

Abstract
Acute exposure of cancer cells to high concentrations of interferon-I (IFN-I) drives growth arrest and apoptosis, whereas chronic exposure to low concentrations provides important pro-survival advantages. Tyrosine-phosphorylated IFN-stimulated gene factor 3 (ISGF3) drives acute deleterious responses to IFN-I, whereas U-ISGF3, lacking tyrosine phosphorylation, drives essential constitutive pro-survival mechanisms. Surprisingly, programmed cell death ligand 1 (PD-L1), often expressed on the surfaces of tumor cells and well recognized for its importance in inactivating cytotoxic T cells, also has important cell-intrinsic pro-tumor activities, including dampening acute responses to cytotoxic high levels of IFN-I and sustaining the expression of the low levels that benefit tumors. More thorough understanding of the newly recognized complex roles of IFN-I in cancer may lead to the identification of novel therapeutic strategies.
Keywords: interferon, STAT2, PD-L1, DNA damage, triple-negative breast cancer
Interferon and cancer
Type I interferon (IFN-I, see Glossary) was discovered as our major defense against virus infections in 1957 [1]. Since those early days, we have continued to learn more and more about this fascinating cytokine, including how it is induced, how it signals to activate new transcription and protein synthesis in target cells, how its complex functions are fine-tuned and, especially relevant for this review, how IFN-I plays important roles in cancer. The pathways of IFN-I synthesis and response are briefly summarized in Figure 1. It has now been 30 years since the discovery of the JAK-STAT pathway, which powers responses to IFN-I and many other cytokines. Much of the huge amount of current information regarding the mechanisms, scope, and therapeutic impact of this pathway is summarized in a recent review (Phillips et al, Cell, in press).
Figure 1. How cancer cells make and respond to IFN-I.
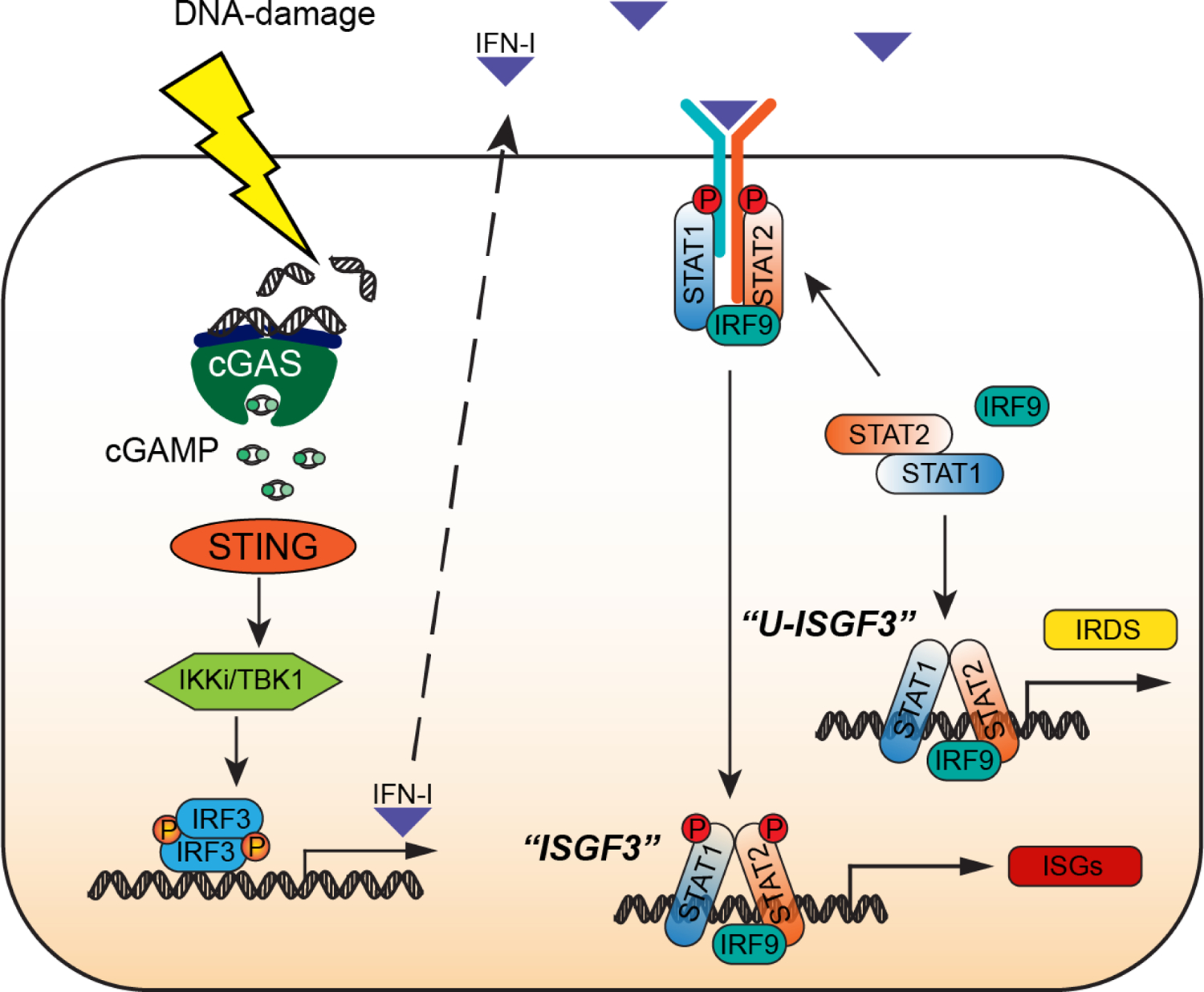
Activation of the cGAS-STING pathway by DNA-damaging chemotherapeutic agents (shown) or in response to endogenous cytoplasmic DNA (not shown) leads to robust production of IFN-I. Newly synthesized autocrine IFN-I drives the expression of interferon-stimulated genes (ISGs) by using the receptor-bound kinases, Janus Kinase 1 (JAK1) and Tyrosine Kinase 2 (TYK2), to phosphorylate and thus activate STAT1 and STAT2 on specific tyrosine residues (Y701 for STAT1, Y690 for STAT2), followed by the formation of tyrosine phosphorylated STAT1:STAT2 heterodimers that associate with IRF9 to form ISGF3, the major transcription factor driving the response to IFN-I. STAT1, STAT2, and IRF9 are themselves ISGs, so that these three proteins are present at high levels in cells that are exposed to IFN-I constitutively, forming an ISGF3 complex that lacks tyrosine phosphorylation (U-ISGF3) which, in turn, drives the expression of a subset of ISGs, the Interferon-Related DNA Damage Resistance Signature (IRDS). Since the conformation of U-ISGF3 is not yet known, the representation in the figure should be regarded as schematic only.
The IFN-I response has two distinct phases. In the first, ISGF3, formed from IRF9 and tyrosine-phosphorylated STATs 1 and 2, drives expression of the full set of interferon-stimulated genes (ISGs). Since the maximal antiviral effects of the initially induced proteins cannot be sustained by cells for very long without causing damage, powerful negative regulatory mechanisms, especially those due to the IFN-I-induced protein SOCS1, suppress the continued phosphorylation of STATs 1 and 2 within about a day. However, STAT1, STAT2, and IRF9 are themselves ISGs, leading to a prolonged steady-state in which these three proteins, now highly expressed but lacking tyrosine phosphorylation, form U-ISGF3, which continues to drive the expression of about a quarter of the initially induced ISGs for many days [2]. The encoded proteins, well tolerated by the cells, continue to provide a substantial level of antiviral protection, albeit much less than in the initial, transient antiviral state.
The ability of cancer cells to induce, tolerate, and sustain the expression of the proteins that are driven by U-ISGF3 is a major manifestation of the effects of IFN-I in cancer. A breakthrough in understanding the complex roles of IFN-I in cancer came from the results of Ralph Weichselbaum and his colleagues [3], who described the expression of relatively high levels of a subset of ISGs in many different types of cancer, and correlated this phenotype with the ability of these cells to resist being killed by ionizing radiation [4]. The subset of ISGs in this “Interferon-Related DNA Damage Resistance Signature” (IRDS) coincides almost exactly with the subset that is induced in response to U-ISGF3 during the antiviral response [3–5]. As explained in more detail later, tumors are exposed to low levels of IFN-I in steady state, produced not only in the tumor microenvironment (TME) but importantly also by the tumor cells themselves, leading to constitutive expression of the IRDS subset of ISGs and substantial protection from DNA damage [2, 6]. In contrast to the benefit that chronic exposure to low levels of IFN-I provides to tumors, it has long been appreciated that acute exposure to high levels of IFN-I, delivered as therapy or induced in response to extensive DNA damage, such as that caused by ionizing radiation, is cytotoxic [7].
Herein we discuss recent evidence that is key to understanding the complex roles of IFN-I in cancer, including the vital role of programmed cell death ligand 1 (PD-L1) in regulating the synthesis of and responses to IFN-I through mechanisms that are intrinsic to cancer cells. Furthermore, U-STAT2 has novel functions in cancer, including its ability to potently inhibit the activity of STING, whose activation by cytoplasmic DNA leads to the synthesis of IFN-I. However, illustrating the complexity, in some tumors IFN-I is an important inhibitor of the epithelial-mesenchymal transition, whereas oncostatin M has the opposite activity, and these two cytokines negatively regulate each other. We focus on the production of IFN-I by cancer cells themselves, on how their responses to different levels of IFN-I are regulated, and on the consequences of these responses for their survival. We do not review the importance of IFN-I as a regulator of the immune system in cancer. This fascinating topic has been well reviewed by others [8, 9].
Cytotoxic versus protective effects of IFN-I in cancer
IFN-I has an important role in regulating responses to DNA damage in cancer cells. The synthesis of IFN-I is induced in response to extensive damage and the resulting acute response contributes to cell killing [7, 10, 11]. Anthracyclines (e.g. doxorubicin) stimulate IFN-I production by activating Toll-like receptor 3 (TLR3), promoting cancer cell death through the expression of the chemokine C-X-C motif ligand 10 (CXCL10) [11]. Cytotoxicity induced by ionizing radiation is correlated with IFN-I expression, and inhibiting the response to IFN-I by using neutralizing antibodies or knocking out the IFN-I receptor decreases the ability of ionizing radiation to induce cell death [7]. ISGs that encode cytotoxic proteins are induced in the acute phase of signaling [12].
On the other hand, chronic stimulation with low doses of IFN-I contributes to resistance to DNA damage. The chronic response to interferon-β (IFNβ) increases the levels of U-ISGF3, which induces the expression of about a quarter of the ISGs, the IRDS subset (Figure 2) [2]. Chronic IFN-I responses and elevated IRDS expression are often observed in cancer cells exposed to repeated or prolonged radiation or chemotherapy, which correlates with acquired resistance to the therapy [7, 10, 13]. Some intrinsic factors also induce chronic IFN-I responses in cancer cells, thus aiding their intrinsic resistance to DNA damage. For example, dysfunction of the Ataxia-telangiectasia mutated (ATM) gene, which encodes a central component of the DNA repair machinery, results in constitutive IFNβ expression through the cytosolic DNA-sensing STING pathway [14], and loss of tumor suppressor p53 function leads to IFNβ expression through a double stranded RNA (dsRNA)-dependent pathway, reducing the cytotoxicity of the DNA damaging agent doxorubicin [15]. In summary, the effects of IFN-I on tumors are determined by the strength and duration of stimulation; strong and acute IFN-I responses are cytotoxic, whereas weak and chronic responses promote cell survival [16].
Figure 2. IFN-I-dependent pathways that affect the growth and survival of cancer cells.
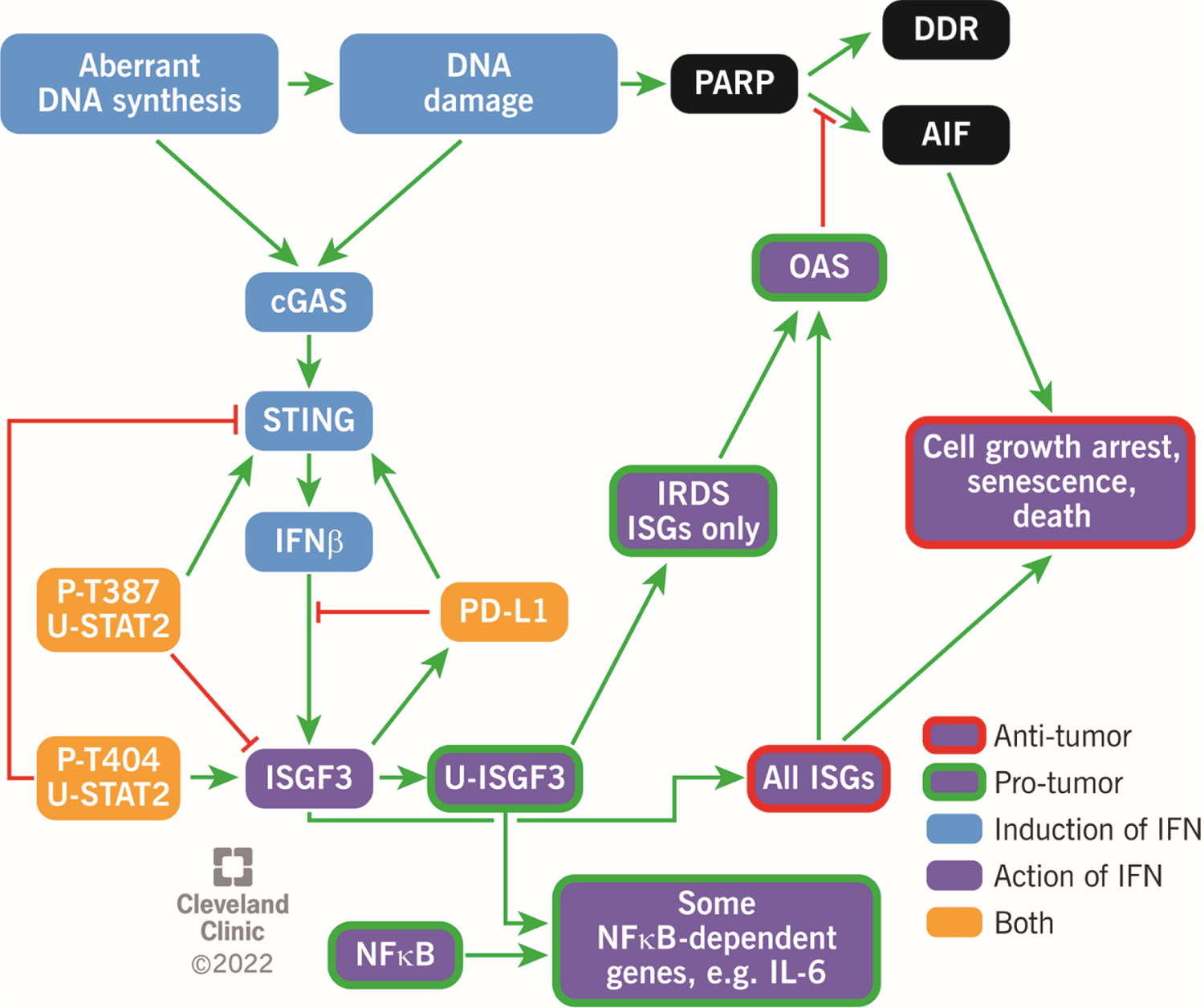
Light blue boxes; aberrant DNA synthesis and inherent DNA damage in cancer cells leads to self-synthesis of IFNβ. In addition to the cGAS-STING pathway shown, double stranded RNA (dsRNAs) produced from endogenous retroviral elements also have a role, by activating the MAVS (mitochondrial antiviral-signaling protein), RIG-I (retinoic acid-inducible gene I), MDA5 (melanoma differentiation-associated protein 5) pathway (not shown). Black boxes; DNA damage activates poly (ADP) ribose polymerase (PARP). Whereas a moderate level of poly (ADP) ribosylation facilitates repair of DNA damage (DDR), excessive poly (ADP) ribosylation activates apoptosis inducing factor (AIF), leading to a form of cell death called parthanotos (an anti-tumor effect). Dark blue boxes; ISGF3 activates the expression of all ISGs, leading to increased expression of many genes whose products inhibit cancer cells. However, strong induction of the STAT1, STAT2, and IRF9 genes by ISGF3 leads to persistent high levels of U-ISGF3, which drives pro-tumorigenic activities. Yellow boxes; PD-L1 and each of the two threonine-phosphorylated forms of U-STAT2 affect the synthesis and actions of IFN-I in opposite directions, helping to suppress but not eliminate the effects of IFN-I in ways that are beneficial to cancer cells.
Contrasting roles of STAT2 in cancer
The canonical function of STAT2, executed by the tyrosine-phosphorylated protein, predominantly mediates the antiproliferative and apoptotic actions of IFN-I, implying that it may be a tumor suppressor [17, 18], and STAT2 does have IFN-I dependent tumor-suppressive activity in a mouse model of brain tumors [19]. The progesterone receptor, a well-known prognostic biomarker in breast cancer, promotes the degradation of STAT2 to inhibit the IFN response [20]. However, emerging evidence has identified a contrasting role of STAT2 in different cancers. For example, STAT2-deficient mice are protected from chemically induced skin and colon cancers [21, 22], and breast cancer cells with a high level of STAT2 have a more aggressive, mesenchymal/stem-like phenotype [23–25]. In pancreatic cancer, STAT2 is found to be highly expressed and associated with poor prognosis [26].
Research from the past decade has revealed additional functions of STAT2 that are independent of tyrosine phosphorylation (U-STAT2) [5, 27–29]. Unlike other members of the STAT family, STAT2 does not form a homodimer to recognize a DNA target [30]. Formation of the heterotrimeric ISGF3 complex is a unique characteristic among STAT-dependent pathways. Nan et al. define as non-canonical the role of the U-STAT2:IRF9 complex in driving gene expression by binding to a subset of ISREs that are present in the promoters of about a quarter of all ISGs [31], and also present in regions upstream of the promoters of a subset of NF-κB-driven genes, for example, IL-6. High expression of STAT2 benefits cancer cells in several ways. The U-STAT2:IRF9 complex enhances the expression of tumor-promoting cytokines and chemokines, especially IL-6, a multifunctional cytokine that activates the pro-tumorigenic tyrosine phosphorylation of STAT3 and, in lung cancer, high levels of STAT2 are associated with a worse prognosis [27]. Two threonine residues in STAT2, T387 and T404, are crucial for its IFN-dependent and IFN-independent functions. T387 phosphorylation is catalyzed by cyclin-dependent kinases, which are highly active in most types of cancer. Phosphorylation of T387 inhibits ISGF3 transcriptional activity, in concordance with IFN resistance in some cancer cells [32]. By contrast, the phosphorylation of T404 promotes ISG expression in response to IFN-I [33].
U-STAT2 binds to STING, inhibiting its ability to drive the synthesis of IFN-I in response to DNA damage, thus helping cancer cells avoid the enhancement of cell death that is due to the production of high levels of endogenous IFN-I. T404 phosphorylation promotes this inhibitory interaction, not only enhancing resistance to DNA damage in tumors, but also compromising the induction of antitumoral cytokines and chemokines in the tumor microenvironment (Wang et al. unpublished). Much more work needs to be done to understand how the phosphorylation of T387 and T404 modulates the functions of immune cells. The phosphorylation of these two threonine residues provides a new perspective in understanding how STAT2 and IFN-I participate in tumor progression and resistance to therapy.
IFN-I produced by cancer cells and IRDS expression
It is important to stress that high levels of U-STAT1, U-STAT2 and IRF9 are needed to drive substantial expression of the IRDS genes [2]. Normal cells do not express high levels of these three proteins or the IRDS proteins, but they are often highly expressed in cancer [3, 24]. Their expression is induced by chronic exposure of cells to low levels of IFN-I [2]. A recent study shows that a subset of cancers constitutively produces IFNβ in the absence of exogenous stimulation, maintaining high levels of the IRDS proteins [6]. Analyses using data from DNA microarray samples from individual patients revealed that substantial percentages of these samples express high levels of IRDS (37% for head and neck cancer, 48% for lung cancer, 29% for prostate cancer, 46% for breast cancer, and 50% for high-grade gliomas [3]).
Through diverse mechanisms, IFNβ is constitutively activated in a subset of cancer cells. Defective p53 function induces IFNβ by increasing dsRNA expression from repetitive elements in the genome [15, 34]. p53 deficiency alone induces low levels of IFNβ, decreasing the cytotoxicity of doxorubicin, a DNA damaging agent [15]. Co-inactivation of the ARF tumor suppressor in addition to p53 mutation helps cancer cells to produce IFNβ and IRDS proteins, promoting long-term proliferation in vitro and tumorigenesis in vivo [34]. By contrast, a combination of p53 deficiency and 5-aza-2’deoxycytidine-induced DNA demethylation leads to extremely high levels of IFNβ that induce massive cell death [15], showing that strong IFN-I responses contribute to cell killing. The cGAS-STING pathway, stimulated by cytoplasmic DNA, is constitutively activated in a subset of cancer cells, inducing chronic IFNβ expression and IRDS expression [6, 10, 14]. Cancer cells constantly experience endogenous DNA damage, due to the collapse of damaged replication forks and also through damage caused by reactive oxygen species (ROS), leading to the presence of DNA in the cytoplasm [10, 35]. Dysfunction of Ataxia-telangiectasia mutated (ATM), a DNA repair kinase, increases the levels of both cytoplasmic DNA and IFN-I expression [14]. Aging is another factor that increases cytoplasmic DNA, through the activation of long interspersed nuclear elements (LINEs), creating DNA damage in the nucleus and causing LINE cDNA to accumulate in the cytoplasm [36, 37].
IRDS expression, increased by self-produced IFNβ, is a protective mechanism that cancer cells use to enable them to survive endogenous DNA damage, as well as the exogenous damage caused by therapy. Remarkably, some cancer cells are addicted to self-produced IFNβ, initiating spontaneous apoptosis when the expression of IFNβ or IFNAR1 is knocked down [6]. Probably, these cells need constitutive expression of IRDS proteins to resist the deleterious effects of the constitutive presence of DNA in the cytoplasm.
How IRDS proteins modulate responses to DNA damage
Relatively little is known about how specific IRDS proteins promote resistance to DNA damage. The three 2’, 5’-oligoadenylate synthetases (OASs) are among the most highly expressed IRDS genes [3, 4], and recent work has shown that OAS1 has an important role in helping cancer cells to survive damage to their DNA [38]. OAS1, which is localized in the nucleus, promotes tumor cell survival in response to chemotherapy and oxidative stress by adding AMP residues in 2’, 5’ linkage to the ends of poly ADP-ribose (PAR) chains [38]. The addition of PAR to proteins (PARylation) by nuclear PAR polymerase 1 (PARP1) is a major post-translational modification that occurs in response to DNA damage [39, 40]. Moderate PARylation helps complexes of DNA repair proteins to assemble [40, 41], but excessive PARylation induces caspase-independent form of programmed cell death that is called PAR-mediated necroptosis, or parthanatos [42, 43]. The addition of AMP residues in 2’, 5’ linkage to PAR by OAS1 limits PARylation in response to DNA damage, thus inhibiting parthanatos [38].
Some ISG proteins suppress DNA damage-induced cell death by inhibiting cytotoxic acute IFN-I synthesis or IFN-I signaling. A resident RIG-I (retinoid acid-inducible gene1)-like receptor protein, LGP2 (Laboratory of Genetics and Physiology 2), increases radio-resistance by suppressing DNA damage-induced IFNβ synthesis, inhibiting cell death [7]. High levels of programmed death ligand 1 (PD-L1) inhibit cancer cell death in response to ionizing radiation or cisplatin by suppressing acute IFN-I signaling [6]. Both LGP2 and PD-L1 are induced by IFN-I and make cells resistant to DNA damage, defining them as IRDS proteins.
IFN-I responses regulated by PD-L1
As discussed in the previous section, IFN-I responses play contradictory roles in cancer cells, depending on the strength and longevity of the responses; strong and acute responses are cytotoxic, whereas weak and chronic responses are pro-survival [16]. A recent study revealed that PD-L1 is a master regulator of IFN-I responses in cancer cells, making cells more resistant to DNA damage [6]. PD-L1 is well known as an immune checkpoint protein that inhibits anti-cancer immune responses [44], but PD-L1-mediated regulation of IFN-I responses is a cancer cell-intrinsic event that operates independently of the immune system. High levels of PD-L1 in cancer cells inhibit cytotoxic acute IFN-I responses but sustain chronic responses that enhance IRDS levels, preventing cancer cell death in response to ionizing radiation and cisplatin [6]. In response to high doses of IFN-I, PD-L1 inhibits the phosphorylation of STAT1 and STAT2, diminishing cytotoxic ISG expression. On the other hand, PD-L1 sustains the constitutive activation of the cGAS-STING pathway and the chronic expression of IFNβ in cancer cells, resulting in the upregulation of pro-survival IRDS expression. The cGAS-STING pathway, and following IFN-I/IRDS expression, is activated by cytoplasmic DNA, which is present constitutively in cancer cells due to endogenous DNA damage [10, 14]. Importantly, levels of endogenous DNA damage are correlated with IFNβ and IRDS expression only when cancer cells express high levels of PD-L1 [6], suggesting a critical role of PD-L1 in constitutive IFN-I expression through the cGAS-STING pathway in these cells. Among a large number of cancer cell lines (1376 all types of cancer cell lines; 206 lung cancer cell lines), 9.2% of each group express high levels of IFNβ/ IRDS/ PD-L1, suggesting that PD-L1 plays an important role in maintaining constitutive IFNβ and IRDS expression in about 10% of all cancer cells [6]. How PD-L1 facilitates STING activation is not yet known, but considering that STING is on the endoplasmic reticulum, further investigation of intracellular PD-L1, not located on the plasma membrane, will help to reveal this mechanism. There is a correlation between the presence of cytoplasmic PD-L1 and poor prognosis in cancer [45, 46]. Downregulation of PD-L1 by several different mechanisms, including inhibition of cyclin-dependent kinase 5 (CDK5) [47], can sensitize cancer cells to radiation or cisplatin. It remains to be investigated whether CDK5 inhibition affects IFN-I synthesis, signaling, or both to sensitize cells to DNA damage.
IFN-dependent regulation of EMT/cancer stem cells
In the long history of attempts to use IFN-I as a potential cancer therapy, there have been numerous setbacks and conflicting results. In the previous sections, we discussed the IRDS expression that is induced by constitutive activation of the cGAS-STING pathway and IFNβ production, which enhance the resistance of cancer cells to DNA damage. However, the ubiquitous roles of IFN-I in activating anti-tumor immunity and inhibiting cancer cells continues to suggest a future in cancer therapy, by delivering IFNα, IFNβ, or therapies that can activate strong and acute endogenous IFN-I production (such as STING and TLR agonists) [48]. As new molecular mechanisms of IFN-I action are uncovered, novel modes of treatments that use IFN-Is in rational combination with other therapies can be devised. In contrast to the IFN signature induced by cancer cell-intrinsic immune-independent factors, the elevated IFN signature observed in immune-activated tumors is a maker of improved prognosis. For example, an immune active subtype of triple negative breast cancer (TNBC) that is characterized by an elevated IFN/STAT1 signature and an increased presence of Tumor Infiltrating Lymphocytes (TILs) responds better than immune-repressed tumors lacking IFN/STAT1 activity and TILs to DNA damaging chemotherapies, resulting in an improved prognosis [49, 50]. Similarly, IFN-I signaling correlates with a repressed cancer stem cell (CSC) gene signature and ultimately improved patient survival in several breast cancer datasets and experimental models, and treatment of CSCs with recombinant IFN-I induces a more differentiated, epithelial phenotype [11, 25, 51–53]. Comparable results have been observed with IFN-I in glioma stem cells and lung cancer stem cells [54–56], and even in response to the proteins encoded by specific ISGs, which can suppress CSC behavior in cisplatin-resistant ovarian cancer cells and hepatic cancer [57]. Thus, in the context of the tumor microenvironment, robust IFN-I signaling not only improves anti-tumor immunity, but also suppresses the stem-cell program to enhance the action of conventional chemotherapies. Conversely, in more de-differentiated cancer cells, IFN-I activity is markedly repressed [25]. The cytokine oncostatin M, an IL-6 family member present in the tumor microenvironment that promotes epithelial-mesenchymal transition (EMT) and CSC reprogramming, concomitantly represses acute IFN-I production, autocrine P-ISGF3 (phosphorylated ISGF3) activation, and ISG transcription [23]. Together, the data suggest that de-differentiating cancer cells repress IFN-I production to silence the IFN “warning system”, thereby preventing both autocrine and paracrine activities. By re-engaging a robust IFN-I response, EMT and CSC reprogramming may be able to be prevented or reversed to enhance the action of current chemotherapies. Yet, contrasting data in pancreatic cancer suggest that IFNs can also potentiate CSC behavior in certain instances, with increasing IFN-I signatures portending poorer patient outcomes [58]. At the moment, it is not clear why there are discrepancies in IFN-I action in difference cancer types. A weak IFN-I response, as already discussed, may result in dampened P-ISGF3 and robust U-ISGF3 signaling, which can induce therapeutic resistance due to the IRDS [13,14,15]. A greater understanding of additional molecular mechanisms that control P-ISGF3/U-ISGF3 activity in specific cancer types and the immune cells within them is needed.
IFN and the efficacy of oncolytic viruses in cancer therapy
In order to avoid the toxic and cytostatic effects of high levels of IFN-I, many cancers acquire mutations that affect their ability to produce or respond to this family of cytokines [59, 60]. Furthermore, mutations in the JAK-STAT pathway also affect the ability of cancer cells to respond to IFN-γ [61, 62]. In both cases, the resulting defects expose the Achilles heels of these cancers, by sensitizing them to oncolytic viruses. In addition to defects in IFN pathways caused by mutations, as pointed out above, PD-L1 inhibits the anti-viral effects of IFN signaling, sensitizing cancer cells to oncolytic viruses.
Excellent recent reviews have summarized the uses of many different oncolytic viruses in a variety of cancers [63] and the mechanisms they use to kill cancer cells, including the many ways in which defects in the IFN-I system sensitize the cancer cells preferentially [59]. In spite of the attractiveness of this therapeutic approach, careful consideration must be given to the toxicity of many naturally occurring and engineered oncolytic viruses towards normal cells and tissues. A promising approach under current development is to avoid toxicity by using non-pathogenic human enteroviruses [60, 64].
Concluding remarks and future perspectives
Cancer cell-intrinsic IFN-I synthesis and responses can have opposite effects in cancer therapy, depending on the strength and longevity of the responses. Strong and acute IFN-I responses are cytotoxic, whereas weak and chronic responses are pro-survival and inhibit the efficacy of cancer therapy. Many cancer cells consistently produce IFN-I, sustaining chronic IFN-I responses and resistance to cancer therapy, a point that is strikingly illustrated by our observation that some cancer cell lines are addicted to self-produced IFN-I and do not survive if the IFNβ gene is knocked out.
Treatment of various cancers with IFN-I as a single agent has had only very limited success. Recent research suggests that IFN-I can facilitate cell killing in combination with DNA damaging agents, including radiation and chemotherapy. Furthermore, blocking chronic IFN-I synthesis and responses can sensitize cancer cells to DNA damage, since chronic IFN-I promotes resistance to DNA damage.
How to block pro-survival chronic IFN-I responses efficiently without hindering cytotoxic IFN-I responses remains to be investigated (see Outstanding Questions). Reducing the expression of PD-L1, which inhibits cytotoxic IFN-I responses but promotes pro-survival IFN-I responses, is a promising approach that can sensitize cancer cells to DNA damaging therapy as well as immunotherapy.
Outstanding Questions.
How can we block pro-survival chronic IFN-I responses without inhibiting DNA damage-induced cytotoxic IFN-I responses? Can the IRDS be inhibited selectively?
What are the mechanisms by which specific IRDS proteins (in addition to OAS1) increase the resistance of cancer cells to DNA damage?
Can a robust tumor-specific response to IFN-I be activated without systemic IFN-I treatment?
Which cancers will benefit from reactivation of IFN-I synthesis in tumors in which it has been repressed? Which will be negatively impacted?
What are the relative efficacies of cytoplasmic DNA and double-stranded RNA in stimulating IFN-I synthesis in cancer cells?
How widespread is addiction to IFN-I in cancer cells?
How are the phosphorylations of T387 and T404 in STAT2 catalyzed and regulated in cancer cells?
What is the role of PD-L1 in inducing constitutive IFN-I production in response to endogenous DNA damage in cancer cells? What is the impact of this IFN-I on the tumor stroma?
Can anti-PD-L1 antibodies that are now used clinically sensitize cancer cells to DNA damage?
How do high levels of PD-L1 suppress responses to IFN-I?
How do high levels of PD-L1 suppress cytotoxic responses to DNA damage?
How is IFN-I function repressed in stem-like mesenchymal cells?
Highlights.
IFN-I constitutively produced by cancer cells sustains pro-survival responses and resistance to DNA-damaging therapies. Cancer cells addicted to self-produced IFN-I die if the IFN-β gene is knocked out but survive if IFN-β is provided exogenously.
PD-L1 stimulates IFN-I synthesis by cancer cells but inhibits their ability to respond to IFN-I, thus establishing a “Goldilocks-like” state in which the pro-tumor IRDS response is sustained and the response to cytotoxic IFN-I response is inhibited.
U-STAT2 functions, regulated by threonine-phosphorylations on T387 and T404, play important roles in cancer, aiding some NF-κB-dependent gene expression and regulating IFN-I synthesis through the cGAS-STING pathway.
IFN-I and oncostatin-M have opposing effects and negatively regulate each other in TNBC. Oncostatin-M favors a pro-tumor, stem-like, mesenchymal phenotype, whereas IFN does the opposite.
Acknowledgments
This study was supported by National Cancer Institute Grants P01CA062220 (to G.R.S.), R21CA252387 (to H.C.), R03CA215941 (to H.C.), and R01CA252224 (to M.W.J.); Department of Defense Breast Cancer Research Program Grants W81XWH-20-1-0464 (to M.W.J.) and W81XWH-18-1-0552 (to M.W.J).
Glossary
- cGAS-STING pathway
a mechanism by which interferon β (IFNβ) expression is induced in response to cytosolic DNA. Cyclic GMP-AMP Synthase (cGAS) detects cytosolic DNA, followed by the synthesis of cyclic GAMP, which activates Stimulator of Interferon Genes (STING) protein, triggering the induction of IFNβ gene expression
- IFN-I
type I Interferon (Interferon-I). A group of cytokines that bind to the IFN-α receptor (IFNAR), which consists of IFNAR1 and IFNAR2. IFN-Is include the IFNα subtypes and IFNβ
- IRDS
IFN-Related DNA damage resistance Signature. A subset of IFN-stimulated genes (ISGs), the levels of which are correlated with the resistance of cancer cells to DNA-damaging cancer therapy, including ionizing radiation and certain types of chemotherapy
- ISGF3
IFN-Stimulated Gene Factor 3. A complex of STATs 1 and 2, which are phosphorylated on specific tyrosine residues (Y701 of human STAT1, Y690 of human STAT2) and Interferon Response Factor 9 (IRF9). ISGF3 is formed in response to IFN-I or IFN-III (type III interferon, IFNλ)
- ISGs
IFN-Stimulated Genes. A group of genes that are induced in response to various types of interferons
- PD-L1
Programmed cell Death-Ligand 1. A protein expressed on the surfaces of cancer cells and immune cells (eg. dendritic cells, macrophages), where it binds to PD-1 expressed on activated T cells, suppressing their function. PD-L1 is also observed in the cytoplasm and nuclei of cancer cells
- STATs
Signal Transducers and Activators of Transcription. STATs 1–6 mediate signals in response to many different cytokines, including interferons. In response to IFN-I, a STAT1-STAT2 heterodimer combines with IRF9 to form ISGF3
- TME
Tumor Micro-Environment. The TME includes the surrounding blood vessels, immune cells, fibroblasts, cytokines, and extracellular matrix
- TNBC
Triple-Negative Breast Cancer. TNBC is a highly aggressive subtype of breast cancer in which estrogen receptor (ER) and progesterone receptor (PR) are not expressed and in which human epidermal growth factor receptor 2 (HER2) is not over expressed
- U-ISGF3
Unphosphorylated ISGF3. U-ISGF3 consists of IRF9 and STATs 1 and 2 lacking phosphorylation of specific tyrosine residues (Y701 of human STAT1 and Y690 of human STAT2). U-ISGF3 play a critical role in inducing a subset of ISGs, including the IRDS genes
- U-STAT2
Unphosphorylated STAT2. U-STAT2 lacks phosphorylation on tyrosine residue Y690
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors have no conflicts of interest to declare.
References
- 1.Lindenmann J et al. (1957) Studies on the production, mode of action and properties of interferon. Br J Exp Pathol 38 (5), 551–562. [PMC free article] [PubMed] [Google Scholar]
- 2.Cheon H et al. (2013) IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J 32 (20), 2751–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weichselbaum RR et al. (2008) An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A 105 (47), 18490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khodarev NN et al. (2004) STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A 101 (6), 1714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheon H et al. (2013) IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. The EMBO Journal 32 (20), 2751–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheon H et al. (2021) PD-L1 sustains chronic, cancer cell-intrinsic responses to type I interferon, enhancing resistance to DNA damage. Proc Natl Acad Sci U S A 118 (47), e2112258118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Widau RC et al. (2014) RIG-I-like receptor LGP2 protects tumor cells from ionizing radiation. Proc Natl Acad Sci U S A 111 (4), E484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snell LM et al. (2017) Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol 38 (8), 542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu R et al. (2022) Type I interferon-mediated tumor immunity and its role in immunotherapy. Cell Mol Life Sci 79 (3), 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erdal E et al. (2017) A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev 31 (4), 353–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sistigu A et al. (2014) Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 20 (11), 1301–9. [DOI] [PubMed] [Google Scholar]
- 12.Cheon H and Stark GR (2009) Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc Natl Acad Sci U S A 106 (23), 9373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaston J et al. (2016) Intracellular STING inactivation sensitizes breast cancer cells to genotoxic agents. Oncotarget 7 (47), 77205–77224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartlova A et al. (2015) DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 42 (2), 332–343. [DOI] [PubMed] [Google Scholar]
- 15.Leonova KI et al. (2013) p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc Natl Acad Sci U S A 110 (1), E89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheon H et al. (2014) Interferons and their stimulated genes in the tumor microenvironment. Semin Oncol 41 (2), 156–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burnette BC et al. (2011) The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res 71 (7), 2488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunn GP et al. (2005) A critical function for type I interferons in cancer immunoediting. Nature Immunology 6 (7), 722–729. [DOI] [PubMed] [Google Scholar]
- 19.Wang J et al. (2003) Dysregulated Sonic hedgehog signaling and medulloblastoma consequent to IFN-alpha-stimulated STAT2-independent production of IFN-gamma in the brain. J Clin Invest 112 (4), 535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walter KR et al. (2020) Progesterone receptor promotes degradation of STAT2 to inhibit the interferon response in breast cancer. Oncoimmunology 9 (1), 1758547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gamero AM et al. (2010) STAT2 contributes to promotion of colorectal and skin carcinogenesis. Cancer Prev Res (Phila) 3 (4), 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang M et al. (2019) Expression profile and prognostic values of STAT family members in non-small cell lung cancer. Am J Transl Res 11 (8), 4866–4880. [PMC free article] [PubMed] [Google Scholar]
- 23.Doherty MR et al. (2019) The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res 21 (1), 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogony J et al. (2016) Interferon-induced transmembrane protein 1 (IFITM1) overexpression enhances the aggressive phenotype of SUM149 inflammatory breast cancer cells in a signal transducer and activator of transcription 2 (STAT2)-dependent manner. Breast Cancer Res 18 (1), 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doherty MR et al. (2017) Interferon-beta represses cancer stem cell properties in triple-negative breast cancer. Proc Natl Acad Sci U S A 114 (52), 13792–13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pang C et al. (2018) Several genes involved in the JAK-STAT pathway may act as prognostic markers in pancreatic cancer identified by microarray data analysis. Medicine (Baltimore) 97 (50), e13297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nan J et al. (2018) IRF9 and unphosphorylated STAT2 cooperate with NF-κB to drive IL6 expression. Proceedings of the National Academy of Sciences 115 (15), 3906–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang W et al. (2017) Unphosphorylated ISGF3 drives constitutive expression of interferon-stimulated genes to protect against viral infections. Science Signaling 10 (476), eaah4248. [DOI] [PubMed] [Google Scholar]
- 29.Testoni B et al. (2011) Chromatin dynamics of gene activation and repression in response to interferon alpha (IFN(alpha)) reveal new roles for phosphorylated and unphosphorylated forms of the transcription factor STAT2. J Biol Chem 286 (23), 20217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bluyssen HA and Levy DE (1997) Stat2 is a transcriptional activator that requires sequence-specific contacts provided by stat1 and p48 for stable interaction with DNA. J Biol Chem 272 (7), 4600–5. [DOI] [PubMed] [Google Scholar]
- 31.Nan J et al. (2018) IRF9 and unphosphorylated STAT2 cooperate with NF-kappaB to drive IL6 expression. Proc Natl Acad Sci U S A 115 (15), 3906–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y et al. (2017) Negative regulation of type I IFN signaling by phosphorylation of STAT2 on T387. EMBO J 36 (2), 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y et al. (2020) A virus-induced conformational switch of STAT1-STAT2 dimers boosts antiviral defenses. Cell Res 31 (2), 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forys JT et al. (2014) ARF and p53 coordinate tumor suppression of an oncogenic IFN-beta-STAT1-ISG15 signaling axis. Cell Rep 7 (2), 514–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wyman C and Kanaar R (2006) DNA double-strand break repair: all’s well that ends well. Annu Rev Genet 40, 363–83. [DOI] [PubMed] [Google Scholar]
- 36.Gasior SL et al. (2006) The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol 357 (5), 1383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simon M et al. (2019) LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab 29 (4), 871–885 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kondratova AA et al. (2020) Suppressing PARylation by 2’,5’-oligoadenylate synthetase 1 inhibits DNA damage-induced cell death. EMBO J 39 (11), e101573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo X and Kraus WL (2012) On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 26 (5), 417–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gupte R et al. (2017) PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 31 (2), 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Sullivan J et al. (2019) Emerging roles of eraser enzymes in the dynamic control of protein ADP-ribosylation. Nat Commun 10 (1), 1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barkauskaite E et al. (2015) Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Mol Cell 58 (6), 935–46. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y et al. (2016) A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 354 (6308). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun C et al. (2018) Regulation and Function of the PD-L1 Checkpoint. Immunity 48 (3), 434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chowdhury S et al. (2016) Programmed death-ligand 1 overexpression is a prognostic marker for aggressive papillary thyroid cancer and its variants. Oncotarget 7 (22), 32318–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tu X et al. (2019) PD-L1 (B7-H1) Competes with the RNA Exosome to Regulate the DNA Damage Response and Can Be Targeted to Sensitize to Radiation or Chemotherapy. Mol Cell 74 (6), 1215–1226 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De S et al. (2021) The ubiquitin E3 ligase FBXO22 degrades PD-L1 and sensitizes cancer cells to DNA damage. Proc Natl Acad Sci U S A 118 (47), e2112674118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arico E et al. (2019) Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers (Basel) 11 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burstein MD et al. (2015) Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res 21 (7), 1688–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang X et al. (2014) Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 25 (1), 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bidwell BN et al. (2012) Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med 18 (8), 1224–31. [DOI] [PubMed] [Google Scholar]
- 52.Slaney CY et al. (2013) The role of Type I interferons in immunoregulation of breast cancer metastasis to the bone. Oncoimmunology 2 (1), e22339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miranda A et al. (2019) Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci U S A 116 (18), 9020–9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du Z et al. (2017) The effects of type I interferon on glioblastoma cancer stem cells. Biochem Biophys Res Commun 491 (2), 343–348. [DOI] [PubMed] [Google Scholar]
- 55.Castiello L et al. (2018) Disruption of IFN-I Signaling Promotes HER2/Neu Tumor Progression and Breast Cancer Stem Cells. Cancer Immunol Res 6 (6), 658–670. [DOI] [PubMed] [Google Scholar]
- 56.Zhan X et al. (2020) Glioma stem-like cells evade interferon suppression through MBD3/NuRD complex-mediated STAT1 downregulation. J Exp Med 217 (5), e20191340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang JL et al. (2022) Targeting Cancer Stem Cells through Epigenetic Modulation of Interferon Response. J Pers Med 12 (4), 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Espinet E et al. (2021) Aggressive PDACs Show Hypomethylation of Repetitive Elements and the Execution of an Intrinsic IFN Program Linked to a Ductal Cell of Origin. Cancer Discov 11 (3), 638–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kooti W et al. (2021) Oncolytic Viruses and Cancer, Do You Know the Main Mechanism? Front Oncol 11, 761015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matveeva OV and Chumakov PM (2018) Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev Med Virol 28 (6), e2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaufman HL (2021) Can Biomarkers Guide Oncolytic Virus Immunotherapy? Clin Cancer Res 27 (12), 3278–3279. [DOI] [PubMed] [Google Scholar]
- 62.Nguyen TT et al. (2021) Mutations in the IFNgamma-JAK-STAT Pathway Causing Resistance to Immune Checkpoint Inhibitors in Melanoma Increase Sensitivity to Oncolytic Virus Treatment. Clin Cancer Res 27 (12), 3432–3442. [DOI] [PubMed] [Google Scholar]
- 63.Yang L et al. (2021) Oncolytic Virotherapy: From Bench to Bedside. Front Cell Dev Biol 9, 790150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Le TH et al. (2020) [The State of The Jak/Stat Pathway Affects the Sensitivity of TumorCells to Oncolytic Enteroviruses]. Mol Biol (Mosk) 54 (4), 634–642. [DOI] [PubMed] [Google Scholar]