

Abstract
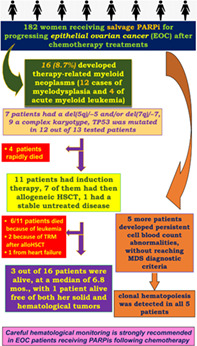
Inhibitors of poly(ADP‐ribose) polymerase (PARPi) are increasingly employed as salvage therapy in epithelial ovarian cancer (EOC), but cytotoxic drug exposure along with PARP inhibition may favor development of hematological disorders. In our study, of 182 women with EOC treated with PARPi, 16 (8.7%) developed therapy‐related myeloid neoplasms (t‐MNs), with 12 cases of myelodysplasia and 4 of acute myeloid leukemia. All experienced persistent cytopenia after PARPi discontinuation. Seven patients had del(5q)/−5 and/or del(7q)/−7, nine had a complex karyotype and TP53 mutations, recently reported as risk factor for t‐MNs in EOC post‐PARPi, were found in 12 out of 13 tested patients. Four patients had a rapid and fatal outcome, one had stable disease, eleven underwent induction therapy, followed by allogeneic hematopoietic cell transplantation in seven. Three of these 11 patients experienced refractory disease, and 8 had complete remission. During a 6.8 months (range 2.3‐49) median observation time, 3 out of 16 patients were alive, with one surviving patient free of both solid and hematological tumors. Ten patients died because of leukemia, two because of transplant‐related events, one from heart failure. Five more patients experienced persistent cell blood count abnormalities following PARPi discontinuation, without reaching MDS diagnostic criteria. A customized Myelo‐panel showed clonal hematopoiesis in all five patients. These findings confirm the actual risk of t‐MNs in EOC patients after chemotherapy and prolonged PARPi therapy. The management of these patients is complex and outcomes are extremely poor. Careful diagnostic procedures are strongly recommended whenever unusual cytopenias develop in patients receiving PARPi therapy.
Keywords: epithelial ovarian cancer, mutations, next generation sequencing, PARP‐inhibitors, therapy‐related myeloid neoplasms
What's new?
PARP inhibitors are increasingly used as salvage therapy in epithelial ovarian cancer. However, cytotoxic drug exposure along with PARP inhibition may favor the development of hematological disorders. Our study of 182 epithelial ovarian cancer patients confirms that the risk of secondary myeloid neoplasms after chemotherapy and prolonged PARPi cannot be minimized, with an incidence reaching 8.7%. Moreover, the authors show that management of these patients is complex, with poor outcomes despite intensive treatment regimens, likely due to the genetic complexity of the myeloid neoplasms. They recommend careful diagnostic procedures whenever unusual cytopenias develop in patients receiving PARPi.
Abbreviations
- AL
acute leukemia
- allo‐HSCT
allogeneic hematopoietic stem cell transplantation
- AML
acute myeloblastic leukemia
- BM
bone marrow
- CBCs
cell blood counts
- CCAUS
clonal cytogenetic abnormalities of undetermined significance
- CCUS
clonal cytopenia of undetermined significance
- CHIP
clonal hematopoiesis of indeterminate (clinical) potential
- CR
complete response
- cyCR
cytogenetic CR
- EB
excess of blasts
- EOC
epithelial ovarian cancer
- FISH
fluorescent in situ hybridization
- GVHD
graft‐vs‐host disease
- HGSOC
high‐grade serous ovarian carcinoma
- HR
homologous recombination
- ICE
idarubicin‐etoposide‐cytarabine
- ICUS
idiopathic cytopenia of undetermined significance
- IDUS
idiopathic dysplasia of unknown significance
- IEO
European Institute of Oncology
- IPSS‐R
revised international prognostic scoring system
- mCR
morphological CR
- MDS
myelodysplasia
- MLD
multilineage dysplasia
- NGS
next generations sequencing
- PARP
Poly‐ADP‐Ribose polymerase
- PARPi
PARP‐inhibitors
- PB
peripheral blood
- PDTL
posttransplant lymphoproliferative disorder
- SNP
single nucleotide polymorphisms
- t‐MNs
therapy‐related myeloid neoplasms
- VUS
variants of uncertain significance
- WHO
World Health Organization
1. INTRODUCTION
Recent insights into the molecular and genetic characteristics of high‐grade serous ovarian carcinoma (HGSOC) have allowed for the development of novel targeted therapeutic strategies. In particular, around 50% of all HGSOC are characterized by a deficiency in homologous recombination (HR) DNA repair. 1 This defect is generally due to BRCA1/2 mutations. In some cases, genomic alterations and/or epigenetic silencing of other genes involved in the pathway, including ATR, ATM, RAD51/54, CHK1/2, NBS1, PTEN and PALB2, 1 , 2 , 3 , 4 may confer a so‐called “BRCAness” profile, in which affected cells cannot correctly repair DNA double‐strand breaks, similarly to a BRCA deficiency. The family of poly(ADP‐ribose) polymerases (PARPs) is involved in single‐strand break repair, and their inhibition under conditions of HR repair deficiency can be leveraged to force double‐strand break lethality in cancer cells. 1 , 5 , 6 , 7 The molecular characteristics of HGSOC have led to the use of PARP inhibitors (PARPi) for this purpose, which have shown efficacy against epithelial ovarian cancer (EOCs) in several clinical trials with significant survival improvements when used as frontline therapy or for recurrence. 8
PARP inhibitors effects in inhibiting cellular DNA repair, however, extend to normal cells, particularly those undergoing rapid cell division, such as hematopoietic cells, 9 and hematologic toxicities have been reported under PARPi therapy. One meta‐analysis showed incidences of 32.9% for grade 3 or 4 neutropenia, 15.9% for thrombocytopenia and 9.1% for anemia in patients receiving PARPi. 10 These side effects frequently arise during the first months of therapy and are usually managed with temporary treatment interruptions or dose delay or reduction.
Preliminary data on the use of PARPi in recurrent disease indicate an incidence of hematologic disorders of 1% to 2%. 11 , 12 A recent meta‐analysis, based on 18 randomized clinical trials, showed that PARPi significantly increased the risk of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) compared to placebo treatment. 8 Of note, this meta‐analysis included trials carried out both in frontline and in recurrent disease. Subgroup analyses of the risk for therapy‐related (tr)‐MDS and AML with PARPi yielded odds ratios of 1.93 (0.68‐5.49) with front‐line maintenance and 4.79 (1.11‐20.63) with second line and beyond compared to controls. 8 These data suggest that hematologic disorders related to therapy may be influenced by both previous platinum‐based treatments and salvage PARPi. A double‐blind, randomized, placebo‐controlled phase 3 trial (SOLO2/ENGOT‐Ov21) confirmed the efficacy of the PARPi olaparib in platinum‐sensitive disease with BRCA1/2 mutations. The final analysis from this trial has documented that MDS or AML occurred in 16 (8%) out of 195 patients in the olaparib group. 12 Similarly, 9 of 130 (6.9%) consecutive patients treated with PARPi after platinum‐based chemotherapy developed tr‐myeloid neoplasms (t‐MNs) at a median 22.8 months of PARPi exposure, according to a preliminary analysis from the European Institute of Oncology (IEO) of Milan. 9 That study was extended and here we report the actual incidence of patients developing hematological disorders after salvage PARPi from a large series of EOC cases. In addition, we describe the peculiar cytogenetic and molecular characteristics and clinical outcomes of the patients who developed t‐MN. Outcomes were quite poor in spite of intensive treatments that often include allogeneic hematopoietic stem cell transplantation (allo‐HSCT).
2. MATERIALS AND METHODS
2.1. Patients
The present study included 182 consecutive patients at IEO treated with PARPi for EOC between 2010 and 2021. Most patients (n = 103, 57%) received PARPi through open clinical trials, 24 (13%) received PARPi as compassionate use and 55 (30%) after regular approval of the drug. One patient received olaparib within the SOLO2 trial and rucaparib as compassionate use. Among patients enrolled in clinical trials, 63 (61.5%) received PARPi as monotherapy, whereas 40 (38.5%) received the cediranib‐olaparib combination, as part of a randomized phase II study for patients with recurrent platinum‐resistant ovarian cancer. 13 Four patients from double‐blind clinical trials were included in the present analysis after unblinded procedures revealed that they had received the PARPi treatment. The main patient characteristics are reported in Table 1.
TABLE 1.
Main clinical features of 182 patients with epithelial ovarian cancer (EOC) with and without therapy‐related myeloid neoplasms (t‐MN) following poly(ADP‐ribose) polymerase inhibitors (PARPi) and previous poly‐chemotherapy
| Characteristics | Patients without t‐MN (n = 166) | Patients with t‐MN (n = 16) |
|---|---|---|
| Median age at EOC diagnosis | 54 (range 33‐78) | 52 (range 39‐69) |
| Histology, n (%) | ||
| Serous carcinoma | 144 (87%) | 15 (94%) |
| Endometrioid carcinoma | 7 (4%) | 1 (6%) |
| Clear cell carcinoma | 5 (3%) | 0 |
| Mixed carcinoma | 5 (3%) | 0 |
| NOS carcinoma | 5 (3%) | 0 |
| Grade, n (%) | ||
| High | 156 (94%) | 15 (94%) |
| Intermediate | 4 (2%) | 1 (6%) |
| Unknown | 6 (4%) | 0 |
| FIGO stage, n (%) | ||
| I | 7 (4.5%) | 0 |
| II | 7 (4.5%) | 2 (13%) |
| III | 119 (71%) | 13 (81%) |
| IV | 33 (20%) | 1 (6%) |
| Germline BRCA1/2 status, n (%) | ||
| Mutated | 79 (47.5%) | 9 (56%) |
| Wild type/VUS | 68 (41%) | 5 (31%) |
| Unknown a | 19 (11.5%) | 2 (13%) |
| Prior chemotherapy lines before PARPi, n (%) | ||
| 1‐2 | 64 (38%) | 8 (50%) |
| 3‐4 | 64 (38%) | 5 (31%) |
| ≥5 | 38 (24%) | 3 (19%) |
| Type of PARPi, n (%) | ||
| Olaparib b | 100 (60%) | 9 (56%) |
| Niraparib | 63 (38%) | 6 (38%) |
| Rucaparib b | 4 (2%) | 1 (6%) |
| PARPi therapy, n (%) | ||
| Treatment | 56 (34%) | 1 (6%) |
| Maintenance | 111 (66%) | 15 (94%) |
| Duration of PARPi therapy, n (%) | ||
| Median (months) | 7.4 (range 0.8‐110.8) | 18.5 (range 4.1‐42.2) |
| ≤6 months | 74 (45%) | 3 (19%) |
| 7‐17 months | 55 (33%) | 3 (19%) |
| ≥18 months | 37 (22%) | 10 (62%) |
| Presents status, n (%) | ||
| Dead | 54 (32%) | 12 (75%) |
| Alive | 94 (57%) | 4 (25%) |
| Lost at follow‐up | 18 (11%) | 0 |
Abbreviations: BRCA, breast cancer gene; FIGO, International Federation of Gynecology and Obstetrics; NOS, not otherwise specified; VUS, variant of uncertain significance.
Patients were enrolled in a 2010 clinical trial that did not require assessment of the BRCA status.
A single patient received both olaparib and rucaparib as maintenance.
2.2. PARP inhibitor treatments
Three different types of PARPi were employed and delivered orally: olaparib at 800 mg/day (capsule formulation) or 600 mg/day (tablet formulation); niraparib at 300 mg/day or rucaparib at 1200 mg/day. Most patients had been heavily pretreated, and 110 (60%) of them had received three or more lines of chemotherapy. In most cases, PARPi was administered as maintenance treatment after partial response or complete response (CR) was achieved after platinum‐based chemotherapy; in the remaining 57 (31%) patients, PARPi was given as therapy for recurrent disease. PARPi was interrupted in case of disease progression or unacceptable toxicity. Patients receiving PARPi for ≥18 months were identified as long responders (Table 1). Treatment toxicities were defined and graded according to the Common Terminology Criteria for Adverse Events. 14
2.3. Hematological and molecular investigations
Peripheral blood (PB) samples were collected prior to administration of a PARPi, then at monthly intervals and at the beginning of every new cycle. In addition to complete cell blood counts (CBCs), patients displaying persistent and/or marked hematological abnormalities underwent bone marrow (BM) evaluation including morphological, immunophenotypic, cytogenetic and fluorescent in situ hybridization (FISH) analysis. Identification of mutations or alterations in gene expression for routine clinical analysis was performed using the diagnostic Oncomine Myeloid Research Assay, a commercially available NGS assay investigating mutations and fusion transcripts in the 69 genes related with most frequency to myeloid malignancies (Table S1). Analysis was performed with IonReporter software applying the last release of Myeloid workflow (ThermoFisher).
To identify mutations for research purposes, we used a custom gene panel, the Myelo‐panel. The purpose of this analysis was to identify germline variants from buccal DNA and somatic variants from PB that are associated with a predisposition to developing myeloid neoplasms, and to stratify patients for possible targeted therapies. DNA was extracted from buccal swabs and from PB using the QIAamp DNA mini kit (Qiagen) following the manufacturer's instructions. Our custom Myelo‐panel analyzes 255 cancer‐predisposing genes (Table S2), including: susceptibility genes, which are those most frequently associated with the risk of development of hematological tumors; AML drivers, which play a key role in leukemogenesis; actionable genes, defined as genes that have a concrete clinical manageability for which either a US Food and Drug Administration‐approved or clinical trial drug is available; and pharmacogenomics single nucleotide polymorphisms (SNPs), allelic variants associated with susceptibility to certain drugs. The sources for our 164 actionable genes and 127 pharmacogenomics SNPs were the Alleanza Contro il Cancro (ACC) Database, based on DGiDb (15); the dGene (annotation tool for cancer genome sequencing data 16 ; PharmGKB (The Pharmacogenomics Knowledgebase, www.pharmgkb.org/); and literature mining. 17 Targeted capture was carried out according to manufacturer's protocols using the AmpliSeq kit and Ion Torrent sequencing. Variant analysis was performed with IonReporter software (ThermoFisher Scientific). Identified variants were annotated in terms of pathogenicity, using different computational tools, including ClinVar, Varsome and RENOVO (see Supporting Information Materials and Methods in Appendix S1 for details). The sequencing coverage and quality statistics for each sample are summarized in Tables S3 and S4.
The World Health Organization (WHO) 2016 Classification was used to define myelodysplastic syndrome (MDS) and acute leukemias (AL) subtypes. 18 The Revised International Prognostic Scoring System (IPSS‐R) 19 was employed to define the MDS prognostic score, and the European Leukemia Network (ELN) was used for AML. 20 Pre‐MDS conditions included idiopathic cytopenia of undetermined significance (ICUS), idiopathic dysplasia of unknown significance (IDUS), clonal hematopoiesis of indeterminate (clinical) potential (CHIP), clonal cytopenia of unknown significance (CCUS) and clonal cytogenetic abnormalities of undetermined significance (CCAUS). These conditions can be divided into: (a) cytopenic states (ICUS and CCUS) or noncytopenic states (IDUS, CHIP, CCAUS) and (b) states with an unknown or negative mutation status (ICUS and IDUS) or cases with known (documented and relevant) somatic mutations/cytogenetic abnormalities (CHIP, CCUS and CCAUS). All of these conditions can persist without clinical manifestation or progression, but can develop into overt MDS. 21 , 22 Hematological procedures were performed according to internal institutional diagnostic and clinical guidelines.
2.4. Statistical methods
Only descriptive statistics were used to report data related to patient demographics, clinico‐pathological characteristics and treatments in Table 1.
3. RESULTS
3.1. Therapy‐related myeloid neoplasms: Diagnosis and clinical presentation
Overall, 16 of 182 (8.7%) patients with EOC treated with PARPi as salvage after chemotherapy developed t‐MNs. As shown in Table 2, the occurrence of t‐MDS was preceded by one or more hematopoietic‐line cytopenia of varying duration, from a few days to 10 months after PARPi discontinuation. In particular, five patients developed a t‐MDS with multilineage dysplasia (MLD), and seven had a MDS with excess of blasts (EB) (four EB1 with BM blasts 5%‐9%, three EB2 with BM blasts 10%‐19%). The four cases of t‐AML evolved from cytopenias within 60 days after PARPi withdrawal.
TABLE 2.
Main clinical and molecular features of 16 patients developing therapy‐related myeloid neoplasms and their treatments
| Type of t‐MNs | EOC status at t‐MN | Type and cycle of PARPi | Hematologic Toxicity | Duration (months) | Karyotype | Molecular biology a | Treatment administration | Type of CT administered | Allo‐HSCT | CR duration (months) | Relapse | Salvage CT | Present status (dead /alive) | Cause of death | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | EB‐2 | NED | Olaparib (41 cycles) | N G2‐3 | 2 | 46,XX, del(5)(q12q33), +13, −16, del(17)(p12pter)[cp12]/46,XX[6] |
Mut. TP53 p.(Cys242Tyr) VAF 52% |
Y | ICE → RD FLAG‐Ida → CR | Y | 5.4 | Y | Decitabine + Venetoclax → RD → phase I trial | Dead | t‐MN PD |
| 2 | AML | NED | Niraparib (10 cycles) | T G1 and N G2 | 1 | 79‐98,XXXX, +3‐4mar[cp11] |
Mut. TP53 p.(His193Leu) VAF 91% |
Y | Vyxeos | N | na | na | na | Dead | t‐MN PD |
| 3 | EB‐1 | NED | Niraparib (25 cycles) | A G2, T G2 and N G3 | 1 | 46,XX, del(5)(q22q35)[3]/45,XX, del(5)(q22q35), −7, +8, +mar[2]/45,XX, del(5)(q22q35), −7[1]/45,XX, del(5)(q22q35), −7, +8, −17[1]/46,XX[3] |
Mut. TP53 p.(Arg175His) VAF 20.05% |
Y | Vyxeos | Y | 4.7 | Y | nd | Dead | t‐MN PD |
| 4 | EB‐1 | CR | Olaparib (24 cycles) | A G2, T G2 and N G2 | 1 | 46,XX, del(5)(q22q35), −7, +8, +8, −17[10]/46,XX, del(5)(q22q35), −17[3]/46,XX[2] |
mut. TP53 p.(Arg249Thr) VAF 53.8% |
Y | Vyxeos | N | 2.8 | N | na | Dead | Heart failure |
| 5 | AML M6 | SD | Olaparib (29 cycles) | A G2 and T G3 | 1 | 46,XX[2] |
Mut. TP53 p.(His179Arg) VAF 9.6% |
Y | ICE → RD FLAG‐Ida → RD decitabine+ven | N | na | NA | Decitabine + ven → RD | Dead | t‐MN PD |
| 6 | EB‐2 | CR | Niraparib (19 cycles) | A G2, T G1, N G1 | 2 | 43,XX, t(1;12)(p21;p13), −7, −13, −16, −18, +mar[12]/46,XX[1] |
Mut. TP53 p.(Val216Met) VAF 40.55% |
Y | ICE ARA‐C 6 g/mq | Y | 4.7 | Y | Ven → RD | Dead | t‐MN PD |
| 7 | AML | SD | Rucaparib (26 cycles) | A G2, T G2, N G4 | 2 | 46,XX, del(13)(q12q14)[2]/46,XX[20] | Mut. TP53 p.(Arg273Cys) VAF 8% | Y | ICE ARA‐C 6 g/mq | Y | 49.3 | N | na | Alive | na |
| 8 | EB‐1 | NED | Olaparib (21 cycles) | A G1 T G2 | 10 | 48‐52,XX, +1, −5, −6, +8, +11, der(17)t(17;2)(p11.2;2), +19 + 22, +2 mar[10] |
Mut. TP53 p.(Pro151Arg) VAF 53.3% |
Y | ICE | Y | 15.6 | N | na | Dead | TRM (PTLD) |
| 9 | EB‐2 | SD | Niraparib (21 cycles) | Pancytopenia and blasts | Immediate hematologic consult | 46,XX,t(3;21)(q26;q22)[3]/46,XX[17] |
mut. SH2B3 tras fus RUNX1(4)‐MECOM(2 |
Y | Vyxeos | Y | 14.3 | N | na | dead | TRM (GVHD) |
| 10 | EB‐1 | CR | Olaparib (33 cycles) | T G1 and A G2 | 10 | 44XX −7, del5, t(5;18)(q11;2); del5(q11),‐18[11/46xx] | nd | Y | 5‐aza | Y | 48.7 | N | na | Alive | na |
| 11 | AML | SD | Olaparib (18 cycles) | A G2, T G2 and N G1 | 1 | 43‐45,XX, −3, del(5)(q22q35),der(7) t(3;7)(2q21;2p21), −17, add(21)(q22), −22, +2mar[3]/46,XX[12] |
Mut.TP53 p.(Met246Val) VAF 6.65% mut, TP53 p.(Ser241Tyr) VAF 33.4% |
Y | 5‐aza + ven → RD | N | na | na | na | Dead | t‐MN PD |
| 12 | MSD‐MLD | CR | Rucaparib (27 cycles) | T G1 | 2 | 46,XX, del(5)(q12q33)[11]/46,XX[1] |
Mut.TET2mut. TP53 p.(Ser99Glufs*48) VAF 24.69% |
N | — | N | na | na | na | Alive | na |
| 13 | MSD‐MLD | PD | Niraparib (11 cycles) Olaparib(3 cycles) | A G3, T G2, N G2 | 9 | 45,XX, del(5)(q213;q234), −7[4]/46,XX[16] |
Mut. U2AF1 mut. TP53 p.(Leu194Hisfs*14 VAF 61.47% |
N | — | N | na | na | na | Dead | Sudden death |
| 14 | MSD‐MLD | SD | Niraparib (11 cycles) | T G4, A G2, N G1 | 5 | 45‐43,XX, del(5)(q22;q35), −7, +15[13]/39‐44,XX, del(5)(q22;q35), −7, +mar[3]/46,XX[4] |
Mut.TP53 p.(Gly244Ser) VAF 52.20% |
N | — | N | na | na | na | Dead | EOC PD |
| 15 | MSD‐MLD | PR | Olaparib (5 cycles) | N G2, T G1 | 2 | 45,XX, t(3;3)(q21;q26), −7[13]/46,XX[2] | nd | N | — | N | na | na | na | Dead | EOC PD |
| 16 | MSD‐MLD | SD | Olaparib (24 cycles) | T G4 | 2 | 46,XX[20]; in FISH del 5q | nd | N | — | N | na | na | na | Dead | Cerebral bleed |
Abbreviations: 5‐aza, 5‐azacytidine 75 mg/smq days 1‐7 q28; A, anemia; allo‐HSCT, allogeneic hematopoietic stem cell transplantation; AML M6, acute erythroblastic leukemia; AML, acute myeloid leukemia; CR, complete remission; CT, chemotherapy; PTDL, posttransplant lymphoproliferative disease; EB1, myelodysplastic syndrome with excess blasts type 1; EB2, myelodysplastic syndrome with excess blasts type 2; EOC, epithelial ovarian cancer; FLAI, Idarubicin 12 mg/smq day 1/day 3/day 5 + Fludarabine 30 mg/smq days 1‐5 + Cytarabine 2000 mg/smq days 1‐5 + GCSF from day 0; ICE, idarubicin 12 mg/smq days 1‐d3 + Cytarabine 100 mg/smq days 1‐7 + Etoposide 100 mg/smq days 1‐5; MSD‐MLD, myelodysplastic syndrome with multilineage dysplasia; Mut., mutation; N, neutropenia; N, no; na, not applicable; nd, not done, no proper material was available for analysis; NED, no evidence of disease; PARPi, poly(ADP‐ribose) polymerase inhibitors; PD, progressing disease; PR, partial remission; RD, refractory disease; SD, stable disease; T, thrombocytopenia; t‐MNs, therapy‐related myeloid neoplasms; VAF, variant allele frequency; ven, venetoclax; Vyxeos Liposomal, daunorubicin 44 mg + cytarabine 100 mg (ratio 5:1); Y, yes.
Next generation sequencing analysis of PB DNA using the Oncomine Myeloid Research Assay and the Ion Torrent S5 technology (ThermoFisher).
All 16 patients developing t‐MNs were characterized cytogenetically and 13 of them were sequenced with the Oncomine Myeloid Research Assay. Table 2 details our cytogenetic and molecular findings. Seven patients had del(5q)/−5 and/or del(7q)/−7, nine had a complex karyotype. An example of complex karyotype with both del(5q) and loss of chromosome 17 is given in Figure 1. A TP53 mutated gene was found in 12 out of 13 patients tested with the Oncomine Myeloid Research Assay.
FIGURE 1.
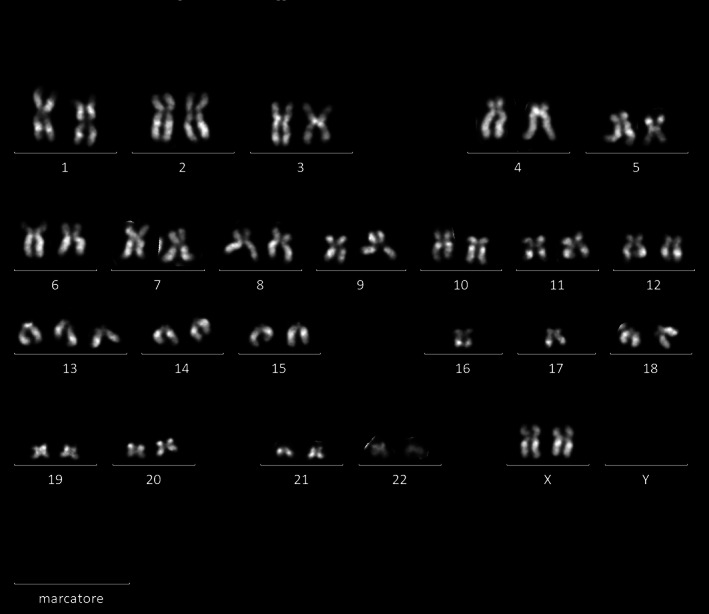
Karyotype (QFQ‐banding) of patient ID #1 (Table 2). Karyotype equals 46, XX, del(5)(q12q33), +13, −16, −17; an example of deletion of chromosome 5 and loss of chromosome 17
3.2. Therapy‐related myeloid neoplasms: Management
PARPi was discontinued in all patients developing t‐MNs. Four patients were unable to receive any specific treatment for their t‐MNs because of a rapid and fatal outcome. In detail, one patient had a sudden cardiac death while at home, one patient died from a massive brain hemorrhage and the two remaining patients died from rapid EOC progression. A fifth patient, with t‐MDS‐MLD and low IPSS‐R score, is currently being monitored through a watch‐and‐wait approach, with excellent control of both the EOC and the hematological malignancy thus far.
Eleven patients underwent induction therapy, seven of whom then underwent allogeneic hematopoietic cell transplantation (allo‐HSCT) (Tables 2 and 3). Treatment included intensive induction chemotherapy according to the ICE (idarubicin‐etoposide‐cytarabine) scheme in five patients or with Vyxeos liposomal therapy in four patients (Table 2). The remaining two patients received low‐intensity treatment, one with 5‐azacitidine alone and the other with 5‐azacitidine plus venetoclax.
TABLE 3.
Allogeneic hematopoietic stem cell transplantation: Details of the procedure and outcomes
| t‐MNs | t‐MN status at PBSCT | EOC status | MAC/RIC | Conditioning regimen | Source SC | HLA compatibility | GVHD prophylaxis | Number of CD34+ cells infused/kg | Graft failure | Acute GvHD | Chronic GvHD | MN best response | EOC best response | Relapse | Cause of death | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | EB‐2 | mCR | NED | MAC | Treo‐Flu a | PBSC | Haploidentical | CTX post reinfusion + MMF + TAC b | 4.4 × 106 | No | No | No | CR | NED | Yes | t‐MN PD |
| 2 | AML | cyCR | NED | MAC | Treo‐Flu a | PBSC | Haploidentical | CTX post reinfusion + MMF + TAC b | 4.6 × 106 | No | Yes (G2) | No | CR | NED | Yes | t‐MN PD |
| 3 | EB‐2 | mCR | CR | MAC | Treo‐Flu a | PBSC | Haploidentical | CTX post reinfusion + MMF + TAC b | 5.3 × 106 | No | No | No | RELAPSE | SD | Yes | t‐MN PD |
| 4 | AML | cyCR | SD | MAC | Treo‐Flu a | PBSC | Identical | MTX + CSA c | 5.3 × 106 | No | Yes (G2) | Yes | CR | CR | No | Alive |
| 5 | EB‐1 | mCR | NED | MAC | Treo‐Flu a | PBSC | Haploidentical | CTX post reinfusion + MMF + TAC b | 5.6 × 106 | No | No | No | CR | NED | No | TRM (PDTL) |
| 6 | EB‐2 | cyCR | SD2 | MAC RIC |
Treo‐Flu a Flu‐CTX‐TBI d |
PBSC PBSC |
Haploidentical Haploidentical |
CTX post reinfusion + MMF + TAC b CTX post reinfusion + MMF + TAC b |
4.8 × 106 6.4 × 106 |
Yes Yes |
Yes (G2) Yes (G4) |
No No |
CR CR |
SD SD |
No | TRM (GVHD) |
| 7 | EB‐1 | cyCR | CR | MAC | Thio‐Flu‐Busulfan e | PBSC | Haploidentical | CTX post reinfusion + MMF + TAC b | 3.9 × 106 | No | No | Yes | CR | CR | No | Alive |
Abbreviations: AML, acute myeloid leukemia; CR, complete response; cyCR, cytogenetic complete remission; EB1, myelodysplastic syndrome with excess blasts type 1; EB2, myelodysplastic syndrome with excess blasts type 2; EOC, epithelial ovarian cancer; GVHD, graft‐vs‐host disease; HLA, human leukocyte antigen; MAC, myeloablative conditioning; mCR, morphological complete remission; NED, no evidence of disease; PBSC, peripheral blood stem cell; PD, progression disease; PTDL, posttransplant lymphoproliferative disease; RIC, reduced intensity conditioning; SC, stem cells; SD, stable disease; t‐MNs, therapy related myeloid neoplasms; TRM, transplant‐related mortality.
Treosulfan 14 g/smq day (−6; −5; −4) and Fludarabine 30 mg/smq day (−6; −5; −4; −3; −2).
Cyclophosphamide (CTX) 50 mg/kg day (+3; +4) and mycophenolate mofetil (MMF) 15 mg/kg t.i.d. up to day +35 + Tacrolimus (TAC) from day +5 to day +100 (target Tacrolimus level 5‐15 ng/mL) or Cyclosporine (CSA) from day −1 to day +100 (range 150‐300 mg).
Methotrexate 15 g/kg day +1, methotrexate 10 mg/kg day (+3; +6) and Cyclosporine from day −1 to day +100 (range 150‐300 mg).
Cyclophosphamide (CTX) 14.5 mg/kg day −6 → day −5, TBI 200 cGy.
Thiotepa (Thio) 5 mg/kg day(−7; −6) and Fludarabine (Flu) 50 mg/smq day (−5; −4; −3) and Busulfan 3.2 mg/kg day(−5; −4; −3); Fludarabine 30 mg/smq day −6 → day −2.
Three of eleven patients (27%) had primary refractory disease and died from leukemia progression, whereas eight patients experienced CR, including five (46%) who achieved cytogenetic CR (cyCR) after Vyxeos liposomal therapy (three patients), ICE (one patient) or 5‐azacitidine (one patient). The remaining three patients (27%) experienced morphological CR (mCR) with persistence of cytogenetic alterations.
One patient died from heart failure before allo‐HSCT and while in cyCR, and the remaining seven patients underwent allo‐HSCT from haplo‐identical family donors (six patients) or from an HLA identical donor. At the time of allo‐HSCT, four patients were in cyCR, three in mCR.
At present, two patients in cyCR are alive in persistent remission at 3 and 4 years, respectively, since allo‐HSCT. Both of them experienced chronic graft‐vs‐host disease (cGVHD): one patient had a limited lung cGVHD that resolved after steroids, but the other patient had extensive skin cGVHD, requiring a variety of immunosuppressive treatments for more than 3 years. This patient recently experienced disease progression of her solid tumor, with an initial response after retreatment with platinum‐based chemotherapy.
Five patients died after allo‐HSCT. One patient in cyCR had relapse at 150 days after allo‐HSCT, and the second patient in cyCR experienced a graft failure, was retransplanted and died of extensive acute GVHD 30 days after the second procedure. Of the three patients in mCR, two died because of leukemia relapse (300 and 179 days after allo‐HSCT, respectively) and one of a posttransplant lymphoproliferative disorder (PDTL) at +90 days.
At a median observation of 6.8 months (range 2.3‐49), 3 of 16 patients (19%) were alive and 13 had died, 10 because of leukemia, 2 from transplant‐related events and 1 from heart failure.
3.3. Persistent cytopenia and clonal hematopoiesis
Five additional patients were referred to hematological consultancy for persistent CBC abnormalities after PARPi discontinuation. PB NGS (using the Oncomine Myeloid Research Assay) disclosed TP53 mutations, in the absence of immunophenotypic abnormalities in two patients, whereas no myeloid neoplasm‐related mutations were detectable in the remaining three patients. Patients harboring a TP53 mutation (Table 4, IDs 1 and 2) underwent further hematological investigations that led to a diagnosis of CCAUS in one patient and CCUS in the second patient. The three patients with a negative NGS assay were classified as having ICUS (Table 4, IDs 3‐5), with inadequate tolerance to PARPi, because of prolonged cytopenias after a short PARPi retreatment.
TABLE 4.
Characteristics and management of five patients with pre‐MDS hematological disorders
| Hematological abnormalities | PARPi (no cycles) | Karyotype analysis | Mutations (VAF) a | Diagnosis | Morphology (WHO criteria) | PARPi management | |
|---|---|---|---|---|---|---|---|
| ID1 | Erythroblastosis with H‐Jolly bodies | Olaparib (14) | 46,XX[8]/46,XX, inv(17)(q21q25) | TP53 (6.10%) | CCAUS | Normal | Stop |
| ID2 | N G3 and T G2 | Olaparib (26) | 45,XX, del(5)(q22q35), −17[6]/46,XX[15] | TP53 (12%) | CCUS | Normal | Stop |
| ID3 | A G3 and T G2 | Niraparib (4) | nd b | Negative | ICUS (transient) | Normal | Continued |
| ID4 | N G2 | Olaparib (6) | nd b | Negative | ICUS (transient) | Normal | Continued |
| ID5 | A G2 | Olaparib (13) | nd b | Negative | ICUS (transient) | Normal | Continued |
Abbreviations: A, anemia; CCAUS, clonal cytogenetic abnormalities of undetermined significance; CCUS, clonal cytopenia of undetermined significance; G, grade according to Common Terminology Criteria for Adverse Events; ICUS, idiopathic cytopenia of undetermined significance; N, neutropenia; PARPi, poly(ADP‐ribose) polymerase inhibitors; T, thrombocytopenia; transient, recovery after PARPi interruption and recurring after PARPi resumption; VAF, variant allele frequency.
Next generation sequencing analysis of PB DNA using Oncomine Myeloid Research Assay and the Ion Torrent S5 technology (ThermoFisher).
Nd, not done, due to only transient blood counts abnormalities and no detectable mutations by NGS.
All five patients were also investigated using our customized Myelo‐panel, which targets 255 cancer predisposing genes, as described in the Section 2. We analyzed buccal DNA for identification of germlines variants and PB for identification of somatic variants. As detailed in Table 5, we identified several variants of uncertain significance (VUS) in all patients. Patient ID1 (CCAUS) carried a BRCA1 germline pathogenic mutation and two somatic variants (in AML driver genes IGFN1 and NBEAL2 23 ), in addition to the known TP53 mutation. In patient ID2 (CCUS), we identified a BRCA1 germline mutation and a somatic variant in EPH4, in addition to the known TP53 mutation. In patients ID3 and ID5 (both ICUS), somatic mutations in GNA11 and NOTCH1 genes, respectively, were detected in the absence of germline abnormalities. In Patient ID4 (ICUS), we identified a pathogenic germline variant in BCR and four somatic mutations in EPHB2, NOTCH1, SLTM and BRCA1, all with an elevated variant allele frequency. Finally, in four patients, we identified mutations in “actionable” genes, although at low frequency (Table 5).
TABLE 5.
Germline and somatic mutations identified in patients with persistent cytopenia and clonal hematopoiesis by analysis with the custom Myelo panel
| ID | Germline mutations a | Somatic mutations a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Type | VAF (%) | AA substitution | Variant annotation b | Gene | Type | VAF (%) | AA substitution | Variant annotation b | Gene classification c | |
| 1 | BRCA1 | INDEL | 51.4 | p.Leu1679TyrfsTer2 | Pathogenic | IGFN1 | SNV | 17.9 | p.Gly1565Asp | VUS | AML Driver |
| NBEAL2 | SNV | 3.1 | p.Leu1093Val | VUS | AML driver | ||||||
| TP53 | SNV | 7.2 | p.Cys238Tyr | Pathogenic | AML driver/susceptibility | ||||||
| 2 | BRCA1 | INDEL | 51.6 | p.Asn363IlefsTer11 | Pathogenic | EPHA4 | SNV | 2.5 | p.Cys191Arg | VUS | Actionable |
| TP53 | SNV | 4.8 | p.Cys176Ser | Pathogenic | AML driver/susceptibility | ||||||
| 3 | NONE | GNA11 | SNV | 3.2 | p.Leu40Phe | Likely pathogenic | Actionable | ||||
| 4 | BCR | INDEL | 56.8 | p.Lys754AsnfsTer34 | Pathogenic | EPHB2 | SNV | 52.9 | p.Gln626Arg | Likely pathogenic | Actionable |
| NOTCH1 | SNV | 40.5 | p.Thr2090Met | VUS | Actionable | ||||||
| SLTM | SNV | 52.2 | p.Gln54Arg | Likely pathogenic | Actionable | ||||||
| BRCA1 | SNV | 45.3 | p.Arg1495Met | Pathogenic | Susceptibility/actionable | ||||||
| 5 | NONE | NOTCH1 | SNV | 3.2 | p.Asn104Thr | VUS | Actionable | ||||
Abbreviations: AA, aminoacidic; AML, acute myeloid leukemia; INDEL, insertion/deletion; SNV, single nucleotide variant; VAF, variant allele frequency; VUS, variant of uncertain significance.
Next generation sequencing analysis using custom Myelo panel and the Ion Torrent S5 technology (ThermoFisher).
Annotation of variants for pathogenicity by ClinVar, Varsome, RENOVO.
Gene classification based on our gene lists in the customized Myelo panel; for definitions, see Section 2.3.
4. DISCUSSION
The present report describes the occurrence of t‐MNs in a series of 182 women receiving salvage PARPi for progressing EOC after chemotherapy treatments at our Milan Center during the last 11 years. Overall, 16 patients (8.7%) developed t‐MNs after a median exposure time to PARPi of about 2 years. At a median observation time of 6.8 months (range 2.3‐49), 3 of 16 (19%) patients were alive, two after allo‐HSCT and one under watch‐and‐wait management for a low‐risk t‐MDS. Ten patients died because of leukemia‐related adverse events, two more from transplant‐related events and one from heart failure. The study confirms that the risk of secondary myeloid neoplasms after chemotherapy and then PARPi cannot be minimized and indicates the poor prognosis for women developing t‐MNs after salvage PARPi, despite early diagnosis and the use of effective therapies supplemented with allo‐HSCT. The genetic complexity observed in patients developing hematological disorders related to therapy can explain to a certain extent these very poor outcomes.
A previous report by our group raised warnings about the risk of hematological disorders after salvage PARPi treatment. 9 In that study, we documented a markedly higher incidence of t‐MNs compared to observations from large multicenter trials with PARPi thus far reported. We have extended our original study by close monitoring of all women referred to our center for salvage PARPi treatment for refractory/relapsed EOC. The extended analysis confirms the significant occurrence of t‐MNs and the overall incidence of 8.7% is comparable to the 6.9% originally reported in our previous study. These numbers are in line with the results of two recent large studies with reported t‐MNs incidences of 4.79% and 8%, respectively. 8 , 12 Thus, the occurrence of t‐MNs is a serious and relevant complication in women with EOC exposed to PARPi after previous chemotherapy treatments.
Three patients in our study had a rapid and fatal outcome, and one patient continues with stable disease. The remaining 12 patients received adequate cytoreductive treatments, according to the most effective approaches, including the ICE schedule or the recently developed combined drug Vyxeos or demethylating agents with or without Venetoclax. 24 , 25 Most patients who experienced a response also underwent allo‐HSCT, taking advantage of expanded donor availability using either identical or haploidentical donor sources. 26 Despite these prompt and quite effective treatments, 3 of 16 patients survived to the last follow‐up, with one patient still free of solid and hematological tumors. This low survival indicates the particularly adverse prognosis of t‐MNs occurring after salvage PARPi and the need to seek for treatment strategies specifically designed for these peculiar hematological neoplasms.
In this regard, it must be also underlined that PARP inhibitors currently used in the clinic target both PARP‐1 and PARP‐2 indiscriminately; however, despite some functional redundancy, it has been recently suggested that the tumorigenic role of the two proteins is related to the type of cancer. Indeed, in prostate cancers only PARP‐2 is selectively upregulated and associated to prostate cancer growth and aggressiveness. 27 In a model of cMyc‐driven lymphomagenesis, loss of PARP‐2 protects from development of c‐Myc‐dependent lymphomas while loss of PARP‐1 accelerates lymphomagenesis. However, no differences were observed on other models of leukemia such as Nocth1‐dependent T‐ALL, where either loss of PARP‐1 or PARP‐2 had no effect on mouse survival. 28 These data underline the critical issue of developing selective PARPi to increase effectiveness and reduce possible side effects. In this perspective, AstraZeneca has developed a PARP‐1‐specific inhibitor, currently in Phase I clinical trial, that in BRCA‐mutated breast cancer models, has shown higher anti‐tumor efficacy compared to Olaparib (AstraZeneca's Petra Study, available at ClinicalTrials.gov‐Identifier: NCT04644068).
Our t‐MN patients were extensively investigated for genetic lesions, and various chromosomal alterations were found, including chromosome 5q deletion, partial or complete loss of chromosome 7 or a complex karyotype. These cytogenetic features are typical of t‐MNs secondary to alkylating agents. 29 In addition, molecular testing identified TP53 mutations in 12 of 13 tested patients. These TP53 mutations frequently co‐occurred with partial deletion, complete loss or rearrangement of chromosome 17 (Table 2), suggesting loss of heterozygosity (LOH) at the TP53 locus in several patients of our cohort developing t‐MNs. Recently, preexisting TP53‐mutated clonal hematopoiesis was reported to be a risk factor for t‐MNs in high‐grade EOC patients treated with rucaparib, and an expansion of the TP53 clone has been observed in patients developing MDS/AML. Furthermore, the presence of HR repair gene mutations in the tumor, either germline or somatic, including BRCA1 and BRCA2, has been associated with increased incidence of MDS/AML. 30 It seems quite likely that the high frequency of TP53 mutations and the numerous additional genetic lesions are responsible for the poor response to treatments, including allo‐HSCT, observed in our patients with t‐MNs after PARPi salvage therapy. 31
It is not possible to establish which factor contributes most to the development of t‐MNs, among the PARPi, prior exposure to chemotherapy (mainly alkylating agents) and the combination of these multiple cytotoxic treatments. 32 It is noteworthy that in our series, patients placed on PARPi maintenance after multiple prior lines of chemotherapy had the highest risk of t‐MNs, suggesting that cumulative prior chemotherapy exposure may be a relevant factor for developing MDS/AML with prolonged salvage PARPi. 8 PARPi therapy has been recently approved for early use after first‐line chemotherapy in newly diagnosed metastatic EOC. This should allow for a better understanding of the role of PARPi in leukemogenesis. 33 Both PARP1 and TP53 can bind the ends of broken DNA, acting as potential sensors and signaling molecules for the detection of double‐strand breaks. PARP1‐dependent poly‐ADP ribosylation is increased in response to DNA damage, and TP53 mutants bind with enhanced efficiency to broken DNA ends. As a consequence, PARP1 inhibition in the presence of mutated TP53 may enhance genetic instability and drive leukemogenesis. 34 Exposure to cytotoxic drugs along with the inhibition of PARP activity and the possible presence of TP53‐mutated clones may interact and contribute to the development of t‐MNs.
The presence of genetic lesions predisposing and/or predicting t‐MNs was supported by the deep analysis of five patients developing pre‐MDS features. With our customized Myelo‐panel, we found “actionable” mutated genes in all but one patient. Moreover, the finding of clonal hematopoiesis in the three “transient” ICUS patients allowed us to re‐classify them within the CCUS subgroup. Patients harboring multiple somatic variants, with high allelic burden, need close follow‐up because of the high risk of evolution into an overt t‐MN. In such cases, pre‐emptive treatment with target drugs might be an appealing option, and additional studies should focus on this issue. Careful diagnostic procedures are strongly recommended whenever “transient” cytopenias are observed during PARPi treatment. As recently advised, even minor hematological abnormalities might be an early safety signal, suggesting the need for attention and possible PARPi discontinuation if required. 35 Furthermore, in the near future, PARPi will be used for broader indications in tumors for which a synthetic lethality approach is expected to be effective. In fact, olaparib has been recently approved for the treatment of BRCA1/2‐mutated, HER2‐negative breast cancer after courses of anthracyclines and taxanes. 36 , 37 Consequently, establishing a preventive measure for identification of gene mutations that may raise risk for t‐MNs in patients treated with PARP inhibitors alone or as a salvage therapy after chemotherapy treatment is urgent and mandatory. In this view, a prospective biological study is ongoing among the gynecological, hematological and molecular research units at our center, aimed at identifying possible genetic abnormalities, associated with an increased risk for t‐MNs after PARPi treatment, including mutations analysis in genes involved in CHIP.
In summary, our study confirms the actual risk of t‐MN in EOC patients treated with chemotherapy and subsequently exposed to prolonged PARPi therapy. The management of these patients is quite complex and the outcome is extremely poor. In this context, a preventive approach based on the identification of gene mutations that may raise risk for t‐MNs and “actionable” gene mutations is warranted and could be crucial for protecting these women from a second devastating and often incurable neoplasm.
AUTHOR CONTRIBUTIONS
The work reported in the article has been performed by the authors, unless clearly specified in the text. Conceptualization: Elisabetta Todisco and Corrado Tarella; Resources: Federica Gigli, Chiara Ronchini, Viviana Amato, Simona Sammassimo, Rocco Pastano, Gabriella Parma, Maria Teresa Lapresa, Francesco Bertolini, Chiara Corsini, Giuliana Gregato, Claudia Poletti; Formal analysis: Elisabetta Todisco, Chiara Ronchini, Giuliana Gregato; Supervision: Elisabetta Todisco, Nicoletta Colombo, Pier Giuseppe Pelicci, Myriam Alcalay, Francesco Bertolini and Corrado Tarella; Writing ‐ original draft: Elisabetta Todisco, Federica Gigli, Chiara Ronchini and Corrado Tarella; Writing ‐ review & editing: Elisabetta Todisco, Federica Gigli, Chiara Ronchini, Myriam Alcalay and Corrado Tarella.
CONFLICT OF INTEREST
F. Bertolini reports research support from Menarini, Gilead, Pierre Fabre and Arqule. N. Colombo reports personal fees from AstraZeneca, Novartis, Clovis Oncology, MSD, GSK, Roche, Pfizer, Immunogen, Mersana, Eisai, OncXerna and Nuvation Bio. All other authors report no conflict of interest.
ETHICS STATEMENT
The study was authorized by the Clinical Trial Office of IEO (UID 2368). All patients gave written informed consent to their participation to the diagnostic and treatment program, according to the IEO ethical committee approval.
Supporting information
Appendix S1 Supporting Information.
ACKNOWLEDGEMENTS
We thank AIRC (Associazione Italiana Ricerca sul Cancro) and the Italian Ministry of Health for support of our research. We thank San Francisco Edit for editing of our article. Open access funding enabled and organized by Projekt DEAL.
Todisco E, Gigli F, Ronchini C, et al. Hematological disorders after salvage PARPi treatment for ovarian cancer: Cytogenetic and molecular defects and clinical outcomes. Int J Cancer. 2022;151(10):1791‐1803. doi: 10.1002/ijc.34162
DATA AVAILABILITY STATEMENT
The data that support the findings of our study are available from the corresponding author upon request.
REFERENCES
- 1. Morgan RD, Clamp AR, Evans DGR, Edmondson RJ, Jayson GC. PARP inhibitors in platinum‐sensitive high‐grade serous ovarian cancer. Cancer Chemother Pharmacol. 2018;81(4):647‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tomao F, Bardhi E, Di Pinto A, et al. Parp inhibitors as maintenance treatment in platinum sensitive recurrent ovarian cancer: an updated meta‐analysis of randomized clinical trials according to BRCA mutational status. Cancer Treat Rev. 2019;80:101909. [DOI] [PubMed] [Google Scholar]
- 3. Cancer Genome Atlas Research Network . Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP‐ribose) polymerase inhibition. Cancer Res. 2006;66:8109‐8115. [DOI] [PubMed] [Google Scholar]
- 6. Liu X, Shi Y, Maag DX, et al. Iniparib nonselectively modifies cysteine‐containing proteins in tumor cells and is not a bona fide PARP inhibitor. Clin Cancer Res. 2012;18:510‐523. [DOI] [PubMed] [Google Scholar]
- 7. Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588‐5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morice PM, Leary A, Dolladille C, et al. Myelodysplastic syndrome and acute myeloid leukaemia in patients treated with PARP inhibitors: a safety meta‐analysis of randomised controlled trials and a retrospective study of the WHO pharmacovigilance database. Lancet Haematol. 2021;8:e122‐e134. [DOI] [PubMed] [Google Scholar]
- 9. Todisco E, Gigli F, Mantiero M, et al. Clinical presentation, diagnosis and management of therapy‐related hematological disorders in women with epithelial ovarian cancer treated with chemotherapy and poly‐ADP‐ribose polymerase inhibitors: a single‐center experience. Int J Cancer. 2021;148(1):170‐177. [DOI] [PubMed] [Google Scholar]
- 10. Zhou J, Feng L, Zhang X. Risk of severe hematologic toxicities in cancer patients treated with PARP inhibitors: a meta‐analysis of randomized controlled trials. Drug Des Dev Ther. 2017;11:3009‐3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pujade‐Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum‐sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT‐Ov21): a double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274‐1284. [DOI] [PubMed] [Google Scholar]
- 12. Poveda A, Floquet A, Ledermann JA, et al. SOLO2/ENGOT‐Ov21 investigators. Olaparib tablets as maintenance therapy in patients with platinum‐sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT‐Ov21): a final analysis of a double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Oncol. 2021;22(5):620‐631. [DOI] [PubMed] [Google Scholar]
- 13. Colombo N, Tomao F, Benedetti Panici P, et al. Randomized phase II trial of weekly paclitaxel vs. cediranib‐olaparib (continuous or intermittent schedule) in platinum‐resistant high‐grade epithelial ovarian cancer. Gynecol Oncol. 2022;164(3):505‐513. [DOI] [PubMed] [Google Scholar]
- 14. Trotti A, Colevas AD, Setser A, et al. CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol. 2003;13(3):176‐181. [DOI] [PubMed] [Google Scholar]
- 15. Griffith M, Griffith OL, Coffman AC, et al. DGIdb: mining the druggable genome. Nat Methods. 2013;10(12):1209‐1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar RD, Chang LW, Ellis MJ, Bose R. Prioritizing potentially druggable mutations with dGene: an annotation tool for cancer genome sequencing data. PLoS One. 2013;8(6):e67980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Somaiah N, Simon NG, Simon GR. A tabulated summary of targeted and biologic therapies for non‐small‐cell lung cancer. J Thorac Oncol. 2012;7:S342‐S368. [DOI] [PubMed] [Google Scholar]
- 18. Barbui T, Thiele J, Gisslinger H, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in‐depth discussion. Blood Cancer J. 2018;8(2):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454‐2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gurnari C, Fabiani E, Falconi G, et al. From clonal hematopoiesis to therapy‐related myeloid: the silent way of cancer progression. Biology. 2021;10(2):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tang G, Medeiros LJ, Wang SA. How I investigate clonal cytogenetic abnormalities of undetermined significance. Int J Lab Hematol. 2018;40:385‐391. [DOI] [PubMed] [Google Scholar]
- 23. Tyner JW, Tognon CE, Bottomly D, et al. Functional genomic landscape of acute myeloid leukemia. Nature. 2018;562(7728):526‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lancet JE, Uy GL, Cortes JE, et al. CPX‐351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol. 2018;36(26):2684‐2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383:617‐629. [DOI] [PubMed] [Google Scholar]
- 26. Ciurea SO, Cao K, Fernandez‐Vina M, et al. The European Society for Blood and Marrow Transplantation (EBMT) consensus guidelines for the detection and treatment of donor‐specific anti‐HLA antibodies (DSA) in haploidentical hematopoietic cell transplantation. Bone Marrow Transplant. 2018;53(5):521‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gui B, Gui F, Takai T, et al. Selective targeting of PARP‐2 inhibits androgen receptor signaling and prostate cancer growth through disruption of FOXA1 function. Proc Natl Acad Sci. 2019;116(29):14573‐14582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Galindo‐Campos MA, Lutfi N, Bonnin S, et al. Distinct roles for PARP‐1 and PARP‐2 in c‐Myc‐driven B‐cell lymphoma in mice. Blood. 2022;139(2):228‐239. [DOI] [PubMed] [Google Scholar]
- 29. McNerney ME, Godley LA, Le Beau MM. Therapy‐related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer. 2017;17:513‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kwan TT, Oza AM, Tinker AV, et al. Preexisting TP53‐mutated clonal hematopoiesis as a risk factor for the development of secondary myeloid malignancies in high‐grade ovarian cancer patients treated with the poly(ADP‐ribose) polymerase inhibitor rucaparib. JAMA Oncol. 2021;7(12):1772‐1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Della Porta MG, Gallì A, Bacigalupo A, et al. Clinical effects of driver somatic mutations on the outcomes of patients with myelodysplastic syndromes treated with allogeneic hematopoietic stem‐cell transplantation. J Clin Oncol. 2016;34(30):3627‐3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Korach J, Turner S, Milenkova T, et al. Incidence of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) in patients (pts) with a germline (g) BRCA mutation (m) and platinum‐sensitive relapsed ovarian cancer (PSR OC) receiving maintenance olaparib in SOLO2: impact of prior lines of platinum therapy. J Clin Oncol. 2018;36:5548. [Google Scholar]
- 33. Di Silvestro P, Colombo N, Scambia G, et al. Efficacy of maintenance olaparib for patients with newly diagnosed advanced ovarian cancer with a BRCA mutation: subgroup analysis findings from the SOLO1 trial. Clin Oncol. 2020;38(30):3528‐3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alvarez‐Gonzalez R, Mendoza‐Alvarez H, Frey M, Zentgraf H. Upregulation of two distinct p53‐DNA binding functions by covalent poly(ADP‐ribosyl)ation: transactivating and single strand break sensing. Cancer Invest. 2013;31(9):563‐570. [DOI] [PubMed] [Google Scholar]
- 35. Morice P‐M, Chrétien B, Da Silva A, Dolladille C, Alexandre J. Occurrence of pancytopenia among patients with cancer treated with poly(adenosine diphosphate‐ribose) polymerase inhibitors: a pharmacoepidemiologic study. JAMA Oncol. 2021;7(12):1899‐1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379:753‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tutt ANJ, Garber JE, Kaufman B, et al. Adjuvant olaparib for patients with BRCA1‐ or BRCA2‐mutated breast cancer. N Engl J Med. 2021;384:2394‐2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information.
Data Availability Statement
The data that support the findings of our study are available from the corresponding author upon request.