

Abstract
Background:
Eosinophilic esophagitis (EoE) is a chronic allergic disease associated with type 2 inflammation and epithelial barrier dysfunction. The etiology is unknown, however, genetic heritability studies suggest environmental factors play a key role in pathogenesis. Detergents, like sodium dodecyl sulfate (SDS), are common ingredients in household products like dish soap and toothpaste. We hypothesized detergent exposure decreases epithelial barrier function and induces esophageal inflammation.
Methods:
Immortalized esophageal epithelial cells (EPC2) were cultured in air-liquid interface (ALI) and exposed to SDS. Barrier function/activity was assessed by transepithelial electrical resistance (TEER), FITC-dextran flux, and RT-PCR. Additionally, SDS-treated mouse esophageal organoids were evaluated for morphology. To investigate the effects of SDS in vivo, mice were treated with 0.5% SDS in drinking water for 14 days. Esophagi were assessed by gross morphology, histopathology, protein expression, and bulk RNA sequencing.
Results:
When EPC2 cells were exposed to SDS (5 μg/ml) for 96hrs,TEER decreased (p=0.03) and FITC-dextran flux increased (p=0.0002). mRNA expression of IL-33 increased 4.5-fold (p=0.02) at 6hrs and DSG1 decreased (p<0.0001) by 72hrs. Disrupted epithelial integrity was noted in SDS-treated esophageal organoids. When mice were exposed to SDS, they showed increased esophageal width, chemokine, and metalloprotease levels. Mice treated with SDS also showed increased IL-33 protein expression, basal zone hyperplasia, CD4+ cell infiltration, and esophageal eosinophilia. RNA sequencing revealed upregulation of immune response pathway genes.
Conclusion:
Exposure to SDS decreases esophageal barrier integrity, stimulates IL-33 production, and promotes epithelial hyperplasia and tissue eosinophilia. Detergents may be a key environmental trigger in EoE pathogenesis.
Keywords: detergent, epithelium, eosinophilic esophagitis, IL-33
Graphical Abstract
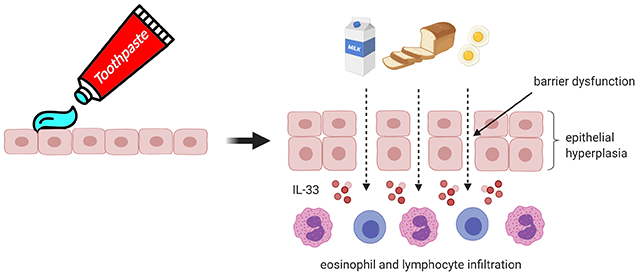
Household products (e.g., toothpaste) contain detergents [e.g., sodium lauryl sulfate, also known as sodium dodecyl sulfate (SDS)].
SDS induces IL-33 production and alters esophageal epithelial integrity, associatedwith remodeling and eosinophilic inflammation.
Detergents may serve as key environmental exposures contributing to increases in eosinophilic esophagitis.
Abbreviations: ALI, air-liquid interface; DSG-1, desmoglein 1; EPC2, esophageal epithelial cells; FITC, fluorescein isothiocyanate; SDS, sodium dodecyl sulfate; TEER, transepithelial electrical resistance
Introduction:
Eosinophilic esophagitis (EoE) is a chronic type 2 inflammatory disorder with clinical manifestations including dysphagia, food impaction, and failure to thrive. Diagnosis requires endoscopic esophageal biopsies that reveal histopathologic features including mucosal eosinophilic infiltrates, basal zone hyperplasia (BZH), spongiosis, and subepithelial fibrosis. BZH represents disruption of the homeostatic differentiation gradient of the stratified squamous epithelium of the esophagus due to an expansion of proliferative basal cells (keratinocytes). EoE is thought to stem from disruption of the epithelial barrier leading to local innate signals and alteration of food or aeroallergen antigen presentation.1 Subsequent exposures to food antigens perpetuate type 2 inflammatory responses that further impair mucosal barrier function.2 BZH, protease dysregulation, and fibrosis represent ineffectual and maladaptive wound repair and tissue remodeling responses resulting in disease pathology.3–5 Currently, there are no FDA-approved therapies. Treatment options include proton pump inhibitors (PPIs), steroids, and food elimination diets.6
The etiology of EoE is unknown; however, 81% of phenotypic variation is due to environmental factors.7 Previous epidemiologic studies suggest that early life exposures such as antibiotics, cesarean delivery and antacids use may predispose to EoE.8–10 These factors may exert influence on both the microbiome and the mucosal epithelium. Recently, Pothoven and Schleimer proposed the epithelial barrier hypothesis, which postulates that epithelial dysfunction precedes the development of allergic sensitization.11 Epithelial barrier integrity is determined by multiple factors including mucus proteins, antimicrobial peptides, tight junctions, adherens junctions, desmosomes, tissue protease activity and the inflammatory microenvironment. Akdis extended the barrier hypothesis by suggesting environmental factors, such as laundry detergents, are driving epithelial barrier dysfunction inducing microinflammation and dysbiosis.12 Due to the importance of environmental factors in EoE pathogenesis, exposures that initiate epithelial barrier dysfunction serve as a logical starting point for investigating the origins of EoE.
The first case series of EoE were documented in the 1990’s13,14 and population-based studies suggest an approximately 20-fold increase in EoE in recent decades for reasons that remain unclear.15,16 Early life exposures and microbial colonization patterns have been identified as potential risk factors for disease development.8,9 EoE is primarily a disease of industrialized nations and commonly presents in the third and fourth decades of life.17 Combined with evidence from twin studies implicating environmental factors7, these clues point to a possible environmental factor changing around the 1950’s.18 Coincident to the appearance of EoE, synthetic surfactants have been increasingly incorporated into household products as detergents and into food as emulsifiers.19,20 In vitro and in vivo studies suggest that detergents found in common household products (e.g. sodium dodecylbenzene sulfonate (SDBS) and sodium dodecyl sulfate (SDS)) can disrupt airway and skin epithelial barriers and promote type 2 inflammation.21–23 Specifically, these studies have shown that SDS and SDBS affect bronchial epithelium and keratinocytes by decreasing epithelial junction integrity and promoting interleukin-33 (IL-33) release.24,25 IL-33 is an epithelial alarmin that is released in response to damage or stress, initiates type 2 inflammation, and is increased in the basal epithelium of EoE subjects.26 We hypothesized that detergents in household products like dish soap and toothpaste might exert similar effects on the esophagus and serve as environmental factors contributing to EoE pathogenesis.
Herein, we examine the effects of SDS in vitro on human esophageal epithelium and in vivo on the mouse esophagus. We demonstrate that SDS increases barrier permeability and promotes IL-33 production from the human esophageal epithelium in vitro. Furthermore, using a mouse model of oral detergent exposure, we show that SDS elicits eosinophilic inflammation and remodeling responses reminiscent of EoE.
Methods:
Submerged and Air-Liquid Interface (ALI) Esophageal Epithelial Cell Cultures
Extensively characterized telomerase-immortalized normal human esophageal epithelial cell line EPC2 27–29, was cultured in keratinocyte-SFM (KSFM) medium (17005-042 Thermo Fisher) as submerged or ALI cultures as previously described.3 Briefly, submerged EPC2 cell cultures were seeded at 8x104/300 μL/well in a 48-well plate and incubated with the detergent SDS (at 1, 6.25, 12.5, 25, and 50 μg/mL for 2 hrs or at 10, 100, 1,000, 5,000, 10,000 ng/mL for 24, 48, 72, and 96 hrs after seeding at 1x105/1 mL/well in a 12-well plate). Conditioned media were assessed for IL-33 by ELISA (DY3625B, R&D, Minneapolis, MN). LIVE/DEAD cellular viability and cytotoxicity were also assessed (L3224, Invitrogen, Waltham, MA). To establish ALI culture, on day 0, 1.0x105 EPC2 cells were seeded on 0.4-μm pore size permeable Transwell inserts (12 mm diameter in 12-well plates) (3460, Corning, NY) in KSFM containing 0.09 mmol/L [Ca2+]. On day 3, ALI cultures were switched to high-calcium KSFM ([Ca2+] = 1.8 mmol/L) to induce stratification of the epithelial cells and post-mitotic terminal differentiation with tight junction formation. On day 7, medium was removed from the upper side of the Transwell to induce differentiation and epithelial stratification. On day 10, differentiated EPC2 cells were exposed to KSFM containing different concentrations of SDS (436143, Sigma) in the basolateral media and assessed for transepithelial electrical resistance (TEER) and FITC-dextran permeability. Media was changed every 2 days. TEER was assessed using an EVOM2 Epithelial Volt/Ohm Meter (World Precision Instruments, Sarasota, FL). TEER was measured on day 10 (before SDS treatment) through day 14. Resistance readings of blank Transwell inserts (no cells) were subtracted from resistance readings of the experimental sample, then multiplied by the area of the ALI culture. Each TEER value is the average from 4 independent Transwell inserts. On day 14, after TEER measurement, cells were briefly washed with 1X HBSS and 250 μL of 2 mg/ml FITC dextran (46944, Sigma, St. Louis, MO) was added on the upper side of the Transwell inserts, 750 μL 1X HBSS was added on the bottom side of Transwell inserts. After 4 hrs incubation at 37°C, 100 μL of 1X HBSS from the bottom side of the Transwell inserts was collected and fluorescence intensity (ex. 485nm, em. 530nm) was measured with a Cytation5 (Agilent, Santa Clara, CA). For real-time reverse-transcription polymerase chain reaction (RT-PCR) assays, cells were harvested at indicated time points to extract total RNA with RNeasy Mini Kit (74104, Qiagen, Hilden, Germany) and synthesize cDNA with SuperScript III First Strand Synthesis System (18080051, Invitrogen, Waltham, MA). Real time RT-PCR was performed on LightCycler 480 (Roche, Basel, Switzerland). Following SDS exposure, mRNA expression was assessed by comparative CT method as previously described30 at 6h and 72h time points for IL33 and DSG1 with ACTB (β-actin) as an internal control utilizing the following primers:
DSG1
L 5’ AACCCAATCGCCAAAATTCACT
R 5’ ACCTCTCGATCAACTATGGATGT
IL33
L 5’ GTGACGGTGTTGATGGTAAGAT
R 5’ AGCTCCACAGAGTGTTCCTTG
ACTB
L 5’ AGAGCTACGAGCTGCCTGAC
R 5’ AGCACTGTGTTGGCGTACAG
Organoid Esophageal Epithelial Cell Cultures
3D Organoids were established from tissue biopsy collected from esophagi of C57BL/6J (The Jackson Laboratory, Bar Harbor, ME) as previously described28 and passaged 2-3 times to generate sufficient organoids for experimental purposes. Organoids were dissociated into single cells using 0.05% Trypsin-EDTA (Thermo 25300120) and live cells were identified by Trypan Blue Exclusion assay and quantified using the CountessTm Automated Cell Counter. 2000 single cells were plated in 50 μl of 75% Matrigel Basement Membrane Matrix (Corning 354234) and 25% growth media (advanced DMEM/F12 (Thermo 12634010) supplemented with 1x GlutaMAX (35050061), 10 mM HEPES (15630080), 1x N-2 supplement (Thermo 17502048), 1x B-27 supplement (Thermo 17504044, 1 mM N-acetyl-L-cysteine (NAC)(Sigma A9165), 50 ng/mL murine epidermal growth factor (Peprotech 315-09), 2% Noggin/R-Spondin conditioned media, and 10 μM Y27632 (Selleck Chemicals). Organoids were treated from day 5 to day 9 with 200 ng/mL SDS and collected for histology on day 11 following a dose response assay to measure the effect of different concentrations of SDS on organoid morphology. 200 ng/mL SDS exerted the greatest effect on the morphology of organoids. Organoids were allowed to recover for two days following treatment to demonstrate that the observed morphological changes persist in organoid culture following withdrawal of SDS.
Exposure of mice to SDS and immunologic and molecular analyses
C57BL/6J 8-week-old mice were exposed to SDS [0.5%] vs. untreated drinking water ad libitum for 14 days. Esophagi were harvested on day 14 and assessed by histology, bead-based multiplex for cytokines and proteases (Eve Technologies, Calgary, Canada), and RNA-Seq (Genewiz, South Plainfield, NJ). Due to a variable treatment response, only mice with eosinophilic esophageal inflammation were included in the SDS-treatment group used for the RNA-Seq dataset. Raw counts were imported into R (4.1.0) and differentially expressed genes were quantified using edgeR.31,32 Pathway analysis was performed using the g:Profiler webserver.33 All data visualization was performed in Python (3.8.8) using Matplotlib (3.4.2), Seaborn (0.11.1), and Pandas (1.3.0). All protocols and procedures for the handling of mice were reviewed and approved by Mayo Clinic Institutional Animal Care and Use Committee.
Histology
Formalin fixed paraffin-embedded tissue sections were dewaxed, rehydrated, and stained with hematoxylin and eosin (H&E), as well as chloroacetate esterase (CAE). For eosinophil peroxidase (EPX) IHC, staining was performed as previously described by Protheroe et al.34 with minor modifications. Specifically, slides were subjected to protease treatment with pepsin solution (00-3009; Invitrogen) for 10 minutes, and treatment with Rodent Block M (RBM961H; BioCare Medical) for 30 minutes. For CD4, IL-33, Ki-67, and myeloperoxidase (MPO) IHC, deparaffinized and rehydrated tissue sections were exposed to heat induced epitope retrieval conditions as described previously.35 The slides were then blocked for endogenous peroxidases with Dual Endogenous Enzyme Blocker (S2003; Dako) for 10 minutes, followed by additional blocking for 1 hr using 2.5% sera from the species in which the secondary antibody was raised in. Primary antibodies were applied onto the slides including: monoclonal rat anti-CD4 (2.5 ug/mL; 14-9766-82, Invitrogen), polyclonal goat anti- IL-33 (0.13 ug/mL; AF3626-SP; R&D), monoclonal rabbit anti-Ki-67 (0.16 ug/mL; MA5-14520; Invitrogen), and polyclonal goat anti-MPO (0.6 ug/mL; AF3667; R&D). Slides stained for CD4 were incubated in primary antibody for 1.5 hrs at room temperature while slides stained for IL-33, Ki-67, and MPO were incubated overnight at 4⁰C. Secondary antibodies were applied onto the slides including: goat anti-rat HRP (MP-7444; Vector) for CD4 staining, goat anti-rabbit HRP (MP-7451; Vector) for Ki-67 staining, and horse anti-goat HRP (MP-7405; Vector) for IL-33 and MPO staining. All slides were incubated in their respective secondary antibody for 30 minutes. Specific CD4 and Ki-67 staining were visualized with a 10-minute incubation using DAB substrate-chromogen (SK-4100; Vector) while IL-33 and MPO staining were visualized with a 5-minute incubation using NovaRed substrate-chromogen (SK-4800; Vector). Slides were then counterstained with either a methyl green (H-3402-500; Vector) or hematoxylin background (S3309; Dako).
Statistical Analysis
Pairwise comparisons were performed using parametric (student’s t-test) or non-parametric tests (Mann-Whitney U) depending on the distribution of the data. Two-tailed statistical tests were performed for each comparison.
Results:
Detergent alters barrier function in human esophageal epithelium ALI cultures and promotes IL-33 production
SDS, also known as sodium lauryl sulfate (SLS), is a detergent found in many household products, including toothpaste where it may be used at concentrations up to approximately 3% w/v (30 mg/mL).36 To examine the effects of detergent on human esophageal epithelium, we reconstructed the stratified squamous epithelia in EPC2 ALI cultures and exposed them to various concentrations of SDS. We predict the esophageal epithelium is typically exposed to lower concentrations than the 3% found in toothpaste (given that toothpaste would be diluted in saliva among other factors), and accordingly we examined serial dilutions of SDS starting from 10,000 ng/mL (i.e. 1:3,000 of toothpaste) to 10 ng/mL. At 5,000 ng/mL, TEER temporarily increased by 24 hrs, followed by a decline up to 96 hrs (p = 0.03), suggesting SDS increased epithelial permeability (Figure 1A). Lower concentrations of SDS demonstrated intermediate effects. To examine the barrier function of EPC2 cells further, we performed FITC-dextran 4kDa permeability assays. SDS increased FITC-dextran 4kDa passage through the ALI cultures (Figure 1B) in a concentration-dependent manner with significant increases in permeability at 5,000 ng/mL (p = 0.0002). To investigate the potential mechanisms involved in epithelial barrier changes, we examined expression of an epithelial junction molecule downregulated in patients with active EoE.37 We found that expression of DSG1 (desmoglein-1) is suppressed significantly when cells are exposed to 5,000 ng/mL or 10,000 ng/mL SDS for 72 hrs (p < 0.0001). Notably, we also found early (6 hrs) increases in IL-33 expression in the cells treated with 5,000 ng/mL and 10,000 ng/mL (p < 0.05) (Figure 1C). To examine the cytotoxic effects of SDS on EPC2 cells we performed submerged cultures exposed to various concentrations of SDS. We found that marked cell death was induced by 25 μg/mL after 2 hrs and by 10 μg/mL after 24 hrs (Supplemental Figure 1A, C, D). Notably, IL-33 release was increased after 2 hrs in response to SDS at 6.25 μg/mL (Supplemental Figure 1B).
Figure 1. SDS induces epithelial barrier dysfunction in vitro.
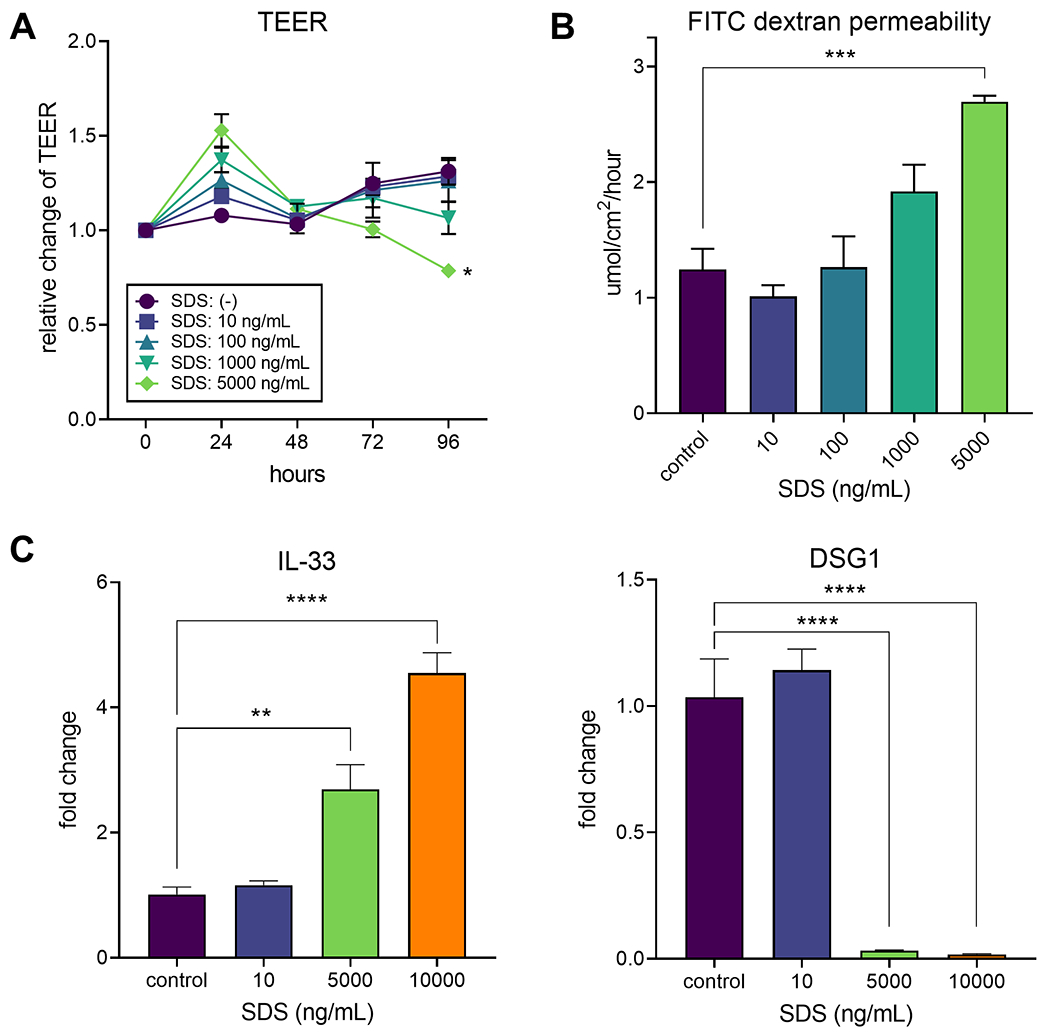
(A) Kinetic changes in TEER in EPC2 ALI cultures exposed to serial dilutions of SDS are shown. Data are presented as mean±SEM of 3 samples and are a representative of 4 experiments. *p < 0.05 compared to the cells with no SDS. (B) EPC2 ALI cultures were exposed to SDS for 96 hrs and permeability to FITC-dextran was examined. Data are presented as mean±SEM of 4 samples. ***p < 0.0001 between the groups indicated by a horizotal line. (C) EPC2 cells were exposed to SDS for 6 hrs (for IL-33) or 72 hrs (for DSG1), and the expression of mRNA for IL-33 and DSG1 was examined by real-time RT-PCR. Data are presented as mean±SEM of 3-4 samples. *p < 0.05, **p < 0.01, ****p < 0.0001 between the groups indicated by horizontal lines.
SDS induces epithelial alterations in esophageal organoid cultures
To explore the effects of SDS on epithelial cell morphology and differentiation, we established mouse esophageal organoid cultures. The organoids were exposed to a low concentration of SDS (200 ng/mL) for 5 days during the 11-day culture of the organoids. The control organoids showed several layers of stratified epithelial cells and organized layers of tissue matrix. In contrast, the organoids exposed to SDS showed disrupted layers of undifferentiated basal cells and irregular tissue matrix (Figure 2).
Figure 2. SDS alters epithelium in esophageal organoids.
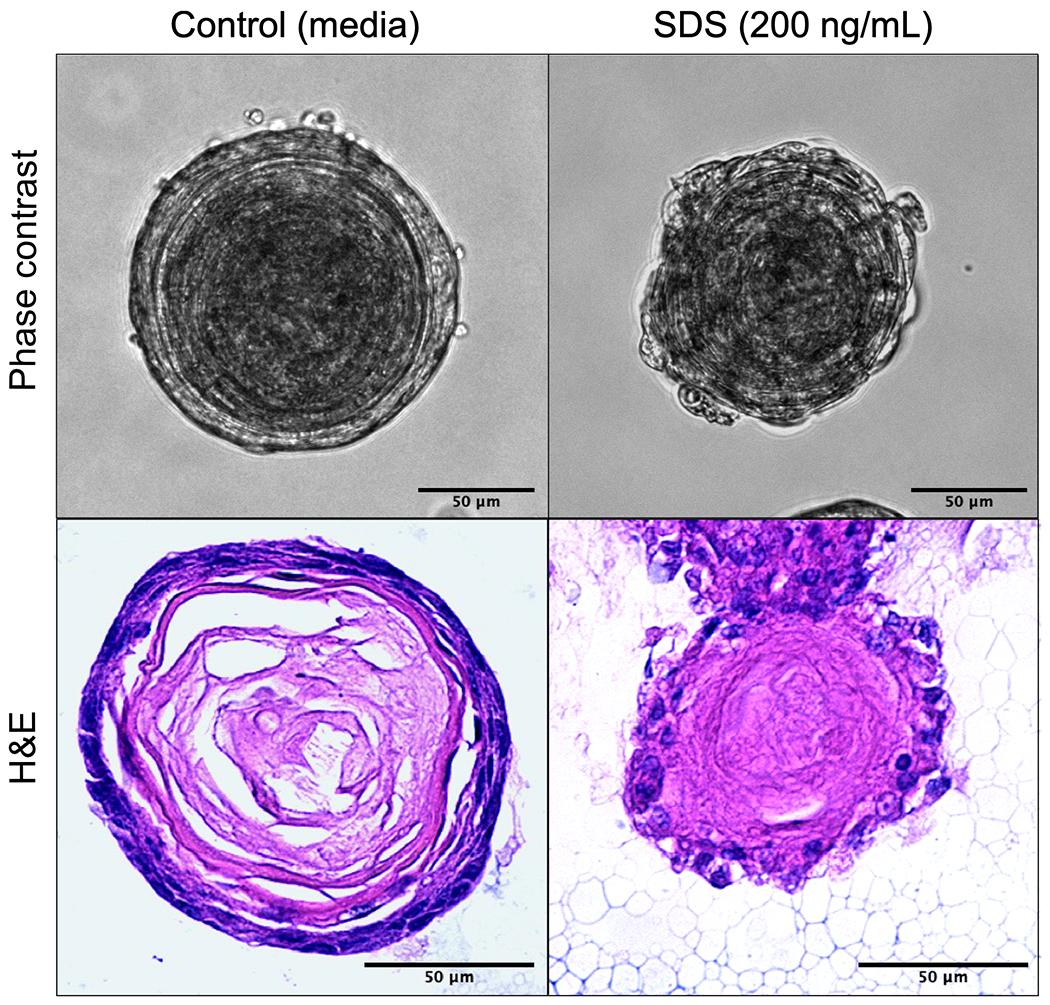
Esophageal organoid cultures were generated ex vivo from naïve wild-type mice. Organoids (n ≥ 10) were exposed to SDS (200 ng/mL) during culture (days 5-9) or untreated and collected on culture day 11 for histology. Representative organoids shown with phase contrast, H&E staining. Scale bar = 50um.
Oral SDS exposure elicits esophageal inflammation and BZH in mice
To explore the in vivo activities of SDS on the esophagus, we administered 0.5% SDS in the drinking water of naive adult C57BL/6J mice ad libitum for 14 days; mice kept on untreated drinking water were used as a control. The esophagi of mice exposed to SDS were enlarged and thickened as compared to those of control mice (Supplemental Figure 2). Pathologic examination showed patchy eosinophilic esophageal inflammation throughout the esophagus (i.e. proximal to distal) in mice exposed to SDS (Figure 3). Esophageal inflammation was commonly observed in 50-100% of mice, depending on cohort, in 9 independent experiments (n=5-30). Eosinophilic abscesses and spongiosis were also observed (Supplemental Figure 3). H&E and EPX staining demonstrated esophageal pathology including BZH, and infiltration of inflammatory cells, including eosinophils, in the submucosa. Notably, increased expression of IL-33 in the mucosal layer was observed by IHC staining. Staining with anti-CD4 antibody revealed infiltration of CD4+ lymphocytes in the submucosa (Figure 3). BZH was corroborated by increased Ki-67 staining specific to the basal cells of the epithelium (Figure 3). Chemotactic factors for eosinophils (eotaxin-1) and T-cells (CCL-21) were elevated in homogenates from whole esophagi of mice exposed to SDS (Supplemental Figure 4). By CAE staining, no differences were observed in the number of mast cells between control mice and those exposed to SDS (data not shown). Neutrophilia was detected by MPO staining (Supplemental Figure 5). The levels of metalloproteinases, including pro-MMP-9 and MMP-8, were highly elevated in the esophagi of mice treated with SDS (Supplemental Figure 6), consistent with tissue inflammation and remodeling.
Figure 3. SDS induces eosinophilic inflammation, CD4 lymphocyte infiltration, BZH, and IL-33 expression.
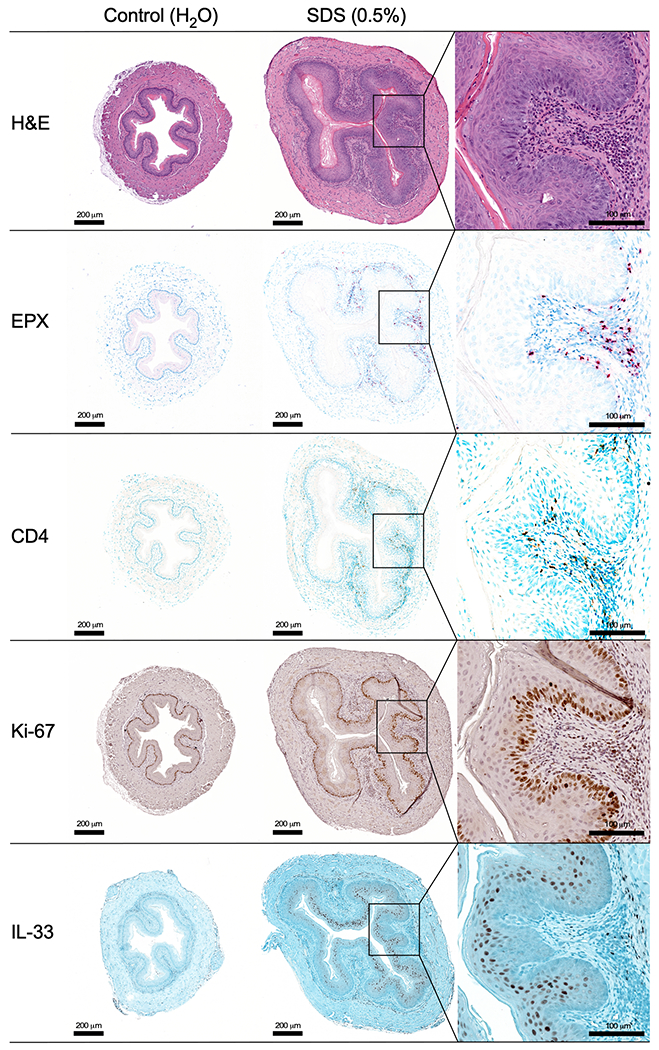
Esophagus was collected from mice exposed to 0.5% SDS in drinking water and compared with regular drinking water controls. Cross-sections were stained with H&E, EPX, CD4, Ki-67, and IL-33. Insets are included for detail for SDS-treated mice. Serial sections are shown from representative mice (n=5 treatment, 5 controls). Scale bars = 200 μm. Inset scale bars = 100 μm.
SDS promotes immune response gene expression in the mouse esophagus
To explore the observed inflammation and remodeling we examined whole esophagus homogenates from SDS exposed mice and controls by RNA-Seq analysis. The analysis revealed upregulation of genes involved in the activation of acute inflammatory and remodeling pathways (Figure 4, Supplemental Table 1). While a number of genes are consistently upregulated in SDS exposed mice compared to controls, considerable variability was observed among the 3 mice. A volcano plot of differentially expressed genes showed increased expression of Rnase2b, Il1b, Zap70, CD3d, CCR7, and Il4ra suggesting infiltration of inflammatory cells and T cells. Krt16 and Defb3 were also upregulated, consistent with remodeling of esophageal epithelium. Indeed, pathway analysis revealed upregulation of molecules involved in innate and adaptive immune responses. In contrast, molecules associated with tissue development and muscle contraction were decreased. All together, these data suggest that exposure of naïve mice to SDS triggers inflammatory immune responses and dysregulation of esophageal tissue homeostasis.
Figure 4. SDS alters gene expression in the mouse esophagus.
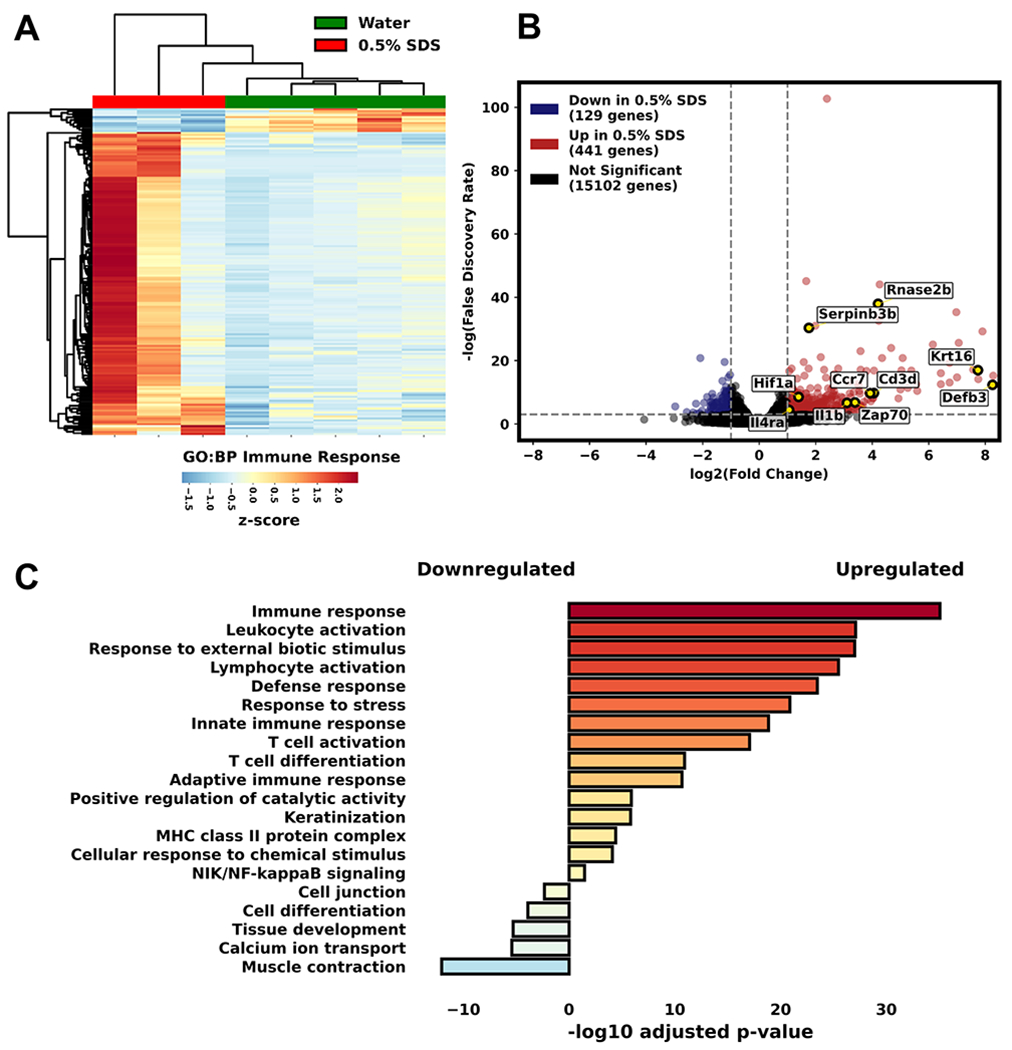
RNA-Seq of whole esophagus homogenates from mice treated with 0.5% SDS in drinking water for 14 days vs. regular drinking water. (A) Heatmap of immune response gene expression. (B) Volcano plot of differentially expressed genes showed changes in barrier- (epithelial) and inflammation-associated (eosinophil, T cell) genes (highlighted yellow). (C) Pathway analysis showing upregulated and downregulated gene expression associated with inflammatory and remodeling pathways. (n=3-5 mice).
Discussion:
In this study, we found that exposure to SDS compromises barrier integrity and activates expression of the pro-inflammatory molecule IL-33 in esophageal epithelial cells in vitro. Oral exposure to SDS in vivo induced inflammation, immune response, and remodeling of the esophageal mucosa. First, we examined the effects of SDS on a human esophageal cell line, EPC2. We found that SDS altered epithelial barrier function in ALI cultures and decreased expression of barrier function associated gene DSG1. Further, SDS increased expression of IL-33 by EPC2 cells. Next, we found that SDS induces basal epithelial disruption in ex vivo mouse organoid cultures. We then demonstrated in vivo that oral administration of SDS to naïve mice promotes eosinophilic inflammation of the esophagus, including abscess formation, and increased infiltration of CD4+ T cells. Moreover, hyperplasia and increased expression of IL-33 in esophageal epithelium were also observed. These results are consistent with previous studies examining the response of lung epithelium and skin keratinocytes to common household detergents.24,25 Together, this study and previous studies suggest a broad effect of common household detergents on epithelial barrier dysfunction and pro-inflammatory and immunogenic responses.
One of the major findings of this study is the biological effects of detergents at very low concentrations. We found that, similar to studies of the effects of laundry detergent on lung epithelium25, very low concentrations of SDS elicited esophageal organoid disruption (200 ng/mL) and IL-33 production (5,000 ng/mL) in ALI culture. Epithelial barrier integrity as examined by TEER and FITC dextran permeability was also reduced with 5,000 ng/mL SDS. Although ALI cultures appeared viable by light microscopy at this duration and concentration of exposure, cytotoxicity was observed after 96 hours at 5,000 ng/mL in submerged cultures. Given potential differences in ALI and submerged cultures it is unclear if, or to what extent, cytotoxicity may be contributing to disruption of barrier function. Notably, due to inherent differences in the culture systems employed (submerged, ALI, organoid) we noted minor differences in detergent concentration and duration of exposure required to demonstrate pathologic changes. Ultimately, low concentrations of detergents can induce these changes in a variety of model systems. In the previous study25, the concentrations of SDBS that compromise the airway epithelial cell barrier were estimated to be around 5,000 ng/mL (after 1:10 dilution of residual liquid), which are similar to the observations with esophageal epithelial cells in this study. The results of 0.5% oral SDS exposure were particularly striking as toothpaste, for example, contains SDS at up to 3% w/v (i.e. 30 mg/mL). Further investigation is needed to understand how the duration, frequency, and concentration of SDS exposure influence pathology. Notably, in contrast to mice, humans lack a protective keratin layer in the esophagus and may be susceptible to even lower concentrations of detergent. Finally, ALI cultures were exposed to SDS from the basolateral area which may not be representative of oral exposure in the esophagus and represents a limitation of these studies.
Consistent with the in vitro models, the in vivo studies examining oral SDS exposure reveal a prominent inflammatory response and remodeling in the mouse esophagus. On the other hand, this model may not replicate typical human exposure as humans may experience chronic, intermittent exposure to numerous household detergents, including during early development, along with other potential environmental factors.8,9 Importantly, previous studies suggest SDS may act as an adjuvant to promote development of immune responses to exogenous antigens.23,38 Based on our experiments, detergent can mediate pathology with similarities to EoE, but it is still unknown whether oral detergents promote an allergic sensitization to food or environmental allergens alone or in combination with other environmental factors. Increased expression of IL-33 and infiltration of eosinophils and CD4+ T cells to the esophagus in our study suggest possible sensitization. Eotaxin-1 and CCL-21, which promote tissue infiltration of eosinophils and lymphocytes, are also upregulated in SDS-treated mice. Further studies will be necessary to specifically address the potential adjuvant activity of SDS for oral and even respiratory antigens. In addition, future investigations are needed to better replicate human exposure conditions, including doses of detergents and frequency of exposure as humans may not continually encounter the concentrations of SDS utilized in the models described herein. Finally, whether genetic susceptibility factors may influence the response to detergent exposure requires investigation.
The effects of oral SDS exposure have previously been examined in rats.39 The direct effects on the esophagus were not examined; however, SDS and other alcohol sulfates consistently and particularly induced irritation of the forestomach (comprised of squamous epithelium like the human esophagus). Indeed, the 1939 study by Epstein et al.40 was prophetic. The authors note that if “alkyl sulfates (SDS) are to be used in dentifrices as detergents, it is imperative to consider not only their cleansing properties, but also their potential injurious and toxic effects.” They find considerable mortality in rats orally exposed to low concentrations of SDS over a three-month period (e.g. roughly 50% mortality in the 0.5% SDS group) relative to similar concentrations of traditional soap (no deaths in the 0.5% traditional soap group) in drinking water. They also note that among those that died, “evidence of gastrointestinal irritation, especially of the esophagus and the rumen (i.e. forestomach). There was edema of the submucosa, with a loss of keratin in some places, and leukocytic infiltration, all of which was indicative of a low-grade inflammatory reaction.” While the average time to death in this study was 7 weeks, and our protocol ran only 2 weeks, we similarly saw evidence of esophageal thickening and leukocytic infiltration (including eosinophils) in SDS-exposed mice after a relatively short period of exposure.
Finally, we observed antimicrobial and neutrophilic responses in the esophagi of detergent-treated mice suggesting dysbiosis. Detergents have been shown to have antimicrobial properties which may alter physiological microbial populations.41 Moreover, the barrier dysfunction linked with detergent exposure may facilitate changes in the location (i.e. tissue penetration) of resident bacteria.42 Existing models to explain the rise in allergic disease share a common theme of dysbiosis. Further investigation is needed to understand how the esophageal microbiome is altered by oral detergent exposure, if this may be contributing to the acute inflammatory responses observed in our model, and, more broadly if/how oral detergent exposure may alter the microbiome of the entire gastrointestinal tract potentially contributing to a host of diseases.43 One of the limitations of our study is that we did not directly assess barrier dysfunction in vivo. Further investigation into microbial changes in location and esophageal barrier dysfunction are required to understand the possible contribution to the observed pathologies in this model.
In conclusion, we have shown that the common household detergent, SDS, alters esophageal integrity inducing barrier permeability. In addition, we have demonstrated that SDS elicits type 2 inflammatory signals and eosinophilic inflammation in the esophagus in vivo. We hypothesize that IL-33 acts on multiple cell types, including ILC2s, resulting in skewing towards type 2 immunity. Persistence and penetration of antigen in this microenvironment likely promotes antigen-specific Th2 responses. Taken together with existing literature 24,25,44 these findings point to a potential role for detergents, which are included in oral household products, in epithelial barrier disruption and inflammation. It is unclear at this time if detergents alone may be sufficient to cause inflammatory changes in the esophagus, mediate the key step of promoting an adaptive immune response in the esophagus, or both. Together, our experiments suggest detergents may serve as key environmental determinants contributing to the pathogenesis of EoE.
Supplementary Material
Acknowledgments:
We thank Sahiti Marella and Simon Hogan (University of Michigan) for their guidance on ALI cultures. We thank Elizabeth Jacobsen (Mayo Clinic) for providing EPX antibody. We appreciate the efforts of the Mayo Clinic research histology core and small animal facility. We are grateful for support from the American Partnership for Eosinophilic Disorders (APFED), Mayo Clinic Foundation, Phoenix Children’s Hospital Foundation, the Roubos Family Fund, and the Donald R. Levin Family Foundation.
Declaration of Funding:
A grant from the American Partnership for Eosinophilic Disorders (APFED), Roubos Family Fund, Donald R. Levin Family Foundation, Phoenix Children’s Hospital Foundation, and Mayo Clinic Foundation. BLW also reports funding from the Consortium of Eosinophilic Gastrointestinal Disease Researchers U54AI117804 (CEGIR), which is part of the Rare Disease Clinical Research Network (RDCRN), an initiative of the Office of Rare Disease Research (ORDR). CEGIR is also supported by patient advocacy groups including American Partnership for Eosinophilic Disorders (APFED), Campaign Urging Research for Eosinophilic Diseases (CURED), and Eosinophilic Family Coalition (EFC). As a member of the RDCRN, CEGIR is also supported by its Data Management and Coordinating Center (DMCC) (U2CTR002818). SF and HN have received NIH grants L30CA264714 (SF), R01DK114436, and R01AA026297. HK has received grants from the NIH, R37AI71106, R01AI128729, and R01HL117823, and from Mayo Foundation.
Abbreviations Used:
- ALI
air liquid interface
- EoE
eosinophilic esophagitis
- eos/hpf
eosinophils per high power field
- FITC
fluorescein isothiocyanate
- H&E
hematoxylin and eosin
- IHC
immunohistochemistry
- TEER
transepithelial electrical resistance
- SDBS
sodium dodecylbenzene sulfonate
- SDS
sodium dodecyl sulfate
Footnotes
Conflicts of Interest: The author have no conflicts of interest to report.
References
- 1.Rochman M, Azouz NP, Rothenberg ME. Epithelial origin of eosinophilic esophagitis. J Allergy Clin Immunol. 2018;142(1):10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis BP, Rothenberg ME. Mechanisms of Disease of Eosinophilic Esophagitis. Annu Rev Pathol. 2016;11:365–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanoni S, Zeng C, Marella S, et al. Identification of anoctamin 1 (ANO1) as a key driver of esophageal epithelial proliferation in eosinophilic esophagitis. J Allergy Clin Immunol. 2020;145(1):239–254 e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azouz NP, Klingler AM, Pathre P, et al. Functional role of kallikrein 5 and proteinase-activated receptor 2 in eosinophilic esophagitis. Sci Transl Med. 2020;12(545). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shoda T, Wen T, Caldwell JM, et al. Loss of Endothelial TSPAN12 Promotes Fibrostenotic Eosinophilic Esophagitis via Endothelial Cell-Fibroblast Crosstalk. Gastroenterology. 2022;162(2):439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muir A, Falk GW. Eosinophilic Esophagitis: A Review. JAMA. 2021;326(13):1310–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alexander ES, Martin LJ, Collins MH, et al. Twin and family studies reveal strong environmental and weaker genetic cues explaining heritability of eosinophilic esophagitis. J Allergy Clin Immunol. 2014;134(5):1084–1092 e1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dellon ES, Shaheen O, Koutlas NT, et al. Early life factors are associated with risk for eosinophilic esophagitis diagnosed in adulthood. Dis Esophagus. 2021;34(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jensen ET, Kappelman MD, Kim HP, Ringel-Kulka T, Dellon ES. Early life exposures as risk factors for pediatric eosinophilic esophagitis. J Pediatr Gastroenterol Nutr. 2013;57(1):67–71. [DOI] [PubMed] [Google Scholar]
- 10.Jensen ET, Kuhl JT, Martin LJ, Rothenberg ME, Dellon ES. Prenatal, intrapartum, and postnatal factors are associated with pediatric eosinophilic esophagitis. J Allergy Clin Immunol. 2018;141(1):214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pothoven KL, Schleimer RP. The barrier hypothesis and Oncostatin M: Restoration of epithelial barrier function as a novel therapeutic strategy for the treatment of type 2 inflammatory disease. Tissue Barriers. 2017;5(3):e1341367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akdis CA. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat Rev Immunol. 2021;21(11):739–751. [DOI] [PubMed] [Google Scholar]
- 13.Attwood SE, Smyrk TC, Demeester TR, Jones JB. Esophageal eosinophilia with dysphagia. A distinct clinicopathologic syndrome. Dig Dis Sci. 1993;38(1):109–116. [DOI] [PubMed] [Google Scholar]
- 14.Straumann A, Spichtin HP, Bernoulli R, Loosli J, Vogtlin J. [Idiopathic eosinophilic esophagitis: a frequently overlooked disease with typical clinical aspects and discrete endoscopic findings]. Schweiz Med Wochenschr. 1994;124(33):1419–1429. [PubMed] [Google Scholar]
- 15.Dellon ES, Erichsen R, Baron JA, et al. The increasing incidence and prevalence of eosinophilic oesophagitis outpaces changes in endoscopic and biopsy practice: national population-based estimates from Denmark. Aliment Pharmacol Ther. 2015;41(7):662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prasad GA, Alexander JA, Schleck CD, et al. Epidemiology of eosinophilic esophagitis over three decades in Olmsted County, Minnesota. Clin Gastroenterol Hepatol. 2009;7(10):1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dellon ES, Hirano I. Epidemiology and Natural History of Eosinophilic Esophagitis. Gastroenterology. 2018;154(2):319–332 e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347(12):911–920. [DOI] [PubMed] [Google Scholar]
- 19.Falbe J. Surfactants in Consumer Products. 1st ed: Springer-Verlag; 1987. [Google Scholar]
- 20.Singer MM, Tjeerdema RS. Fate and effects of the surfactant sodium dodecyl sulfate. Rev Environ Contam Toxicol. 1993;133:95–149. [DOI] [PubMed] [Google Scholar]
- 21.Wilhelm KP, Freitag G, Wolff HH. Surfactant-induced skin irritation and skin repair. Evaluation of the acute human irritation model by noninvasive techniques. J Am Acad Dermatol. 1994;30(6):944–949. [DOI] [PubMed] [Google Scholar]
- 22.Davenport HW. Destruction of the gastric mucosal barrier by detergents and urea. Gastroenterology. 1968;54(2):175–181. [PubMed] [Google Scholar]
- 23.Clausen SK, Sobhani S, Poulsen OM, Poulsen LK, Nielsen GD. Study of adjuvant effect of model surfactants from the groups of alkyl sulfates, alkylbenzene sulfonates, alcohol ethoxylates and soaps. Food Chem Toxicol. 2000;38(11):1065–1074. [DOI] [PubMed] [Google Scholar]
- 24.Xian M, Wawrzyniak P, Ruckert B, et al. Anionic surfactants and commercial detergents decrease tight junction barrier integrity in human keratinocytes. J Allergy Clin Immunol. 2016;138(3):890–893 e899. [DOI] [PubMed] [Google Scholar]
- 25.Wang M, Tan G, Eljaszewicz A, et al. Laundry detergents and detergent residue after rinsing directly disrupt tight junction barrier integrity in human bronchial epithelial cells. J Allergy Clin Immunol. 2019;143(5):1892–1903. [DOI] [PubMed] [Google Scholar]
- 26.Travers J, Rochman M, Caldwell JM, Besse JA, Miracle CE, Rothenberg ME. IL-33 is induced in undifferentiated, non-dividing esophageal epithelial cells in eosinophilic esophagitis. Sci Rep. 2017;7(1):17563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harada H, Nakagawa H, Oyama K, et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res. 2003;1(10):729–738. [PubMed] [Google Scholar]
- 28.Kasagi Y, Chandramouleeswaran PM, Whelan KA, et al. The Esophageal Organoid System Reveals Functional Interplay Between Notch and Cytokines in Reactive Epithelial Changes. Cell Mol Gastroenterol Hepatol. 2018;5(3):333–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whelan KA, Muir AB, Nakagawa H. Esophageal 3D Culture Systems as Modeling Tools in Esophageal Epithelial Pathobiology and Personalized Medicine. Cell Mol Gastroenterol Hepatol. 2018;5(4):461–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- 31.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40(10):4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raudvere U, Kolberg L, Kuzmin I, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47(W1):W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Protheroe C, Woodruff SA, de Petris G, et al. A novel histologic scoring system to evaluate mucosal biopsies from patients with eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2009;7(7):749–755 e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nazaroff CD, LeSuer WE, Masuda MY, Pyon G, Lacy P, Jacobsen EA. Assessment of Lung Eosinophils In Situ Using Immunohistological Staining. Methods Mol Biol. 2021;2223:237–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Company Colgate-Palmolive. Colgate Toothpaste Material Safety Data Sheet. 2019; https://www.medline.com/media/catalog/Docs/MSDS/MSD_SDSD94656.pdf. Accessed March 17, 2022, 2022.
- 37.Sherrill JD, Kc K, Wu D, et al. Desmoglein-1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal Immunol. 2014;7(3):718–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nilzen A, Wikstrom K. The influence of lauryl sulphate on the sensitization of guineapigs to chrome and nickle. Acta Derm Venereol. 1955;35(4-5):292–299. [PubMed] [Google Scholar]
- 39.HERA Substance Team. Human & Environmental Risk Assessment (HERA) on ingredients of European household cleaning products: Alcohol Sulphates Human Health Risk Assessment. 2002; p. 122. Available at: https://www.heraproject.com/files/3-HH-04-%20HERA%20AS%20HH%20web%20wd.pdf. Accessed March 17, 2022.
- 40.Epstein S, Throndson AH, Dock W, Tainter ML. Possible deleterious effects of using soap substitutes in dentrifices. J Am Dent Assoc. 1939;26:1461–1471. [Google Scholar]
- 41.Leoty-Okombi S, Gillaizeau F, Leuillet S, et al. Effect of Sodium Lauryl Sulfate (SLS) Applied as a Patch on Human Skin Physiology and Its Microbiota. Cometics. 2021;1(1):12. [Google Scholar]
- 42.Knoop KA, McDonald KG, Kulkarni DH, Newberry RD. Antibiotics promote inflammation through the translocation of native commensal colonic bacteria. Gut. 2016;65(7):1100–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manfredo Vieira S, Hiltensperger M, Kumar V, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science. 2018;359(6380):1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Putterman G, Wolejsza NF, Laden K. The effect of detergents on swelling of stratum corneum. J Soc Cosmset Chem. 1977;Soc(REF17):521–532. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.