criteria. Leukemia. 2012;26(4):716–9. [DOI] [PubMed] [Google Scholar]
- 9. Barbui T, Thiele J, Gisslinger H, Kvasnicka HM, Vannucchi AM, Guglielmelli P, Orazi A, Tefferi A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in‐depth discussion. Blood Cancer J 2018;8(2):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, Ruggeri M, et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol. 2011;29(23):3179–84. [DOI] [PubMed] [Google Scholar]
- 11. Barbui T, De Stefano V, Carobbio A, Iurlo A, Alvarez‐Larran A, Vannucchi AM, et al. Direct oral anticoagulants for myeloproliferative neoplasms (MPN‐DOACs): results from an international study on 442 patients. Blood. 2020;136:42–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaifie A, Kirschner M, Wolf D, Maintz C, Hänel M, Gattermann N, et al. Bleeding, thrombosis, and anticoagulation in myeloproliferative neoplasms (MPN): analysis from the German SAL‐MPN‐registry. J Hematol Oncol. 2016;9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Appelmann I, Kreher S, Parmentier S, Wolf H‐H, Bisping G, Kirschner M, et al. Diagnosis, prevention, and management of bleeding episodes in Philadelphia‐negative myeloproliferative neoplasms: recommendations by the hemostasis working Party of the German Society of hematology and medical oncology (DGHO) and the Society of Thrombosis and Hemostasis Research (GTH). Ann Hematol. 2016;95(5):707–18. [DOI] [PubMed] [Google Scholar]
- 14. Stein BL, Martin K. From Budd‐Chiari syndrome to acquired von Willebrand syndrome: thrombosis and bleeding complications in the myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. 2019;2019(1):397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gisslinger H, Gotic M, Holowiecki J, Penka M, Thiele J, Kvasnicka HM, et al. Anagrelide compared with hydroxyurea in WHO‐classified essential thrombocythemia: the ANAHYDRET study, a randomized controlled trial. Blood. 2013;121(10):1720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harrison CN, Campbell PJ, Buck G, Wheatley K, East CL, Bareford D, et al. Hydroxyurea compared with anagrelide in high‐risk essential thrombocythemia. N Engl J Med. 2005;353(1):33–45. [DOI] [PubMed] [Google Scholar]
- 17. Budde U, van Genderen PJ. Acquired von Willebrand disease in patients with high platelet counts. Semin Thromb Hemost. 1997;23(5):425–31. [DOI] [PubMed] [Google Scholar]
- 18. Leupin L, Beck EA, Furlan M, Bucher U. Hemostasis disorders with reduced activity of the von Willebrand factor in myeloproliferative syndromes. Schweiz Med Wochenschr. 1983;113(19):713–6. [PubMed] [Google Scholar]
- 19. Langer AL, Connell NT. Acquired von Willebrand syndrome. Hematol Oncol Clin North Am. 2021;35(6):1103–16. [DOI] [PubMed] [Google Scholar]
- 20. Rottenstreich A, Kleinstern G, Krichevsky S, Varon D, Lavie D, Kalish Y. Factors related to the development of acquired von Willebrand syndrome in patients with essential thrombocythemia and polycythemia vera. Eur J Intern Med. 2017;41:49–54. [DOI] [PubMed] [Google Scholar]
- 21. Sucker C, Michiels JJ, Zotz RB. Causes, etiology and diagnosis of acquired von Willebrand disease: a prospective diagnostic workup to establish the most effective therapeutic strategies. Acta Haematol. 2009;121(2–3):177–82. [DOI] [PubMed] [Google Scholar]
- 22. Fabris F, Casonato A, Grazia del Ben M, De Marco L, Girolami A. Abnormalities of von Willebrand factor in myeloproliferative disease: a relationship with bleeding diathesis. Br J Haematol. 1986;63(1):75–83. [DOI] [PubMed] [Google Scholar]
- 23. Sacco M, Ranalli P, Lancellotti S, Petrucci G, Dragani A, Rocca B, et al. Increased von Willebrand factor levels in polycythemia vera and phenotypic differences with essential thrombocythemia. Res Pract Thromb Haemost. 2020;4(3):413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Budde U, Dent JA, Berkowitz SD, Ruggeri ZM, Zimmerman TS. Subunit composition of plasma von Willebrand factor in patients with the myeloproliferative syndrome. Blood. 1986;68(6):1213–7. [PubMed] [Google Scholar]
- 25. Mohri H, Tanabe J, Yamazaki E, Yoshida M, Harano H, Matsuzaki M, et al. Acquired type 2A von Willebrand disease in chronic myelocytic leukemia. Hematopathol Mol Hematol. 1996;10(3):123–33. [PubMed] [Google Scholar]
- 26. Tiede A, Rand JH, Budde U, Ganser A, Federici AB. How I treat the acquired von Willebrand syndrome. Blood. 2011;117(25):6777–85. [DOI] [PubMed] [Google Scholar]
- 27. Trotti C, Sant'Antonio E, Vanni D, Casetti IC, Borsani O, Pietra D, et al. Acquired von Willebrand syndrome in myeloproliferative neoplasms with extreme thrombocytosis. Hematol Oncol. 2021;39(4):589–92. [DOI] [PubMed] [Google Scholar]
- 28. Bertozzi I, Bogoni G, Biagetti G, Duner E, Lombardi AM, Fabris F, et al. Thromboses and hemorrhages are common in MPN patients with high JAK2V617F allele burden. Ann Hematol. 2017;96(8):1297–302. [DOI] [PubMed] [Google Scholar]
- 29. Budde U, Schaefer G, Mueller N, Egli H, Dent J, Ruggeri Z, et al. Acquired von Willebrand's disease in the myeloproliferative syndrome. Blood. 1984;64(5):981–5. [PubMed] [Google Scholar]
- 30. Michiels JJ, Budde U, van der Planken M, van Vliet HH, Schroyens W, Berneman Z. Acquired von Willebrand syndromes: clinical features, aetiology, pathophysiology, classification and management. Best Pract Res Clin Haematol. 2001;14(2):401–36. [DOI] [PubMed] [Google Scholar]
- 31. Laffan MA, Lester W, O'Donnell JS, Will A, Tait RC, Goodeve A, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre doctors organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol. 2014;167(4):453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Genderen PJ, Prins FJ, Lucas IS, van de Moesdijk D, van Vliet HH, van Strik R, et al. Decreased half‐life time of plasma von Willebrand factor collagen binding activity in essential thrombocythaemia: normalization after cytoreduction of the increased platelet count. Br J Haematol. 1997;99(4):832–6. [DOI] [PubMed] [Google Scholar]
- 33. van Genderen PJ, Budde U, Michiels JJ, van Strik R, van Vliet HH. The reduction of large von Willebrand factor multimers in plasma in essential thrombocythaemia is related to the platelet count. Br J Haematol. 1996;93(4):962–5. [DOI] [PubMed] [Google Scholar]
- 34. Federici AB, Rand JH, Bucciarelli P, Budde U, van Genderen PJ, Mohri H, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost. 2000;84(2):345–9. [PubMed] [Google Scholar]
- 35. Tefferi A, Barbui T. Personalized management of essential thrombocythemia‐application of recent evidence to clinical practice. Leukemia. 2013;27(8):1617–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2015 update on diagnosis, risk‐stratification and management. Am J Hematol. 2015;90(2):162–73. [DOI] [PubMed] [Google Scholar]
- 37. Mohri H, Motomura S, Kanamori H, Matsuzaki M, Watanabe S, Maruta A, et al. Clinical significance of inhibitors in acquired von Willebrand syndrome. Blood. 1998;91(10):3623–9. [PubMed] [Google Scholar]
- 38. Lancellotti S, Dragani A, Ranalli P, Petrucci G, Basso M, Tartaglione R, et al. Qualitative and quantitative modifications of von Willebrand factor in patients with essential thrombocythemia and controlled platelet count. J Thromb Haemost. 2015;13(7):1226–37. [DOI] [PubMed] [Google Scholar]
- 39. Tefferi A, Smock KJ, Divgi AB. Polycythemia vera‐associated acquired von Willebrand syndrome despite near‐normal platelet count. Am J Hematol. 2010;85(7):545. [DOI] [PubMed] [Google Scholar]
- 40. Mital A, Prejzner W, Bieniaszewska M, Hellmann A. Prevalence of acquired von Willebrand syndrome during essential thrombocythemia: a retrospective analysis of 170 consecutive patients. Pol Arch Med Wewn. 2015;125(12):914–20. [DOI] [PubMed] [Google Scholar]
- 41. Mital A, Prejzner W, Hellmann A. Acquired von Willebrand syndrome during the course of Myelofibrosis: analysis of 32 cases. Adv Clin Exp Med. 2015;24(6):1001–6. [DOI] [PubMed] [Google Scholar]
- 42. Raife TJ, Cao W, Atkinson BS, Bedell B, Montgomery RR, Lentz SR, et al. Leukocyte proteases cleave von Willebrand factor at or near the ADAMTS13 cleavage site. Blood. 2009;114(8):1666–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–61. [DOI] [PubMed] [Google Scholar]
- 44. James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–8. [DOI] [PubMed] [Google Scholar]
- 45. Singh I, Themistou E, Porcar L, Neelamegham S. Fluid shear induces conformation change in human blood protein von Willebrand factor in solution. Biophys J. 2009;96(6):2313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang X, Halvorsen K, Zhang CZ, Wong WP, Springer TA. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. 2009;324(5932):1330–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ruggeri M, Rodeghiero F, Tosetto A, Castaman G, Scognamiglio F, Finazzi G, et al. Postsurgery outcomes in patients with polycythemia vera and essential thrombocythemia: a retrospective survey. Blood. 2008;111(2):666–71. [DOI] [PubMed] [Google Scholar]
- 48. Tiede A. Diagnosis and treatment of acquired von Willebrand syndrome. Thromb Res. 2012;130(Suppl 2):S2–6. [DOI] [PubMed] [Google Scholar]
- 49. Franchini M, Mannucci PM. Acquired von Willebrand syndrome: focused for hematologists. Haematologica. 2020;105(8):2032–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Federici AB, Budde U, Rand JH. Acquired von Willebrand syndrome 2004: international registry‐‐diagnosis and management from online to bedside. Hamostaseologie. 2004;24(1):50–5. [DOI] [PubMed] [Google Scholar]
- 51. Mannucci PM, Lombardi R, Bader R, Horellou MH, Finazzi G, Besana C, et al. Studies of the pathophysiology of acquired von Willebrand's disease in seven patients with lymphoproliferative disorders or benign monoclonal gammopathies. Blood. 1984;64(3):614–21. [PubMed] [Google Scholar]
- 52. Mohri H, Noguchi T, Kodama F, Itoh A, Ohkubo T. Acquired von Willebrand disease due to inhibitor of human myeloma protein specific for von Willebrand factor. Am J Clin Pathol. 1987;87(5):663–8. [DOI] [PubMed] [Google Scholar]
- 53. Colella MP, Duarte GC, Marques JF Jr, De Paula EV. Haemostatic management of extreme challenges to haemostasis in acquired von Willebrand syndrome. Haemophilia. 2012;18(3):e188–91. [DOI] [PubMed] [Google Scholar]
- 54. Karger R, Weippert‐Kretschmer M, Budde U, Kretschmer V. Diagnosis and therapeutic management in a patient with type 2B‐like acquired von Willebrand syndrome. Blood Coagul Fibrinolysis. 2011;22(2):144–7. [DOI] [PubMed] [Google Scholar]
- 55. Landolfi R, Marchioli R, Kutti J, Gisslinger H, Tognoni G, Patrono C, et al. Efficacy and safety of low‐dose aspirin in polycythemia vera. N Engl J Med. 2004;350(2):114–24. [DOI] [PubMed] [Google Scholar]
- 56. Wille K, Sadjadian P, Becker T, Kolatzki V, Horstmann A, Fuchs C, et al. High risk of recurrent venous thromboembolism in BCR‐ABL‐negative myeloproliferative neoplasms after termination of anticoagulation. Ann Hematol. 2019;98(1):93–100. [DOI] [PubMed] [Google Scholar]
- 57. De Stefano V, Ruggeri M, Cervantes F, Alvarez‐Larrán A, Iurlo A, Randi ML, et al. High rate of recurrent venous thromboembolism in patients with myeloproliferative neoplasms and effect of prophylaxis with vitamin K antagonists. Leukemia. 2016;30(10):2032–8. [DOI] [PubMed] [Google Scholar]
- 58. Hernández‐Boluda JC, Arellano‐Rodrigo E, Cervantes F, Alvarez‐Larrán A, Gómez M, Barba P, et al. Oral anticoagulation to prevent thrombosis recurrence in polycythemia vera and essential thrombocythemia. Ann Hematol. 2015;94(6):911–8. [DOI] [PubMed] [Google Scholar]
- 59. An update on the bleeding risks associated with DOACs. Drug Ther Bull. 2017;55(11):129–32. [DOI] [PubMed] [Google Scholar]
- 60. Rocca B, Tosetto A, Betti S, Soldati D, Petrucci G, Rossi E, et al. A randomized double‐blind trial of 3 aspirin regimens to optimize antiplatelet therapy in essential thrombocythemia. Blood. 2020;136(2):171–82. [DOI] [PubMed] [Google Scholar]
- 61. How J, Story C, Ren S, Neuberg D, Rosovsky RP, Hobbs GS, et al. Practice patterns and outcomes of direct oral anticoagulant use in myeloproliferative neoplasm patients. Blood Cancer J. 2021;11(11):176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. De Stefano V, Carobbio A, Di Lazzaro V, Guglielmelli P, Iurlo A, Finazzi MC, et al. Benefit‐risk profile of cytoreductive drugs along with antiplatelet and antithrombotic therapy after transient ischemic attack or ischemic stroke in myeloproliferative neoplasms. Blood Cancer J. 2018;8(3):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood. 2013;122(13):2176–84. [DOI] [PubMed] [Google Scholar]
- 64. Falanga A, Marchetti M. Thrombotic disease in the myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. 2012;2012:571–81. [DOI] [PubMed] [Google Scholar]
- 65. van Genderen PJ, Michiels JJ. Erythromelalgic, thrombotic and haemorrhagic manifestations of thrombocythaemia. Presse Med. 1994;23(2):73–7. [PubMed] [Google Scholar]
- 66. McMahon B, Stein BL. Thrombotic and bleeding complications in classical myeloproliferative neoplasms. Semin Thromb Hemost. 2013;39(1):101–11. [DOI] [PubMed] [Google Scholar]
- 67. Cronberg S, Wallmark E, Söderberg I. Effect on platelet aggregation of oral administration of 10 non‐steroidal analgesics to humans. Scand J Haematol. 1984;33(2):155–9. [DOI] [PubMed] [Google Scholar]
- 68. Avram S, Lupu A, Angelescu S, Olteanu N, Mut‐Popescu D. Abnormalities of platelet aggregation in chronic myeloproliferative disorders. J Cell Mol Med. 2001;5(1):79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Moore SF, Hunter RW, Harper MT, Savage JS, Siddiq S, Westbury SK, et al. Dysfunction of the PI3 kinase/Rap1/integrin α(IIb)β(3) pathway underlies ex vivo platelet hypoactivity in essential thrombocythemia. Blood. 2013;121(7):1209–19. [DOI] [PubMed] [Google Scholar]
- 70. Matsuura S, Thompson CR, Belghasem ME, Bekendam RH, Piasecki A, Leiva O, et al. Platelet dysfunction and thrombosis in JAK2V617F−mutated primary Myelofibrotic mice. Arterioscler Thromb Vasc Biol. 2020;40(10):e262–e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lamrani L, Lacout C, Ollivier V, Denis CV, Gardiner E, Ho Tin Noe B, et al. Hemostatic disorders in a JAK2V617F‐driven mouse model of myeloproliferative neoplasm. Blood. 2014;124(7):1136–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Eaton N, Subramaniam S, Schulte ML, Drew C, Jakab D, Haberichter SL, et al. Bleeding diathesis in mice lacking JAK2 in platelets. Blood Adv. 2021;5(15):2969–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Michiels JJ, Berneman Z, Schroyens W, Finazzi G, Budde U, van Vliet HH. The paradox of platelet activation and impaired function: platelet‐von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in essential thrombocythemia and polycythemia vera. Semin Thromb Hemost. 2006;32(6):589–604. [DOI] [PubMed] [Google Scholar]
- 74. van Genderen PJ, Michiels JJ, van Strik R, Lindemans J, van Vliet HH. Platelet consumption in thrombocythemia complicated by erythromelalgia: reversal by aspirin. Thromb Haemost. 1995;73(2):210–4. [PubMed] [Google Scholar]
- 75. Karakantza M, Giannakoulas NC, Zikos P, Sakellaropoulos G, Kouraklis A, Aktypi A, et al. Markers of endothelial and in vivo platelet activation in patients with essential thrombocythemia and polycythemia vera. Int J Hematol. 2004;79(3):253–9. [DOI] [PubMed] [Google Scholar]
- 76. Michiels JJ, Berneman Z, Schroyens W, Koudstaal PJ, Lindemans J, van Vliet HH. Platelet‐mediated thrombotic complications in patients with ET: reversal by aspirin, platelet reduction, and not by coumadin. Blood Cells Mol Dis. 2006;36(2):199–205. [DOI] [PubMed] [Google Scholar]
- 77. Michiels JJ, Berneman Z, Van Bockstaele D, van der Planken M, De Raeve H, Schroyens W. Clinical and laboratory features, pathobiology of platelet‐mediated thrombosis and bleeding complications, and the molecular etiology of essential thrombocythemia and polycythemia vera: therapeutic implications. Semin Thromb Hemost. 2006;32(3):174–207. [DOI] [PubMed] [Google Scholar]
- 78. Sakemura R, Hefazi M, Siegler EL, Cox MJ, Larson DP, Hansen MJ, et al. Targeting cancer‐associated fibroblasts in the bone marrow prevents resistance to CART‐cell therapy in multiple myeloma. Blood. 2022. 10.1182/blood.2021012811 [DOI] [PubMed] [Google Scholar]
- 79. Rumi E, Pietra D, Ferretti V, Klampfl T, Harutyunyan AS, Milosevic JD, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014;123(10):1544–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hauschner H, Bokstad Horev M, Misgav M, Nagar M, Seligsohn U, Rosenberg N, et al. Platelets from Calreticulin mutated essential thrombocythemia patients are less reactive than JAK2 V617F mutated platelets. Am J Hematol. 2020;95(4):379–86. [DOI] [PubMed] [Google Scholar]
- 81. Beer PA, Campbell PJ, Scott LM, Bench AJ, Erber WN, Bareford D, et al. MPL mutations in myeloproliferative disorders: analysis of the PT‐1 cohort. Blood. 2008;112(1):141–9. [DOI] [PubMed] [Google Scholar]
- 82. Boddu P, Falchi L, Hosing C, Newberry K, Bose P, Verstovsek S. The role of thrombocytapheresis in the contemporary management of hyperthrombocytosis in myeloproliferative neoplasms: a case‐based review. Leuk Res. 2017;58:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Orlin JB, Berkman EM. Improvement of platelet function following plateletpheresis in patients with myeloproliferative diseases. Transfusion. 1980;20(5):540–5. [DOI] [PubMed] [Google Scholar]
- 84. Tatarskiĭ AR, Alieva KM, Buriachkovskaia LI, Petrova LE, Bobkov EV, Chuchalin AG. The morphological properties of the blood thrombocytes in bronchial asthma patients before and after thrombocytapheresis. Ter Arkh. 1995;67(3):39–40. [PubMed] [Google Scholar]
- 85. Goldfinger D, Thompson R, Lowe C, Kurz L, Belkin G. Long‐term plateletpheresis in the management of primary thrombocytosis. Transfusion. 1979;19(3):336–8. [DOI] [PubMed] [Google Scholar]
- 86. Weisbach V, Friedlein H, Glaser A, Zingsem J, Zimmermann R, Eckstein R. The influence of automated plateletpheresis on systemic levels of hematopoietic growth factors. Transfusion. 1999;39(8):889–94. [DOI] [PubMed] [Google Scholar]
- 87. Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol. 2012;87(3):285–93. [DOI] [PubMed] [Google Scholar]
- 88. Harrison CN, Bareford D, Butt N, Campbell P, Conneally E, Drummond M, et al. Guideline for investigation and management of adults and children presenting with a thrombocytosis. Br J Haematol. 2010;149(3):352–75. [DOI] [PubMed] [Google Scholar]
- 89. Alberto A‐L, Arturo P, Paola G, Juan Carlos H‐B, Eduardo A‐R, Francisca F‐M, et al. Antiplatelet therapy versus observation in low‐risk essential thrombocythemia with a CALR mutation. Haematologica. 2016;101(8):926–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Barbui T, Finazzi G, Carobbio A, Thiele J, Passamonti F, Rumi E, et al. Development and validation of an international prognostic score of thrombosis in World Health Organization‐essential thrombocythemia (IPSET‐thrombosis). Blood. 2012;120(26):5128–33. quiz 252. [DOI] [PubMed] [Google Scholar]
- 91. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double‐blind, placebo‐controlled trial of Ruxolitinib for Myelofibrosis. N Engl J Med. 2012;366(9):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Harrison CN, Vannucchi AM, Kiladjian JJ, Al‐Ali HK, Gisslinger H, Knoops L, et al. Long‐term findings from COMFORT‐II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30(8):1701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia Vera. N Engl J Med. 2015;372(5):426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pardanani A, Tefferi A, Masszi T, Mishchenko E, Drummond M, Jourdan E, et al. Updated results of the placebo‐controlled, phase III JAKARTA trial of fedratinib in patients with intermediate‐2 or high‐risk myelofibrosis. Br J Haematol. 2021;195(2):244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mesa RA, Kiladjian JJ, Catalano JV, Devos T, Egyed M, Hellmann A, et al. SIMPLIFY‐1: a phase III randomized trial of Momelotinib versus Ruxolitinib in Janus kinase inhibitor‐Naïve patients with Myelofibrosis. J Clin Oncol. 2017;35(34):3844–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Harrison CN, Vannucchi AM, Platzbecker U, Cervantes F, Gupta V, Lavie D, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open‐label, phase 3 trial. Lancet Haematol. 2018;5(2):e73–81. [DOI] [PubMed] [Google Scholar]
- 97. Mesa RA, Vannucchi AM, Mead A, Egyed M, Szoke A, Suvorov A, et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST‐1): an international, randomised, phase 3 trial. Lancet Haematol. 2017;4(5):e225–e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mascarenhas J, Hoffman R, Talpaz M, Gerds AT, Stein B, Gupta V, et al. Pacritinib vs best available therapy, including Ruxolitinib, in patients with Myelofibrosis: a randomized clinical trial. JAMA Oncol. 2018;4(5):652–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Verstovsek S, Mesa RA, Gotlib J, Gupta V, DiPersio JF, Catalano JV, et al. Long‐term treatment with ruxolitinib for patients with myelofibrosis: 5‐year update from the randomized, double‐blind, placebo‐controlled, phase 3 COMFORT‐I trial. J Hematol Oncol. 2017;10(1):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analysed in this review.