

Abstract
A method for the synthesis of functionalized alternating copolymers by reversible deactivation radical polymerization was developed. Copolymerization by reversible addition–fragmentation chain transfer of hexenyl vinyl ether with a novel fluorinated divinyl monomer yields alternating cyclopolymers that can be chemoselectively modified by three distinct orthogonal functionalization reactions. Along the thiol‐ene click reaction and amidation, a third functionalization was achieved via NHC‐catalyzed transesterification or acylation resulting in a small library of ABC‐type alternating terpolymers.
Keywords: Alternating Polymerization, Orthogonal Functionalization, Postmodification, RAFT Polymerization, Radical Polymerization
The alternating radical copolymerization of divinyl monomer A and hex‐5‐enyl vinyl ether (HVE) using reversible addition–fragmentation chain transfer (RAFT) is reported. The obtained cyclopolymer B can chemically be further modified by three orthogonal functionalization reactions i.e. thiol‐ene click reaction, amidation and transesterification or acylation to provide access to various alternating ABC‐type terpolymers C.

Introduction
Polymers found in nature (i.e. proteins, DNA and polysaccharides) exhibit a broad range of possible structures and show defined and unique functions. The control of structure and function is mainly attributed to the uniformity and perfect arrangement of functional building blocks (secondary and tertiary structure) in a defined sequence (primary structure). In contrast to the sequence‐defined biopolymers, synthetic polymers are far less complex and mostly consist of only a few repeating building blocks with disperse chain lengths. Although possible, the construction of monodisperse, sequence‐defined synthetic macromolecules with methods like an iterative multistep approach is often time consuming and requires great synthetic effort. But even in copolymers consisting of only two different monomers the controlled incorporation of the two monomeric units is often challenging and, in many cases, disperse polymers with a statistical distribution of the individual entities along the polymer chain result. Despite great efforts and progress, the synthesis of polymers with precisely defined structures remains one of the biggest challenges of modern polymer chemistry. [1]
Considering radical polymerization, various methods for reversible deactivation radical polymerization (RDRP) have been developed over the last two decades. These methods allow the control of molecular weight, chain‐end structure and the polymer architecture to some degree. [2] Moreover, radical polymerization can be conducted with a wide range of monomers like styrenes, (meth)acrylates, (meth)acrylamides which carry various functional pendant groups. Considering radical copolymerization, the differentiation of two monomers by the reactive growing polymeric radical is difficult and hence often not selective providing random copolymers. However, alternating radical copolymerization is feasible for electronically divergent monomer pairs. [3] In these systems the monomer reactivity ratios are close to zero, therefore favoring crosspropagation over homopropagation. [4] Other methods to achieve an alternating sequence include the copolymerization of monomers with bulky pendant groups [5] or the elegant selective cyclopolymerization of divinyl monomers with cleavable pendant groups. [6] With the latter two approaches, the alternating copolymerization of comonomers that show similar reactivity has been realized. [7] The obtained sequence‐controlled polymers may show a high degree of alternation, but lack the uniformity in terms of chain length and thus are different to the above‐mentioned sequence‐defined polymers.
Inspired by our previous studies on the alternating copolymerization of two electronically distinct monomers i.e. hexafluoroisopropyl acrylate and various vinyl ethers, [8] we decided to implement the divinyl monomer 1 with a cleavable linker moiety as a cyclizing building block leading to an electron‐poor growing C‐radical in combination with vinyl ether 2 which leads upon incorporation to an electron‐rich C‐radical ensuring an alternating sequence with these two building blocks. As polymerization technique, we selected reversible addition–fragmentation chain transfer (RAFT). Owing to the nature of the pendant groups, copolymer 3 can be chemically further modified by three distinct orthogonal functionalization reactions i.e. thiol‐ene click reaction, [9] amidation [10] and transesterification[ 11 , 12 ] or acylation [13] to yield various ABC‐type alternating terpolymers 4 (Scheme 1). [14]
Scheme 1.
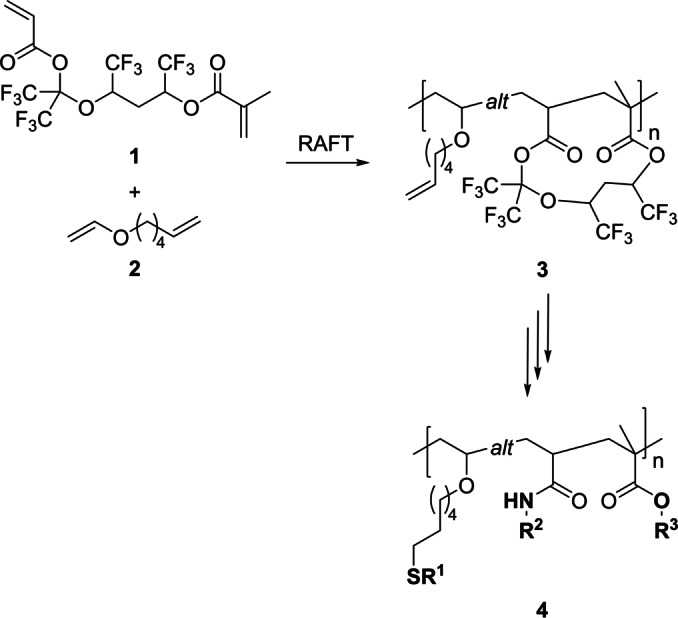
Synthesis of alternating cyclopolymer 3 and subsequent threefold orthogonal functionalization leading to alternating ABC‐type terpolymers.
Results and Discussion
Divinyl monomer 1 was prepared as a diastereoisomeric mixture (dr≈1:1, racemic) starting from commercially available 1,1,1,5,5,5‐hexafluoropentan‐2,4‐dione in a three‐step procedure on a multigram scale with 14 % overall yield (see the Supporting Information). We started our investigations by cyclocopolymerization of divinyl monomer 1 (1.0 equiv) with n‐butyl vinyl ether (NBVE, 1.0–8.0 equiv) as the electron‐rich comonomer and identified RAFT polymerization as the RDRP method of choice, as it is compatible with (meth)acrylates and vinyl ethers. [15] Polymerizations were conducted in sealed tubes (70 °C, 2–27 h) with dimethyl malonyl benzodithioate (5) as the chain transfer agent (CTA, 1–15 mol %) and α,α′‐azobisisobutyronitrile (AIBN) as the initiator (0.5–1 mol %) in trifluorotoluene under diluted conditions to suppress potential cross‐linking reactions between the growing polymer chains (Scheme 2, Table 1). The crude product was purified by size‐exclusion chromatography (SEC) in CH2Cl2 and the obtained polymer 6 was analysed by 1H NMR spectroscopy and ESI or MALDI mass spectrometry (see Supporting Information). In addition, the average molecular weight and the polydispersity index were approximated by single detection direct calibration GPC against PMMA standards (the obtained polymers differ considerably from the used standards in terms of complexity).
Scheme 2.

Alternating copolymerization of divinyl monomer 1 with NBVE.
Table 1.
Conditions for copolymerization of divinyl monomer 1 and various vinyl ethers. Reactions were carried out with PhCF3 as the solvent and 1 mol % AIBN as the initiator at 70 °C.
|
Entry |
CTA Reagent |
Vinyl ether |
Monomer ratio of 1/vinyl ether |
t [h] |
c [mol L−1] of 1 |
Incorporation ratio of 1/vinyl ether[a] |
DPn,1 |
M n,exp [g mol−1][b] |
PDI[b] |
|---|---|---|---|---|---|---|---|---|---|
|
1 |
5 (2 mol %) |
NBVE |
1 : 1 |
2 |
0.1 |
55 : 45 |
9 |
5800[a] |
–[c] |
|
2 |
5 (2 mol %) |
NBVE |
1 : 2 |
2 |
0.1 |
53 : 47 |
12 |
7200 |
1.3 |
|
3 |
5 (2 mol %) |
NBVE |
1 : 4 |
2 |
0.1 |
51 : 49 |
11 |
7000 |
1.2 |
|
4 |
5 (2 mol %) |
NBVE |
1 : 8 |
2 |
0.1 |
50 : 50 |
16 |
9600[a] |
–[c] |
|
5 |
5 (10 mol %) |
NBVE |
1 : 8 |
18 |
0.1 |
48 : 52 |
9 |
5500 |
1.2 |
|
6 |
5 (15 mol %) |
NBVE |
1 : 8 |
27 |
0.05 |
50 : 50 |
5 |
3100[a] |
–[c] |
|
7 |
5 (2 mol %) |
NBVE |
1 : 8 |
5 |
0.05 |
50 : 50 |
41 |
25 000 |
1.2 |
|
8[d] |
5 (1 mol %) |
NBVE |
1 : 8 |
4 |
0.05 |
50 : 50 |
96 |
56 800 |
1.2 |
|
9 |
– |
NBVE |
1 : 8 |
2 |
0.05 |
50 : 50 |
646 |
880 000 |
2.5 |
|
10 |
5 (2 mol %) |
OVE |
1 : 8 |
16 |
0.05 |
51 : 49 |
41 |
28 800 |
1.8 |
|
11 |
11 (2 mol %) |
OVE |
1 : 8 |
15 |
0.05 |
51 : 49 |
40 |
26 000 |
1.5 |
|
12 |
11 (2 mol %) |
DVE |
1 : 8 |
15 |
0.05 |
51 : 49 |
28 |
19 500 |
1.5 |
|
13 |
11 (2 mol %) |
HVE |
1 : 8 |
15 |
0.05 |
51 : 49 |
36 |
22 500 |
1.4 |
|
14 |
11 (2 mol %) |
HVE |
1 : 8 |
24 |
0.05 |
51 : 49 |
49 |
30 600 |
1.4 |
|
15 |
– |
HVE |
1 : 8 |
3 |
0.05 |
51 : 49 |
148 |
93 000 |
5.4 |
[a] Determined by 1H NMR spectroscopy. [b] Approximated by GPC at 25 °C using THF as the eluent against PMMA standards. [c] Not determined. [d] 0.5 mol % AIBN used.
We first investigated the copolymerization with equimolar amounts of both comonomers for 2 h with a concentration of 0.1 mol L−1 and obtained the copolymer 6 with a 1/NBVE incorporation ratio of 55 : 45, as determined by 1H NMR spectroscopy (Table 1, entry 1). Doubling the amount of NBVE enhanced the incorporation of the latter monomer resulting in a ratio of 53 : 47 (entry 2) and with a fourfold NBVE‐excess, copolymer 6 was obtained with an incorporation ratio of 51 : 49 (entry 3). Pleasingly, an equimolar incorporation of both comonomers based on NMR analysis was achieved with a monomer feed ratio of 1 : 8 (entry 4). Under these conditions we also prepared a low molecular weight copolymer using a larger amount of CTA (10 mol %) to investigate the chain composition in more detail by mass spectrometry (entry 5). Unfortunately, polymer chains containing a larger fraction of the vinyl ether entity were also detected. Since NBVE is not able to undergo homopolymerization under the selected conditions for electronic reasons,[ 15 , 16 ] we assumed that the additional NBVE units present must have been incorporated between the acrylate and methacrylate moiety. Obviously, radical cyclization of the intermediate α‐ester C‐radical cannot fully compete with intermolecular addition to NBVE. To favour the cyclization of the growing polymer α‐ester C‐radical, we further decreased the concentration to 0.05 mol L−1 (entry 6), which indeed increased the degree of alternation, but complete suppression of the unwanted side reaction could not be achieved (see Supporting Information). Mass spectrometry analysis also revealed, that all of the polymer chains carry a dimethyl malonate moiety (from reinitiation by 5) as the headgroup and most copolymers terminate with NBVE units at both ends of the chain as indicated in the structure 6.
With the optimized conditions in hand, we targeted larger molecular weight cyclocopolymers by increasing the reaction time and by further decreasing the CTA loading from 2 to 1 mol % (entries 7 and 8). High conversion was achieved in both cases (>90 %). The obtained polymers show a narrow PDI of 1.2 and copolymers 6 with an M n of 25 000 and 57 000 g mol−1 were obtained. Without addition of the CTA the obtained polymer also shows an equimolar incorporation ratio of both monomers, but the high average molecular weight of 880 000 g mol−1 and the increased PDI of 2.5 indicate the expected loss of control over the polymerization (entry 9). In addition, we were able to use the prepared poly(1‐alt‐NBVE) 6 as a macroRAFT reagent to control the polymerization of methyl methacrylate (625 equiv, PhCH3, 70 °C, 20 h) yielding block copolymer poly(1‐alt‐NBVE)‐block‐poly(methyl methacrylate) (97 000 g mol−1, PDI=1.4) demonstrating a high degree of livingness of the cyclocopolymerization process (see Supporting Information).
To check whether the addition of α‐alkoxy radicals, that are generated as growing polymeric radicals during the copolymerization, to the divinyl building block 1 occurs regioselectively, we reacted 1‐butoxyethyl phenyl selenide as a precursor of an α‐butoxy C‐radical with 1 under reductive conditions with tin hydride in a model radical addition/cyclization cascade to give 7 a as a mixture of diastereoisomers (Scheme 3). To simplify NMR‐analysis, this reaction was conducted using diastereoisomerically pure divinyl monomer syn‐1 (racemic). Note that in the cyclocopolymerisation no change in polymerization efficiency was noted upon switching from the isomerically pure divinyl monomer syn‐1 to a diastereoisomeric mixture of 1. Pleasingly, C‐radical addition to syn‐1 occurred with complete regioselectivity to the vinyl group of the acrylate moiety indicating a high degree of sequence‐control along the polymer chain in the corresponding cyclocopolymerization process. The regioisomeric product 7 b was not formed. The high regioselectivity observed is caused by electronic effects as the two CF3‐groups and the acetal functionality further increase the electrophilicity of the acrylate‐type double bond in 1.
Scheme 3.

Reductive addition/cyclization of 1‐butoxyethyl phenyl selenide to syn‐1 as a model reaction.
Next, the reactivity ratios of the comonomer pair 1 and NBVE were determined. Polymerizations were carried out with different monomer feeds and stopped at low conversions. The incorporation ratios were plotted and the Mayo–Lewis copolymerization equation was fitted to the resulting graph using the least square method (see Supporting Information). [17] With r1 =0.14 and r2=0 the values are either very close to or are at zero, which further supports our findings that copolymerization occurs in an alternating mode.
We continued the studies by investigating the chemoselective postfunctionalization of poly(1‐alt‐NBVE) 6 (Scheme 4). The hexafluoroacetone acetal ester moiety in 6 is known to react with nucleophilic amines to the corresponding amides. [18] Unfortunately, the benzodithioate group of 6 was found to exhibit a similar reactivity towards amines generating free thiol groups, that can form disulfides or undergo backbiting at esters to form thiolactones. [19] As both side reactions would alter the polymer architecture, we had to first remove the benzodithioate group applying a known reaction with Et3B/air. [20]
Scheme 4.

Orthogonal postmodification of poly(1‐alt‐NBVE) 6 via desulfuration, amidation and subsequent transesterification or acylation.
The desulfurized copolymers derived from 6 (M n=25 000 mol L−1, PDI=1.2, 1.0 equiv) were then reacted with various amines (2.0–4.0 equiv) in a mixture of THF and DMF (1 : 1) at room temperature for 16 h (Table 2). After purification by SEC in CH2Cl2, polymers of type 8 were obtained. A high degree of functionalization (>99 %) was achieved in the amidation reaction, as confirmed by 1H‐, 19F NMR and mass spectrometry analysis for linear, α‐branched and benzylic primary amines (see Supporting Information). Morpholine could also be employed, albeit reaction time had to be extended to 72 h (functionalization >95 %). Importantly, the second type of ester moiety that is sterically shielded by the α‐quaternary C‐centre and electronically less activated remained untouched under these conditions and hence the resulting polymeric products 8 still carry activated α‐trifluoromethylalkyl ester entities with additional free hydroxyl groups, which opens the possibility for a second postfunctionalization by either transesterification at the activated esters or acylation of the free alcohols.
Table 2.
Orthogonal difunctionalization of 6 to give either 9 or 10.
|
Entry |
Amine |
Alcohol or acyl halide |
M n,exp [a] [g mol−1] |
PDI[a] |
Yield[b,c] [%] (Product) |
|---|---|---|---|---|---|
|
1 |
|
|
21 800 |
1.2 |
>99 (9 a) |
|
2 |
|
|
20 600 |
1.2 |
>99 (9 b) |
|
3 |
|
|
20 900 |
1.2 |
71 (9 c) |
|
4 |
|
|
25 500 |
1.3 |
>99 (10 a) |
|
5 |
|
|
27 800 |
1.3 |
98 (10 b)[d] |









[a] Approximated by GPC at 25 °C using THF as the eluent against PMMA standards. [b] Isolated yield after two functionalization steps. [c] The degree of functionalization was >98 % for both postmodification reactions. [d] The degree of functionalization for the amidation was >98 % and 96 % for the second postmodification.
Inspired by our studies on the NHC‐catalyzed transesterification of polytrifluoroethyl acrylate, transesterification of 8 was investigated. [12] N,N‐di(tert‐butyl)imidazol‐2‐ylidene (I t Bu) was selected as the NHC catalyst with 1,4‐dioxane as the solvent at 100 °C and we were pleased to achieve full conversion to polymers of type 9 after 3 d for linear and benzylic alcohols (15 equiv, 9 a–9 c) in good to excellent yields. In addition, we also conducted acylation reactions on 8 with acyl halides (1.5 equiv) in the presence of triethylamine (1.5 equiv) in CH2Cl2 for 16 h and were able to isolate polymers of type 9 in excellent yields (9 a and 9 b). The high degree of postfunctionalization was confirmed by 1H‐ and 19F NMR analysis (see Supporting Information). For low molecular weight polymers, mass spectrometry can also be used to analyse reaction outcome (see Supporting Information). GPC analysis of product polymers 9 and 10 after purification by SEC revealed that the average molecular weight slightly decreased (20 600 to 21 800 g mol−1) while the PDI (1.2) remained narrow for 9. The molecular weight (25 500 and 27 800 g mol−1) and the PDI (1.3) slightly increased for polymers 10. The different M n measured can be explained by different GPC behaviour of polymer 6 in comparison to 9 and 10.
Finally, in order to apply the introduced strategy to the preparation of alternating polymers with three orthogonally addressable functionalities, we replaced NBVE by oct‐7‐enyl vinyl ether (OVE) (8 equiv of vinyl ether, c 1 =0.05 mol L−1, PhCF3, 1 mol % AIBN) and copolymerization with 1 was achieved with CTA 5 under similar conditions (incorporation ratio of 1 to OVE was 49 : 51, see Table 1, entry 10), indicating a high degree of alternation. The reaction time had to be increased (16 h), as the polymerization with OVE proceeded slower. Unfortunately, the PDI increased to 1.8. We therefore switched to dimethyl malonyl 4‐cyanobenzodithioate (11) as the CTA that provided the targeted copolymer with a PDI of 1.5 (entry 11). With dec‐9‐enyl vinyl ether (DVE) as the comonomer a similar result was obtained (entry 12), while with hex‐5‐enyl vinyl ether (HVE, 2) the PDI further decreased and copolymer 3 with a PDI of 1.4 resulted (entry 13). In both cases, the polymerization proceeded slower as compared to the NBVE process, but the incorporation ratio of 1 with respect to the vinyl ether stayed consistently at 51 : 49. Copolymerization of 1 with HVE for 24 h resulted in 3 with an average molecular weight of 30 600 g mol−1 and a PDI of 1.4, that was used as a model substrate for the subsequent postfunctionalization steps (entry 14, Scheme 5). The polymerization could also be conducted without the addition of the CTA resulting in the same ratio of 51 : 49, but again with an increased average molecular weight (93 000 g mol−1) and PDI of 5.4 (entry 15). We also determined the reactivity ratios for the 1/HVE pair, with r 1=0.15 and r 2=0 they are similar to the 1/NBVE system indicating an alternating copolymerization process.
Scheme 5.
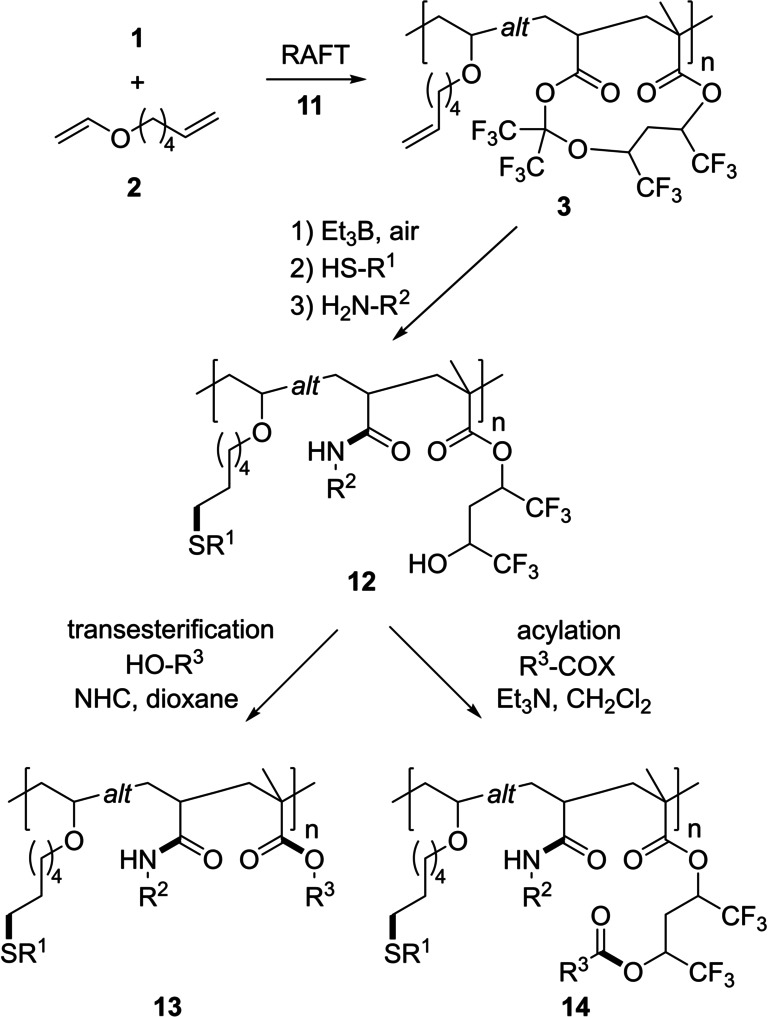
Synthesis of alternating cyclopolymer 3 and subsequent threefold orthogonal functionalization.
First, the removal of the reactive end group with Et3B/air proceeded smoothly without degradation of the terminal double bonds of 3 (see Supporting Information). The thiol‐ene click reaction was conducted with an excess of thiol (5.0 equiv) and 2,2‐dimethoxy‐2‐phenylacetophenone (0.2 equiv) as the photoinitiator in THF under UV light (365 nm) irradiation for 3 h. The degree of functionalization was >99 % for all tested thiols and was confirmed by NMR spectroscopy. A low molecular weight sample was also characterized by mass spectrometry and no partially unreacted polymer side chains were detected. Subsequent amidation of the product copolymer was conducted in analogy to the modification of poly(1‐alt‐NBVE) to afford copolymers of type 12, which were further functionalized with the established conditions for either transesterification to 13 or acylation to 14. For the transesterification a larger excess of alcohol (30 equiv) was needed for full conversion. A variety of different thiols, amines, alcohols and acyl halides were successfully utilized for the sequential chemoselective threefold modification of poly(1‐alt‐HVE) 3 (Table 3). The employed thiol component could bear fluorinated alkyl groups (entry 1 and 6), α‐branched alkyl groups (entries 2–4 and 7), an ester moiety (entry 5), a benzylic position (entry 8) or free hydroxyl groups (entry 9). For the second step, benzylic (entry 1) as well as propargylic amines (entry 2) engaged in the reaction and alkoxy groups (entry 3, 4 and 6), cycloalkyl moieties (entry 5), imidazoyl (entry 7) and hydroxyl groups (entry 8 and 9) were also introduced by the amidation process. Alternating ABC‐type terpolymers 12 a–12 c carry functional groups, that would also be addressed in the third functionalization step, so the modification was stopped after amidation. Some of the other polymers were subjected to the final transesterification with aliphatic alcohols (13 a,c). Aminoalcohols could be used (13 b) and ethylene glycol also reacted well (13 d) without crosslinking of the polymer chains. Considering acylation as the final step, we were able to introduce a highly fluorinated side chain (14 a) and a thiophene moiety (14 b).
Table 3.
Used substrates for the orthogonal threefold functionalization of 11.
|
Entry |
Thiol |
Amine |
Alcohol or Acyl halide |
M n,exp [a] [g mol−1] |
PDI[a] |
Degree of Functionalization [%][b] |
Yield [%][c] |
Product |
|---|---|---|---|---|---|---|---|---|
|
1 |
|
|
|
37 100 |
1.4 |
>98 |
73 |
13 a |
|
2 |
|
|
|
[d] |
[d] |
98 |
88 |
13 b |
|
3 |
|
|
|
26 100 |
1.6 |
>98 |
84 |
13 c |
|
4 |
|
|
|
23 400 |
1.5 |
97 |
81 |
13 d |
|
5 |
|
|
|
[d] |
[d] |
96 |
94 |
14 a |
|
6 |
|
|
|
36 200 |
1.4 |
>98 |
77 |
14 b |
|
7 |
|
|
|
[d] |
[d] |
>98 |
>99 |
12 a |
|
8 |
|
|
|
33 700 |
1.5 |
>98 |
>99 |
12 b |
|
9 |
|
|
|
26 700 |
1.5 |
>98 |
93 |
12 c |
























[a] Approximated by GPC at 25 °C using THF as the eluent against PMMA standards. [b] Determined by 1H NMR or 19F NMR spectroscopy. [c] Isolated yield after two or three functionalization steps. [d] No GPC data was acquired due to poor solubility in THF.
Conclusion
In summary, we have developed a new divinyl monomer 1 for the alternating copolymerization with vinyl ethers by RAFT polymerization. The divinyl monomer 1 was carefully designed so that electronic effects steer the control of the alternating radical cyclopolymerization. Moreover, electronics build into this building block are also of key importance for the subsequent orthogonal chemical functionalization. Hence, the obtained cyclopolymers were chemoselectively modified in up to three distinct and orthogonal functionalization reactions yielding alternating ABC‐type terpolymers. Copolymers were analysed by 1H, 19F NMR, GPC and MS. Subsequent sequential functionalization of poly(1‐alt‐NBVE) 6 by amidation and transesterification or acylation was achieved with high efficiency (>95 %). With HVE 2 as the vinyl ether monomer component, poly(1‐alt‐HVE) (3) was obtained that contains a terminal alkene moiety as an additional addressable chemical functionality. Various thiols, amines and alcohols or acyl halides were used to synthesize a small library of different types of functionalized alternating ABC‐type terpolymers 4.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft DFG (SFB 1459) for supporting our work. Open Access funding enabled and organized by Projekt DEAL.
P. Gerdt, A. Studer, Angew. Chem. Int. Ed. 2022, 61, e202206964; Angew. Chem. 2022, 134, e202206964.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1.
- 1a. Lutz J.-F., Ouchi M., Liu D. R., Sawamoto M., Science 2013, 341, 1238149; [DOI] [PubMed] [Google Scholar]
- 1b. Sequence-controlled polymers (Ed.: Lutz J.-F.), Wiley-VCH, Weinheim, 2018; [Google Scholar]
- 1c. Meier M. A. R., Barner-Kowollik C., Adv. Mater. 2019, 31, 1806027; [DOI] [PubMed] [Google Scholar]
- 1d. Solleder S. C., Schneider R. V., Wetzel K. S., Boukis A. C., Meier M. A. R., Macromol. Rapid Commun. 2017, 38, 1600711. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Audran G., Bagryanskaya E. G., Marque S. R. A., Postnikov P., Polymer 2020, 12, 1481; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Pan X., Fantin M., Yuan F., Matyjaszewski K., Chem. Soc. Rev. 2018, 47, 5457; [DOI] [PubMed] [Google Scholar]
- 2c. Grubbs R. B., Grubbs R. H., Macromolecules 2017, 50, 6979; [Google Scholar]
- 2d. Discekici E. H., Anastasaki A., Read de Alaniz J., Hawker C. J., Macromolecules 2018, 51, 7421; [Google Scholar]
- 2e. Perrier S., Macromolecules 2017, 50, 7433; [Google Scholar]
- 2f. Mishima E., Yamago S., J. Polym. Sci. Part A 2012, 50, 2254. [Google Scholar]
- 3.
- 3a. Tesch M., Kudruk S., Letzel M., Studer A., Chemistry 2017, 23, 5915; [DOI] [PubMed] [Google Scholar]
- 3b. Niu S., Ding M., Chen M., Feng T., Zhang L., Wei L., Cheng Z., Zhu X., J. Polym. Sci. Part A 2013, 51, 5263; [Google Scholar]
- 3c. Mishima E., Yamago S., Macromol. Rapid Commun. 2011, 32, 893; [DOI] [PubMed] [Google Scholar]
- 3d. Kırcı B., Lutz J.-F., Matyjaszewski K., Macromolecules 2002, 35, 2448; [Google Scholar]
- 3e. Kametani Y., Ouchi M., Polym. Chem. 2020, 11, 6505; [Google Scholar]
- 3f. Chen G.-Q., Wu Z.-Q., Wu J.-R., Li Z.-C., Li F.-M., Macromolecules 2000, 33, 232; [Google Scholar]
- 3g. Cunningham R. D., Kopf A. H., Elenbaas B. O. W., Staal B. B. P., Pfukwa R., Killian J. A., Klumperman B., Biomacromolecules 2020, 21, 3287; [DOI] [PubMed] [Google Scholar]
- 3h. Banerjee S., Domenichelli I., Ameduri B., ACS Macro Lett. 2016, 5, 1232; [DOI] [PubMed] [Google Scholar]
- 3i. Benoit D., Hawker C. J., Huang E. E., Lin Z., Russell T. P., Macromolecules 2000, 33, 1505. [Google Scholar]
- 4.
- 4a. Huang J., Turner S. R., Polymer 2017, 116, 572; [Google Scholar]
- 4b. Rzaev Z., Prog. Polym. Sci. 2000, 25, 163. [Google Scholar]
- 5. Oh D., Furuya Y., Ouchi M., Macromolecules 2019, 52, 8577. [Google Scholar]
- 6.
- 6a. Kubota H., Ouchi M., Macromolecules 2022, 55, 4025; [Google Scholar]
- 6b. Kametani Y., Nakano M., Yamamoto T., Ouchi M., Sawamoto M., ACS Macro Lett. 2017, 6, 754; [DOI] [PubMed] [Google Scholar]
- 6c. Ouchi M., Polym. J. 2021, 53, 239; [Google Scholar]
- 6d. Ouchi M., Nakano M., Nakanishi T., Sawamoto M., Angew. Chem. Int. Ed. 2016, 55, 14584; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14804; [Google Scholar]
- 6e. Ouchi M., Sawamoto M., Polym. J. 2018, 50, 83; [Google Scholar]
- 6f. Pasini D., Nitti A., Eur. Polym. J. 2020, 122, 109378; [Google Scholar]
- 6g. Pasini D., Takeuchi D., Chem. Rev. 2018, 118, 8983. [DOI] [PubMed] [Google Scholar]
- 7. Nishimori K., Ouchi M., Chem. Commun. 2020, 56, 3473. [DOI] [PubMed] [Google Scholar]
- 8. Tesch M., Hepperle J. A. M., Klaasen H., Letzel M., Studer A., Angew. Chem. Int. Ed. 2015, 54, 5054; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5142. [Google Scholar]
- 9.
- 9a. Zhang Y., Chu C.-W., Ma W., Takahara A., ACS Omega 2020, 5, 7488; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Lowe A. B., Polym. Chem. 2014, 5, 4820; [Google Scholar]
- 9c. Kharash M. S., Read J., Mayo F. R., J. Soc. Chem. Ind. 1938, 57, 752; [Google Scholar]
- 9d. Flores J. D., Treat N. J., York A. W., McCormick C. L., Polym. Chem. 2011, 2, 1976; [Google Scholar]
- 9e. Chen G., Amajjahe S., Stenzel M. H., Chem. Commun. 2009, 1198; [DOI] [PubMed] [Google Scholar]
- 9f. Campos L. M., Killops K. L., Sakai R., Paulusse J. M. J., Damiron D., Drockenmuller E., Messmore B. W., Hawker C. J., Macromolecules 2008, 41, 7063. [Google Scholar]
- 10.
- 10a. Gevrek T. N., Degirmenci A., Sanyal R., Klok H.-A., Sanyal A., ACS Appl. Polym. Mater. 2021, 3, 2507; [Google Scholar]
- 10b. Kakuchi R., Matsubara K., Fukasawa K., Amii H., Macromolecules 2021, 54, 6204; [Google Scholar]
- 10c. Lai H., Ouchi M., ACS Macro Lett. 2021, 10, 1223; [DOI] [PubMed] [Google Scholar]
- 10d. Ulrich S., Sadeghpour A., Rossi R. M., Bruns N., Boesel L. F., Macromolecules 2018, 51, 5267. [Google Scholar]
- 11.
- 11a. Easterling C. P., Kubo T., Orr Z. M., Fanucci G. E., Sumerlin B. S., Chem. Sci. 2017, 8, 7705; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Fleischmann C., Anastasaki A., Gutekunst W. R., McGrath A. J., Hustad P. D., Clark P. G., Laitar D. S., Hawker C. J., J. Polym. Sci. Part A 2017, 55, 1566; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Grasa G. A., Güveli T., Singh R., Nolan S. P., J. Org. Chem. 2003, 68, 2812; [DOI] [PubMed] [Google Scholar]
- 11d. Ito D., Ogura Y., Sawamoto M., Terashima T., ACS Macro Lett. 2018, 7, 997; [DOI] [PubMed] [Google Scholar]
- 11e. Samanta R. C., de Sarkar S., Fröhlich R., Grimme S., Studer A., Chem. Sci. 2013, 4, 2177; [Google Scholar]
- 11f. Samanta S. R., Cai R., Percec V., Polym. Chem. 2015, 6, 3259. [Google Scholar]
- 12. Hepperle J., Samanta R., Studer A., Synlett 2013, 24, 1233. [Google Scholar]
- 13.
- 13a. Brantley E. L., Jennings G. K., Macromolecules 2004, 37, 1476; [Google Scholar]
- 13b. Cheng G., Böker A., Zhang M., Krausch G., Müller A. H. E., Macromolecules 2001, 34, 6883; [Google Scholar]
- 13c. Huang W., Kim J.-B., Bruening M. L., Baker G. L., Macromolecules 2002, 35, 1175; [Google Scholar]
- 13d. Laschewsky A., Rekaï E. D., Wischerhoff E., Macromol. Chem. Phys. 2001, 202, 276; [Google Scholar]
- 13e. Wang B., Liu T., Chen H., Yin B., Zhang Z., Russell T. P., Shi S., Angew. Chem. Int. Ed. 2021, 60, 19626; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 19778. [Google Scholar]
- 14.
- 14a. Das A., Theato P., Chem. Rev. 2016, 116, 1434; [DOI] [PubMed] [Google Scholar]
- 14b. Blasco E., Sims M. B., Goldmann A. S., Sumerlin B. S., Barner-Kowollik C., Macromolecules 2017, 50, 5215. [Google Scholar]
- 15. Sugihara S., Kawamoto Y., Maeda Y., Macromolecules 2016, 49, 1563. [Google Scholar]
- 16.
- 16a. Kamachi M., Tanaka K., Kuwae Y., J. Polym. Sci. Part A 1986, 24, 925; [Google Scholar]
- 16b. Matsumoto A., Nakana T., Oiwa M., Makromol. Chem. Rapid Commun. 1983, 4, 277; [Google Scholar]
- 16c. Sugihara S., Yoshida A., Kono T.-A., Takayama T., Maeda Y., J. Am. Chem. Soc. 2019, 141, 13954. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Mayo F. R., Lewis F. M., J. Am. Chem. Soc. 1944, 66, 1594; [Google Scholar]
- 17b. Buback M., Feldermann A., Barner-Kowollik C., Lacík I., Macromolecules 2001, 34, 5439. [Google Scholar]
- 18. Spengler J., Böttcher C., Albericio F., Burger K., Chem. Rev. 2006, 106, 4728. [DOI] [PubMed] [Google Scholar]
- 19. Moad G., Rizzardo E., Thang S. H., Polym. Int. 2011, 60, 9. [Google Scholar]
- 20. Alagi P., Hadjichristidis N., Gnanou Y., Feng X., ACS Macro Lett. 2019, 8, 664. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.