

Abstract
Interleukin (IL)‐38 is a member of the IL‐1 cytokine family with reported anti‐inflammatory activity. The highest constitutive IL‐38 expression is detected in the skin, where it is mainly produced by differentiating keratinocytes. However, little data are available regarding its biological functions. In this study, we investigated the role of IL‐38 in skin physiology. We demonstrate here that dermal fibroblasts and epithelial cells of skin appendages, such as eccrine sweat glands and sebaceous glands, also express IL‐38. Next, using two‐ and three‐dimensional cell cultures, we show that endogenous expression of IL‐38 correlates with keratinocyte differentiation and its ectopic overexpression inhibits keratinocyte proliferation and enhances differentiation. Accordingly, immunohistochemical analysis revealed downregulation of IL‐38 in skin pathologies characterized by keratinocyte hyperproliferation, such as psoriasis and basal or squamous cell carcinoma. Finally, intracellular IL‐38 can shuttle between the nucleus and the cytoplasm and its overexpression modulates the activity of the transcription regulators YAP and ID1. Our results indicate that IL‐38 can act independently from immune system activation and suggest that it may affect the epidermis directly by decreasing proliferation and promoting differentiation of keratinocytes. These data suggest an important role of keratinocyte‐derived IL‐38 in skin homeostasis and pathologies characterized by epidermal alterations.
Keywords: differentiation, IL‐17A, IL‐22, IL‐38, keratinocyte, nuclear localization, proliferation, psoriasis
1. INTRODUCTION
Interleukin (IL)‐38, encoded by the IL‐1F10 gene, is a cytokine of the IL‐1 family, which was first described in 2001. 1 IL‐38 shares high amino acid sequence homology with the anti‐inflammatory receptor antagonists IL‐1 receptor antagonist (IL‐1Ra; 41% similarity) and the IL‐36 receptor antagonist (IL‐36Ra; 43% similarity), suggesting that IL‐38 might itself exhibit anti‐inflammatory properties. 2 The identity of the IL‐38 receptor(s) remains, however, controversial. Indeed, IL‐38 has been proposed to act as an antagonist for the IL‐1 receptor (IL‐1R1) or the IL‐36 receptor, or to transmit anti‐inflammatory signals through IL‐1R accessory protein like 1 (IL‐1RAPL1). 3 , 4 , 5
Anti‐inflammatory effects of IL‐38 have been observed in many in vitro studies. 3 , 4 , 6 However, the cytokine was found also to induce proinflammatory responses in certain conditions, 3 , 4 , 7 depending on its form, concentration, additional stimuli and target cells. Some discrepancies thus still need to be elucidated. 8 Meanwhile, numerous studies have reported altered IL‐38 levels in many inflammatory diseases, including systemic lupus erythematosus, rheumatoid arthritis, inflammatory bowel disease, primary Sjögren's syndrome and coronary heart disease. 9
Although IL‐38 mRNA is detected in various tissues and organs such as heart, placenta, foetal liver, spleen, thymus and tonsils, 2 its highest constitutive expression is observed in the skin, with epidermal keratinocytes being the main producing cells. 1 , 10 IL‐38 levels significantly increase during keratinocyte differentiation in vitro. 10 , 11 Consistently, IL‐38 expression was found to be substantially reduced in psoriatic epidermis, as compared to healthy skin. 12 IL‐38 was also downregulated in cultured keratinocytes exposed to psoriasis‐related inflammatory cytokines known to impact cell differentiation, such as IL‐17A and IL‐22. 11 Interestingly, IL‐38 was shown in turn to limit IL‐17 and IL‐22 expression in peripheral blood mononuclear cells, 3 suggesting a mutual regulation of these cytokines. Finally, during imiquimod‐induced skin inflammation, expression of the differentiation markers keratin 10 (KRT10) and loricrin (LOR) was lower in IL‐38 knockout compared to wild type mice, indicating that IL‐38‐deficiency further inhibited keratinocyte differentiation. 5
Two decades after its discovery, IL‐38 remains the least understood cytokine of the IL‐1 family, as little is known about its subcellular localization and intracellular activity, the mechanisms of its release or secretion and its biological functions. In this study, we investigated the role of IL‐38 in epidermal homeostasis and pathology. First, we identified dermal fibroblasts and cells of sweat and sebaceous glands as additional sources of IL‐38 in the skin. Next, we demonstrated the dependence of endogenous IL‐38 expression on keratinocyte differentiation in 2D and 3D cell culture models. Furthermore, we showed that IL‐38 overexpression directly promotes the differentiation of keratinocytes, while inhibiting their proliferation. Finally, we described the subcellular localization of IL‐38 in healthy and pathologic human keratinocytes and its potential role in IL‐38 function.
2. METHODS
See the Supplementary Materials and Methods in Appendix S1 for more experimental methods and details.
2.1. Human skin samples
Skin biopsies were taken from patients presenting at the Department of Plastic and Reconstructive Surgery and Division of Clinical Pathology of the Geneva University Hospitals in Switzerland, or children undergoing surgery at the Polish‐American Children's Hospital, Krakow, Poland. This study was conducted according to the Declaration of Helsinki and approved by local ethical committee of the University Hospitals of Geneva, Switzerland, and the Jagiellonian University, Poland. Written informed consent was obtained for each individual.
2.2. Monolayer cell culture
Primary human keratinocytes were isolated from foreskin biopsies after enzymatic digestion and subsequently cultured in serum free KGM™ Gold Keratinocyte Growth Medium (Lonza).
The human N/TERT keratinocyte cell line (N/TERT‐1) was obtained from Prof. Ellen H. van den Bogaard Laboratory (Radboud University Medical Center, Nijmegen, The Netherlands). N/TERT‐1 cells were cultured in keratinocyte‐serum free medium (K‐SFM, Gibco) until 50% confluency and then switched to experiment medium composed of 50% K‐SFM and 50% DF‐K as previously described by 13 and. 14 Stimulation with Th17 cytokines (IL‐17A (100 ng/ml) and IL‐22 (50 ng/ml), R&D Systems, Abingdon, UK), was performed at 80% cell confluency. Cells were harvested at 1, 4, 8 and 24 h post stimulation for quantitative gene expression analysis.
The Normal Human Keratinocyte (NHK) cell line, 15 as well as NHK cells inducibly overexpressing human IL‐38 (NHK/38 cells) or E. Coli β‐galactosidase (NHK/β‐Gal cells) were cultured as previously described. 16 NHK cells inducibly overexpressing the IL‐1 receptor antagonist (NHK/IL‐1Ra cells) were obtained by stable transfection of tetracycline repressor expressing NHK cells 16 with plasmid pcDNA4/TO/IL‐1Ra, containing the human full‐length IL‐1Ra coding sequence (transcript variant 3, GenBank accession NM_000577.5). 17 To induce IL‐38, β‐galactosidase or IL‐1Ra overexpression, NHK/38, NHK/β‐Gal and NHK/IL‐1Ra cells were treated with doxycycline (Dox) at 1 μg/ml for 24 h. Empty pZeoSV2 vector transfected NHK cells (NHK Ø) were generated using the Neon transfection system as described. 16
For keratinocyte differentiation, 50 000 cells per well were plated in 12 multi‐well plates and 2 mM CaCl2 was added into the medium when cells reached 80% confluency. Cell lysates were collected or coverslips fixed at desired time points (day 1, 3, 5 and 7).
2.3. In vitro reconstructed human epidermis (RHE) model
Reconstructed human epidermis were generated using N‐TERT‐1 keratinocytes as previously described. 18 After 10 days of culture at the air‐liquid interface, RHE were stimulated with cytokines for 24 h or 72 h for mRNA and protein analysis, respectively. The concentrations of cytokines were as follows: IL‐17 (30 ng/ml), IL‐22 (30 ng/ml) (both from R&D Systems), TNF‐α (5 ng/ml), IFN‐γ (5 ng/ml), IL‐4 (10 ng/ml) and IL‐13 (10 ng/ml; all from Peprotech).
2.4. Antibodies
The following primary antibodies were used: mouse monoclonal anti‐IL‐38 (H127C, eBioscience), polyclonal goat anti‐IL38 antibody (AF2427, R&D Systems), rabbit monoclonal anti‐loricrin (EPR7148(2)(B), Abcam), rabbit monoclonal anti‐keratin 7 (EPR17078, Abcam), rabbit monoclonal anti‐keratin 10 (EP1607IHCY, Abcam), rabbit monoclonal anti‐keratin 14 (LL002, Bio SB), rabbit monoclonal anti‐vimentin (EPR3776, Abcam), rabbit monoclonal anti‐carbonic anhydrase 2/CA2 (EPR5195, Abcam), mouse IgG2b kappa monoclonal isotype control (AB18421, Abcam), mouse monoclonal anti‐YAP (63.7, sc‐101 199, Santa Cruz), rabbit monoclonal anti‐ID1 (195–14, Biocheck), mouse monoclonal anti‐alpha tubulin (DM1A, Abcam).
2.5. Immunohistochemistry
Skin tissue samples or RHE were fixed in 4% formaldehyde, embedded in paraffin, and 5‐μm sections were cut. Antigen retrieval was performed in 10 mmol/L citrate buffer pH 6.0 for 30 min at 95°C. The activity of endogenous peroxidase was quenched with Bloxall blocking solution (Vector Laboratories). Samples were stained using the R.T.U. Vectastain Kit with the ImmPACT AMEC Red Substrate Kit (Vector Laboratories) according to the manufacturer's protocol. Sections were counterstained with Mayer's haematoxylin solution, mounted with Glycerol Mounting Medium (Dako). Double staining was made using the ImmPRESS Duet Double Staining Polymer Kit, according to the manufacturer's protocol (Vector laboratories).
For immunofluorescence, sections were blocked with 3% BSA and 5% normal goat serum in PBS for 1 h. Next, sections were incubated for 2 h with primary antibodies followed by 1 h incubation with Alexa Fluor 488‐conjugated donkey anti‐rabbit and Alexa Fluor 555‐conjugated donkey anti‐mouse IgG secondary antibodies (Invitrogen) and mounted with DAPI Fluoromount‐G (SouthernBiotech).
2.6. Immunocytochemistry
Coverslips with cells were fixed with 4% paraformaldehyde in PBS buffer at room temperature for 15 min and permeabilized with 0.1% Triton X‐100 (Sigma‐Aldrich) in PBS buffer for 5 min. Cells were stained for 1 h with primary antibodies followed by Alexa Fluor 568‐conjugated donkey anti‐mouse IgG (Invitrogen) and Alexa Fluor 488‐conjugated donkey anti‐rabbit IgG secondary antibodies (Invitrogen) and mounted with DAPI Fluoromount‐G (SouthernBiotech).
2.7. Statistical analysis
Data are represented as mean ± SEM. Statistical comparison between two groups was performed using the paired Student t‐test. For multiple comparisons two‐way analysis of variance (ANOVA) followed by appropriate correction was used. For correlation analysis Pearson's correlation test was applied. Data were analysed with the GraphPad prism version 8 software. p < 0.05 was considered to be of statistical significance.
3. RESULTS
3.1. IL‐38 is expressed in multiple cell types in human skin
As previously reported, 11 , 16 we observed that IL‐38 is produced by keratinocytes of all epidermal layers in normal human skin with a slight accumulation in the granular layer (Figure S1a). Although keratinocytes are the main producers of IL‐38 in the skin, the cytokine is also detected in dermal fibroblasts identified by co‐expression of vimentin (Figure S1b). Additionally, we observed that epithelial cells of skin appendages also produce IL‐38. In ducts of eccrine sweat glands, IL‐38 was present in both basal and suprabasal cell layers, the latter showing stronger labelling (Figure S1c). Moreover, IL‐38 was specifically expressed by dark cells of the secretory coils of eccrine sweat glands, which were identified by carbonic anhydrase II (CAII) co‐staining (Figure S1d). Finally, sebocytes of sebaceous glands were also found to produce IL‐38 (Figure S1e). The specificity of IL‐38 detection on skin sections was confirmed using an isotype‐matched control antibody (Figure S2), as validated and described previously. 16 These data indicate that different cell populations produce IL‐38 in normal human skin.
3.2. IL‐38 expression correlates with keratinocyte differentiation
It has been previously shown that IL‐38 mRNA expression is increased in differentiated keratinocytes. 10 Thus, we investigated the correlation between IL‐38 expression and keratinocyte differentiation in more detail. Our results showed that IL‐38 mRNA and protein levels increased during differentiation of primary keratinocytes and of the N/TERT‐1 keratinocyte cell line. IL‐38 mRNA expression reached its peak at day 7 or day 5 of calcium‐induced differentiation in primary and N/TERT‐1 cells, respectively (Figure 1A,B). IL‐38 mRNA expression levels exhibited a stronger positive correlation over time with the expression levels of LOR mRNA (R 2 = 0.765; p = 0.004), a marker of granular layer and the stratum corneum transition, than with KRT10, a marker of the spinous layer (R 2 = 0.69; p = 0.013; Figure 1A,B). IL‐38 protein expression increased between days 0 and 5 of N/TERT‐1 cell differentiation, with an almost exclusive intracellular localization (Figure 1C). The levels of IL‐38 in supernatants remained below the detection limit, even in differentiated cells.
FIGURE 1.
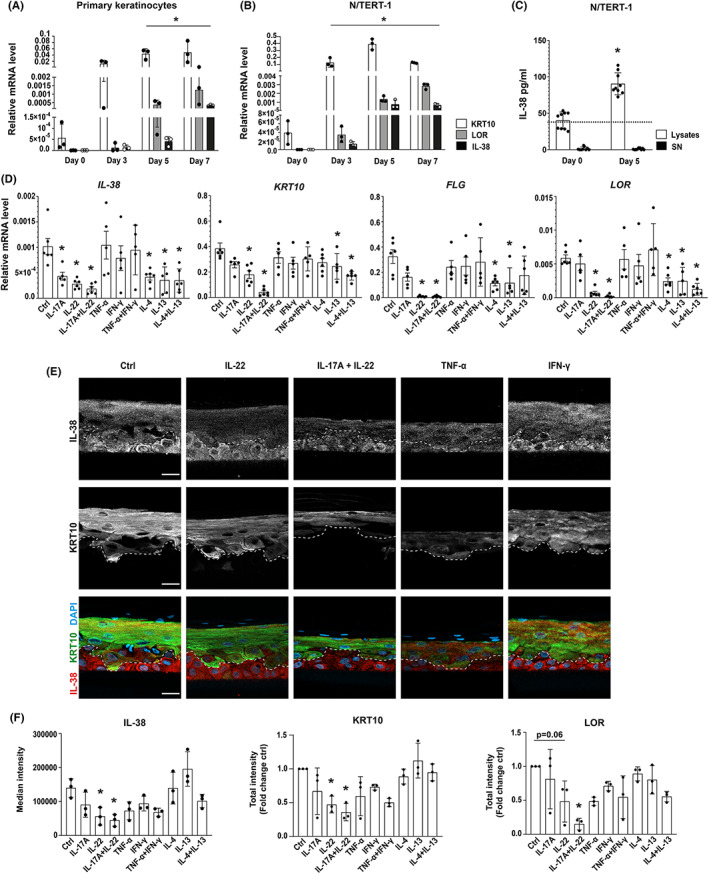
Regulation of IL‐38 expression in differentiating keratinocytes and in RHE stimulated with inflammatory cytokines. IL‐38, KRT10, and LOR mRNA levels, relative to 18S rRNA, assessed by RTqPCR in (A) primary keratinocytes and (B) the N/TERT‐1 cell line differentiated in monolayer cultures for 0, 3, 5 and 7 days. Data are means ± SEM (n = 3). (C) IL‐38 protein levels measured by ELISA in cell lysates and supernatants of N/TERT‐1 keratinocytes differentiated for 0 and 5 days. The dotted line represents the lower detection limit of the ELISA. Data are means ± SEM (n = 9). (D) IL‐38, KRT10, FLG and LOR mRNA levels, relative to the mean of 18S rRNA and GAPDH mRNA levels, assessed by RTqPCR in RHE stimulated with indicated cytokines for 24 h. Data are means ± SEM (n = 5–6). (E) Immunofluorescent staining for IL‐38 (upper panels, white; lower panels, red) and KRT10 (middle panels, white; lower panels, green) in RHE cultured in the presence of indicated cytokines for 72 h. Nuclei were counterstained with DAPI. The broken line represents the boundary between basal and suprabasal layers, based on KRT10 staining in suprabasal cells. Representative pictures chosen from two experiments out of three are shown. Scale bar, 50 μm. (F) Quantification of IL‐38, KRT10 and LOR immunofluorescent staining, shown as median intensity (IL‐38) or as total intensity in a 1000 μm2 area (KRT10 and LOR). Data are means ± SEM (n = 3). Data are (A, B, D, F) biological replicates or (C) technical triplicates of biological triplicates. *p < 0.05 as compared to (A, B, C) day 0 or (D, F) control. p values were calculated using a paired t‐test (C) or two‐way ANOVA with Dunnet's correction (a, B, D and F). KRT10, keratin 10; LOR, loricrin, FLG, filaggrin.
Next, we employed a 3D reconstituted human epidermis (RHE) model to analyse the regulation of endogenous IL‐38 expression by inflammatory cytokines known to impact epidermal differentiation. For that, we used N/TERT‐1 cells that share many features of primary keratinocytes, including their ability to differentiate into RHE. 14 24 h stimulation with the Th17 cytokines IL‐17A and IL‐22, which strongly inhibit keratinocyte differentiation, 19 significantly decreased the expression of mRNAs encoding IL‐38 and all analysed differentiation markers (KRT10, filaggrin [FLG] and LOR; Figure 1D). A similar, although slightly less pronounced decrease was observed with the Th2 cytokines IL‐4 and IL‐13, alone or in combination (Figure 1D). TNF‐⍺ and IFN‐γ, on the other hand, had no impact on mRNA expression levels of IL‐38 or the differentiation markers after 24 h (Figure 1D). To elaborate on these findings, we analysed by immunofluorescence the protein expression of IL‐38 and of selected differentiation markers in RHEs stimulated with cytokines for 72 h (Figure 1E,F, Figure S3a). In control RHE, IL‐38 was expressed in all cell layers. KRT10 expression was observed in differentiating, suprabasal cells (Figure 1E) and LOR was selectively expressed in differentiated cells of the granular layer (Figure S3). IL‐22 alone or in combination with IL‐17A significantly decreased IL‐38, as well as KRT10 and LOR protein expression. Interestingly, the decrease in IL‐38 protein expression induced by IL‐22 was mostly observed in suprabasal differentiating cells (Figure 1E) and not in proliferating basal cells. Additionally, reduced IL‐38 and differentiation marker protein levels were observed after 72 h stimulation with TNF‐α, which significantly affected RHE architecture (Figure 1E). Stimulation with Th2 cytokines for 72 h did not decrease staining intensity for IL‐38, KRT10 or LOR proteins (Figure 1E,F). Finally, to confirm that decreased IL‐38 levels are related to inhibited cell differentiation, rather than to a direct effect of Th17 cytokines on IL‐38 gene expression, we stimulated N/TERT‐1 monolayer cell culture with IL‐17A and IL‐22. Short‐term stimulation did not reduce IL‐38 mRNA levels, and the progressive decrease in differentiation marker expression, which was observed over 24 h, preceded a slower reduction in IL‐38 levels (Figure S3b). Altogether, correlation between IL‐38 and differentiation marker expression was observed for all tested cytokines.
3.3. IL‐38 overexpression inhibits proliferation and promotes keratinocyte differentiation
Since in our experimental system IL‐38 was mainly detected intracellularly (Figure 1C), we used an immortalized normal human keratinocyte (NHK) cell line with doxycycline (Dox)‐inducible IL‐38 overexpression (NHK/38) to investigate a potential impact of IL‐38 on the function of its producing cells. As previously described, 16 NHK/38 cells already produced higher levels of IL‐38 mRNA and protein in the absence of Dox as compared to control NHK cells, reflecting a leaky expression from the inducible system at baseline. Nevertheless, addition of Dox for 24 h strongly enhanced IL‐38 expression (Figure 2A). Using WST‐1 and BrdU assays, we observed that IL‐38 overexpression significantly decreased the metabolic activity and the proliferation of NHK cells, as compared to empty vector transfected NHK cells (NHK Ø) and β‐Galactosidase overexpressing NHK cells (NHK/β‐Gal), used as controls (Figure 2B,C). At the same time, we did not observe any cytotoxic effect of IL‐38 overexpression (Figure 2D). We next investigated the effect of IL‐38 overexpression on cell differentiation. Expression of mRNA encoding cyclin D1 (CCND1), a central regulator of cell cycle progression, decreased during the differentiation of NHK/38, but not control NHK cells (Figure 2E). Furthermore, NHK/38 Dox + cells upregulated the expression of both early and late differentiation markers, such as KRT10, FLG and LOR, while at the same time downregulating keratin 14 (KRT14), a marker of proliferating cells (Figure 2F). Although there was no difference in proliferation rates of NHK/38 Dox‐ and NHK/38 Dox + cells, enhanced differentiation was selectively observed in Dox‐treated NHK/38 cells (Figure 2F). Overall, these data define IL‐38 as an important regulator of keratinocyte proliferation and differentiation in vitro. Finally, we examined the effect of IL‐38 overexpression on inflammatory responses in NHK cells. Using IL‐36γ‐ or IL‐1α–induced IL‐8 secretion as a read‐out, we did not observe any significant difference between NHK/38 and NHK/β‐Gal cells (Figure S4). In contrast, overexpression of IL‐1Ra, used as a positive control, dose‐dependently inhibited IL‐1α‐induced IL‐8 production (Figure S4).
FIGURE 2.
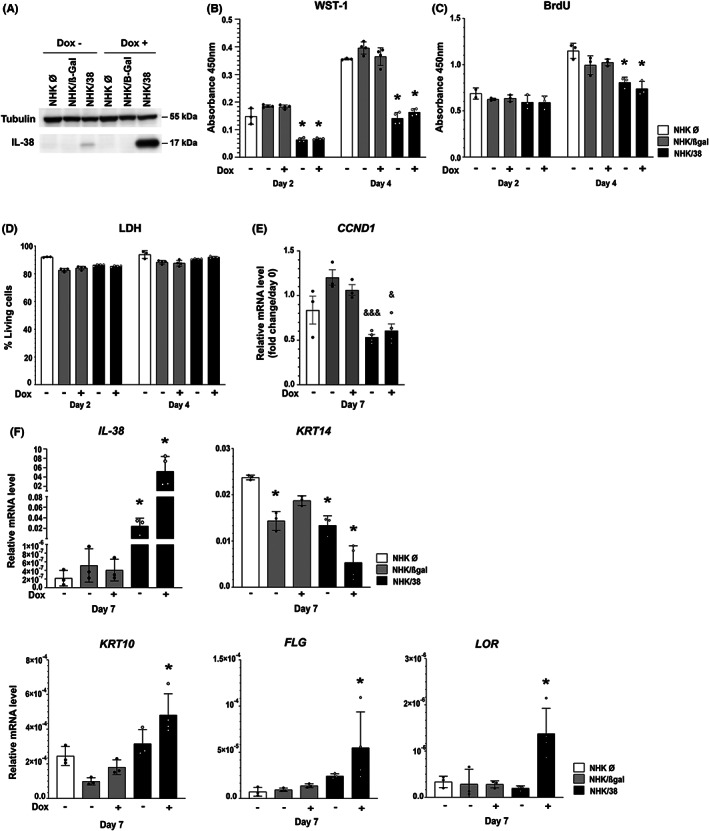
Effects of IL‐38 overexpression on NHK cells. (A) Western blot analysis of IL‐38 protein expression in empty vector transfected NHK cells (NHK Ø), and in NHK/β‐Gal and NHK/38 cells without and with Dox induction for 24 h. Tubulin levels are shown as a loading control. (B) Metabolic activity, assessed by WST‐1 colorimetric assay, in NHK Ø, NHK/β‐Gal and NHK/38 cells cultured for 2 and 4 days after induction without or with Dox. Data are means ± SEM (n = 3–4). (C) Cell proliferation assessed by BrdU assay in NHK Ø, NHK/β‐Gal and NHK/38 cells cultured for 2 and 4 days after induction without or with Dox. Data are means ± SEM (n = 3). (D) Cell viability, assessed by measuring lactate dehydrogenase (LDH) activity in supernatants of NHK Ø, NHK/β‐Gal and NHK/38 cells incubated during 2, 4 or 6 days after induction without or with Dox. Results are shown as the percentage of living cells. Data are means ± SEM (n = 3–4). (E) Fold change in CCND1 mRNA levels, normalized to 18S rRNA, relative to day 0, assessed by RTqPCR in NHK Ø, NHK/β‐Gal and NHK/38 cells differentiated for 7 days after induction without or with Dox. Data are means ± SEM (n = 3–4). (F) IL‐38, KRT14, KRT10, FLG and LOR mRNA levels, relative to 18S rRNA, assessed by RTqPCR in NHK Ø, NHK/β‐Gal and NHK/38 cells differentiated for 7 days after induction without or with Dox. Data are means ± SEM (n = 3–5). Data are (B‐D) technical triplicates or technical duplicates of biological duplicates, or (E, F) biological replicates. *p < 0.05 as compared to NHK Ø cells, & p < 0.05, &&& p < 0.001 as compared to NHK/β‐Gal cells, p values were calculated using a two‐way ANOVA with Tukey's multiple comparison.
3.4. IL‐38 is downregulated in hyperproliferative skin disorders
Next, we analysed IL‐38 expression in skin disorders characterized by increased keratinocyte proliferation, such as psoriasis, atopic dermatitis, non‐melanoma skin cancers and benign skin tumors. Immunohistochemical analysis revealed that IL‐38 expression is decreased in all analysed pathologies when compared to healthy skin (Figure 3A). Moreover, the same trend was observed in sebaceous and eccrine sweat gland‐derived tumors, including eccrine carcinoma, eccrine poroma and sebaceoma. In disorders, where lesions arise from the uncontrolled growth of basal cells, like psoriasis, atopic dermatitis and basal cell carcinoma, we observed nuclear localization of IL‐38 in some cells (Figure 3A), identified as keratinocytes by KRT10 staining (Figure 3B). The specificity of IL‐38 detection by immunohistochemistry on human skin sections was confirmed using an isotype‐matched control antibody (Figure S5a), as well as by staining with a different anti‐IL‐38 antibody, which produced a similar staining pattern (Figure S5b). Interestingly, in normal human epidermis, IL‐38 could also be detected in the nuclei of basal keratinocytes, while in differentiated cells of suprabasal layers IL‐38 was mainly cytoplasmic (Figure 3A). In line with these observations, the localization of IL‐38 was mostly cytoplasmic in disorders characterized by accelerated growth of squamous cells, such as differentiated squamous cell carcinoma, seborrheic keratosis and verrucae, as well as in eccrine poroma and sebaceoma (Figure 3A). We hypothesized that nuclear localization of IL‐38 may be related to the undifferentiated state of the cells.
FIGURE 3.
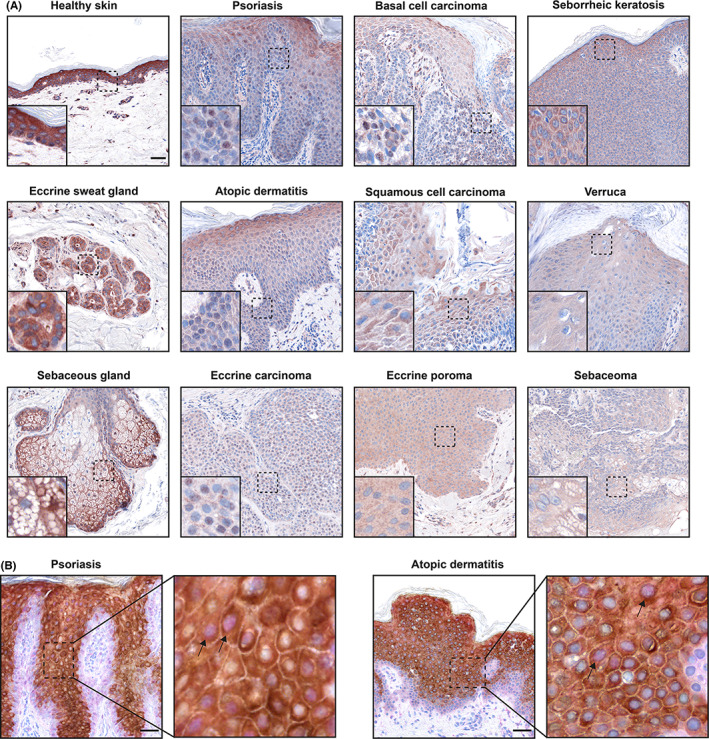
IL‐38 expression in human skin pathologies. (A) Immunohistochemical analysis of IL‐38 expression in human skin. (B) Double immunohistochemical staining of IL‐38 (pink) and KRT10 (brown) in psoriasis and atopic dermatitis. The insets demonstrate higher magnification of the highlighted areas. The arrows point to examples of IL‐38 nuclear localization. Scale bar, 50 μm. One representative donor of one to three is shown.
3.5. IL‐38 translocates from the nucleus to the cytoplasm during cell differentiation
To test the hypothesis that a nuclear localization of IL‐38 is preferentially observed in proliferating, rather than differentiating keratinocytes, we analysed the subcellular localization of IL‐38 in NHK/38 cells during calcium‐induced differentiation. By comparing nucleus‐to‐cytoplasm ratios of fluorescence intensity in immunostained samples, we found increased IL‐38 nuclear translocation in non‐differentiated proliferating cultures, as compared to differentiating cells (Figure 4A,B). These data suggest that intracellular IL‐38 can shuttle between the nucleus and cytoplasm and that progressive keratinocyte differentiation correlates with its nuclear exclusion.
FIGURE 4.
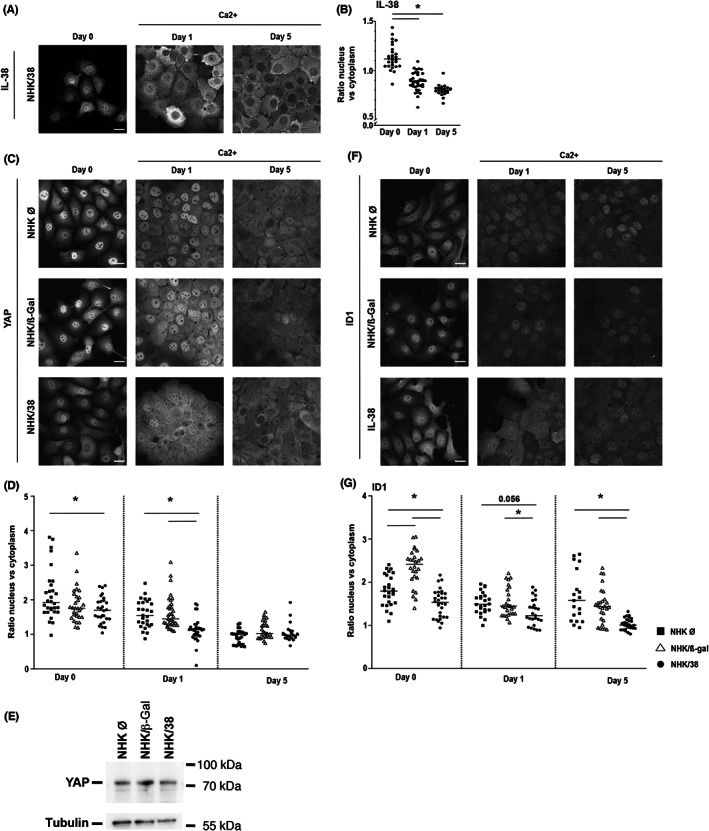
Localization of IL‐38, YAP and ID1 in NHK cells. (A) IL‐38 protein localization assessed by immunofluorescence in Dox‐treated NHK/38 cells cultured without (=day 0) or with 2 mM Ca2+ for 1 or 5 days. Scale bar 20 μm (B) IL‐38 protein localization quantified as the ratio of nuclear/cytoplasmic staining intensity on days 0, 1 and 5. Data are individual values and means (n ≥ 21). (C) YAP protein localization assessed by immunofluorescence in Dox‐treated NHK Ø, NHK/β‐Gal and NHK/38 cells cultured without (=day 0) or with 2 mM Ca2+ for 1 or 5 days. (D) YAP protein localization quantified as the ratio of nuclear/cytoplasmic staining intensity on days 0, 1 and 5. Data are individual values and means (n ≥ 21). (E) YAP expression assessed by Western blotting in Dox‐treated NHK Ø, NHK/β‐Gal and NHK/38 cells cultured without Ca2+ (=day 0). Tubulin levels are shown as loading control. (F) ID1 protein localization assessed by immunofluorescence in Dox‐treated NHK Ø, NHK/β‐Gal and NHK/38 cells cultured without (=day 0) or with 2 mM Ca2+ for 1 or 5 days. (G) ID1 protein localization quantified as the ratio of nuclear/cytoplasmic staining on days 0, 1 and 5. Data are individual values and means (n ≥ 19). Data are values for individual cells (n ≥ 19) from biological triplicates. (B, D and G) *p < 0.05. p values were calculated using a two‐way ANOVA with Tukey's multiple comparison.
3.6. IL‐38 negatively regulates YAP and ID1 during keratinocyte differentiation
To further investigate the mechanisms involved in the regulation of cell proliferation and differentiation by IL‐38, we examined the subcellular localization of the transcriptional regulators YAP and ID1, which are implicated in the regulation of cell proliferation and differentiation of various cell types, including keratinocytes. 20 , 21 , 22 , 23 , 24 , 25 We observed that the nuclear translocation of YAP progressively decreased during cell differentiation (Figure 4C,D), which corresponds to its function. This decrease in the nucleus‐to‐cytoplasm ratio of YAP staining occurred more rapidly in IL‐38 overexpressing than in control cells (Figure 4C,D). Global YAP expression levels were comparable in NHK cells without or with IL‐38 overexpression (Figure 4E). Similarly, IL‐38 overexpressing cells showed a decrease in the nuclear localisation of ID1 that persisted over 5 days of differentiation (Figure 4F,G). These data indicate that IL‐38 overexpression is associated with reduced nuclear translocation of YAP and ID1. Conversely, treatment of NHK/38 cells with cerivastatin, which causes nuclear exclusion of YAP, 26 also reduced the nucleus‐to‐cytoplasm ratio of the IL‐38 staining, without affecting IL‐38 expression levels (Figure S6).
4. DISCUSSION
IL‐1 family cytokines play an important role in host defense, initiating innate and adaptive inflammatory responses in the skin. Among the four anti‐inflammatory IL‐1 family members, IL‐1Ra, IL‐36Ra, IL‐37 and IL‐38, which are all constitutively expressed in epidermal keratinocytes, IL‐38 is the least known and understood. 8 So far, most studies assessed the effects of IL‐38 on immune cells and its impact on inflammatory responses. The hypothesis of IL‐38 exhibiting additional effects related to the function of the epidermis has only started to emerge. 10 Our study expands on this topic and reveals that IL‐38 significantly affects keratinocyte biology and thus potentially epidermal homeostasis.
An important finding of our study is that IL‐38 is expressed by multiple cell types in normal human skin. Previous studies demonstrated its expression by keratinocytes and immune cells, for example macrophages. Here we show that dermal fibroblasts and epithelial cells of skin appendages, such as eccrine sweat glands and sebaceous glands, also express IL‐38. These observations further suggest a role of IL‐38 in skin immunity, as it is well known that both sweat and sebum play an important role in antimicrobial functions of the epidermis and may modulate local inflammation. 27 Our results confirm and extend a previous study, in which IL‐38 expression was detected by immunohistochemistry in the epidermis, as well as in dermal fibroblasts and endothelial cells of normal human skin. 11 Validation of immunostaining specificity is critical, but also difficult, and non‐specific antibody binding may lead to inaccurate conclusions. In this study, we used two different anti‐IL‐38 antibodies, the specificity of which we previously validated on transfected, IL‐38 expressing cells and by preadsorption with recombinant IL‐38, 16 and which we compared to their respective isotype controls. In addition, the expression pattern that we observed for the IL‐38 protein by immunostaining in human skin is consistent with in situ hybridization and single cell RNA‐seq data from mouse skin, showing Il‐38 expression in keratinocytes, as well as in hair follicles. 5 , 28 , 29 IL‐38 mRNA expression was also reported in bulk RNA‐seq experiments in microdissected human eccrine sweat glands. 30 Overall, these correlations between mRNA expression and immunostaining data support the specificity of IL‐38 protein detection.
Next, we explicitly demonstrate that IL‐38 expression strongly depends on the differentiation state of keratinocytes. In our experiments, keratinocytes upregulated IL‐38 mRNA and protein production during calcium‐induced differentiation in monolayer culture, in line with previous reports 10 , 11 and with RNA‐seq data linking increased IL‐38 expression to human keratinocyte differentiation in several studies. 31 , 32 , 33 , 34 To further explore the correlation between keratinocyte differentiation and IL‐38 expression, we stimulated RHE with a panel of inflammatory cytokines. We showed that the Th17 cytokines IL‐17A and IL‐22 significantly reduced IL‐38 mRNA and protein levels, along with the expression of differentiation markers KRT10, LOR and FLG. A similar correlation was observed, although only at the mRNA level, after stimulation with the Th2 cytokines IL‐4 and IL‐13, which are also known to impact keratinocyte differentiation. 35 , 36 , 37 We could not observe an effect of Th2 cytokines on IL‐38, KRT10 and LOR protein levels at the 72 h time point investigated. The reason for this differential effect on mRNA and protein expression is unclear, but relative protein stability might explain that only major changes in mRNA expression, such as the ones induced by Th17 cytokines, lead to a sizable reduction in protein levels. Consistent with our data, it has been previously reported that IL‐17A and IL‐22 downregulate IL‐38 expression in monolayer human keratinocyte cultures. 11
Our next question was whether IL‐38 itself could alter basic functions of keratinocytes, such as proliferation and differentiation. To address this issue, we used an inducible IL‐38 overexpression system in the NHK keratinocyte cell line. Here, we show for the first time that IL‐38 may directly inhibit the proliferation of its producing cells. Interestingly, the expression of CCND1, which contributes to cell cycle progression, decreased during the differentiation of IL‐38 overexpressing, but not control NHK cells. The expression of mRNA encoding KRT14, a marker of proliferating keratinocytes, was also reduced in IL‐38 overexpressing, as compared to control cells. At the same time, IL‐38 overexpression stimulated keratinocyte differentiation, as Dox‐treated NHK/38 cells significantly upregulated KRT10, FLG and LOR levels, when compared to controls. However, the effect on keratinocyte differentiation in our experimental system required high IL‐38 concentrations, possibly because NHK cells are HPV‐immortalized cells, in which differentiation is notably difficult to induce. 38 Lower amounts of IL‐38 might be sufficient to promote similar cellular changes in the more permissive context of primary keratinocytes differentiating in 3D epidermal structures. Interestingly, previous in vivo studies reported that subcutaneous injections of recombinant IL‐38 reduced the severity of psoriasis‐like inflammation in mice and restored keratinocyte differentiation. 5 , 11 Altogether, our findings suggest a potential positive feedback mechanism where differentiating keratinocytes upregulate IL‐38 expression and high levels of IL‐38 can further block proliferation, while promoting terminal differentiation. Our experiments show that this effect is completely independent of interactions between keratinocytes and immune cells.
Accordingly, we report a global decrease in IL‐38 expression in skin disorders of different aetiology: psoriasis, atopic dermatitis, basal, squamous and eccrine cell carcinoma, seborrheic keratosis, verrucae, eccrine poroma and sebaceoma. In line with this observation, reduced IL‐38 expression has been previously reported in psoriasis. 11 , 12 Interestingly, we observed increased nuclear localization of IL‐38 in lesions involving uncontrolled proliferation of basal cells, such as psoriasis and basal cell carcinoma, whereas the localization of IL‐38 was mostly cytoplasmic in disorders characterized by accelerated growth of differentiated cells, such as squamous cell carcinoma. At the same time, we observed an exclusion of the cytokine from the nucleus during in vitro differentiation of IL‐38 overexpressing cells. The precise mechanism for the nuclear import and export of IL‐38 remains to be defined. The protein contains one predicted nuclear export sequence (NES), but no classical nuclear localisation sequence (NLS), and alternative mechanisms for entering the nucleus might be at play. Therefore, we report a novel phenomenon of IL‐38 shuttling between the nucleus and cytoplasm, with nuclear localization preferentially observed in undifferentiated cells.
IL‐1 family cytokines regulate inflammatory responses by signalling through cell surface receptors belonging to the IL‐1 receptor family, 39 Several cell surface receptors and downstream signalling pathways have been proposed for IL‐38. However, direct binding of IL‐38 to an IL‐1 family receptor still remains to be demonstrated in vivo. 9 An effect of extracellular IL‐38 on cultured keratinocytes has so far been reported in a single study, in which IL‐38 antagonized the production of inflammatory mediators induced by IL‐36γ. 11 In the present study, we did not observe any significant effect of IL‐38 overexpression on IL‐36γ‐ or IL‐1α–induced IL‐8 production in NHK cells. In contrast, overexpression of IL‐1Ra inhibited the effect of IL‐1α as expected.
In addition to their extracellular activity, intracellular functions were previously reported for some IL‐1 family members, including IL‐1α, IL‐1Ra, IL‐33 and IL‐37. 17 , 40 , 41 , 42 , 43 , 44 We hypothesize that IL‐38 may also exert intracellular functions in keratinocytes. A recent study further demonstrated that IL‐38 promotes the production of inflammatory mediators, such as TNF‐α or TGF‐β1, in a cell‐intrinsic manner in macrophages in vitro, suggesting that IL‐38 may have intracellular effects different from its extracellular, anti‐inflammatory functions. 45
In this context, nuclear localization suggests a possible role of IL‐38 in the regulation of gene transcription. Furthermore, our previous study revealed several IL‐38‐interacting molecules, including ID1, which might represent partners for potential intracellular functions of IL‐38 in keratinocytes. 16 ID1 was identified as an inhibitor of helix–loop–helix DNA binding proteins 46 and functions as a negative regulator of differentiation in various cell types. 23 ID1 inhibits differentiation of keratinocytes and has been proposed to be expressed in cells with high proliferative potential, such as keratinocytes of psoriatic skin, and downregulated in cells that undergo terminal differentiation. 24 , 47 Here we observed that IL‐38 overexpression significantly decreased nuclear localization of ID1 at early stages of keratinocyte differentiation. ID1 was previously shown to contribute to keratinocyte cell cycle progression by increasing CCND1 expression. 48 Nuclear exclusion of ID1 might thus promote cell differentiation through reduced CCND1 expression. We analysed another major regulator of the genes involved in cell proliferation, the transcription co‐activator YAP. 22 Similar to ID1, we found increased nuclear exclusion of YAP in IL‐38 overexpressing cells during early differentiation, which is likely to reduce its transcriptional effects. Indeed, nuclear YAP promotes proliferation and maintains keratinocytes in an undifferentiated state, while its shift to the cytoplasm parallels cell differentiation. 49 Global YAP expression levels were comparable with or without IL‐38 overexpression in NHK cells. It remains to be established whether interaction with IL‐38 may directly promote exclusion of ID1 from the nucleus. Similarly, the effect of IL‐38 overexpression on YAP localization might be direct or indirect. Finally, in cerivastatin‐treated NHK/38 cells, both YAP and IL‐38 relocalized to the cytoplasm. IL‐38 expression levels were not affected by cerivastatin treatment. Cerivastatin is an inhibitor of the mevalonate pathway and was shown to interfere with the membrane localization and activation of Rho GTPases through depletion of cellular geranylgeranyl pyrophosphate. This in turn led to YAP phosphorylation, which inhibited its nuclear translocation and activity. 26 It will be interesting to investigate if similar mechanisms might be involved in the control of intracellular IL‐38 localization and if the nuclear/cytoplasmic distribution of IL‐38 is influenced by YAP localization or activity.
In conclusion, we show here that IL‐38, in addition to its reported functions on immune cells, can also act directly on human keratinocytes, leading to decreased proliferation and enhanced differentiation, suggesting that IL‐38 may affect the epidermal homeostasis at large. Accordingly, IL‐38 levels are decreased in hyperproliferative skin disorders. Moreover, we suggest that IL‐38 can have intracellular functions and demonstrate for the first time that IL‐38 can shuttle between the nucleus and the cytoplasm, depending on the differentiation state of the cell. Future studies are needed to elucidate the precise mechanisms by which IL‐38 affects skin biology.
AUTHOR CONTRIBUTIONS
LM, MS, GP and JB involved in conceptualization and visualization. LM, MS, JB, ADB and DTA involved in investigation. LM, JB and GP involved in formal analysis and methodology. WHB and GP involved in project administration. LM, MS and JB wrote the original draft. LM, MS, ADB, JD, WHB, GP and JB reviewed and edited the manuscript. JD, MW, GK and WHB provided resources. JB, WHB and GP acquired the fund. GP and JB involved in supervision. All authors have read and approved the final manuscript.
CONFLICT OF INTEREST
WHB received honoraria as advisor or invited speaker from Abbvie, Almirall, BMS, Celgene, Leo, Lilly, Novartis, UCB. The other authors have no conflict of interest related to the present manuscript to declare.
Supporting information
Appendix S1. Supplementary materials and methods.
Figure S1. IL‐38 expression in healthy human skin.
Figure S2. Specificity of IL‐38 detection by immunofluorescence in human skin.
Figure S3. Regulation of IL‐38 and differentiation marker expression in RHE and N/TERT‐1 cells.
Figure S4. Inflammatory response in NHK cells overexpressing IL‐38.
Figure S5. Specificity of IL‐38 detection by immunohistochemistry in human skin
Figure S6. Localization of YAP and IL‐38 in cerivastatin‐treated NHK/38 cells.
ACKNOWLEDGEMENTS
We are grateful to Nicolò C. Brembilla and Paula Nunes‐Hasler for valuable comments and help with data analysis. We would like to thank Ali Modarressi for providing healthy skin samples used in this study, Bernard Foglia for technical support and Jérôme King for assistance with statistical analysis. This work was supported by grants from the Swiss National Science Foundation (310030_188470 to GP, 310030_175470/1 to WHB), the Rheumasearch Foundation, the Kurt and Senta Herrmann Foundation, the Medicor Foundation, the Ernest Boninchi Foundation, and the Eli Lilly Immunodermatology Award. Open access funding provided by Universite de Geneve.
Mermoud L, Shutova M, Diaz‐Barreiro A, et al. IL‐38 orchestrates proliferation and differentiation in human keratinocytes. Exp Dermatol. 2022;31:1699‐1711. doi: 10.1111/exd.14644
DATA AVAILABILITY STATEMENT
No large datasets were generated or analyzed during the current study. Minimal datasets necessary to interpret and or replicate data in this paper are available upon request to the corresponding author.
REFERENCES
- 1. Lin H, Ho AS, Haley‐Vicente D, et al. Cloning and characterization of IL‐1HY2, a novel interleukin‐1 family member. J Biol Chem. 2001;276:20597‐20602. [DOI] [PubMed] [Google Scholar]
- 2. Bensen JT, Dawson PA, Mychaleckyj JC, Bowden DW. Identification of a novel human cytokine gene in the interleukin gene cluster on chromosome 2q12‐14. J Interf Cytok Res. 2001;21:899‐904. [DOI] [PubMed] [Google Scholar]
- 3. van de Veerdonk FL, Stoeckman AK, Wu G, et al. IL‐38 binds to the IL‐36 receptor and has biological effects on immune cells similar to IL‐36 receptor antagonist. Proc Natl Acad Sci USA. 2012;109:3001‐3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mora J, Schlemmer A, Wittig I, et al. Interleukin‐38 is released from apoptotic cells to limit inflammatory macrophage responses. J Mol Cell Biol. 2016;8:426‐438. [DOI] [PubMed] [Google Scholar]
- 5. Han Y, Mora J, Huard A, et al. IL‐38 ameliorates skin inflammation and limits IL‐17 production from γδ T cells. Cell Rep. 2019;27:835‐846. [DOI] [PubMed] [Google Scholar]
- 6. Rudloff I, Godsell J, Nold‐Petry CA, et al. Brief report: interleukin‐38 exerts Antiinflammatory functions and is associated with disease activity in systemic lupus erythematosus. Arthritis Rheumatol. 2015;67:3219‐3225. [DOI] [PubMed] [Google Scholar]
- 7. Pan Y, Wang M, Chen X, et al. Elevated IL‐38 inhibits IL‐23R expression and IL‐17A production in thyroid‐associated ophthalmopathy. Int Immunopharmacol. 2021;91:107300. [DOI] [PubMed] [Google Scholar]
- 8. Martin P, Goldstein JD, Mermoud L, Diaz‐Barreiro A, Palmer G. IL‐1 family antagonists in mouse and human skin inflammation. Front Immunol. 2021;12:652846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xia HS, Liu Y, Fu Y, Li M, Wu YQ. Biology of interleukin‐38 and its role in chronic inflammatory diseases. Int Immunopharmacol. 2021;95:107528. [DOI] [PubMed] [Google Scholar]
- 10. Lachner J, Mlitz V, Tschachler E, Eckhart L. Epidermal cornification is preceded by the expression of a keratinocyte‐specific set of pyroptosis‐related genes. Sci Rep. 2017;7:17446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mercurio L, Morelli M, Scarponi C, et al. IL‐38 has an anti‐inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti‐IL‐17A treatment. Cell Death Dis. 2018;9:1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boutet MA, Bart G, Penhoat M, et al. Distinct expression of interleukin (IL)‐36alpha, beta and gamma, their antagonist IL‐36Ra and IL‐38 in psoriasis, rheumatoid arthritis and Crohn's disease. Clin Exp Immunol. 2016;184:159‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dickson MA, Hahn WC, Ino Y, et al. Human keratinocytes that express hTERT and also bypass a p16(INK4a)‐enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. 2000;20:1436‐1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Smits JPH, Niehues H, Rikken G, et al. Immortalized N/TERT keratinocytes as an alternative cell source in 3D human epidermal models. Sci Rep. 2017;7:11838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steenbergen RD, Walboomers JM, Meijer CJ, et al. Transition of human papillomavirus type 16 and 18 transfected human foreskin keratinocytes towards immortality: activation of telomerase and allele losses at 3p, 10p, 11q and/or 18q. Oncogene. 1996;13:1249‐1257. [PubMed] [Google Scholar]
- 16. Talabot‐Ayer D, Mermoud L, Borowczyk J, et al. Interleukin‐38 interacts with destrin/Actin‐depolymerizing factor in human keratinocytes. PloS One. 2019;14:e0225782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palmer G, Trolliet S, Talabot‐Ayer D, Mezin F, Magne D, Gabay C. Pre‐interleukin‐1alpha expression reduces cell growth and increases interleukin‐6 production in SaOS‐2 osteosarcoma cells: differential inhibitory effect of interleukin‐1 receptor antagonist (icIL‐1Ra1). Cytokine. 2005;31:153‐160. [DOI] [PubMed] [Google Scholar]
- 18. Borowczyk J, Buerger C, Tadjrischi N, et al. IL‐17E (IL‐25) and IL‐17A differentially affect the functions of human keratinocytes. J Invest Dermatol. 2020;140:1379‐1389.e2. [DOI] [PubMed] [Google Scholar]
- 19. Rabeony H, Petit‐Paris I, Garnier J, et al. Inhibition of keratinocyte differentiation by the synergistic effect of IL‐17A, IL‐22, IL‐1α, TNFα and Oncostatin M. PloS One. 2014;9:e101937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schlegelmilch K, Mohseni M, Kirak O, et al. Yap1 acts downstream of α‐catenin to control epidermal proliferation. Cell. 2011;144:782‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elbediwy A, Vincent‐Mistiaen ZI, Spencer‐Dene B, et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development. 2016;143:1674‐1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tang C, Takahashi‐Kanemitsu A, Kikuchi I, Ben C, Hatakeyama M. Transcriptional co‐activator functions of YAP and TAZ are inversely regulated by tyrosine phosphorylation status of Parafibromin. iScience. 2018;1:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003;13:410‐418. [DOI] [PubMed] [Google Scholar]
- 24. Björntorp E, Parsa R, Thornemo M, Wennberg AM, Lindahl A. The helix–loop–helix transcription factor Id1 is highly expressed in psoriatic involved skin. Acta Derm Venereol. 2003;83:403‐409. [DOI] [PubMed] [Google Scholar]
- 25. Rognoni E, Walko G. The roles of YAP/TAZ and the hippo pathway in healthy and diseased skin. Cells. 2019;8:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sorrentino G, Ruggeri N, Specchia V, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. 2014;16:357‐366. [DOI] [PubMed] [Google Scholar]
- 27. Brogden NK, Mehalick L, Fischer CL, Wertz PW, Brogden KA. The emerging role of peptides and lipids as antimicrobial epidermal barriers and modulators of local inflammation. Skin Pharmacol Physiol. 2012;25:167‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Joost S, Zeisel A, Jacob T, et al. Single‐cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity. Cell Syst. 2016;3:221‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joost S, Annusver K, Jacob T, et al. The molecular anatomy of mouse skin during hair growth and rest. Cell Stem Cell. 2020;26:441‐457. [DOI] [PubMed] [Google Scholar]
- 30. Na CH, Sharma N, Madugundu AK, et al. Integrated transcriptomic and proteomic analysis of human eccrine sweat glands identifies missing and novel proteins. Mol Cell Proteomics. 2019;18:1382‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bao X, Rubin AJ, Qu K, et al. A novel ATAC‐seq approach reveals lineage‐specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol. 2015;16:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones J, Chen Y, Tiwari M, Li J, Ling J, Sen GL. KLF3 mediates epidermal differentiation through the epigenomic writer CBP. iScience. 2020;23:101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones J, Chen Y, Tiwari M, Li J, Ling J, Sen GL. BRD4 is necessary for differentiation downstream of epidermal lineage‐determining transcription factors. J Invest Dermatol. 2020;140:2077‐2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rubin AJ, Barajas BC, Furlan‐Magaril M, et al. Lineage‐specific dynamic and pre‐established enhancer‐promoter contacts cooperate in terminal differentiation. Nat Genet. 2017;49:1522‐1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim BE, Leung DY, Boguniewicz M, Howell MD. Loricrin and involucrin expression is down‐regulated by Th2 cytokines through STAT‐6. Clin Immunol. 2008;126:332‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Omori‐Miyake M, Yamashita M, Tsunemi Y, Kawashima M, Yagi J. In vitro assessment of IL‐4‐ or IL‐13‐mediated changes in the structural components of keratinocytes in mice and humans. J Invest Dermatol. 2014;134:1342‐1350. [DOI] [PubMed] [Google Scholar]
- 37. Howell MD, Fairchild HR, Kim BE, et al. Th2 cytokines act on S100/A11 to downregulate keratinocyte differentiation. J Invest Dermatol. 2008;128:2248‐2258. [DOI] [PubMed] [Google Scholar]
- 38. White EA. Manipulation of epithelial differentiation by HPV oncoproteins. Viruses. 2019;11:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mantovani A, Dinarello CA, Molgora M, Garlanda C. Interleukin‐1 and related cytokines in the regulation of inflammation and immunity. Immunity. 2019;50:778‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Werman A, Werman‐Venkert R, White R, et al. The precursor form of IL‐1alpha is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci USA. 2004;101:2434‐2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Watson JM, Lofquist AK, Rinehart CA, et al. The intracellular IL‐1 receptor antagonist alters IL‐1‐inducible gene expression without blocking exogenous signaling by IL‐1 beta. J Immunol. 1995;155:4467‐4475. [PubMed] [Google Scholar]
- 42. Banda NK, Guthridge C, Sheppard D, et al. Intracellular IL‐1 receptor antagonist type 1 inhibits IL‐1‐induced cytokine production in keratinocytes through binding to the third component of the COP9 signalosome. J Immunol. 2005;174:3608‐3616. [DOI] [PubMed] [Google Scholar]
- 43. Carriere V, Roussel L, Ortega N, et al. IL‐33, the IL‐1‐like cytokine ligand for ST2 receptor, is a chromatin‐associated nuclear factor in vivo. Proc Natl Acad Sci USA. 2007;104:282‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ross R, Grimmel J, Goedicke S, et al. Analysis of nuclear localization of interleukin‐1 family cytokines by flow cytometry. J Immunol Methods. 2013;387:219‐227. [DOI] [PubMed] [Google Scholar]
- 45. Huard A, Do HN, Frank AC, et al. IL‐38 ablation reduces local inflammation and disease severity in experimental autoimmune encephalomyelitis. J Immunol. 2021;206:1058‐1066. [DOI] [PubMed] [Google Scholar]
- 46. Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein id: a negative regulator of helix–loop–helix DNA binding proteins. Cell. 1990;61:49‐59. [DOI] [PubMed] [Google Scholar]
- 47. Schaefer BM, Koch J, Wirzbach A, Kramer MD. Expression of the helix–loop–helix protein ID1 in keratinocytes is upregulated by loss of cell‐matrix contact. Exp Cell Res. 2001;266:250‐259. [DOI] [PubMed] [Google Scholar]
- 48. Hamajima Y, Komori M, Preciado DA, et al. The role of inhibitor of DNA‐binding (Id1) in hyperproliferation of keratinocytes: the pathological basis for middle ear cholesteatoma from chronic otitis media. Cell Prolif. 2010;43:457‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang H, Pasolli HA, Fuchs E. Yes‐associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci USA. 2011;108:2270‐2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplementary materials and methods.
Figure S1. IL‐38 expression in healthy human skin.
Figure S2. Specificity of IL‐38 detection by immunofluorescence in human skin.
Figure S3. Regulation of IL‐38 and differentiation marker expression in RHE and N/TERT‐1 cells.
Figure S4. Inflammatory response in NHK cells overexpressing IL‐38.
Figure S5. Specificity of IL‐38 detection by immunohistochemistry in human skin
Figure S6. Localization of YAP and IL‐38 in cerivastatin‐treated NHK/38 cells.
Data Availability Statement
No large datasets were generated or analyzed during the current study. Minimal datasets necessary to interpret and or replicate data in this paper are available upon request to the corresponding author.