

Purpose
To describe the unique clinical features, determine the genomics, and investigate the metabolic derangement of an extremely rare form of a hereditary lethal kidney cancer syndrome.
Patients and Methods
Three patients with lethal kidney cancer (age 19, 20, and 37 years) exhibiting persistent (1 to 3 months) extremely high levels of blood lactate (> 5 mM) despite normal oxygen perfusion, highly avid tumors on [18F]fluorodeoxyglucose positron emission tomography (PET), and pleomorphic histopathologic features were identified and treated in a single institute. Integrated studies including whole-genome sequencing (WGS), targeted sequencing, immunohistochemistry, cell-based assays, and 18F-glutamine PET imaging were performed to investigate this rare kidney cancer syndrome.
Results
All three patients with kidney cancer were initially given various diagnoses as a result of diverse tumor histopathology and atypical clinical presentations. The correct diagnoses of these SDHB-mutated renal cell carcinomas were first made based on cancer genomics. Genomic studies of the blood and tumors of these patients identified three different kinds of germline loss-of-function mutations in the SDHB gene and the common loss of heterozygosity in the remaining SDHB allele thorough somatic chromosome 1p deletion. In one patient, WGS revealed that a germline mutation of SDHB coupled with loss of heterozygosity was the sole genetic event. Cancer evolution analysis of SDHB tumors based on WGS demonstrated that SDHB in kidney epithelium fulfills the Knudson two-hit criteria as a major tumor suppressor gene. SDHB−/− tumor cells displayed increase in glucose uptake and lactate production, alteration in mitochondrial architecture, and defect in oxidative respiration. 18F-Glutamine PET imaging studies demonstrated increased glutamine metabolism.
Conclusion
SDHB-deficient metastatic renal cell carcinoma is a rare, aggressive form of kidney cancer that manifests with clinical evidence of a severe Warburg effect, and genomic studies demonstrated two genetic hits at SDHB genes during kidney tumorigenesis.
INTRODUCTION
Renal cell carcinoma (RCC) is a heterogeneous group of malignancies arising from renal epithelium.1-3 Major RCC subtypes include clear cell RCC (approximately 75%),4 papillary RCC (approximately 15%),5 chromophobe RCC (approximately 5%),6 and unclassified RCC (4% to 6%).7-9 The remaining kidney cancer subtypes are rare (< 1%), among which succinate dehydrogenase (SDH) –deficient RCC is the newest recognized subtype.1 The SDH enzyme consists of A, B, C, and D subunits.10,11 Germline SDH mutations are common in patients with hereditary paraganglioma and pheochromocytoma syndrome and GI stromal tumors.12-15 Recent studies have begun to elucidate SDH mutations in kidney cancer with fewer than 50 patients with SDH-deficient RCCs reported thus far (0.05% to 0.2%).16,17 The most commonly mutated gene in SDH-RCC is SDHB (83%).18-20 As a result of its rarity and recent designation, clinical and molecular features associated with aggressive SDHB-deficient RCC are unknown.
The aerobic glycolysis Warburg effect in cancer was first recognized by Otto Warburg,21,22 who described a phenomenon where glucose undergoes lactic acid fermentation in lieu of aerobic respiration despite normal oxygen tension, providing adaptive advantages for proliferating cancer cells.23-26 Direct investigation of the Warburg effect in human cancer remains a challenge.27,28 The increased activity detected by [18F]fluorodeoxyglucose (FDG) positron emission tomography (PET) reflects glucose uptake and thereby renders indirect evidence of aerobic glycolysis. In normal human subjects, lactate produced from glucose fermentation is quickly removed from the blood by liver. Hence, clinically relevant lactic acidosis, defined as low blood pH (≤ 7.35) with high plasma lactate (≥ 5 mM), commonly denotes a serious compromise in tissue oxygenation, namely type A lactic acidosis.29-32 Rare cases of lactic acidosis despite adequate tissue oxygen, namely type B lactic acidosis, have been reported in highly proliferative, hematologic malignancies and anecdotally in solid tumors.33-35
The occurrence of the Warburg effect in cancers has long been considered a consequence of cancer progression.36 However, the discovery of FH and SDH mutations, key enzymes of the tricarboxylic acid (TCA) cycle, as primary underlying genetic events of rare hereditary human cancers posits that deregulated metabolism could function as oncogenic driver and offers the opportunity to study the Warburg effect in humans.10,37-40 Of note, kidney cancers of all types are considered to be metabolic diseases2 in which deregulated metabolism plays an important role in the pathobiology.41-44 Here, we present clinical and genomic features of three young patients with metastatic SDHB-deficient RCC whose tumors displayed high avidity in [18F]FDG-PET and whose blood exhibited persistent hyperlactatemia. These patients were initially incorrectly diagnosed and were later found to have SDHB-deficient RCC based on genomics; all three patients eventually died despite various systemic treatments.
PATIENTS AND METHODS
Patients
Blood draws, biopsies, and sample collection were all done as part of standard of care or under institutional review board (IRB) –approved protocols. All off-label use of US Food and Drug Administration–approved medications was done with patient informed consent. The glutaminase inhibitor CB-839 was administered under a single-patient investigational new drug application with IRB and US Food and Drug Administration approvals. CB-839 was provided by Calithera Biosciences (South San Francisco, CA).
Germline DNA Analysis
Germline DNA was purified from blood and subjected to sequencing on all coding exons of the SDHB, SDHC, SDHD, and AF2 genes by GeneDx (Elmwood Park, NJ).45
Histopathology and Immunohistochemistry
Immunohistochemistry for SDHB was performed using anti-SHDB antibody (ab14714, clone 21A11; ABCAM, Cambridge, United Kingdom).20
Whole-Genome Sequencing
Whole-genome sequencing (WGS) was performed at the New York Genome Center (New York, NY). Genomic DNA libraries of tumor-normal pair samples were prepared from the primary kidney tumor of patient 1 and the primary tumor and metastases of patient 3. Paired-end 2 × 100 base pair sequence reads were performed using Illumina HiSEquation 2500 instruments (Illumina, San Diego, CA), mapped using the Burrows-Wheeler Aligner, and processed with the Genome Analysis Toolkit. Genome-wide detection and analysis of somatic mutations, copy number alterations, and structural variants were performed.46
Clonality Analysis
For each mutation, we calculated cancer cell fractions as previously described,47 using mutant allele burden, tumor purity, and locus-specific copy number in tumor and matched normal samples. Subclones were identified by clustering cancer cell fractions using Dirichlet process–based clustering.46
Targeted Exome Sequencing
DNA from tumors and matched normal tissue was analyzed using the MSK-IMPACT assay (Memorial Sloan Kettering Cancer Center, New York, NY), which provides ultra-deep sequencing coverage (mean > 500×). Target-specific probes for hybrid selection were designed to capture all protein-coding exons of 341 cancer genes.48
Metabolic Assessment of Patient-Derived SDHB-Deficient Cancer Cells
Short-term primary cell cultures were established from patient 1’s tumor (SDHB−/− tumor), patient 1’s tumor-adjacent tissue (SDHB+/− cells), and an unrelated patient’s tumor-adjacent normal kidney tissues (normal kidney). Extracellular acidification rate (ECAR) and oxygen consumption rate were evaluated using Seahorse XFe96 Extracellular Flux Analyzer (Seahorse Biosciences, Billerica, MA). Oxygen consumption rate and ECAR were normalized to protein concentration of plated cells per manufacturer protocol. Lactate and glucose levels were assayed using YSI 2300 STAT Plus Glucose and Lactate Analyzer (YSI, Life Sciences, Yellow Springs, OH). Intracellular metabolite levels were measured by gas chromatography–mass spectrometry. Metabolites were extracted by 80% methanol, and count values were normalized to cell number.
18F-Glutamine PET
A glutamine-labeled PET scan was done under an IRB-approved protocol, which used [18F]4-l-fluoroglutamine (2S,4R) as a nontherapeutic medical imaging agent to investigate the pharmacokinetics, metabolism, and biodistribution. Radiotracer synthesis and PET imaging were performed as described.49
RESULTS
Clinical History
Patient 1 was a 19-year-old white man who presented with a palpable right upper quadrant mass. Imaging studies revealed a 12 × 11 × 13 cm [18F]FDG-PET–avid right kidney mass along with metastatic lesions involving axial and appendicular skeleton, lungs, and lymph nodes (maximum standardized uptake value [SUV] 48.0; Fig 1), which is different from clear cell RCC that has variable [18F]FDG-PET positivity (30% to 60%).50-52 The patient underwent cytoreductive radical nephrectomy, and the pathology reported high-grade unclassified RCC.53,54 The patient was first treated on a clinical trial with concurrent bevacizumab and everolimus,55 followed by off-label use of vandetanib and dasatinib, and finally treatment with sunitinib without any objective responses. Follow-up 18FDG-PET demonstrated disease progression with extensive metastatic lesions (Data Supplement). The patient died 9 months after cancer diagnosis.
Fig 1.
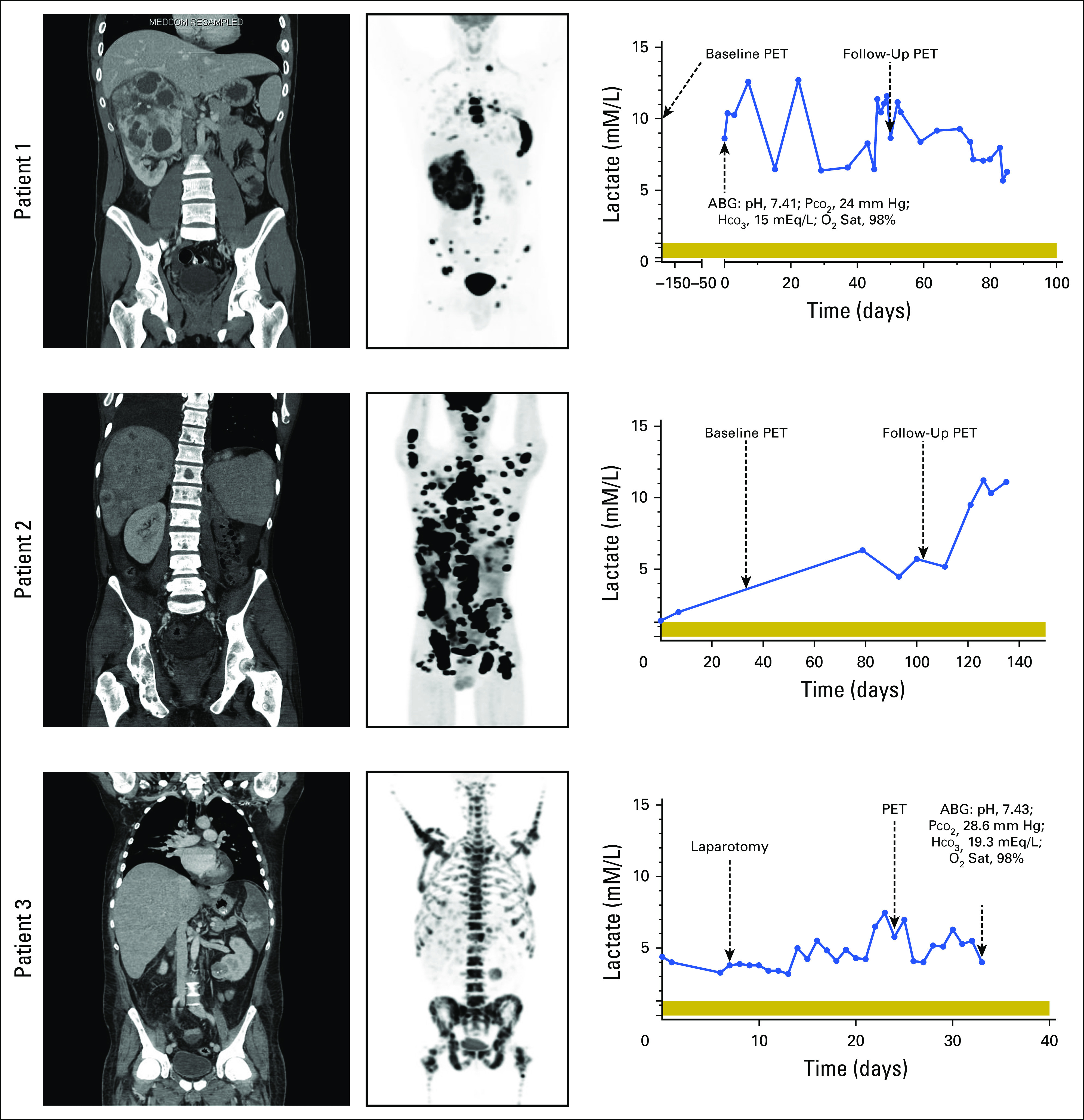
Computed tomography (CT), [18F]fluorodeoxyglucose (FDG) positron emission tomography (PET), and blood lactate levels. Left panels are baseline body CT scans and center panels are baseline [18F]FDG-PET scans of patients 1, 2, and 3. Right panels are corresponding blood lactate levels over the monitoring period for patients 1, 2, and 3. Normal range of lactate level is 0.3 to 1.3 mM/L, as indicated by the horizontal shading. ABG, arterial blood gas; HCO3, bicarbonate; O2 Sat, oxygen saturation; Pco2, partial pressure of carbon dioxide.
Patient 2 was a 20-year-old white man who presented with extensive skull masses, underwent frontotemporal-parietal craniectomy, and was later found to have a 4.4 × 4.7 × 6.6 cm left kidney mass with metastases to lymph nodes, liver, and axial and appendicular skeleton (Fig 1). The initial pathology favored metastatic translocation–type RCC. [18F]FDG-PET confirmed extensive metastatic lesions and noted additional involvement of the parietal lobe and cranium (maximum SUV, 44.1; Fig 1). The patient subsequently underwent cytoreductive nephrectomy, and the diagnosis was amended to mucinous tubular and spindle cell RCC with sarcomatoid transformation. Over a 3-year course, the patient was treated with sunitinib, interleukin-2, cabozantinib, axitinib, temsirolimus, and a phase I glutaminase inhibitor CB-839. Despite therapy, the patient experienced disease progression (Data Supplement) and died 39 months after cancer diagnosis.
Patient 3 was a 37-year-old white man who presented with right shoulder pain and left-sided abdominal pain. On computed tomography scan, he was found to have a 4.5 × 3.9 × 4.0 cm left renal mass and metastases involving the right scapula, left rib, and axial spine (maximum SUV, 33; Fig 1). Biopsy of scapular and renal masses revealed high-grade, poorly differentiated cancer consistent with metastatic RCC. As a result of rapid progression, he was admitted and treated with one cycle of doxorubicin and gemcitabine. His hospital course was complicated by multiple unexplainable hemolytic and thrombotic events, including pulmonary emboli, numerous cerebral vascular events, and infarction of the superior mesenteric artery necessitating exploratory laparotomies for small bowel and colon resections. The patient died 2 months after cancer diagnosis, and tissues from the primary renal mass and multiple metastatic sites were obtained at autopsy.
Persistent Hyperlactatemia Without Acidemia or Hypoxia
Patient 1 was admitted for tachycardia and exertional dyspnea and found to have an elevated plasma lactate of 9.6 mM/L (normal values, 0.3 to 1.3 mM/L) with an arterial blood pH of 7.41, partial pressure of carbon dioxide of 24 mm Hg, bicarbonate of 15 mEq/L, and oxygen saturation of 98% (Fig 1). Studies revealed no overt ischemia, and the patient continued to maintain adequate oxygenation. Subsequent lactate measurements showed persistent hyperlactatemia (5 to 13 mM) without dyspnea or pH changes, consistent with adequate metabolic and respiratory compensation. Patient 2 initially exhibited a slightly greater than normal lactate level of 1.4 mM/L (Fig 1), which gradually increased to 5.5 mM/L and peaked at 11.2 mM/L. The increasing blood lactate was associated with increasing tumor burden. Patient 3 had persistently elevated lactate levels with a baseline of 4.4 mM/L and a peak of 7.5 mM/L (Fig 1). After the patient’s superior mesenteric artery infarction and subsequent resection, lactate levels remained between 3.2 and 5 mM/L for nearly 2 weeks before death. As a result of the high blood lactate level and suspicion of SDHB-deficient RCC based on FDG-PET images, an arterial blood gas was obtained (pH, 7.43; partial pressure of carbon dioxide, 28.6 mm Hg; bicarbonate, 19.3 mEq/L; and oxygen saturation, 98%; Fig 1).
Diverse Histopathology of SDHB-Deficient RCC
The histopathologic features varied significantly among our patients with SDHB-deficient cancer, making the initial diagnosis challenging. The kidney tumor from patient 1 was heterogeneous, with areas of low-grade and high-grade cancer and ossification (Fig 2A). The kidney tumor from patient 2 exhibited high-grade features with areas of pleomorphic spindle cells and increased mitotic activity (Fig 2A). The kidney tumor, scapula metastasis, and liver metastasis from patient 3 revealed exclusively high-grade carcinoma (Fig 2A). Common histology described for SDHB-deficient renal tumors, such as bubbly eosinophilic cytoplasm and cytoplasmic inclusions,56 was detected only in patient 1’s primary tumor but not in the tumors of patients 2 and 3.
Fig 2.
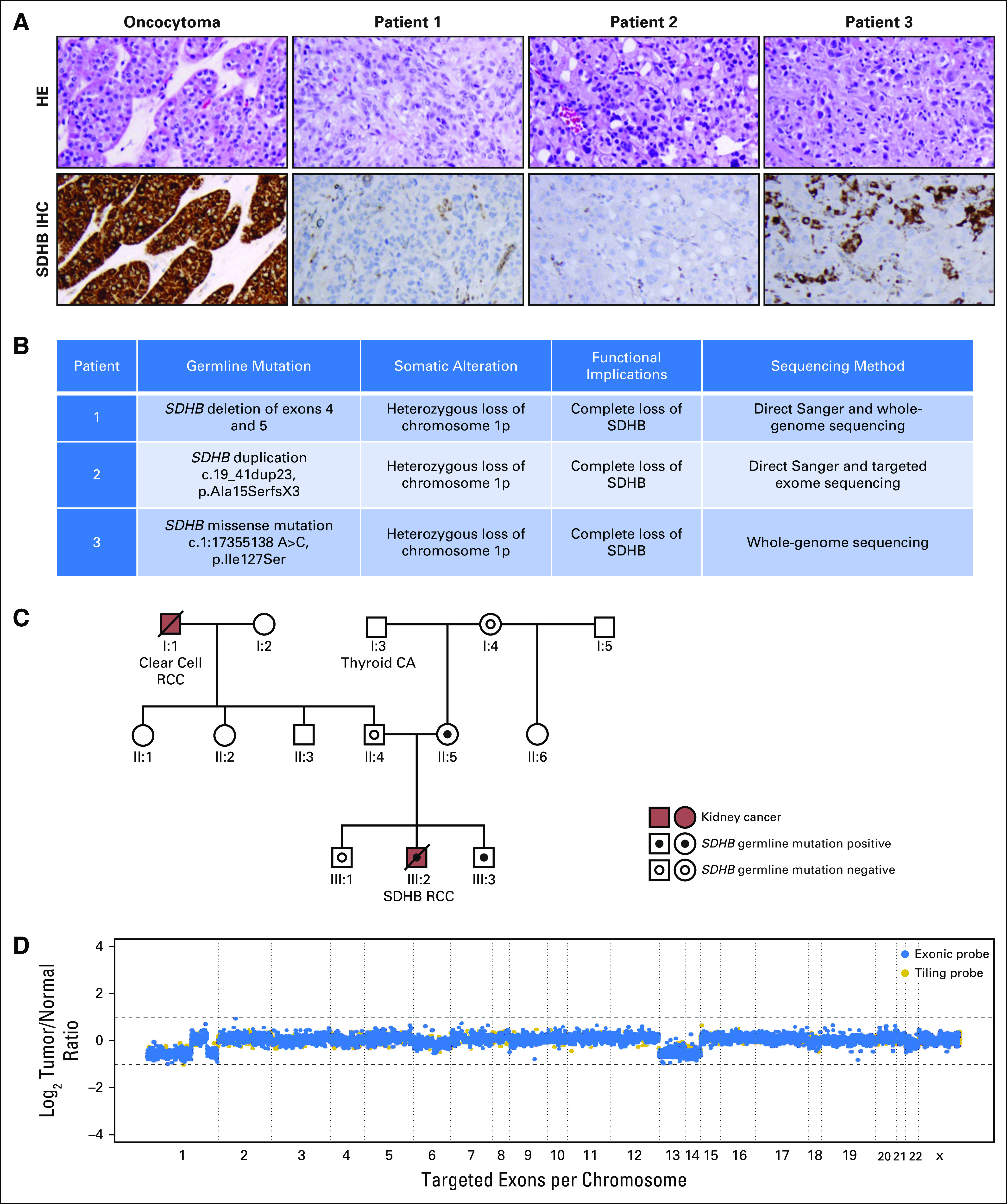
Histologic and genomic features of metastatic SDHB-deficient renal cell carcinoma (RCC). (A) Histopathology and SDHB immunohistochemistry (IHC). Top panels are representative hematoxylin and eosin (HE) images of the primary kidney tumor of patient 1, primary kidney tumor of patient 2, and liver metastasis of patient 3. Bottom panels are SDHB IHC of tumors of patients 1, 2, and 3. SDHB staining was lost in the tumor cells but retained in the intervening stromal cells or hepatocytes. (B) Summary of relevant germline and somatic mutations in all three primary tumors. The c.19_41dup23 mutation of patient 2’s germline SDHB gene caused a frameshift starting with codon alanine 15, changed this amino acid to serine, and created a premature stop codon at positive 3 of the reading frame denoted pAla15SerfsX3. (C) Genogram of patient 1’s family. (D) Copy number plot of patient 2’s primary kidney tumor. CA, cancer.
Loss of SDHB Staining in Tumors
As a result of unusual clinical and histopathologic presentations, SDHB-deficient RCC was suspected in these patients. Germline and somatic mutations of the SDHB gene of all three patients were confirmed by genomic studies. Immunohistochemistry stain for SDHB protein was subsequently established at Memorial Sloan Kettering Cancer Center, demonstrating the loss of SDHB staining in cancer but not adjacent non-neoplastic cells (Fig 2A). As a control, oncocytoma, a kidney tumor with wild-type SDHB, showed strong SDHB staining (Fig 2A).
Patient 1’s Family Pedigree
The young age of patient 1 and the unusual histopathologic features of his tumor were concerning for a hereditary RCC. Accordingly, germline DNA analysis was performed by direct sequencing of the SDHB, SDHC, SDHD, and AF2 genes,45 which revealed a germline exon 4-5 deletion within the SDHB gene in patient 1 (Fig 2B). This alteration was also detected in this patient’s mother and younger brother (Fig 2C), confirming maternal inheritance. Both the patient’s mother and younger brother are not known to have any malignancy; however, baseline lactate from the patient’s mother and younger brother were normal (1.2 mM/L) and elevated (1.7 mM/L), respectively (normal, 0.3 to 1.3 mM/L), which could reflect constitutive haploinsufficiency of SDHB and be of surveying value.
Sequencing of Patient 2’s Germline and Tumor DNA
Sequencing of patient 2’s blood DNA revealed duplication within the SDHB gene, resulting in a frameshift mutation (Fig 2B). Ultra-deep targeted sequencing of his tumor DNA was performed (mean coverage, 774×). The copy number profile showed broad copy number losses on chromosomes 13 and 14 and chromosome arms 1p and 1q (Fig 2D). Germline duplication in SDHB and somatic loss of 1p, where SDHB resides (1p36), resulted in complete genetic loss of SDHB in patient 2’s tumor (Fig 2B).
WGS of Patient 1’s Primary Tumor
To determine genetic events contributing to the pathogenesis of SDHB-deficient RCC, WGS of patient 1’s tumor and matched normal tissue was performed to mean haploid coverage of approximately 90× with greater than 96% of the genome at ≥ 30×. A total of 339 somatic alterations consisting of 260 single nucleotide substitutions, 72 indels, and seven rearrangements were identified (Fig 3 and Data Supplement). WGS identified only a single nonsynonymous somatic substitution (SREBF1 p.D329Y) with no significant impact based on functional prediction.57 Copy number analysis of the tumor revealed few genomic alterations including loss of the short arm of chromosome 1 where SDHB resides (Fig 3). Furthermore, close examination of the SDHB locus in the retained allele found that both tumor and normal tissue have the same deletion spanning exons 4 and 5 of the SDHB coding sequence (chr1:17,353,986 to 17,359,321; Fig 3), resulting in a frameshift mutation. Altogether, a germline intragenic deletion in SDHB and a somatic loss of 1p resulted in complete loss of SDHB in the tumor (Fig 2B).
Fig 3.
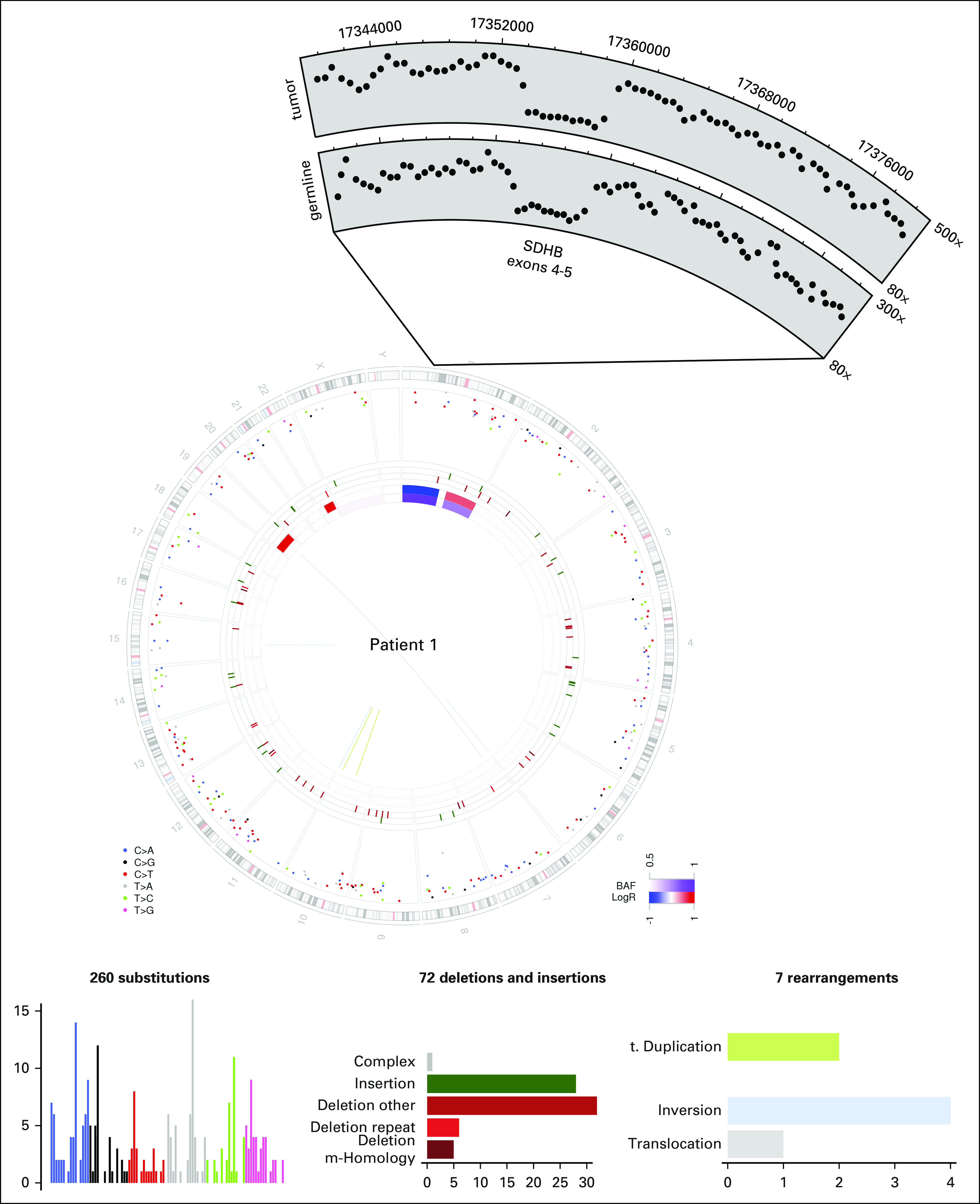
Whole-genome sequencing (WGS) of patient 1’s primary kidney tumor. Circos plot of patient 1’s WGS. The two innermost tracks depict the copy number changes (red, increase; blue, decrease; purple, allelic imbalance, one allele amplified or deleted) in the tumor when compared with the normal for patient 1. The outermost track shows the intermutation distance for substitutions each plotted according to the type of nucleotide change. The middle track shows the genomic positions of the small insertion (green) and deletion (red) along the genome. Rearrangements are plotted as arcs inside the Circos plot. Also shown is a zoomed-in view of the coverage over the SDHB locus in both tumor and matched normal samples, demonstrating the same deletion of exons 4 and 5 in both samples.
WGS and Clonal Evolution of Patient 3’s Primary and Metastatic Tumors
WGS was performed on patient 3’s primary kidney tumor as well as his metastatic liver and scapula bone lesions (Fig 4 and Data Supplement). Copy number profiles of all samples demonstrated extensive aberrations, and the majority of these events were shared. All tumor samples had the same heterozygous nonsynonymous coding mutation in the SDHB gene (chr1: 17355138 A>C, c.380T>G, p.Ile127Ser), whereas the other allele was lost by an arm-level event on chromosome 1p (Fig 4). Of note, the same SDHB mutation has been reported in patients with GI stromal tumor and paraganglioma.58 Detailed characterization of the subclonal structure using the substitution data established a long trunk with 5,198 of 8,047 substitutions in 100% of cells across tumor sites. This subclone also contained mutations in MTOR and NF2 that might have functional impact (Fig 4). The second largest subclone supported by 820 substitutions was present in approximately 55% and 100% of cells in the liver and bone metastases, respectively, likely reflecting tumor heterogeneity.46 Overall, subclonal relationship among the three tumor sites suggests relatively late metastasis during clonal evolution.
Fig 4.
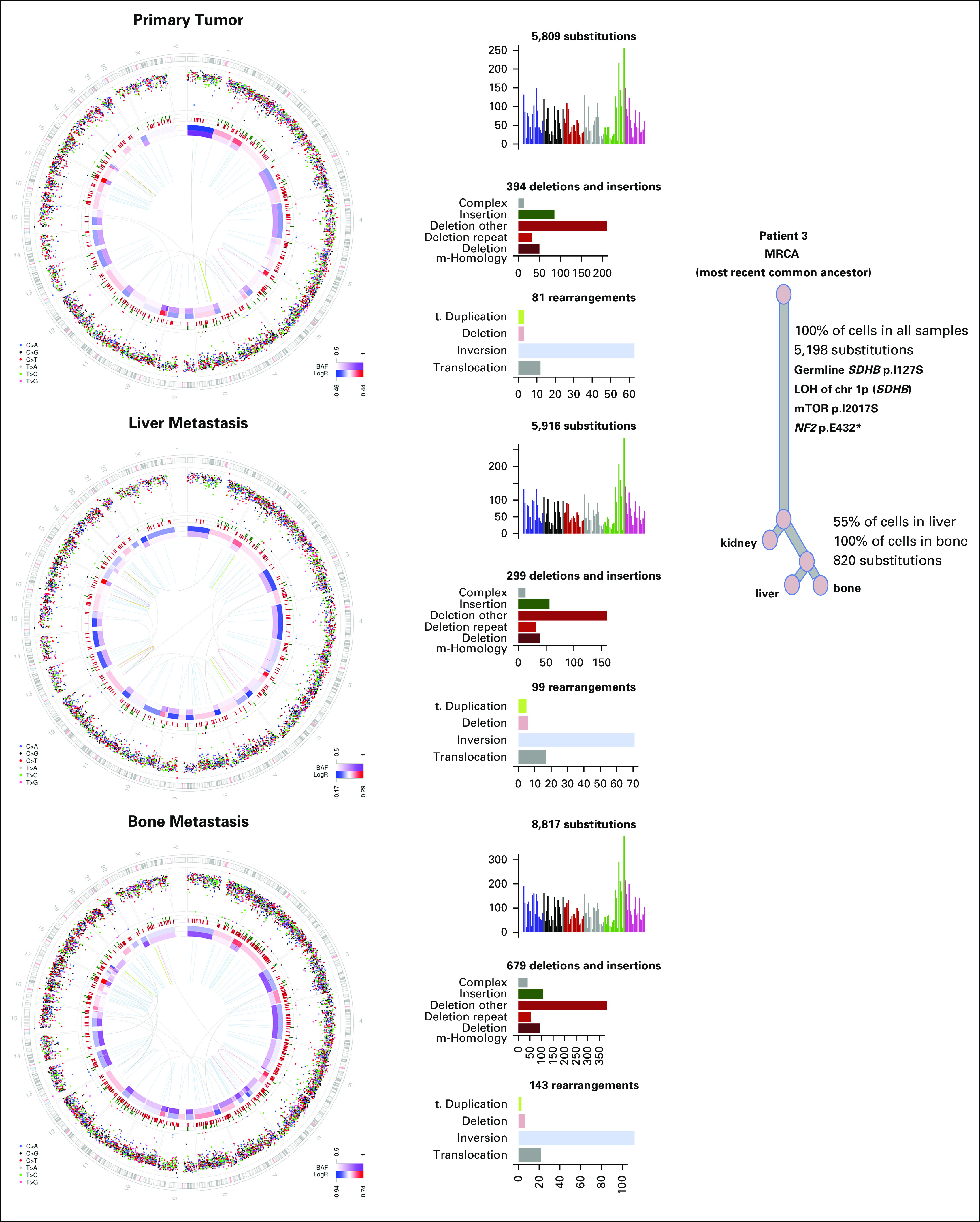
Whole-genome sequencing and clonal evolution analysis of patient 3’s paired primary-metastatic kidney tumors. MRCA denotes the most recent common ancestor cell identified during the cancer evolution of individual tumors.46 LOH, loss of heterozygosity; mTOR, mammalian target of rapamycin.
Disruption of Mitochondrial Architecture in SDHB-Deficient Cancer Cells
Because SDHB encodes a TCA cycle protein, we examined the mitochondria morphology of patient 1’s SDHB-deficient primary tumor cells under electron microscopy. Cells from unrelated normal kidney tissues (SDHB+/+) and nontumor (SDHB+/−) kidney tissues of patient 1 exhibited normal mitochondrial architecture with intact membranes and cristae (Fig 5A). In contrast, mitochondria of SDHB−/− tumor cells contained fewer cristae and less electron-dense matrix (Fig 5A).
Fig 5.

Morphologic and metabolic assessment of SDHB-deficient primary kidney cancer cells. (A) Electron microscopy images of indicated culture cells. Representative fields from transmitting electron microscopy images of the mitochondrial at 15,000× of primary cell cultures derived from an unrelated patient’s normal tissue (normal), tumor tissue from patient 1 (SDHB−/−), and adjacent nontumor tissue from patient 1 (SDHB+/−). (B) Extracellular acidification rates and oxygen consumption rate of indicated culture cells. Error bars indicate standard deviation (n = 8). (C) Glucose consumption and lactate production. Direct measurement of glucose and lactate in surrounding media over a 6-hour period is presented. Results were normalized to cell numbers and showed increased glucose consumption and lactate production in SDHB−/− primary cell cultures. Error bars indicate standard deviation (n = 3). (D) Relative succinate levels. Intracellular succinate was quantified by gas chromatography–mass spectrometry and then normalized to cell number. Error bars indicate standard deviation (n = 3). FCCP, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone.
Metabolic Assessment of SDHB-Deficient Primary Cancer Cells
To assess mitochondrial oxidative phosphorylation and glycolysis, oligomycin, an inhibitor of electron transport chain (ETC) complex V, was used to inhibit mitochondrial oxygen consumption, thereby activating glycolysis, and carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), an uncoupler of the mitochondrial proton gradient, was used to induce maximum mitochondrial oxygen consumption. Normal SDHB+/+ kidney cells and RCC4 cells exhibited similar decreases and increases in oxygen consumption in response to oligomycin (13.3 ± 0.9 pmol/min and 15.1 ± 1.0 pmol/min, respectively) and FCCP treatment (25.0 ± 3.0 pmol/min and 26.3 ± 3.0 pmol/min, respectively), whereas the SDHB−/− tumor cells exhibited minimum changes (3.3 ± 0.4 pmol/min; P < .001 v SDHB+/+ and P < .001 v RCC4), demonstrating a lack of oxidative phosphorylation (Fig 5B). When the basal ECAR was measured, it was much higher in SDHB−/− cancer cells (21 ± 1.1 mpH/min), and there was a minor increase upon oligomycin treatment (3.3 ± 1.1 mpH/min; 16%; Fig 5B). This suggests that these cells may be at near-maximum capacity for lactate production at baseline as a result of the pre-existing defect in oxidative respiration. In contrast, normal SDHB+/+ kidney cells and RCC4 cells had lower basal ECARs (5.0 ± 0.5 mpH/min and 8.1 ± 2.6 mpH/min, respectively) that increased upon oligomycin treatment (9.3 ± 1.3 mpH/min, 186%, P < .001; and 9.9 ± 1.4 mpH/min, 121%, P < .001, respectively; Fig 5B). Direct measurements of glucose and lactate levels in cell culture media confirmed that SDHB−/− cancer cells used more glucose and produced more lactate compared with unrelated primary normal kidney cell cultures and two clear cell RCC cell lines, RCC4 and A498 (Fig 5C). Measurement of intracellular metabolites showed that SDHB−/− kidney cancer cells had increased succinate compared with SDHB+/− cells and RCC4 and A498 (Fig 5D), suggesting that loss of both copies of SDHB resulted in succinate accumulation.
Glutamine Uptake in the SDHB-Deficient Kidney Cancer
To determine whether the use of other carbon sources was altered in SDHB-deficient kidney cancer, an 18F-glutamine PET was used to measure glutamine uptake in patient 2 (Fig 6A). Patient 2’s tumors exhibited elevated 18F-glutamine uptake, implicating increased dependence on glutamine metabolism. Using primary SDHB−/− tumor cells from patient 1, glutamine deprivation resulted in decreases in glutamate and succinate, which are downstream catabolites of glutamine (Fig 6B). In contrast, malate downstream of the metabolic defect was not affected by glutamine deprivation. Similarly, lactate downstream of glucose metabolism was unaffected by glutamine deprivation (Fig 6B). Furthermore, prolonged glutamine deprivation in SDHB−/− tumor cells resulted in increased cell death (Fig 6C).
Fig 6.
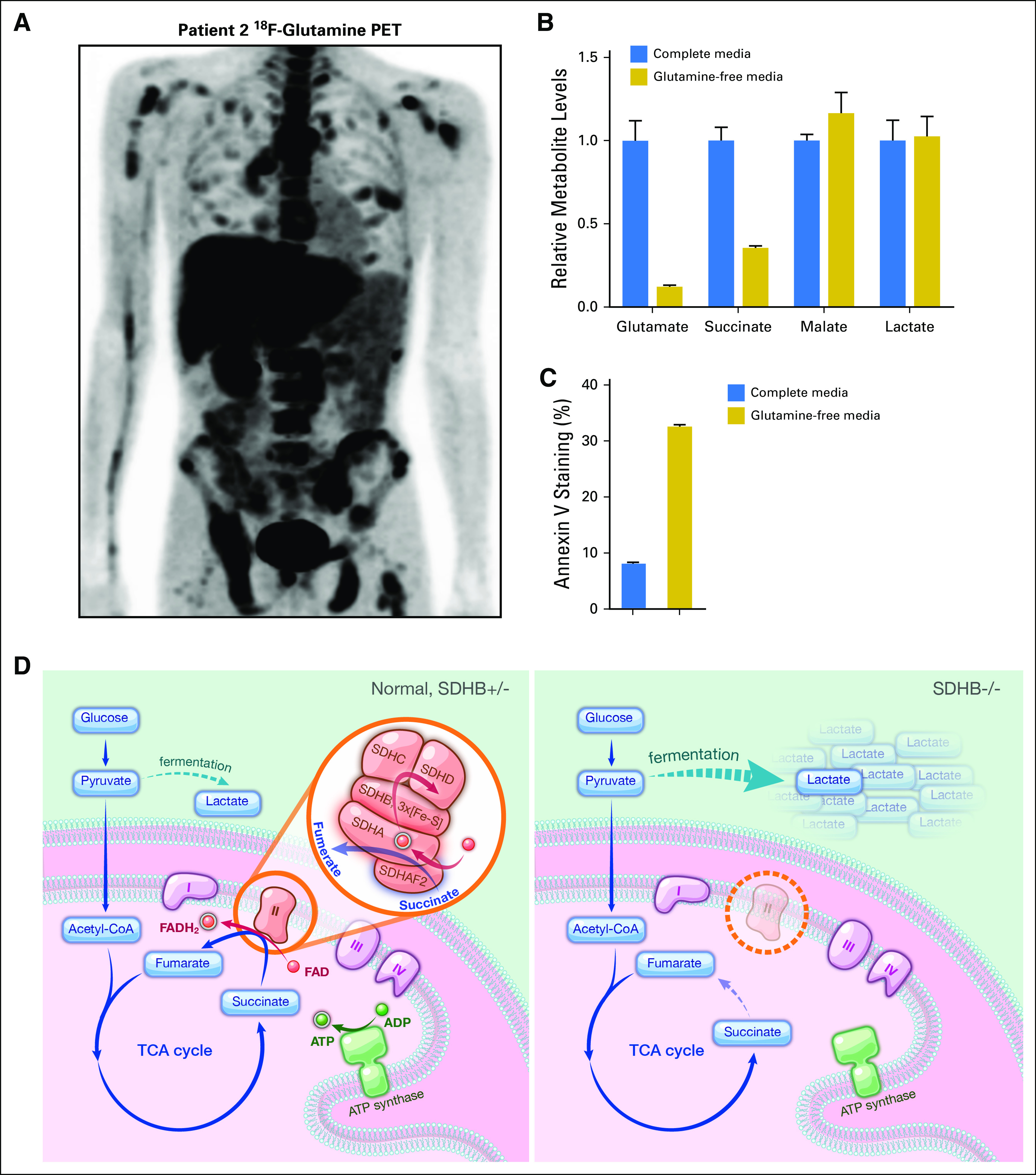
Glutamine metabolism and model of lactate production in SDHB-deficient human kidney cancer. (A) The 18F-glutamine positron emission tomography (PET) scan of patient 2. (B) Levels of intracellular metabolites. Relative metabolite levels were measured in SDHB−/− primary cell cultures after 6 hours of glutamine starvation. Counts are normalized to cell numbers, and results from full complete media are denoted at 1. Error bars indicate standard deviation (n = 3). (C) Measurement of apoptosis. Annexin V staining of apoptotic cells was determined in SDHB−/− primary cell culture after 72 hours of glutamine deprivation. Error bars indicate standard deviation (n = 3). (D) Model depicts the increased glucose flux and lactate production in SDHB-deficient cancer cells. CoA, coenzyme A; FAD, flavin adenine dinucleotide; TCA, tricarboxylic acid.
DISCUSSION
Here we report three patients with SDHB-deficient lethal RCC. WGS identified a novel exon 4-5 deletion of the SDHB gene in the germline as the first genetic event of patient 1 and demonstrated chromosome 1p loss in this patient’s tumor as the second genetic event, leading to the homozygous loss of SDHB at 1p36. All three individuals carry distinct germline mutations in one copy of the SDHB gene, and through chromosome 1p loss, the remaining copy of SDHB is lost in their tumors, consistent with the Knudson two-hit hypothesis.59
Mitochondrion generates ATP and provides biosynthetic intermediates via two interlinked processes (ie, ETC and TCA cycle). Unlike the other dedicated TCA cycle enzymes, the SDH enzyme also functions as complex II in ETC, coupling oxidation of succinate to fumarate in the TCA cycle with electron transfer to ubiquinone in the ETC. SDH enzyme is highly conserved and consists of A, B, C, and D subunits, with SDHA and SDHB as the catalytic subunits extending into the mitochondrial matrix and anchored to its inner membrane with SDHC and SDHD (Fig 6D). Recent studies elucidated a direct causal relationship between the loss of SDHB, SDHC, or SDHD and the development of rare hereditary kidney cancer. Of note, homozygous mutations in SDHA cause severe neurologic defects in infants, known as Leigh syndrome, characterized by subacute necrotizing encephalomyelopathy, failure to thrive, ataxia, seizures, and severe lactic acidosis.37 To our knowledge, the association between chronic hyperlactatemia and overt metastatic SDHB kidney cancer has never been reported.
Metabolic assessment of patient 1's SDHB−/− tumor cells demonstrated the uncoupling between ETC and TCA, manifested by the lack of oxidative phosphorylation, the overproduction of lactate, and the dependence on aerobic glycolysis, exemplifying the extreme Warburg effect in human cancer (Fig 6D). The mechanisms by which SDHB loss might contribute to tumorigenesis have been investigated using cell-based assays and mouse paraganglioma models.60-63 These studies highlighted the role of accumulated succinate as an oncometabolite that inhibits α-ketoglutarate–dependent dioxygenases, resulting in metabolic and epigenetic changes.60-63 In fact, our genomic study of these patients demonstrated that the complete loss of SDHB constitutes the first event in the pathogenesis of SDHB-deficient kidney cancer.
From 2011 to 2015, 4,328 patients were diagnosed with RCC at Memorial Sloan Kettering Cancer Center, among whom eight (0.18%) have SDHB-deficient RCC. Limited clinical data presented in our small series suggested varied treatment benefit with vascular endothelial growth factor and possibly mammalian target of rapamycin inhibitors, and the use of glutaminase inhibitor in this rare disease deserves further investigation. Altogether, we wish to raise awareness of the demographic, radiographic, biochemical, histologic, and immunohistochemical features associated with lethal human SDHB-deficient kidney cancers.
Supplementary Material
{kind=link}
ACKNOWLEDGMENT
We dedicate this article to the patients and their families and are forever grateful for their inspirational courage instilled upon us when fighting against this deadly disease.
Footnotes
See accompanying editorial doi:https://doi.org/10.1200/PO.17.00037
Supported by the Bryan Fund for SDHB kidney cancer; the Tuttle Fund for the cure of rare kidney cancer; the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research; and Memorial Sloan Kettering Cancer Center Cancer Center Support Grant/Core Grant (No. P30 CA008748). C.-H.L. and G.G. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Chung-Han Lee, William Lee, Ying-Bei Chen, Emily H. Cheng, Mark P. Dunphy, W. Marston Linehan, James J. Hsieh
Financial support: Mark P. Dunphy, James J. Hsieh
Administrative support: Dayna Oschwald, Mark P. Dunphy, James J. Hsieh
Provision of study material or patients: Ramaprasad Srinivasan, Mark P. Dunphy, James J. Hsieh
Collection and assembly of data: Chung-Han Lee, William Lee, Ying-Bei Chen, Justin R. Cross, Yiyu Dong, Almedina Redzematovic, Roy Mano, Elizabeth Y. Wei, Dayna Oschwald, A. Ari Hakimi, Mark P. Dunphy, W. Marston Linehan, James J. Hsieh
Data analysis and interpretation: Chung-Han Lee, Gunes Gundem, William Lee, Ying-Bei Chen, Justin R. Cross, Almedina Redzematovic, Emily H. Cheng, Ramaprasad Srinivasan, Dayna Oschwald, Mark P. Dunphy, Elli Papaemmanuil, James J. Hsieh
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or po.ascopubs.org/site/ifc.
Chung-Han Lee
Consulting or Advisory Role: Exelixis
Research Funding: Pfizer (Inst), Eisai (Inst)
Gunes Gundem
No relationship to disclose
William Lee
Employment: Helix OpCo
Leadership: Helix OpCo
Stock and Other Ownership Interests: Helix OpCo
Travel, Accommodations, Expenses: Helix OpCo
Ying-Bei Chen
No relationship to disclose
Justin R. Cross
No relationship to disclose
Yiyu Dong
No relationship to disclose
Almedina Redzematovic
No relationship to disclose
Roy Mano
No relationship to disclose
Elizabeth Y. Wei
No relationship to disclose
Emily H. Cheng
No relationship to disclose
Ramaprasad Srinivasan
No relationship to disclose
Dayna Oschwald
No relationship to disclose
A. Ari Hakimi
No relationship to disclose
Mark P. Dunphy
No relationship to disclose
W. Marston Linehan
No relationship to disclose
Elli Papaemmanuil
No relationship to disclose
James J. Hsieh
Honoraria: Novartis, Pfizer, Chugai, Eisai, Cancer Genomics
Consulting or Advisory Role: Novartis, Pfizer, Chugai, Eisai, Cancer Genomics
Research Funding: Novartis, Pfizer, Chugai, Eisai, Cancer Genomics
REFERENCES
- 1.Moch H, Cubilla AL, Humphrey PA, et al. : The 2016 WHO classification of tumours of the urinary system and male genital organs—Part A: Renal, penile, and testicular tumours. Eur Urol 70:93-105, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Linehan WM, Srinivasan R, Schmidt LS: The genetic basis of kidney cancer: A metabolic disease. Nat Rev Urol 7:277-285, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. doi: 10.1016/j.semcdb.2016.09.002. Hsieh JJ, Manley BJ, Khan N, et al: Overcome tumor heterogeneity-imposed therapeutic barriers through convergent genomic biomarker discovery: A braided cancer river model of kidney cancer. Semin Cell Dev Biol 10.1016/j.semcdb.2016.09.002 [epub ahead of print on September 8, 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network : Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499:43-49, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network, Linehan WM, Spellman PT, et al. : Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med 374:135-145, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis CF, Ricketts CJ, Wang M, et al. : The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 26:319-330, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zisman A, Chao DH, Pantuck AJ, et al. : Unclassified renal cell carcinoma: Clinical features and prognostic impact of a new histological subtype. J Urol 168:950-955, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Beltran A, Scarpelli M, Montironi R, et al. : 2004 WHO classification of the renal tumors of the adults. Eur Urol 49:798-805, 2006 [DOI] [PubMed] [Google Scholar]
- 9.Chen YB, Xu J, Skanderup AJ, et al. : Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun 7:13131, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pollard PJ, Wortham NC, Tomlinson IP: The TCA cycle and tumorigenesis: The examples of fumarate hydratase and succinate dehydrogenase. Ann Med 35:632-639, 2003 [DOI] [PubMed] [Google Scholar]
- 11. Gill AJ: Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology 44:285-292, 2012. [DOI] [PubMed]
- 12.Bardella C, Pollard PJ, Tomlinson I: SDH mutations in cancer. Biochim Biophys Acta 1807:1432-1443, 2011 [DOI] [PubMed] [Google Scholar]
- 13.Eng C: SDHB: A gene for all tumors? J Natl Cancer Inst 100:1193-1195, 2008 [DOI] [PubMed] [Google Scholar]
- 14.Gill AJ, Pachter NS, Clarkson A, et al. : Renal tumors and hereditary pheochromocytoma-paraganglioma syndrome type 4. N Engl J Med 364:885-886, 2011 [DOI] [PubMed] [Google Scholar]
- 15.Shuch B, Ricketts CJ, Metwalli AR, et al. : The genetic basis of pheochromocytoma and paraganglioma: Implications for management. Urology 83:1225-1232, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricketts C, Woodward ER, Killick P, et al. : Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 100:1260-1262, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Vanharanta S, Buchta M, McWhinney SR, et al. : Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet 74:153-159, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ricketts CJ, Forman JR, Rattenberry E, et al. : Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat 31:41-51, 2010 [DOI] [PubMed] [Google Scholar]
- 19. doi: 10.1038/gim.2014.162. Evenepoel L, Papathomas TG, Krol N, et al: Toward an improved definition of the genetic and tumor spectrum associated with SDH germ-line mutations. Genet Med 17:610-620, 2015. [DOI] [PubMed] [Google Scholar]
- 20.Williamson SR, Eble JN, Amin MB, et al. : Succinate dehydrogenase-deficient renal cell carcinoma: Detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol 28:80-94, 2015 [DOI] [PubMed] [Google Scholar]
- 21.Warburg O, Wind F, Negelein E: The metabolism of tumors in the body. J Gen Physiol 8:519-530, 1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koppenol WH, Bounds PL, Dang CV: Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 11:325-337, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Owen OE, Kalhan SC, Hanson RW: The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277:30409-30412, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Vander Heiden MG, Cantley LC, Thompson CB: Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 324:1029-1033, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee DC, Sohn HA, Park ZY, et al. : A lactate-induced response to hypoxia. Cell 161:595-609, 2015 [DOI] [PubMed] [Google Scholar]
- 26.Lu J, Tan M, Cai Q: The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett 356:156-164, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fraley DS, Adler S, Bruns FJ, et al. : Stimulation of lactate production by administration of bicarbonate in a patient with a solid neoplasm and lactic acidosis. N Engl J Med 303:1100-1102, 1980 [DOI] [PubMed] [Google Scholar]
- 28.Kreisberg RA: Lactate homeostasis and lactic acidosis. Ann Intern Med 92:227-237, 1980 [DOI] [PubMed] [Google Scholar]
- 29.Kraut JA, Madias NE: Lactic acidosis. N Engl J Med 371:2309-2319, 2014 [DOI] [PubMed] [Google Scholar]
- 30.Khosravani H, Shahpori R, Stelfox HT, et al. : Occurrence and adverse effect on outcome of hyperlactatemia in the critically ill. Crit Care 13:R90, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huckabee WE: Abnormal resting blood lactate: I. The significance of hyperlactatemia in hospitalized patients. Am J Med 30:840-848, 1961 [DOI] [PubMed] [Google Scholar]
- 32.Madias NE: Lactic acidosis. Kidney Int 29:752-774, 1986 [DOI] [PubMed] [Google Scholar]
- 33.Friedenberg AS, Brandoff DE, Schiffman FJ: Type B lactic acidosis as a severe metabolic complication in lymphoma and leukemia: A case series from a single institution and literature review. Medicine (Baltimore) 86:225-232, 2007 [DOI] [PubMed] [Google Scholar]
- 34.de Groot R, Sprenger RA, Imholz AL, et al. : Type B lactic acidosis in solid malignancies. Neth J Med 69:120-123, 2011 [PubMed] [Google Scholar]
- 35.Martinez-Outschoorn UE, Whitaker-Menezes D, Valsecchi M, et al. : Reverse Warburg effect in a patient with aggressive B-cell lymphoma: Is lactic acidosis a paraneoplastic syndrome? Semin Oncol 40:403-418, 2013 [DOI] [PubMed] [Google Scholar]
- 36.Hanahan D, Weinberg RA: Hallmarks of cancer: The next generation. Cell 144:646-674, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Eng C, Kiuru M, Fernandez MJ, et al. : A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer 3:193-202, 2003 [DOI] [PubMed] [Google Scholar]
- 38.Grubb RL III, Franks ME, Toro J, et al. : Hereditary leiomyomatosis and renal cell cancer: A syndrome associated with an aggressive form of inherited renal cancer. J Urol 177:2074-2079, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Frezza C, Zheng L, Folger O, et al. : Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 477:225-228, 2011 [DOI] [PubMed] [Google Scholar]
- 40.Linehan WM, Rouault TA: Molecular pathways: Fumarate hydratase-deficient kidney cancer—Targeting the Warburg effect in cancer. Clin Cancer Res 19:3345-3352, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hakimi AA, Reznik E, Lee CH, et al. : An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell 29:104-116, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei EY, Hsieh JJ: A river model to map convergent cancer evolution and guide therapy in RCC. Nat Rev Urol 12:706-712, 2015 [DOI] [PubMed] [Google Scholar]
- 43.Wettersten HI, Hakimi AA, Morin D, et al. : Grade-dependent metabolic reprogramming in kidney cancer revealed by combined proteomics and metabolomics analysis. Cancer Res 75:2541-2552, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu J, Pham CG, Albanese SK, et al. : Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J Clin Invest 126:3526-3540, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ricketts CJ, Shuch B, Vocke CD, et al. : Succinate dehydrogenase kidney cancer: An aggressive example of the Warburg effect in cancer. J Urol 188:2063-2071, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gundem G, Van Loo P, Kremeyer B, et al. : The evolutionary history of lethal metastatic prostate cancer. Nature 520:353-357, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bolli N, Avet-Loiseau H, Wedge DC, et al. : Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun 5:2997, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Voss MH, Hakimi AA, Pham CG, et al. : Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin Cancer Res 20:1955-1964, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venneti S, Dunphy MP, Zhang H, et al. : Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med 7:274ra17, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyakita H, Tokunaga M, Onda H, et al. : Significance of 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) for detection of renal cell carcinoma and immunohistochemical glucose transporter 1 (GLUT-1) expression in the cancer. Int J Urol 9:15-18, 2002 [DOI] [PubMed] [Google Scholar]
- 51.Majhail NS, Urbain JL, Albani JM, et al. : F-18 fluorodeoxyglucose positron emission tomography in the evaluation of distant metastases from renal cell carcinoma. J Clin Oncol 21:3995-4000, 2003 [DOI] [PubMed] [Google Scholar]
- 52.Ho CL, Chen S, Ho KM, et al. : Dual-tracer PET/CT in renal angiomyolipoma and subtypes of renal cell carcinoma. Clin Nucl Med 37:1075-1082, 2012 [DOI] [PubMed] [Google Scholar]
- 53.Srigley JR, Delahunt B, Eble JN, et al. : The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. Am J Surg Pathol 37:1469-1489, 2013 [DOI] [PubMed] [Google Scholar]
- 54.Sankin A, Hakimi AA, Hsieh JJ, et al. : Metastatic non-clear cell renal cell carcinoma: An evidence based review of current treatment strategies. Front Oncol 5:67, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. doi: 10.1200/JCO.2016.67.9084. Voss MH, Molina AM, Chen YB, et al: Phase II trial and correlative genomic analysis of everolimus plus bevacizumab in advanced non-clear cell renal cell carcinoma. J Clin Oncol 34:3846-3853, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gill AJ, Pachter NS, Chou A, et al. : Renal tumors associated with germline SDHB mutation show distinctive morphology. Am J Surg Pathol 35:1578-1585, 2011 [DOI] [PubMed] [Google Scholar]
- 57.Reva B, Antipin Y, Sander C: Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res 39:e118, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collins N, Dietzek A: Contiguous bilateral head and neck paragangliomas in a carrier of the SDHB germline mutation. J Vasc Surg 55:216-219, 2012 [DOI] [PubMed] [Google Scholar]
- 59.Knudson AG: Two genetic hits (more or less) to cancer. Nat Rev Cancer 1:157-162, 2001 [DOI] [PubMed] [Google Scholar]
- 60.Letouzé E, Martinelli C, Loriot C, et al. : SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23:739-752, 2013 [DOI] [PubMed] [Google Scholar]
- 61.Selak MA, Armour SM, MacKenzie ED, et al. : Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7:77-85, 2005 [DOI] [PubMed] [Google Scholar]
- 62.Xiao M, Yang H, Xu W, et al. : Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26:1326-1338, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cardaci S, Zheng L, MacKay G, et al. : Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat Cell Biol 17:1317-1326, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]