

Abstract
The importance of studying the mechanistic aspects of long non-coding RNAs is being increasingly emphasized as more and more regulatory RNAs are being discovered. Non-coding RNA sequences directly associate with generic RNA-binding proteins as well as specific proteins, which cooperate in the downstream functions of the RNA and can also be dysregulated in various physiologic states and/or diseases. While current methods exist for identifying RNA–protein interactions, these methods require high quantities of input cells or use pooled capture reagents that may increase non-specific binding. We have developed a method to efficiently capture specific RNAs using less than one million input cells. One single oligonucleotide is used to pull down the target RNA of choice and oligonucleotide selection is driven by sequence accessibility. We perform thermal elution to specifically elute the target RNA and its associated proteins, which are identified by mass spectrometry. Ultimately, two target and control oligonucleotides are used to create an enrichment map of interacting proteins of interest.
This protocol was validated in: eLife (2021), DOI: 10.7554/eLife.68263
Keywords: Pull down , Mass spectrometry , RNA–protein interactions , Temperature elution , Non-coding RNA
Background
The interplay between an organism’s two essential macromolecules, RNA and proteins, dictate much of the cell’s functions ( Zhu, 2010 ). Messenger RNAs contain instructions for protein synthesis and may contain regulatory sequences that affect the levels of the encoded protein ( Mayr, 2017 ). As a complement, proteins can bind to RNA and modulate expression, function, and stability ( Moore, 2005 ). Perturbation or loss of physical interactions can lead to loss of genomic stability, aberrant cellular signaling, or global changes in gene expression (Allerson et al., 1999; Batista and Chang, 2013 ; Lee et al., 2016). It has been shown that at least 5% of the human proteome is capable of binding to RNA, and our catalogue of RNA binding domains is incredibly incomplete ( Castello et al., 2012 ). Of particular recent interest are a class of non-protein-coding RNA transcripts called long non-coding RNAs (lncRNAs) that have been shown to hold regulatory roles in embryonic stem cell (ESC) differentiation and can be dysregulated in cancer ( Guttman et al., 2011 ; Bergmann et al., 2015 ; He et al., 2017 ). In addition, lncRNAs have also been shown to serve as therapeutic molecular targets, and the ease of designing antisense oligonucleotides to target them for degradation or inhibition is a large untapped field of therapeutics ( Liang et al., 2017 ; Crooke et al., 2017 ; Zong et al., 2015 ; Arun et al., 2016 , 2018; Chang et al., 2020 ; Yu et al., 2021 ).
However, as many more functional lncRNAs are being discovered, their molecular mechanisms of action are largely unknown. Elucidation of RNA–protein interactions have not progressed as quickly, due to the lack of streamlined and efficient methods. Current methods include crosslinking cells, needing an incredibly high quantity of starting material, or requiring expensive and long modified capture reagents ( Hacisuleyman et al., 2014 ; Chen et al., 2016 ; Chu et al., 2011 ). Multiple oligonucleotides used for antisense capture also can result in possible off-target effects ( Chu et al., 2015 ). In addition, the final elution conditions may not necessarily be compatible with mass spectrometry (MS). Because the ultimate goal is to identify proteins, the final elution buffer used must be compatible with downstream applications in order to avoid additional buffer exchange or purification steps, which could lead to loss of material and cause potential biases.
We have developed a method to efficiently capture RNAs without the need to use a high quantity of input or pooled capture reagents to identify the associated proteins. One single oligonucleotide is used to pull down the target RNA of choice and oligonucleotide selection is driven by sequence accessibility. Because the pull down is mediated by one single oligonucleotide, temperature can be used as an option for elution. The melting point of the oligonucleotide-to-RNA interaction can be empirically determined and thus an optimal elution temperature can be found. This allows for a very high degree of selectivity and discrimination, as tighter interactors that elute at higher temperatures are more likely to be specific. An additional step of quality control is possible with this method as well, as the full elution of the target RNA can be quantified using qRT-PCR on the eluate and the beads. In addition, elution can be performed in a buffer that is completely compatible with any downstream application, such as digestion or peptide-labelling for MS. Elution of the RNA–protein complex from the capture oligonucleotide is thermally controlled and measurable by qRT-PCR, thus allowing for quality control and specificity of protein hits. We call this method S ingle O ligonucleotide C apture of R NA A nd T emperature E lution S eries, or SOCRATES .
Graphical abstract
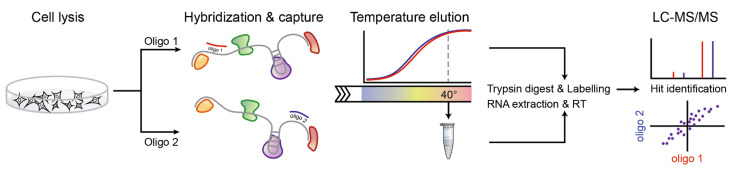
Schematic representation of the SOCRATES workflow. SOCRATES utilizes a single 20-mer oligonucleotide for RNA pull down followed by a temperature elution series and liquid chromatography–mass spectrometry (LC-MS)/MS to identify specific RNA–protein interactions.
Materials and Reagents
10 cm sterile cell culture dish (e.g., Corning, Falcon ® , catalog number: 353003)
15 mL and 50 mL conical tubes (e.g., Crystalgen, catalog number: 23-2265; Corning, Falcon ® , catalog number: 352098)
Sterile pipette tips (e.g., Corning)
0.2 mL PCR strip tubes (e.g., Corning, Axygen ® , catalog number: PCR-0208-CP-C)
Low adhesion microcentrifuge (Eppendorf) tube (e.g., USA Scientific, catalog number: 1415-2690)
96-well reaction plates (e.g., Thermo Fischer Scientific, Applied Biosystems TM , catalog number: 4346907)
Optical adhesive film (e.g., Thermo Fischer Scientific, Applied Biosystems TM , catalog number: 4311971)
200 proof ethyl alcohol (e.g., UltraPure, catalog number: 200CSPTP)
Sterile water
Ice
1× DPBS (e.g., Thermo Fisher Scientific, Gibco TM , catalog number: 14190250)
Dynabeads TM MyOne TM streptavidin C1 (Invitrogen, catalog number: 65001)
IP lysis buffer (e.g., Pierce IP Lysis Buffer; Thermo Fisher Scientific, catalog number: 87787)
cOmplete TM mini protease inhibitor cocktail (Roche, catalog number: 11836153001)
SUPERase-In RNase inhibitor (Invitrogen, catalog number: AM2694)
Magnetic racks (e.g., Invitrogen DynaMag, catalog number: 12321D for 1.5–2 mL Eppendorf tubes, 12331D for 0.2 mL PCR strip tubes)
BCA assay kit (e.g., Pierce BCA protein assay kit; Thermo Fisher Scientific, catalog number: 23227)
Triethylammonium bicarbonate (TEAB) 1 M for mass spectrometry (Thermo Scientific, catalog number: 90114)
TRIzol (Thermo Fisher Scientific, Invitrogen, catalog number: 15596018)
GlycoBlue (Thermo Fisher Scientific, Invitrogen, catalog number: AM9516)
DNase I, amplification grade, for cDNA synthesis (e.g., Thermo Fisher Scientific, Invitrogen, catalog number: 18068015)
Ethylenediaminetetraacetate acid disodium salt (EDTA) (included in DNase I kit; Thermo Fisher Scientific, catalog number: 18068015)
Nuclease-free water (e.g., Thermo Fisher Scientific, Invitrogen, catalog number: AM9937)
TaqMan reverse transcription kit (Thermo Fisher Scientific, Invitrogen, catalog number: N8080234)
PowerUp SYBR green master mix (Thermo Fischer Scientific, Applied Biosystems, catalog number: A25743)
Chloroform, purified (e.g., Avantor Performance Materials, MACRON, catalog number: 4432-10)
Isopropanol, molecular biology grade (e.g., Fisher Scientific, catalog number: BP2618500)
Cell lysis buffer (see Recipes)
cDNA master mix per reaction (see Recipes)
qPCR master mix (see Recipes)
Equipment
Biological safety cabinet for tissue culture (e.g., NuAire)
Cell culture incubator (e.g., Heracell-I Copper CO 2 incubator, Thermo Fischer Scientific, Thermo Scientific TM , model: Heracell TM 150i and 240i, catalog number: 50116050)
Gentle rotating mixer for Eppendorf tubes (end-over-end rotation e.g., VWR, catalog number: 10136-084)
Centrifuge for 15 and 50 mL tubes (e.g., Eppendorf, model: 5804)
Centrifuge for 1.5 mL reaction tubes with cooling function (e.g., Eppendorf, model: 5427 R)
Vacuum suction
Micro-pipettes (e.g., Gilson, catalog number: F167300)
NanoDrop 2000 UV-Vis spectrophotometer (Thermo Fisher Scientific, Thermo Scientific TM , model: NanoDrop TM 2000, catalog number: ND-2000)
PCR machine (e.g., Applied Biosystems Proflex Thermocycler, Thermo Fisher Scientific, Applied Biosystems TM , catalog number: 4484073)
qPCR machine (e.g., Applied Biosystems StepOne Plus Real-Time PCR system, Thermo Fisher Scientific, Thermo Scientific TM , catalog number: 4376600)
-80 °C freezer (e.g., VWR, catalog number: 10160-728)
Refrigerator (4 °C)
Procedure
-
Design of antisense oligonucleotides (ASOs) for RNA capture
Choose a target gene for pull down. The protocol described here is equally suitable for long non-coding RNAs and protein-coding transcripts.
Design 20-mers complementary to exons of the RNA sequence. Here, 100% sequence identity is required; design at least two ASOs for the target sequence as well as a negative control [e.g., peptidylprolyl isomerase B ( PPIB )]. It is recommended to start with up to ten individual ASOs for new targets tiling the entire length of the mature transcript, as not every ASO will result in an efficient pull down.
Use Nucleotide BLAST ( https://blast.ncbi.nlm.nih.gov/Blast.cgi ) to ensure the ASOs are specific to the intended target only. Stringency is important at this step—even one or two mismatches can result in unwanted off-target pull downs.
ASOs used in pull-down experiments carry a modified 3’ end with biotin to assist in capture. In the United States, 3’ biotin modifications can be ordered, for example, from LGC Biosearch Technologies. We generally prefer a 50 nmol scale with a 3’ biotin, six carbon linker, and reverse-phase cartridge purification.
Upon arrival, resuspend lyophilized ASOs in sterile DPBS to a working stock concentration of 200 µM.
Store at 4 °C for short-term storage (up to one week) or at -20 °C for long-term storage. Aliquot to avoid repeated freeze-thaw cycles.
-
Preparation of cell lysate
Prepare a master mix of cell lysis buffer (see Recipe 1). The volume of each reaction is 500 μL, which can be scaled up if needed.
Obtain a 10 cm plate at 70%–80% confluence of the cell lines of interest. Place on ice and aspirate the medium. Wash twice with 1× DPBS.
Leave the plate tilted for a minute to remove all of the residual PBS. This is to prevent dilution of the lysis buffer.
Lyse the cells on the plate using approximately 1 mL of cell lysis buffer per plate on ice for 10 min; then, harvest cells by scraping and transfer to low adhesion microcentrifuge tubes.
Spin down lysate at 13,000 × g for 10 min at 4 °C to pellet debris.
If harvesting from multiple plates and using multiple Eppendorf tubes, combine and mix supernatant lysate in a 15 mL conical tube before proceeding to the capture step.
-
(Highly recommended) Perform a BCA assay on the cell lysate. Adjust the cell lysate to a concentration of 0.3 mg/mL using cell lysis buffer.
Note: While this step is optional, we highly recommend for it to be performed. We have found that in general, a 0.3 mg/mL protein concentration provides the optimum balance between pull-down efficiency and protein yield for peptide detection, as the cell input for this protocol is very low compared to previously published protocols (see Figure 1 below). As lysate protein concentration increases, we observe that yield decreases as seen in Figure 2 below.
-
Anti-sense capture with ASOs and magnetic beads
From the lysate stock, aliquot 50 μL for 10% RNA input and 50 μL for 10% protein input, if needed. Leave the tubes on ice for the whole duration of the pull down. Do not add TRIzol to the input until the pull-down samples are in TRIzol at the end of this protocol.
-
Aliquot 500 μL of lysate stock per sample and add 100 pmol of oligonucleotide to make a final concentration of 200 nM. This is generally 0.5 μL of the 200 µM stock. Do not vortex.
Note: Extra lysate can be labelled and stored at -80 °C for future validation experiments such as western blot.
-
Incubate at room temperature for 1 h with gentle agitation on the rotator.
Note: As a general note for mass spectrometry and pull-down experiments, it is very important to adhere to the same protocol for each replicate. Any deviation should be recorded in the event of inconsistent results. In addition, fluctuations in room temperature (e.g., 20–28 °C) may also be a source of error that may be compounded throughout the experiment. Take care not to vortex the sample prior to capture with the magnetic beads.
While the samples are incubating, prepare the Dynabeads by aliquoting 100 μL per reaction into a fresh Eppendorf tube for each reaction and place on the magnetic stand for 2 min to capture the beads.
Remove the Dynabeads storage buffer gently and wash twice with 100 µL of lysis buffer, capturing the beads for 2 min in between washes. Basic lysis buffer without protease inhibitor cocktail and RNase inhibitor can be used here as well.
Remove the lysis wash buffer and remove the beads from the magnet. Place on ice until ready to use.
Add samples to the washed Dynabeads and incubate at room temperature for 30 min with the same gentle agitation. Do not vortex.
Once the incubation is done, place the samples on a magnetic stand on ice to capture the beads for 2 min.
Wash the beads three times with 800 µL of basic lysis buffer. Allow 2 min between washes for bead capture on the stand. Take care to remove all buffer, as any remaining will interfere with the elution process.
-
Elution of RNA and protein from capture ASOs
Preheat a thermocycler to 40 °C.
Very carefully resuspend beads in 90 µL of ice cold 100 mM TEAB by pipetting up and down and transfer to a 0.2 mL PCR tube or a PCR tube compatible with the thermocycler.
-
Incubate the sample at 40 °C for exactly 10 min and immediately place on ice afterwards.
Note: In our experience, the vast majority of the oligonucleotides that we have tested achieve >95% elution at 40 °C. However, if elution is not fully achieved at that temperature, another oligonucleotide should be chosen or a temperature curve can be performed in order to identify the optimum elution temperature, as in Figure 3 below.
Allow the samples to cool on ice for 15 s. Perform a quick spin and place the sample on a magnetic stand on ice compatible with PCR tubes.
-
Leave the samples in the magnetic stand for 60 s to capture the beads. Very carefully remove the eluate and transfer to a new Eppendorf tube.
Note: Removing the eluate from the beads is a critical step. After performing temperature elution, do not wait longer than one minute to transfer the eluate. The RNA will rebind to the capture oligonucleotide over time and efficiencies will not be accurate. Process in batches if handling many samples.
Immediately aliquot 30 µL for mass spectrometry, 30 µL for RNA, and the remaining for immunoblots. As TEAB is a volatile compound, take care when pipetting.
-
Add 1 mL of TRIzol to the RNA aliquot as well as the input that was set aside in the beginning of the experiment. Store protein samples at -20 °C until ready to process. Proceed to cDNA synthesis and qPCR analysis with the RNA samples.
Note: Ideally, the samples should be processed within 24 h for consistency, but they are stable in TRIzol for up to one month. Care must be taken in order to have consistent results across replicates.
-
cDNA synthesis and qPCR to analyze pull-down efficiency
Isolate RNA from pull-down samples according to the manufacturer’s instructions. Addition of glycogen (GlycoBlue) is necessary in order to ensure complete extraction of RNA.
Allow the RNA pellet to air dry for 5 min. Resuspend in 8 µL of nuclease-free water.
Perform DNase digestion to remove any potentially co-purified genomic DNA by adding 1 µL of 10× DNase reaction buffer and 1 µL (1 U/µL) of DNase I to the RNA sample (total volume: 10 µL).
Mix reaction by gently pipetting up and down. Spin briefly in a microcentrifuge.
Incubate reaction at 25 °C in a PCR machine for 15 min.
Add 1 µL of 25 mM EDTA.
Mix reaction by gently pipetting up and down. Spin briefly in a microcentrifuge.
Inactivate reaction at 65 °C in a PCR cycler for 10 min.
Transfer samples on ice immediately after the 10 min incubation is completed. Do not leave samples in the PCR machine while ramping down to room temperature.
Prepare cDNA master mix (see Recipe 2).
Add 39 µL of cDNA master mix to samples on ice.
Mix reaction by gently pipetting up and down. Spin briefly in a microcentrifuge.
-
Perform the following incubation in a PCR machine for cDNA synthesis:
25 °C for 10 min
48 °C for 30 min
95 °C for 5 min
cDNA can be stored short-term at 4 °C or long-term at -20 °C.
-
Prepare qPCR master mix (see Recipe 3) containing primers for the target gene as well as qPCR master mix containing primers for an internal control ( housekeeping ) gene. When calculating the quantity of master mix needed, consider that samples will be pipetted in triplicates.
Note: For optimal results, primers for qPCR should not overlap with the capture oligonucleotide binding site, and the capture oligonucleotide should not bind within the qPCR amplified region.
Commonly used genes for internal controls include beta-actin, GAPDH , or ribosomal protein genes. Internal controls should be chosen carefully; their expression level should be consistent and independent of experimental conditions such as ASO treatments. The expression level of an internal control gene should be in the same range as the target gene. General rules regarding qPCR experiments and qPCR primer design can be found in Bustin et al. (2009).
Prepare a 96-well plate on ice.
Add 15 µL of master mix containing either the target gene or internal control gene primers to the wells.
Dilute the prepared cDNA 1:3 by adding 100 µL of nuclease-free water to each reaction.
Pipette 5 µL of diluted cDNA into one well of the 96-well plate. Each sample will be pipetted in triplicate for both the target and the housekeeping gene (= six wells containing 5 µL of cDNA per sample).
Seal the 96-well plate with an optical adhesive film. Avoid touching the film with your gloves, handling carefully by the edges.
Spin down the plate at 100 × g for 1 min at room temperature.
-
Place the plate in the qPCR machine and run the following program:
95 °C for 10 min
40 cycles of 95 °C for 15 s
60 °C for 60 s
Followed by
Melt curve 95 °C → 60 °C, 1 °C/min
-
After pull-down analysis, choose the best two oligonucleotides with the highest pull-down efficiency (ideally >70%) for the target gene and control gene and submit the eluate for digestion, iTRAQ 4-plex labelling, and sequencing at the mass spectrometry facility.
Note: Submission for digestion and labelling should occur within 24 h of obtaining the eluate if possible. The samples can be kept at -20 °C until the pull-down efficiency for the batch is confirmed via qRT-PCR. Lastly, TEAB is highly volatile, so care must be taken when handling this reagent.
Figure 1. SOCRATES requires 150,000 cells as input.

SOCRATES input cell number requirement is much lower than current methods ( West et al., 2014 ; Chu et al., 2015 ; McHugh et al., 2015 ). CHART: Capture Hybridization Analysis for RNA Targets; ChIRP: Chromatin isolation by RNA Purification; RAP-MS: RNA Antisense Purification with Mass Spectrometry.
Figure 2. Lysate protein concentration affects pull-down efficiency.
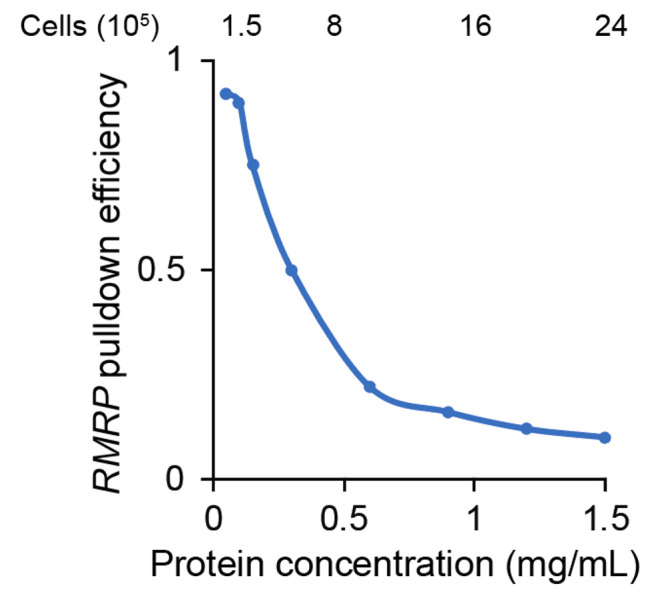
In this case, a protein concentration of 0.1 mg/mL would be optimal to pull down RMRP , a 286 nt transcript.
Figure 3. Temperature elution curve for PHAROH .

PHAROH ’s optimal elution temperature is 40 °C; >95% has been eluted off the beads and only <5% remained on the beads.
Data analysis
Pull-down efficiency
-
Export Ct values from the qPCR machine and analyze using the 2 -ΔΔCt Method ( Livak and Schmittgen, 2001 ). Input should be adjusted to 100% depending on how much was taken from the initial reaction. Pull down should range from 10%–90%, which can vary based on the oligonucleotide position along the RNA. Poor efficiencies may be due to saturated lysate or incorrect temperature for elution. Adherence to the protocol has a large impact on result consistency.
Note: We hypothesize that pull-down efficiency generally correlates with accessibility to the binding site. We have tiled the lncRNA NORAD with 32 oligonucleotides and found that areas of decreased binding correlated with published sites protein occupancy (see Figure 4 ). Regarding RNA length and pull-down efficiency, we have been successful in pull downs of RNAs that range from 286 nt (RMRP) to 5,300 nt (NORAD), with no fragmentation of the RNA detected (see Figure 5 ).
The two oligonucleotides for the target and control gene with the highest efficiency should be chosen for downstream mass spectrometry processing.
Figure 4. Pull-down profile maps accurately to published data.

Smoothed moving average pull-down profile of NORAD and occupancy PAR-CLIP data taken from West et al. (2014), Chu et al. (2015), and Lee et al. (2016); CLIP-seq data taken from Munschauer et al. (2018). The blue bars indicate areas of low pull-down efficiency, which correspond well with published occupancy data.
Figure 5. Full-length NORAD can be pulled down using single oligonucleotides.

Two primers spanning different regions of NORAD that are approximately 2 kb apart yield similar pull-down efficiencies, assayed by primer pairs on the 5’ and 3’ end, implying that NORAD is not fragmented. N = 3, error bars represent standard error of mean.
Mass spectrometry and ranked target acquisition
To identify proteins that bind to the target RNA, calculate the log2 enrichment ratio of target RNA to control RNA for each oligonucleotide. If using two control oligonucleotides, take the average enrichment of the two control oligonucleotides and use the resulting number as the denominator for the enrichment ratio.
Plot the log2 enrichment ratio as a scatterplot with oligonucleotide 1 and oligonucleotide 2 as the x and y coordinates (see Figure 6 adapted from Yu et al., 2021). Quadrant I will contain proteins that bind to both oligonucleotides against the target RNA and quadrant III will be enriched for proteins that bind specifically to the control RNA. Averaging the two enrichment ratios will provide a ranked list of the top protein hits that bind to the target RNA. Proceed to confirmation via immunoblot.
Figure 6. Mass spectrometry target scatterplot.
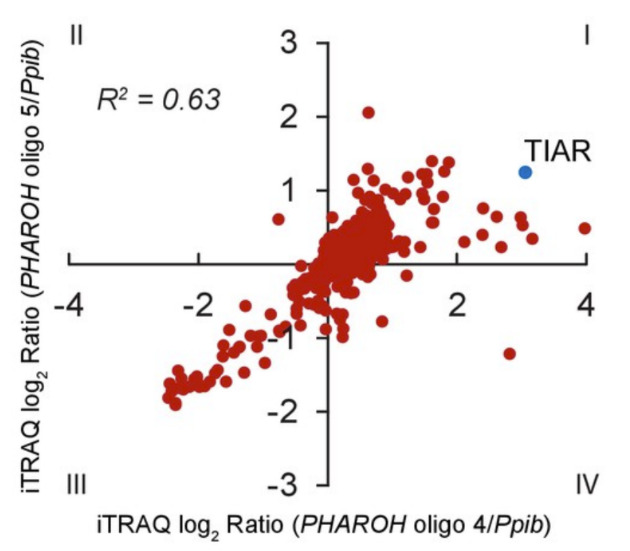
Log2 enrichment ratios plotted by oligonucleotide reveal the interacting proteins with the highest enrichments (adapted from Yu et al., 2021).
Recipes
-
Cell lysis buffer
Pierce IP buffer base containing 150 mM NaCl, 25 mM Tris-Cl pH 7.4, 1% NP-40, 1 mM EDTA, and 5% glycerol
1× cOmplete mini protease inhibitor cocktail: one tablet in 10 mL of lysis buffer
100 U/mL SUPERase-In (Invitrogen)
Note: We have tested the protocol without SUPERase-In as well and found little change to the RNA integrity number.
-
cDNA master mix per reaction
Note: All components, except for nuclease-free H 2 O, are included in the TaqMan reverse transcription kit.
10× TaqMan RT buffer 5 µL
Random hexamers (50 µM) 2.5 µL
MgCl 2 (25 mM) 11 µL
dNTPs (10 mM) 10 µL
RNase inhibitor (20 U/µL) 1 µL
RTase (50 U/µL) 1.25 µL
Nuclease-free H 2 O 8.25 µL
Total volume 39 µL
Use immediately after preparation.
-
qPCR master mix
PowerUp SYBR green master mix 10 µL
Nuclease-free H 2 O 4 µL
Primer mix 1 µL
Total 15 µL
Primer mix consists of forward and reverse primers, each at 10 µM, final concentration 500 nM per well
Use immediately after preparation.
Acknowledgments
We would like to acknowledge the following funding sources: NIGMS 5R35GM131833 (D.L.S.), NCI 5P01CA013106-Project 3 (D.L.S.), NCI 2P3OCA45508, and NCI 5F31CA220997 (A.T.Y.). The original research paper in which this method was used is Yu et al. (2021).
Competing interests
The authors of this manuscript declare that there are no conflicts of interest or competing interests.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
Q&A
Post your question about this protocol in Q&A and get help from the authors of the protocol and some of its users.
References
- 1. Allerson C. R. , Cazzola M. and Rouault T. A. ( 1999 . ). Clinical severity and thermodynamic effects of iron-responsive element mutations in hereditary hyperferritinemia-cataract syndrome . J Biol Chem 274 ( 37 ): 26439 - 26447 . [DOI] [PubMed] [Google Scholar]
- 2. Arun G. , Diermeier S. , Akerman M. , Chang K. C. , Wilkinson J. E. , Hearn S. , Kim Y. , MacLeod A. R. , Krainer A. R. , Norton L. , et al. .( 2016 . ). Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss . Genes Dev 30 ( 1 ): 34 - 51 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arun G. , Diermeier S. D. and Spector D. L. ( 2018 . ). Therapeutic Targeting of Long Non-Coding RNAs in Cancer . Trends Mol Med 24 ( 3 ): 257 - 277 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Batista P. J. and Chang H. Y. ( 2013 . ). Long noncoding RNAs: cellular address codes in development and disease . Cell 152 ( 6 ): 1298 - 1307 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bergmann J. H. , Li J. , Eckersley-Maslin M. A. , Rigo F. , Freier S. M. and Spector D. L. ( 2015 . ). Regulation of the ESC transcriptome by nuclear long noncoding RNAs . Genome Res 25 ( 9 ): 1336 - 1346 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bustin S. A. , Benes V. , Garson J. A. , Hellemans J. , Huggett J. , Kubista M. , Mueller R. , Nolan T. , Pfaffl M. W. , Shipley G. L. , et al. .( 2009 . ). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments . Clin Chem 55 ( 4 ): 611 - 622 . [DOI] [PubMed] [Google Scholar]
- 7. Castello A. , Fischer B. , Eichelbaum K. , Horos R. , Beckmann B. M. , Strein C. , Davey N. E. , Humphreys D. T. , Preiss T. , Steinmetz L. M. , et al. .( 2012 . ). Insights into RNA biology from an atlas of mammalian mRNA-binding proteins . Cell 149 ( 6 ): 1393 - 1406 . [DOI] [PubMed] [Google Scholar]
- 8. Chang K. C. , Diermeier S. D. , Yu A. T. , Brine L. D. , Russo S. , Bhatia S. , Alsudani H. , Kostroff K. , Bhuiya T. , Brogi E. , et al. .( 2020 . ). MaTAR25 lncRNA regulates the Tensin1 gene to impact breast cancer progression . Nat Commun 11 ( 1 ): 6438 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen C. K. , Blanco M. , Jackson C. , Aznauryan E. , Ollikainen N. , Surka C. , Chow A. , Cerase A. , McDonel P. and Guttman M. ( 2016 . ). Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing . Science 354 ( 6311 ): 468 - 472 . [DOI] [PubMed] [Google Scholar]
- 10. Chu C. , Qu K. , Zhong F. L. , Artandi S. E. and Chang H. Y. ( 2011 . ). Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions . Mol Cell 44 ( 4 ): 667 - 678 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chu C. , Zhang Q. C. , da Rocha S. T. , Flynn R. A. , Bharadwaj M. , Calabrese J. M. , Magnuson T. , Heard E. and Chang H. Y. ( 2015 . ). Systematic discovery of Xist RNA binding proteins . Cell 161 ( 2 ): 404 - 416 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crooke S. T. , Baker B. F. , Witztum J. L. , Kwoh T. J. , Pham N. C. , Salgado N. , McEvoy B. W. , Cheng W. , Hughes S. G. , Bhanot S. , et al. .( 2017 . ). The Effects of 2'-O-Methoxyethyl Containing Antisense Oligonucleotides on Platelets in Human Clinical Trials . Nucleic Acid Ther 27 ( 3 ): 121 - 129 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guttman M. , Donaghey J. , Carey B. W. , Garber M. , Grenier J. K. , Munson G. , Young G. , Lucas A. B. , Ach R. , Bruhn L. , et al. .( 2011 . ). lincRNAs act in the circuitry controlling pluripotency and differentiation . Nature 477 ( 7364 ): 295 - 300 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hacisuleyman E. , Goff L. A. , Trapnell C. , Williams A. , Henao-Mejia J. , Sun L. , McClanahan P. , Hendrickson D. G. , Sauvageau M. , Kelley D. R. , et al. .( 2014 . ). Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre . Nat Struct Mol Biol 21 ( 2 ): 198 - 206 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He H. , Wang N. , Yi X. , Tang C. and Wang D. ( 2017 . ). Long non-coding RNA H19 regulates E2F1 expression by competitively sponging endogenous miR-29a-3p in clear cell renal cell carcinoma . Cell Biosci 7 : 65 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee S. , Kopp F. , Chang T. C. , Sataluri A. , Chen B. , Sivakumar S. , Yu H. , Xie Y. and Mendell J. T. ( 2016 . ). Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins . Cell 164 ( 1-2 ): 69 - 80 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liang X. H. , Sun H. , Nichols J. G. and Crooke S. T. ( 2017 . ). RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus . Mol Ther 25 ( 9 ): 2075 - 2092 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Livak K. J. and Schmittgen T. D. ( 2001 . ). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method . Methods 25 ( 4 ): 402 - 408 . [DOI] [PubMed] [Google Scholar]
- 19. Mayr C. ( 2017 . ). Regulation by 3'-Untranslated Regions . Annu Rev Genet 51 : 171 - 194 . [DOI] [PubMed] [Google Scholar]
- 20. McHugh C. A. , Chen C. K. , Chow A. , Surka C. F. , Tran C. , McDonel P. , Pandya-Jones A. , Blanco M. , Burghard C. , Moradian A. , et al. .( 2015 . ). The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC 3. Nature 521 ( 7551 ): 232 - 236 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moore M. J. ( 2005 . ). From birth to death: the complex lives of eukaryotic mRNAs . Science 309 ( 5740 ): 1514 - 1518 . [DOI] [PubMed] [Google Scholar]
- 22. Munschauer M. , Nguyen C. T. , Sirokman K. , Hartigan C. R. , Hogstrom L. , Engreitz J. M. , Ulirsch J. C. , Fulco C. P. , Subramanian V. , Chen J. , et al. .( 2018 . ). The NORAD lncRNA assembles a topoisomerase complex critical for genome stability . Nature 561 ( 7721 ): 132 - 136 . [DOI] [PubMed] [Google Scholar]
- 23. West J. A. , Davis C. P. , Sunwoo H. , Simon M. D. , Sadreyev R. I. , Wang P. I. , Tolstorukov M. Y. and Kingston R. E. ( 2014 . ). The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites . Mol Cell 55 ( 5 ): 791 - 802 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu A. T. , Berasain C. , Bhatia S. , Rivera K. , Liu B. , Rigo F. , Pappin D. J. and Spector D. L. ( 2021 . ). PHAROH lncRNA regulates Myc translation in hepatocellular carcinoma via sequestering TIAR . Elife 10 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu X. ( 2010 . ). Seeing the yin and yang in cell biology . Mol Biol Cell 21 ( 22 ): 3827 - 3828 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zong X. , Huang L. , Tripathi V. , Peralta R. , Freier S. M. , Guo S. and Prasanth K. V. ( 2015 . ). Knockdown of nuclear-retained long noncoding RNAs using modified DNA antisense oligonucleotides . Methods Mol Biol 1262 : 321 - 331 . [DOI] [PubMed] [Google Scholar]