

Summary
Tumor-infiltrating lymphocytes (TILs), especially CD8+ TILs, represent a favorable prognostic factor in high-grade serous ovarian cancer (HGSOC) and other tumor lineages. Here, we analyze the spatial heterogeneity of different TIL subtypes in HGSOC. We integrated RNA sequencing, whole-genome sequencing, bulk T cell receptor (TCR) sequencing, as well as single-cell RNA/TCR sequencing to investigate the characteristics and differential composition of TILs across different HGSOC sites. Two immune “cold” patterns in ovarian cancer are identified: (1) ovarian lesions with low infiltration of mainly dysfunctional T cells and immunosuppressive Treg cells and (2) omental lesions infiltrated with non-tumor-specific bystander cells. Exhausted CD8 T cells that are preferentially enriched in ovarian tumors exhibit evidence for expansion and cytotoxic activity. Inherent tumor immune microenvironment characteristics appear to be the main contributor to the spatial differences in TIL status. The landscape of spatial heterogeneity of TILs may inform potential strategies for therapeutic manipulation in HGSOC.
Keywords: TILs, spatial heterogeneity, multiple lesions, multi-omics, high-grade serous ovarian cancer
Graphical abstract
Highlights
-
•
Quantitative and qualitative immune “cold” in primitive and omental metastatic lesions
-
•
Tumor-specific but exhausted CD8+ T cells enrich in ovarian sites
-
•
Non-tumor-specific bystander T cells enrich in omental metastases
-
•
CD8+ Tex cells develop from an early differentiation state with proliferative ability
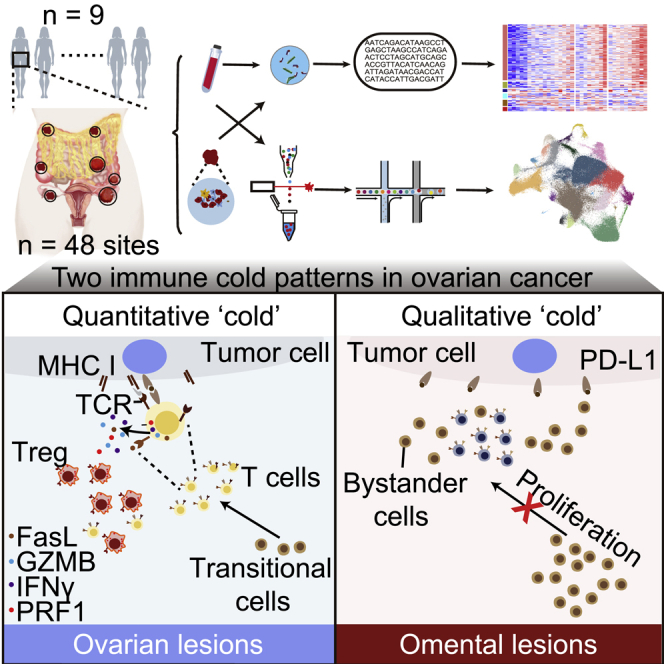
Yang et al. conduct a multi-omics study and identify two immune “cold” patterns in ovarian cancer: ovarian lesions with tumor-specific but exhausted and low-infiltrated T cells and omental lesions with non-tumor-specific bystander T cells. The landscape of spatial heterogeneity of TILs may inform potential strategies for therapeutic manipulation in HGSOC.
Introduction
High-grade serous ovarian cancer (HGSOC) affects 239,000 women worldwide each year and represents the most lethal type of gynecological cancer.1 Almost 80% of patients are diagnosed as stage III or IV disease, and many succumb to primary treatment resistance or relapse within 18 months, leading to a 5-year survival rate of about 30%.2 Unfortunately, the overall survival odds for HGSOC patients have not improved markedly despite years of extensive biological research and clinical trials and the addition of bevacizumab and PARP inhibitors to the therapeutic armamentarium. There is thus a strong need for effective new therapies including ones that induce effective immune engagement potentially through immune checkpoint blockade (ICB), such as programmed cell death (PD-1) or its ligand (PD-L1) antibodies,3 with median response rates lower than 15%.4 The mechanism(s) underlying the lack of response to ICB despite the presence and prognostic impact of T cell infiltration remains largely unknown.
HGSOC often presents with widespread abdominal cavity dissemination with the omentum as the most frequent site of metastasis.5,6 Multi-site studies, albeit controversial,7 indicate that genomic inter-lesional heterogeneity8 is associated with poor survival.9 The effect of spatial immunologic variation, especially in T cell infiltration, recognition, and expansion, across various tumor foci in the ovary (primary) and distant metastatic foci in the peritoneal cavity and their contribution to the limited response to immune therapy in HGSOC remains unexplored.
To provide a detailed analysis of the landscape of heterogeneity of infiltrating T cells in primary and metastatic lesions and their differential characteristics in HGSOC, we performed multi-site sampling and simultaneous RNA sequencing (RNA-seq), whole-genome sequencing (WGS), and bulk T cell receptor (TCR) sequencing as well as single-cell RNA-seq (scRNA-seq) and paired TCR sequencing (scTCR-seq) in 9 patients (48 sites) with untreated primary HGSOC. We identify two different immune patterns in ovarian cancer: (1) ovarian lesions with low infiltration of mainly dysfunctional T cells and immunosuppressive Treg cells. These exhausted CD8 T cells with cytotoxic function are clonally expanded; (2) omental lesions infiltrated with non-tumor-specific bystander cells. Decreased major histocompatibility complex class I (MHC class I) antigen presentation ability and failure of T cell infiltration into omental tumors may contribute to lack of tumor-specific T cells in omental metastasis. Together these observations may partly explain the poor response of ovarian cancer to current immunotherapy approaches.
Results
Differential transcriptomic profiles across multiple sites in HGSOC
We performed WGS and RNA-seq on 48 sites from nine treatment-naive pathological HGSOC patients (Table S1; Figure 1A). Each site in the same patient had a similar proportion of tumor (Table S1; STAR Methods). Copy number variation (CNV) and somatic mutations of these tumors were consistent with known HGSOC genomic patterns10 (Figures S1A and S1B) with the exception of OV001, which, despite high-grade characteristics on histopathology, did not have TP53 mutations and instead with the only tumor with a NF1 aberration. High-level gene amplifications present in ovarian cancer, such as CCNE1, MYC were present in 2/9 (Figure S1B). Eight of the nine HGSOC tumors had somatic TP53 mutations, while 2/9 patients harbored germline BRCA1/2 mutations (Figures S1A and S1B). Importantly, the majority of the CNV and somatic mutation events did not demonstrate spatial genomic heterogeneity among tumor sites (Figure S1B). The detection of TP53 mutations in metastases but not in ovarian sites of OV004 is one key exception. Manual inspection of sequence tracks failed to identify TP53 mutations in ovarian sites. To characterize the relationship between multiple sites in HGSOC, we first performed principal-component analysis (PCA) on transcriptomic profiles of primary ovarian (Ov, while HGSOC originate in the fallopian tube, the ovary represents the most frequent site of initial seeding consistent with the definition of primary), omental (Om), and other metastatic lesions (Ot). PCA demonstrated two drivers of heterogeneity, patient-specific processes with tumors across different sites within a given individual tending to cluster together and tumors within different sites tending to cluster together for different patients (Figure 1B). For example, while ovarian tumors from OV004, OV005, OV006, and OV008 were clearly separated from omental and other sites, ovarian tumors from OV001, OV002, OV003, and OV009 tended to cluster closer to their metastatic sites than to other ovarian tumors. Decomposition of immune cell proportions using ssGSEA analysis of RNA-seq data recapitulated the heterogeneity observed in the PCA with information content specific to patients and also to tumor site (Figure 1C). Figure 1C also demonstrates the robustness and the consistency of the analysis, with tumors from the left and right ovary from the same patient clustering together and multiple different omental lesions from the same patient clustering together. Similar to the PCA, immune cell-based clustering suggested that, while most ovarian tumors were in a single cluster, OV001, OV002, OV003, and OV009 ovarian tumors tended to cluster with their metastatic sites. Interestingly, the estimated proportions of various immune components were low in ovarian tumors (Figure 1C). In contrast, the immune components were markedly higher in most omental sites compared with matched ovarian tumors; with other sites having lower immune content and indeed a subset of the other metastatic sites clustered with the ovarian tumors (Figure 1C). A panel of 159 genes selected based on 6 different characteristics of T cell quantity or spatial distribution11 were used to further characterize the samples demonstrating highest T cell infiltration in omental lesions, with most of the ovarian tumors having low levels, indicating a “desert” T cell phenotype (Figure 1D). In parallel, CD4+ and CD8+ T cell infiltration into the different lesions were evaluated based on immunohistochemistry (IHC) staining (Figure S1C) and flow cytometry analysis (Figure S1D). Consistent with the transcriptional profiling data, the density of CD4+ and CD8+ T cells was much lower in ovarian lesions than in omental lesions (Figures 1E and 1F), indicating that the ovarian lesions are immune “cold” lesions. FAP, a marker of activated stroma, in contrast, did not vary across lesion location (Figure 1E).
Figure 1.

Differential transcriptomic profiles across multiple sites in HGSOC
(A) Overview of the study design.
(B) The PCA plot of mRNA expression.
(C) The abundance of 28 immune cell types (identified by ssGSEA) is shown according to distinct locations.
(D) The gene expression of six immune-related pathways in tumors of different locations.
(E) Quantification of densities of CD4+ and CD8+ cells, and FAP H scores across three sites. Data represent mean ± SEM. p values were determined by Tukey’s multiple comparisons test.
(F) Quantification of all T (CD45+CD3+), CD4+ T (CD45+CD3+CD4+), and CD8+ (CD45+CD3+CD8+) T proportions in tumors from each sample, respectively. Ovarian samples = 8, omental samples = 5. Data represent mean ± SEM. p values were determined by Student’s t test.
Distinct characteristics and differential composition of TILs across different lesions in HGSOC by scRNA-seq
To further detail the landscape of infiltrated T cells and explore the heterogeneity among different lesions, we sorted CD45+CD3+ T cells from single-cell suspensions prepared from 13 ovarian (Ov), 7 omental (Om), 4 other distant metastatic (Ot) sites, and 6 PBMCs of patients OV004, OV005, OV006, OV008, OV009, and OV010, and performed scRNA-seq and matched scTCR-seq using the 10 × 5′ platform (Figures 1A and S2A; Table S1). After removing confounding batch effects and patient-specific variability (see STAR Methods), a total of 227,769 CD45+CD3+ immune cells from all subjects were available for analysis (Table S2).
Using dimension reduction of Uniform Manifold Approximation and Projection (UMAP) with a resolution of 0.2, we identified 22 stable clusters, including 7 clusters for CD4+ and 15 clusters for CD8+ T cells, each with unique signature genes (Figures 2A, 2B, and S2B–S2E). In addition to typical CD8+ and CD4+ T cell clusters, including naive,12 effector (Teff), memory (Memory), mucosal-associated invariant T cells (MAIT) of blood and tissue, conventional regulatory T (Treg), and dysfunctional “exhausted” T cells (Tex), we also identified two proliferative clusters that highly expressed MKI67: CD8_C05-TYMS expressing markers associated with exhaustion (designated as Tex.prol) and CD4_C04-TYMS expressing markers associated with Treg (designated as Treg.prol) (Figures 2B, S2D, and S2E). CD8_C03 (Tex) population showed the highest expression of CXCL13, HAVCR2, and the co-inhibitory receptor PDCD1, as well as increased expression of GZMB, GZMA, and GZMH, indicating that cells in this cluster potentially have cytotoxic activity in addition to exhaustion features. Furthermore, a pre-dysfunctional cluster (CD8_C02, referred to as “transitional”) was defined by high expression of GZMK13 and a progenitor exhaustion cluster (CD8_C07) was defined by higher GPR18314 (a central memory marker) and lower PDCD1 (an exhaustion marker) than CD8_C03 (Tex). We identified additional CD8 positive subsets, including CD8_C04 (NK-like) and CD8_C15 (γδ-like). CD8_C04 (NK-like) highly expressed KLRD1 and NKG7, known markers of NK15/NKT16 cells, and CD8_C15 (γδ-like) highly expressed TRDV2 and TRGV9, known markers of γδ T cells17 (Figures 2B and S2E).
Figure 2.

Distinct characteristics and differential composition of TILs across different lesions in HGSOC by scRNA-seq
(A) Uniform Manifold Approximation and Projection (UMAP) of 227,769 single CD3+ T cells from 6 HGSOC patients, showing the formation of 22 main clusters, including 15 for CD8+ cells (including 9 tumor-infiltrating T cell clusters and 6 T cell clusters from blood), 7 for CD4+ cells (including 5 tumor-infiltrating T cell clusters and 2 T cell clusters from blood).
(B) Violin plots showing marker genes across 22 CD3+ T cell clusters.
(C) Bar plot indicating relative proportions of each cell cluster detected in blood and solid tumor lesions, including ovarian (Ov), omental (Om), and other distant metastatic (Ot).
(D) Boxplot of the relative proportions of each tumor-infiltrating T cell clusters detected in solid tumor lesions, including Ov, Om, and Ot. wilcox.test.
(E) The co-expression of CD8 and PD-1 in the ovarian site was evaluated by opal multiplex IHC. AEC color signals were extracted from each digitized single-marker image by color deconvolution, followed by pseudo-coloring. A representative image is shown. Nuclei (blue), GZMB (red), CD8 (magenta), PD-1 (cyan), and pan-CK (green). Scale bars, 20 μm. White arrow indicates CD8+ PD-1+ T cells.
(F) A representative image of CD8+ T cells in omentum samples is shown. Nuclei (blue), GZMB (red), CD8 (magenta), PD-1 (cyan), and pan-CK (green). Scale bars, 20 μm. Cyan arrows indicate PD-1+ cells and red arrows indicate GZMB+ cells.
(G) Quantification of Treg (CD45+CD3+CD4+CD25+CD127−) and CD8+ Tex (CD45+CD3+CD8+PD-1+/LAG3+) proportions in tumors from each sample, respectively. Ovarian samples = 8, omental samples = 5. Data represent mean ± SEM. p values were determined by Student’s t test.
(H) Quantification of CD4+ central memory T (Tcm) (CD45+CD3+CD4+CD45RA-CCR7+), CD8+ Tcm CD45+CD3+CD8+CD45RA-CCR7+) proportions in tumors from each sample, respectively. Ovarian samples = 8, omental samples = 5. Data represent mean ± SEM. p values were determined by Student’s t test.
We next investigated the relative proportions of different clusters between ovarian, omental, other sites, and blood (Figures 2C, 2D, and S2F–S2I). Interestingly, the proportion of dysfunctional cells, including CD8_C03 (Tex), CD8_C05 (Tex.prol), and CD8_C07 (Tex.prog), and immunosuppression cluster, CD4_C02 (Treg), were significantly enriched in ovarian tumors (Figure 2D). CD4_C03 (Tex) exhibited a trend to increase in ovarian compared with omental or other sites. In contrast, naive, memory, and transition state clusters were enriched in omental sites (Figure 2D). Opal-IHC showed that T cells in ovarian lesions were likely to be exhausted (CD8+PD-1+), while T cells in omental lesions were more likely to be in a non-exhausted state (CD8+PD-1− or CD8+GZMB−) (Figures 2E and 2F). The major cellular composition difference between ovarian and omental lesions was again observed by flow cytometry analysis (Figures 2G, 2H, and S3A; Table S3). Treg and Tex cells were significantly increased in ovarian sites, while central memory T were enriched in omental lesions, and other subsets, including naive, effector memory T, and effector memory re-expressing CD45RA T cells (TEMRA) were comparable in these two sites (Figure S3B). Taken together, increased Tex and Treg is consistent with primary ovarian tumors being immunosuppressed.
Patient-derived TMB is associated with skewed T cell differentiation
Tumor mutation burden (TMB), neoantigen burden, and high genomic instability, including deficient mismatch repair and homologous recombination deficiency (HRD), have been associated with increased T cell infiltration and better response to checkpoint inhibitors in some cancer types.18,19,20 To explore whether heterogeneity in T cell infiltration in different tumor sites or different patients is related to genomic aberrations, TMB,21 HRD score,22 and COSMIC mutational signature23 of each sample were assessed according to previous analysis pipelines (Figures S4A and S4B). Concordant with a previous study in NSCLC,24 the correlation matrix revealed that CD4_C03 (Tex) and CD8_C03 (Tex) clusters correlated with TMB, neoantigen burden, and HRD score, suggesting that CD4_C03 (Tex) and CD8_C03 (Tex) may be antigen-engaged T cell subsets (Figures S4C and S4D). However, the association of TMB, HRD score, and COSMIC mutational signatures with CD4_C03 (Tex) and CD8_C03 (Tex) is observed at the patient level rather than site level within individual patients (Figures S4A and S4B). In addition, we also constructed multi-region evolutionary trees based on somatic single-nucleotide variants and structural variants25 across tumor sites (Figure S4E). Compared with PBMC, spatial genomic heterogeneity among tumors within individual patients is low, especially between ovarian and omental metastatic tumors. Thus, spatial genomic features, including TMB, HRD scores, and COSMIC mutational signature and evolution trajectory fail to explain the differences in T cell infiltration across different lesion sites within patients.
Tumor-specific but exhausted CD8+ cells preferentially infiltrate primary ovarian tumors, while non-tumor-specific bystander cells are enriched in omentum metastases
To further investigate functional differences of CD8+ T cell clusters across locations, we first assessed transcriptional features of terminal exhaustion and effector memory signatures among CD8 T cell clusters by functional scores derived from previous reports26,27 (Figures 3A, 3B, and S5A). As expected, CD8_C03 (Tex) and CD8_C05 (Tex.prol) had the highest terminal exhaustion characteristic (Figure 3B), while CD8_C02 (Tex,trans), CD8_C04 (NK-like), and CD8_C06 (Teff) had features associated with effector and memory (Figure S5A). Given that exhausted T cells are frequently generated as a consequence of persistent antigen exposure,28 we next tested whether CD8_C03 (Tex) and CD8_C05 (Tex.prol) transcriptionally resemble neoantigen-reactive populations using a tumor-specific signature.29 Consistent with the concept that exhausted cells have undergone chronic antigen stimulation, the tumor-specific signature was significantly enriched in these two exhausted T subsets (Figure 3C). Intra-tumoral T cells can also be CD39− bystanders that recognize virus rather than tumor antigens.30,31 Bystander signatures, including virus-specific and CD39− CD69− signatures, were dramatically increased in CD8_C02 (Tex,trans), CD8_C04 (NK-like), and CD8_C06 (Teff) that are enriched in omental tumors (Figures S5B and S5C). Collectively, CD8_C03 (Tex) and CD8_C05 (Tex.prol), which are enriched in ovarian tumors, exhibited high exhaustion, tumor-specific score, and low bystander score, whereas CD8_C02 (Tex,trans) and CD8_C04 (NK-like), which are enriched in omental tumors, exhibited the opposite characteristics (Figures 3D and 3E). Overall, tumor-specific signatures were strongly positively correlated with a terminal exhausted signature and were negatively associated with a bystander signature (Figures 3F and 3G). Spatially, ovarian lesions had profoundly higher tumor-specific and terminal exhaustion scores than omental samples (Figures 3F and 3H). Conversely, omental lesions exhibited markedly higher bystander scores (Figures 3G and 3H). A heatmap of all signature scores showed the same distribution (Figure 3I). Consistently, flow cytometry analysis confirmed more CD8+ tumor-specific T cells, expressing CD39+, were enriched in ovarian lesions, whereas more CD8+ bystander T cells enriched in omental lesions (Figures 3J and S5D).
Figure 3.

Characterization of CD8+ tumor-infiltrating T cells in HGSOC
(A) UMAP of 94,424 single CD8+ tumor-infiltrating cells, showing the formation of 9 main clusters in tumor tissues.
(B and C) UMAP of CD8+ tumor-infiltrating cells colored according to gene signature scores, (B) terminally exhausted CD8+ signature, and (C) tumor-specific CD8+ signature.
(D) Violin plots showing the sorted gene signatures scores (up, terminally CD8+ signature score; down, tumor-specific signature score) across 9 CD8+ tumor-infiltrating cell clusters.
(E) Violin plots showing the sorted bystander CD8+ signature score across 9 CD8+ tumor-infiltrating cell clusters.
(F and G) Correlations between different gene signatures in all CD8+ tumor-infiltrating cells at the sample level, (F) terminally exhausted CD8+ signature score and tumor-specific CD8+ signature score, and (G) bystander signature score and tumor-specific CD8+ signature score, each color represents a different tumor site.
(H) Violin plots showing the gene signatures scores (left, terminally exhausted CD8+ signature score, middle, tumor specific CD8+ signature score, right, bystander CD8+ signature score) in CD8+ tumor-infiltrating cells from different positions, including Ov, Om, and Ot. wilcox.test.
(I) Heatmap showing multi gene signatures and sample positions information at the sample level, arranged from low to high by the terminally CD8+ signature score.
(J) Quantification of CD8+ bystander T (CD45+CD3+CD8+CD39−) and CD8+ tumor-specific T (CD45+CD3+CD8+CD39+) cells proportions in tumors from each sample, respectively. Ovarian samples = 8, omental samples = 5. Data represent mean ± SEM. p values were determined by Student’s t test.
(K) Potential developmental trajectory of CD8+ tumor-infiltrating cells inferred by Monocle2 based on gene expressions, each color represents a different cluster.
(L) Density plot showing the density patterns of cells from different tumor positions, including Ov, Om, and Ot along the pseudotime, each color represents a tumor site.
In addition, we reconstructed CD8 T cell antigen receptor (TCR) sequences from the scTCR-seq data. More than 70% of cells in all the tumor subsets had matched TCR information, with the exception of the NK-like subsets, indicating limited drop out (Figure S5E). Given that peptide-MHC complex are recognized by specific TCRs, neoantigen and associated TCRs should be present in the same tissue.32 Accordingly, we first selected TCRs that had the same distribution as neoantigens and excluded neoantigen/TCR pairs identified in only one sample (Figures S5F and S5G). Peptide motifs in CDR3 are important for defining antigen specificity with a single antigen being recognized by multiple related TCRs. Consequently, clustering of CDR3 sequences is characteristic of an antigen-driven T cell response.33 Thus, we calculated the pairwise similarity of CDR3 sequences between selected TCRs (same distribution as neoantigens) and randomly selected TCRs (Figure S5H). Selected CDR3 had higher similarity in each patient (Figure S5I). Finally, we calculated the proportion of cells corresponding to the selected and unselected TCRs in different clusters (Figure S5J). The proportion of selected cells were highest in CD8_C03 (Tex), followed by CD8_C06 (Teff) and lower in CD8_C02 (Tex,trans) (Figure S5K), which again supports the contention that CD8_C03 (Tex) represent a tumor-specific cluster. We also compared bulk TCR data of each sample with three virus-specific TCR libraries (see STAR Methods), with the results showing that omentum samples contained the highest proportion of virus-specific TCR (Figure S5L), further supporting their bystander T cell features.
More importantly, pseudotime analysis showed that omental TILs tend to be in early to mid-differentiation with continued transit, while TILs in ovarian tumors have limited transit consistent with terminally differentiated exhausted T cells (Figures 3K and 3L). These results collectively indicated that the T cells infiltrating ovarian lesions were characterized by tumor-specific terminal exhaustion, while the T cells in the omentum were non-exhausted but also non-tumor specific.
Exhausted CD8 T cells enriched in primary ovarian tumors exhibit evidence of expansion
As noted above, we identified a proliferative CD8+ cluster (CD8_C05 (Tex.prol)) that highly expresses proliferation marker genes, such as TUBB, STMN1, and MKI67, which is enriched in ovarian tumors (Figures S6A and S6B). To better characterize this cluster, we used label transfer to interrogate the “second best” cluster for each proliferating cell.34 Interestingly, the CD8_C05 (Tex.prol) cells were majorly regrouped into CD8_C03 (Tex) or CD8_C02 (Tex,trans) (Figure 4A). Very few cells were reattributed to naive or effector memory CD8+ T cell populations, suggesting that proliferating cells were transcriptionally closer to late-differentiated T exhaustion cells. Differential expression analyses in the regrouped CD8_C02 (Tex,trans) and CD8_C03 (Tex) cells after label transfer showed that proliferation-related pathways, including G2 M checkpoint, mitotic spindle, DNA-repair, oxidative phosphorylation,35 and E2F target36 pathways were concurrently elevated in this subclass (Figure S6C).
Figure 4.

Exhausted CD8 T cells enriched in primary ovarian tumors are clonally expanded
(A) UMAP showing the label transfer result from the CD8_C05 proliferation cluster, each color represents a different cluster, as in Figure 2A.
(B) Heatmap depicting the mean cluster expression of a panel of T cell-related genes.
(C) The co-expression of GZMB, CD8, and PD-1 was evaluated by opal multiplex IHC. AEC color signals were extracted from each digitized single-marker image by color deconvolution, followed by pseudo-coloring. A representative image is shown. Nuclei (blue), GZMB (red), CD8 (magenta), PD-1 (cyan), and pan-CK (green). Scale bars, 20 μm. The white arrows indicate single-marker cells and the yellow arrow indicates a triple-positive cell.
(D) Correlation of exhaustion signature and effector signature in CD8_C03 (Tex) T cells with or without proliferation, each point represents a T cell, each color represents a different proliferation state, the point size represents the clonal size of the TCR.
(E) Comparison of gene signatures between CD8_C03 (Tex) T cells with proliferation and those without proliferation with shared TCR clone type, each dot represents a TCR clone type, dot size represents the TCR clone size.
(F) Clonal expansion levels of CD8+ T cell clusters quantified by STARTRAC-expa indices for each patient (n = 6).
(G) Fraction of proliferating T cells in CD8_C03 dysfunctional T cells (including the original CD8_C03 cluster and the CD8_C03 cluster label transferred from the CD8_C05 cluster) stratifying cells by their dysfunctional score.
Then we performed differential analysis of functional markers between CD8_C02 (Tex,trans), CD8_C03 (Tex), CD8_C05 (Tex.prol), and CD8_C06 (Teff). As expected, CD8_C02 (Tex,trans) showed increased GZMK, GZMM, and GZMA, which are markers of transition status (Figures 4B and S6D). Compared with CD8_C02 (Tex,trans), CD8_C05 (Tex.prol) had modestly increased levels of co-inhibition and co-stimulation genes (PDCD1, LAG3, TIGIT, CTLA4, TNFRSF4/9/14/18, and ICOS) and transcription factors (TOX, RBPJ, and IRF9) (Figures 4B and S6D), which are necessary and sufficient to induce major features of Tex cells.37 Of note, these co-inhibition, co-stimulation, and transcription factors were most highly expressed in CD8_C03 (Tex) consistent with exhaustion status (Figures 4B and S6D). Cytotoxic markers (GZMB, PRF1, GNLY, and GZMH) were low in both CD8_C05 (Tex.prol) and CD8_C02 (Tex,trans), indicating poor cytotoxic effector function. Notably, although weaker than CD8_C06 (Teff) cells, CD8_C03 (Tex) exhibited moderate GZMB, and PRF1 in the context of high FASLG and IFNG effector genes (Figures 4B and S6D). Consistently, we observed the exhausted but with cytotoxic function T cells (CD8+PD-1+GZMB+) in ovarian samples, but not in omental samples, by using opal multiplex IHC stains from site-matched FFPE sections, indicating the existence of CD8_C03 (Tex) cells exclusively in ovarian lesions and having modest cytotoxic activity (GZMB+), despite the expression of exhaustion markers (PD-1+) (Figure 4C). Furthermore, we found that the CD8_C03 (Tex) gene signature score was associated with better overall survival, longer disease-specific survival, and better predicted response to ICB in TCGA ovarian cancer patients38 (Figures S6E–S6G), which further suggests that the Tex population in ovarian cancer may have cytolytic activity and may contribute to response to ICB and improved outcomes.
To further explore the relationship between exhausted and cytotoxic functions, we calculated effector and exhaustion scores after label transfer. The positive correlation of exhaustion score and effector score in both proliferating and non-proliferating cells suggests that CD8 T cells in HGSOC concurrently exhibit cytotoxic capacity and exhaustion status (Figure 4D). Proliferating T cells displayed lower effector and exhaustion scores, with a clone size that was much smaller than that of non-proliferating T exhausted cells (Figure 4E). The STARTRAC-expansion index14 also showed that the CD8_C05 (Tex.prol) subclass had modest clonal expansion, while the CD8_C03 (Tex) subclass had the highest degree of clonal expansion (Figure 4F). When the dysfunction population was divided into deciles according to exhaustion score, we found that, as exhaustion scores increase, the proportion of proliferating cells first increased slightly, and then decreased sharply (Figure 4G). The most exhausted cells completely lost proliferative ability (Figure 4G). These results together are consistent with the exhausted CD8 T cell subclass developing from an early differentiation state with high proliferative capacity. Remarkably, we found there was a higher proportion of proliferating cells in each interval in ovarian tumors than in omental tumors (Figure 4G).
Exhausted CD8 T cells are a consequence of differentiation
We performed STARTRAC-transition analysis to reveal T cell state transitions among CD8 cells. As expected, the probability of the same TCR being present between CD8_C02 (Tex,trans), CD8_C03 (Tex), and CD8_C05 (Tex.prol) was markedly higher compared with other clusters, indicating their considerable developmental state transitions exist across them (Figures 5A–5D). A UMAP of representative clonal sharing among CD8_C02 (Tex,trans), CD8_C03 (Tex), and CD8_C05 (Tex.prol) is shown in Figure 5E. To further investigate clonal sharing among CD8_C02 (Tex,trans), CD8_C03 (Tex), and CD8_C05 (Tex.prol), we selected the top 30 clonal TCRs shared between CD8_C02 (Tex,trans) and CD8_C03 (Tex) clusters with or without proliferative status (CD8_C05 (Tex.prol)), and calculated the proportion of clonotype in each subclass. Interestingly, most of the top shared clones across the three subclasses were most frequently expressed as CD8_C03 (Tex), especially the top 10 clones to the left of the dotted line (Figure 5F). As the proliferative cells decreased (CD8_C05 (Tex.prol)), T cells in these clone types tend to be more in CD8_C02 (Tex,trans) status (Figure 5F, left). In particular, the vast majority of TCR clones shared between the non-proliferating CD8_C02 (Tex,trans) and CD8_C03 (Tex) clusters were in the CD8_C02 (Tex,trans) cluster (Figure 5F, right). On the whole, these results further support that the exhausted CD8 T cells develop following proliferation and clonal expansion. Label transfer of CD8_C02 (Tex,trans) cells showed that many of these cells were regrouped into CD8_C03 (Tex) (Figures 5G and 5H), supporting the concept that CD8_C02 (Tex,trans) are transiting to the CD8_C03 (Tex) subclass.
Figure 5.

Exhausted CD8 T cells are a consequence of differentiation
(A) Heatmap showing the transition of all CD8+ tumor-infiltrating cells quantified by pSTARTRAC-tran indices for each patient (n = 6).
(B–D) Developmental transition of CD8_C03(Tex) cells (B), CD8_C02 (Tex,trans) (C), and CD8_C05 (Tex.prol) (D) clusters with other CD8+ cluster cells quantified by pSTARTRAC-tran indices for each patient (n = 6), Kruskal-Wallis test.
(E) UMAP distribution of cells bearing a selected TCR of interest (shared among CD8_C02, CD8_C03, and CD8_C05).
(F) Cluster distribution of top 30 shared TCRs and colored by the CD8+ tumor-infiltrating cell clusters. Left, shared among CD8_C02 (Tex,trans), CD8_C03 (Tex), and CD8_C05 (Tex.prol); right, shared only between CD8_C02, and CD8_C03.
(G) Visualization of the silhouette coefficient score on the UMAP of the CD8+ tumor-infiltrating cells. Silhouette coefficient is calculated on the basis of the mean intracluster distance and the mean of the nearest cluster distance for each cell of each cluster.
(H) UMAP showing the label transfer result from the CD8_C02-GZMK cluster, each color represents a different cluster as in Figure 2A.
(I and J) Quantification of each cluster contribution to shared clones. Each dot corresponds to a shared clone between the three clusters: CD8_C02 (Tex,trans), CD8_C03 (Tex), and CD8_C04 (NK-like) in all sites (I), ovarian sites (J) (left), omental sites (J) (middle), and other metastasis sites (J) (right) of CD8+ tumor-infiltrating cells. Dots highlighted in red correspond to clones that are shared with the proliferation cluster (CD8_C05).
The results presented thus far are consistent with the hypothesis that transition between CD8_C02 (Tex,trans) and CD8_C03 (Tex) clusters occurs while cells are proliferating. To test this possibility, we measured the frequency of proliferative cells among clones shared between the CD8_C02 (Tex,trans) and CD8_C03 (Tex) clusters across different sites (Figures 5I and 5J) using CD8_C04 (NK-like) as a comparator. As shown in Figure 5I, clone sharing predominantly occurred between CD8_C02 (Tex,trans) and CD8_C03 (Tex), with most of these shared clones also being present in proliferating cells. Specifically, compared with omental and other tumor sites, primary ovarian sites had a higher frequency of state transitions driven by proliferation between CD8_C02 (Tex,trans) and CD8_C03 (Tex) (Figure 5J). Together this suggests that terminal exhaustion T cell differentiation preferentially occurs in primary ovarian sites.
CD4 Treg suppress the immune microenvironment in primary ovarian tumor sites
For CD4+ T cells (see Figure 6A for a UMAP of CD4 T cells), CD4_C02 (Treg) and CD4_C03 (Tex) were enriched in ovarian tumors (Figures 2E and 2F), while naive, memory, and transition functional state clusters were mainly present in omental tumors (Figures 2E and 2F). We next assessed the expression of tumor-specific and bystander gene signatures in the CD4+ clusters (Figures 6B and 6C). Notably, tumor-specific signature was significantly enriched in CD4_C03 (Tex), followed by a CD4_C02 (Treg) population (Figure 6B), while bystander signature was enriched in other naive and effector/memory clusters, including CD4_C01/C05/C06 (Figure 6C). Similar to CD8+ T cells, CD4+ cells in primary ovarian tumors displayed the highest tumor-specific and terminal exhaustion scores (Figure 6D). Again, similar to CD8+ T cells, CD4+ T cells in omental sites exhibited the highest bystander score (Figure 6E). Furthermore, CD4+, similar to CD8+, exhausted clusters expressed co-inhibitory and co-stimulatory receptor genes, including TIGIT, HAVCR2, CTLA4, PDCD1, and TNFRSF14 (Figure 6F). There were differences with, for example, the co-stimulatory receptors TNFRSF4/18 and the co-inhibitory receptor LAG3 being highly expressed in CD4 Tex cluster, while TNFRSF9 was enriched in the CD8 Tex cluster (Figure 6F). Of note, unlike CD8 Tex cells, almost all cytotoxic makers, including GZMA, GZMB, PRF1, GZMK, GNLY, and CCL5, were absent in CD4_C03 (Tex), indicating a lack of cytotoxic activity (Figure 6F).
Figure 6.

CD4 Treg cells are responsible for suppressing the immune microenvironment in primary ovarian tumor sites
(A) UMAP of 81,385 single CD4+ T cells, showing the formation of 7 main clusters.
(B and C) UMAP of CD4+ cells colored according to gene signatures scores: (B) tumor-specific CD8+ signature score, (C) CD39−CD69− signature score.
(D) Violin plots showing the gene signatures scores (left, terminally exhausted CD8+ signature score; right, tumor-specific CD8+ signature score) in CD4+ tumor-infiltrating cells from different sites, including Ov, Om, and Ot. wilcox.test.
(E) Violin plots showing the bystander gene signatures scores in CD4+ tumor-infiltrating cells from different sites, including Ov, Om, and Ot. wilcox.test.
(F) Heatmap depicting the expression of a panel of T cell-related genes in CD4_C03 and CD8_C03 clusters.
(G) UMAP showing the label transfer result from CD4_C04 proliferation cluster, each color represents a different cluster as in Figure 2A; here, CD4_C04 was mainly label transferred to CD4_C02.
(H) Developmental transition of CD4_C04 (Treg.prol) with other CD4+ cells quantified by pSTARTRAC-tran indices for each patient (n = 6), Kruskal-Wallis test.
(I and J) Potential developmental trajectory of CD8+ tumor-infiltrating cells inferred by Monocle2 based on gene expressions, each color represents a different cluster (I) or lesions sites (J).
(K) Fraction of proliferating T cells in the CD4_C02 cluster (including the original CD4_C02 cluster and the CD4_C02 cluster label transferred from the CD4_C04 cluster) stratifying cells by their Treg score.
A CD4 T cell cluster with proliferation characteristics expressed MKI67 and FOXP3 (Figures 2B and S7A). Unlike proliferative CD8_C05 (Tex.prol), label transfer of CD4_C04 (Treg.prol) showed that these cells are exclusively related to CD4_C02 (Treg) but not exhausted CD4_C03 (Tex) cells (Figure 6G). So, in addition to the lack of cytotoxicity noted above (Figure 6F), CD4_C03 (Tex) did not exhibit proliferative capacity, which was further supported by the relatively small clone size compared with exhausted CD8 T cells (CD8_C03) (Figure S7B). TCR similarity analysis by STARTRAC-transition showed that CD4_C04 (Treg.prol) shared TCRs with CD4_C02 (Treg) rather than CD4_C03 (Tex) (Figures 6H and S7C). Moreover, the transition between CD4_C02 (Treg) and CD4_C04 (Treg.prol) mainly occurred in ovarian tumors, represented by the green line (Figure S7D). Monocle2 reconstructed a trajectory capturing the progression of CD4 reprogramming with a root at the highest naive state (CD4_C07) and ending with two termini (Treg (CD4_C02 and CD4_C04) and Tex (CD4_C03)) corresponding to two distinct reprogramming outcomes (Figure 6I). More importantly, while the terminal differentiated T cell clusters were enriched in ovarian tumors, early differentiated T cells were more frequent in omental tumors (Figure 6J). Meanwhile, we computed the Treg score for each cell in Treg cells and calculated the proportion of proliferating cells in each score interval (Figure 6K). Within the Treg cell pool in ovarian tumors, as the Treg score increased, the proportion of proliferating cells decreases sharply (Figure 6K). More importantly, the proportion of proliferating cells in each interval is higher in ovarian tumors than that in omental tumors (Figure 6K).
CD4 T cells can support effective anti-tumor CD8 function, but their crosstalk within the TME is not well characterized. To investigate molecular links underlying the intercellular communication of CD4+ and CD8+ T cells in HGSOC, CellphoneDB analysis39 was used to identify molecular interactions between ligand-receptor pairs and major cell types to construct cellular communication networks. We found that interactions between Treg clusters, including CD4_C02 (Treg) and CD4_C04 (Treg.prol), and CD8 dysfunctional clusters, such as CD8_C03/05/07 rather than CD8_C01/02/04/06/09 non-dysfunctional subsets, were commonly observed (Figure S7E). We subsequently analyzed detailed reciprocal connections between CD4_C02 (Treg) and all CD8 populations and identified markedly different ligand-receptor pairs between ovarian and omental tumors (Figure S7F). Notably, the KLRC1-HLA-E axis, a novel checkpoint in the TME40 was exclusively enriched in ovarian tumors, whereas ICAM1/ICAM2, which has been characterized as a site for the cellular entry of human rhinovirus41 and production of proinflammatory effects,42 was enriched in omental tumors.
Inherent TME characteristics contribute to spatial differences of TIL status
To explore mechanisms underlying differences in infiltration of the Tex classes in tumor lesions, we performed pairwise STARTRAC-migration analysis of CD8_02/03/05 clusters between different lesions. We did not find evidence for T cell clusters in omental or other tumors preferentially migrating to ovarian tumors or vice versa (Figure 7A). Moreover, migration of T cells between blood and different tumor lesions was extremely low, with no evidence for preference for different tumor sites (Figure S8A). Therefore, spatial-specific migration of individual T cell clusters is limited or absent. We subsequently analyzed the top 10 TCR clones per cluster in blood (Figure 7B, top) or in tumors (ovarian and omental tumors) (Figure 7B, bottom) for potentially transcriptional reprogramming between blood and different tumor foci. Notably, the top 10 TCR clonotypes from CD8_C03 (Tex) and C05 (Tex.prol) exhausted clusters were not observed in blood, consistent with these clones expanding intratumorally. Together the data argue that the preferential infiltration of CD8_C03 (Tex) and C05 (Tex.prol) in ovarian tumors is not due to migration from blood or other tumor sites.
Figure 7.

Inherent TME characteristics contribute to spatial differences of TIL status
(A) Developmental migration of CD8+ tumor-infiltrating cells between every two of the three tumor sites quantified by pSTARTRAC-migr indices for each patient (n = 6), Kruskal-Wallis test.
(B) Top 10 shared clones of blood and tumor (bottom) being shared with tumor and blood, respectively, for each CD8 cluster. This analysis was performed in ovarian (Ov, left), and omental (Om, right) sites, respectively.
(C) The number of nodes of the network diagrams were counted and compared among tumor sites. Tukey’s multiple comparisons test.
(D) Enrichment plots from gene set enrichment analysis (GSEA) showing significantly differentially regulated pathways between ovarian and omental sites at the single-cell level in all CD3+ tumor-infiltrating T cell (left), CD8+ tumor-infiltrating cells (middle), or CD4+ tumor-infiltrating cells (right). NES, normalized enrichment score.
(E) Quantification of MHC class I H scores across three sites. Data represent mean ± SEM. p values were determined by ANOVA.
(F and G) Quantification of the ratio of densities of CD4+ (F) and CD8+ (G) cells in tumor and stromal area among tumor sites. Data represent mean ± SEM. p values were determined by Tukey’s multiple comparisons test.
Intra-tumoral T cell dysfunction has recently been suggested to be associated with reactivity to tumor antigens.13,43 Consistent with this concept, TMB correlated with the proportion of dysfunctional T cells in primary ovarian tumors (Figures S3C and S3D), the differentiation process being associated with neoantigen recognition. We computed the CDR3 sequence similarity to investigate whether this differentiation process is antigen driven (not all patients shown, Figure S8B). Shared CDR3 sequences between transition states (CD8_C02) and exhausted states (CD8_C03 (Tex) and CD8_C05 (Tex.prol)) were significantly elevated compared with unshared CDR3 sequences in different patients but not in different tumor regions (Figures 7C and S8C). This suggests that, while neoantigen may drive differentiation toward exhausted states, this does not explain the differences in exhausted T cells between different tumor sites.
We next performed differential analysis of signaling pathways between primary ovarian tumors and omental tumors on bulk transcriptomic profiles. Compared with omental tumors, proliferation-related pathways (G2 M checkpoint, mitotic spindle, DNA-repair, oxidative phosphorylation, and E2F targets) and interferon signaling were concurrently increased in ovarian tumors (Figure S8D). Consistently, these pathways were enriched in total (Figure 7D, left), CD8+ (Figure 7D, middle), or CD4+ T cells (Figure 7D, right) in primary ovarian tumors compared with omental tumors, indicating that inherent TME characteristics contribute to spatial differences of TIL status. Notably, proliferation-related pathways, oxidative phosphorylation, and glycolysis were all associated with T cell proliferation and function.
In contrast, consistent with the decreased interferon signaling in omental metastasis, MHC class I in tumor area detected by IHC was lower in omental metastasis (Figures 7E and S8E). As interferon increases antigen presentation44 and MHC class I, reflecting antigen presentation ability and providing a marker of inflamed T cell infiltration,11 the results suggested that omental tumors have lower antigen presentation ability. Meanwhile, IHC staining of T cells showed that both CD4 and CD8 T cells were preferentially located in stroma rather than in omental tumors (Figures 7F, 7G, and S8F), indicating that most of the T cells in omental masses are excluded from contact with tumor cells.
Discussion
T cells represent a major contributor to anti-tumor activity, a concept that is supported by the observation that intra-tumoral TILs are associated with an improved outcome in multiple diseases, including ovarian cancer. The major components of the intra-tumoral T cell compartment include naive, effector, memory, Treg, and exhausted or dysfunctional T cells.45 To explore potential mechanisms underlying the limited response to ICB in ovarian cancer, we used scRNA-seq and TCR sequence analysis to determine immune contexture across different tumor sites and across different ovarian cancer patients. We supplemented these platforms with IHC analysis to provide spatial analysis. Together, this study provided a detailed analysis of the immune landscape across different lesions (Figure S9). Importantly, we found that the immune contexture in different tumor sites and in particular the two most common sites of ovarian cancer, the ovary and the omentum, were markedly different. Ovarian tumors were characterized by an immunosuppressive environment consisting of Tregs and three different populations of exhausted CD8+ T cells as well as an exhausted CD4+ T cell population that likely acquired the exhausted phenotypes through interaction with tumor antigens in the local ovarian ecosystem. In contrast, TILs in omental lesions appear to consist primarily of non-tumor-specific bystander cells with little evidence for response to tumor-specific antigens. Differences in TMB or in tissue-specific immune cell migration do not appear to underlie the diversity of TIL lineages in ovarian and omental lesions. Decreased MHC class I levels and antigen presentation could contribute to the low levels of exhausted of T cells and the decreased differentiation of T cells in omental tumors. While the exact underlying mechanisms remain to elucidated, decreased MHC class I antigen presentation and interferon signaling, oxidative phosphorylation, and failure of T cell infiltration into omental tumors may contribute to lack of tumor-specific T cells in omental metastasis and thus immune evasion.46
The exhausted T cell state in ovarian tumors is likely a consequence of antigen stimulation leading to effector T cells eventually becoming exhausted due to prolonged antigen stimulation. We provide evidence for transition between the three types of exhausted T cells in ovarian tumors. Furthermore, the exhausted T cells retain a number of markers that suggest that they could retain some degree of T cell killing activity. This may contribute to an elevated CD8_C03 (Tex) terminal exhausted signature score being associated with better prognosis in TCGA ovarian cohort. This may not be unique to ovarian cancer as Zhang and co-workers47 reported that CD8-CXCL13 and CD4-CXCL13 T cells, which are proposed to represent exhausted T cells, predict effective responses to PD-L1 blockade in breast cancer. However, other studies suggest that dysfunctional T cells can no longer be reversed and activated by PD-1 therapy.48
Recently, Luca et al. developed a machine learning-based algorithm, the EcoTyper, to deconvolve cell states and ecotypes,49 which identified two T cell-associated carcinoma ecotypes (CEs) across many tumor types, CE9 and CE10, wherein CE9-T cells express activation and exhaustion markers, similar to our CD8_C03 (Tex) cluster, and CE10-T cells that express GZMK and other naive and memory markers, similar to our CD8_C02 (Tex,trans) cluster. In agreement with our study, CE9-T cells, characterized by higher immunoreactivity, preferentially infiltrated tumors compared with CE10-T cells and were strongly associated with longer overall survival. In addition, characteristics of T cells identified in our studies are also recapitulated in a recent report, which showed that antigen presentation gene sets, IFN gene sets, and oxidative phosphorylation are enriched in infiltrated compared with excluded tumor cells.50 Similar to the CD8_C03 (Tex) subset in our study, they defined a CD8+ GZMB T subpopulation enriched in T cell-infiltrated tumors that simultaneously exhibited exhaustion and cytotoxic characteristic, such as PRF1, GZMB, LAG3, CTLA4, PDCD1, and HAVCR2. Similar to the CD8_C02 (Tex,trans) cluster in our study, they identified a CD8+ GZMK T cell subpopulation that also lacks CD39 and thus likely represents a bystander population enriched in stroma and likely tumor excluded. Interestingly consistent with this concept, a GZMK/CD8+ ratio, which may represent a bystander signature, was significantly associated with shorter PFS. Both of these studies were based on single-site sampling and thus did not observe the spatial heterogeneity of the immune contexture in ovarian cancer that may contribute to the limited response to immune therapy in HGSOC. Critically, the number of ovarian cancer samples analyzed by single-cell sequencing was limited and thus this study combined with the recently published data provides an extended dataset that will greatly enhance our understanding of immune contexture across lesions in ovarian cancer and potentially contribute to development of effective immune therapy approaches in ovarian cancer.
In line with previous reports,13,51 our data show that the dysfunctional T cell populations in ovarian cancer do not form a discrete cell population but rather develop from a precursor state with proliferative capacity. As cells differentiate into an exhausted state, they lose proliferative capacity. The proliferating precursor population has evidence for replication stress, high DNA repair capacity, and oxidative phosphorylation, properties that have been observed in other tumor lineages.13,52
Although we used multiple complementary approaches (including opal-IHC, flow cytometry analysis, and genomic, bulk, and single-cell transcriptional and TCR data), these approaches are mainly based on computational inference from static molecular snapshots. However, the derivation of the dysfunctional CD8 T cell state is likely the consequence of a dynamic process that occurs during tumor development. Indeed, as all of the tumors in this analysis were late stage with extensive spread, the “molecular snapshot” likely represents an immune status that is permissive for tumor growth, potentially contributing to the extensive exhaustion states. To fully elucidate the underlying mechanism, both a dynamic study and an analysis of tumors at different stages of development will likely be needed. Furthermore, the status of these clusters was inferred by the expression of marker genes rather than by functional assays. Future studies incorporating lineage tracing, and single-cell spatially resolved analysis will be needed to elucidate underlying mechanisms.
One key clinical question will be to determine how to convert the immunosuppressive and exhausted environment to one that favors tumor clearance. In particular it will be important to determine whether the exhausted T cell state can be reversed to a functional state or whether the exhausted T cells are in an irreversible terminal state or trajectory. If this is the case, effective ovarian cancer immunotherapy may require use of modified T cells, such as CAR-T and TCR-T, combined with ICB. The lack of tumor reactive cells in omental tumors will likely require different approaches to induce immune engagement like that in ovarian tumors. Indeed, the marked difference in the immune contexture in ovarian and omental sites may be the major reason for failure of current immunotherapy approaches. Approaches that are effective in ovarian tumors may not have significant activity in omental tumors and vice versa.
Our results, including trajectory analyses, TCR sharing, and cross-tissue comparisons, are most consistent with the final model: (1) ovarian lesions have a tumor immunosuppressive environment with a high proportion of exhausted T cells and Treg. (2) The majority of tumor-specific TILs in ovarian lesions are exhausted as a consequence, with interaction with tumor antigens. (3) TILs in omental lesions were primarily tumor non-specific and non-exhausted. Moreover, the decreased MHC class I antigen presentation and failure of T cell infiltration into tumors may be associated with immune evasion of omental metastases. These results deepen our understanding of the poor response to ICB therapy in ovarian cancer while concurrently providing information that could improve our ability to engage the immune system in ovarian cancer.
Limitations of the study
Our study shows characteristics and differential composition of TILs across different HGSOC sites and identifies two immune “cold” patterns in ovarian cancer. There are, however, limitations of our study. First, lack of functionally validated using neo-antigen peptide pools to determine the tumor specificity, although several methods, including literature-based gene sets, TCRs and neoantigens analysis, cross-alignment with disease-specific TCR repertoires, and CD39+ flow cytometry analysis were used in our study. Second, the samples used in our study were all from treatment-naive patients. Examining changes in the tumor microenvironment both before and after immunotherapy would have made the findings in this manuscript much more impactful. In addition, only infiltrating T cells were investigated in our study. Future studies need to preferentially cover all cells in the microenvironment and perform single-cell spatially resolved analysis to explore cellular interactions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-CD8 | Maxim biotechnologies | Cat#MAB-0021; RRID: AB_2925214 |
| Human Fc blocking antibody | STEMCELL technologies | Cat#60012; RRID: AB_2925215 |
| Rabbit monoclonal anti-CD4 | Maxim biotechnologies | Cat#RMA-0620; RRID: AB_2925216 |
| Rabbit monoclonal anti-FAP | abcam | Cat#ab207178; RRID: AB_2864720 |
| Rabbit monoclonal anti-HLA A | abcam | Cat#ab52922; RRID: AB_881225 |
| Rabbit recombinant monoclonal PD1 antibody | abcam | Cat#ab137132; RRID: AB_2894867 |
| Rabbit polyclonal Granzyme B antibody | abcam | Cat#ab4059; RRID: AB_304251 |
| Mouse polyclonal anti-CK(Pan) | Maxim biotechnologies | Cat#RAB-0050; RRID: AB_2925217 |
| HRP-labeled Goat Anti-Rabbit IgG(H + L) | Beyotime Biotechnology | Cat#A0208; RRID: AB_2892644 |
| HRP-labeled Goat Anti-Mouse IgG(H + L) | Beyotime Biotechnology | Cat#A0216; RRID: AB_2860575 |
| Live/dead dye, Fixable Viability stain 620 | BD Biosciences | Cat#564996; RRID: AB_2869636 |
| APC-H7 Mouse anti-Human CD45 | BD Biosciences | Cat#560178; RRID: AB_1645479 |
| BV786 Mouse anti-Human CD3 | BD Biosciences | Cat#563800; RRID: AB_2744384 |
| PE Mouse Anti-Human CD3 | BD Biosciences | Cat#555340; RRID: AB_395746 |
| APC Mouse Anti-Human CD45 | BD Biosciences | Cat#340943; RRID: AB_400555 |
| BB700 Mouse anti-Human CD4 | BD Biosciences | Cat#566392; RRID: AB_2744421 |
| BV650 Mouse anti-Human CD8 | BD Biosciences | Cat#563821; RRID: AB_2744462 |
| BB515 Mouse anti-Human CD25 | BD Biosciences | Cat#564467; RRID: AB_2744340 |
| Alexa Fluor® 647 Mouse anti-Human CD127 | BD Biosciences | Cat#558598; RRID: AB_647113 |
| BV510 Mouse Anti-Human CD45RA | BD Biosciences | Cat#563031; RRID: AB_2722499 |
| BV421 Mouse Anti-Human CD197 (CCR7)o | BD Biosciences | Cat#562555; RRID: AB_2728119 |
| BV605 Mouse Anti-Human CD279 (PD-1) | BD Biosciences | Cat#563245; RRID: AB_2738091 |
| APC-R700 Mouse Anti-Human LAG-3 (CD223) | BD Biosciences | Cat#565774; RRID: AB_2744329 |
| FITC Mouse Anti-Human CD39 | BD Biosciences | Cat#561444; RRID: AB_10896292 |
| Opal™ 520 Reagent Pack | Perkin Elmer | Cat# FP1487001KT |
| Opal™ 540 Reagent Pack | Perkin Elmer | Cat# FP1494001KT |
| Opal™ 620 Reagent Pack | Perkin Elmer | Cat# FP1495001KT |
| Opal™ 690 Reagent Pack | Perkin Elmer | Cat# FP1497001KT |
| DAPI | Perkin Elmer | Cat# FP1490A |
| Antibody Diluent/Block (100 mL) | Akoya | Cat# ARD1001 |
| Opal Polymer HRP Ms + Rb (50 mL) | Perkin Elmer | Cat# ARH1001EA; AB_2890927 |
| Biological samples | ||
| High grade serous ovarian cancer samples | Tongji Hospital | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Formaldehyde solution | Sigma | 252549-100ML |
| AR9 buffer | Perkin Elmer | Cat# AR900250ML |
| Ficoll-Paque Plus medium | GE Healthcare | N/A |
| Red blood cell lysis | Solarbio life science | Cat# R1010 |
| Critical commercial assays | ||
| ChromiumTM Single Cell 5′ Reagent Version 2 Kit | 10x Genomics | Cat#100265 |
| QIAamp DNA Mini Kit | QIAGEN | Cat. No./ID: 51,304 |
| RNeasy Mini Kit | QIAGEN | Cat. No./ID: 74,104 |
| NEBNext Poly(A) mRNA Magnetic Isolation Module Kit | NEB | Cat#E7490L |
| NEBNext Ultra RNA Library Prep Kit for Illumina | NEB | Cat#E7530L |
| Qubit dsDNA HS Kit | Invitrogen | Cat# Q32854 |
| Multi Tissue Dissociation Kit 1 | Miltenyi Biotec. | Cat#130110201 |
| Deposited data | ||
| Raw data | This paper | https://ngdc.cncb.ac.cn/gsa-human/HRA002767 |
| TCGA bulk RNA-seq of ovarian cancer | TCGA | https://xenabrowser.net/datapages/ |
| Software and algorithms | ||
| ImageJ,v1.8.0 | Schneider et al.53 | https://imagej.nih.gov/ij/ |
| FlowJo, v10.6.2 | BD Biosciences | https://www.flowjo.com/ |
| R | N/A | https:www.r-project.org |
| GSEA | Broad Institute | https://www.gsea-msigdb.org/gsea/index.jsp |
| Inform analysis software | Akoya biosciences | https://www.akoyabio.com/phenoimager/software/inform-tissue-finder/ |
| STAR, v2.7.3 | Dobin et al.54 | https://github.com/alexdobin/STAR |
| RSEM | Li and Dewey55 | https://github.com/deweylab/RSEM |
| BWA | Li and Durbin56 | http://bio-bwa.sourceforge.net/ |
| Mutect2 | Cibulskis et al.57 | https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2 |
| Oncotator | Ramos et al.58 | https://github.com/broadinstitute/oncotator |
| Patchwork | Mayrhofer et al.59 | https://github.com/thomasp85/patchwork |
| SvABA | Wala et al.60 | https://github.com/walaj/svaba |
| R package deconstructSigs | Rosenthal et al.61 | https://github.com/raerose01/deconstructSigs |
| COSMIC | Tate et al.62 | https://cancer.sanger.ac.uk/cosmic |
| HLA-VBSeq v2 | Nariai et al.63 | http://nagasakilab.csml.org/hla/ |
| NetMHCpan 4.0 | Jurtz et al.64 | https://services.healthtech.dtu.dk/service.php?NetMHCpan-4.0 |
| NetMHCIIpan 3.0 | Nielsen and Andreatta65 | https://services.healthtech.dtu.dk/service.php?NetMHCIIpan-3.2 |
| ssGSEA | Hanzelmann et al.66 | https://gseapy.readthedocs.io/en/latest/ |
| IMonitor v1.4.1 | Zhang et al.67 | https://github.com/zhangwei2015/IMonitor |
| VDJdb | Bagaev et al.68; Shugay et al.69 | VDJdb (cdr3.net) |
| McPAS-TCR | Tickotsky et al.70 | http://friedmanlab.weizmann.ac.il/McPAS-TCR/ |
| TBAdb | Sun et al.71 | https://db.cngb.org/pird |
| Cell Ranger Software v3.0.2 | 10x Genomics | https://github.com/10XGenomics/cellranger |
| Seurat v4, R package | Satija Lab | https://github.com/satijalab/seurat |
| R packages survival v3.2-13 | Terry Therneau | https://github.com/therneau/survival |
| R packages survminer v0.4.9 | Alboukadel KASSAMBARA | https://github.com/kassambara/survminer |
| TIDE (Tumor Immune Dysfunction and Exclusion) | Jiang et al.38 | https://github.com/jingxinfu/TIDEpy |
| STARTRAC | Zhang et al.32 | https://github.com/Japrin/STARTRAC |
| clusterProfiler v3.18.0 | Yu et al.72 | https://github.com/YuLab-SMU/clusterProfiler |
| CellPhoneDB v2.1.1 | Efremova et al.39 | https://github.com/ventolab/CellphoneDB |
| Monocle2 v2 2.4.0 | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/monocle.html |
| GraphPad Prism v8.0.2 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Chaoyang Sun (suncydoctor@gmail.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Clinical specimens
This study was reviewed and approved by the Institutional Review Board of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (TJ-IRB20190320). All the enrolled patients signed an informed consent form, and all the blood samples were collected using the rest of the standard diagnostic tests, with no burden to the patients.
According to the pathological results of the intra-operative frozen section and the macroscopic view of the tumor during the operation, fresh samples were surgically resected from the above-described patients. Then we obtained the proportion of tumor area in the whole tissue according to the HE images, and >50% of the samples were included in the analysis. Nine primary, untreated HGSOC patients who were pathologically diagnosed were enrolled in this study. Their ages ranged from 25 to 70 years old. For patients OV001, OV002 and OV003, their tumor tissues across all sites and PBMC isolated from blood were collected for FFPE, WGS, RNA-seq, bulk TCR-seq. For patients OV004, OV005, OV006, OV008, OV009 and OV010, their PBMC isolated from blood and tumor tissues across all sites were obtained for above sequencing and tissue dissociation to sort and obtain CD45+ CD3+ single cell suspension for scRNA-seq (Table S1). Another 8 ovarian and 5 omental samples from 5 HGSOC patients were collected for flow cytometry analysis (Table S3).
Method details
Single cell collection
Peripheral blood mononuclear cells (PBMCs) were isolated from fresh peripheral blood by Ficoll-Paque Plus medium (GE Healthcare) according to the manufacturer’s instructions. Briefly, 4 mL of fresh peripheral blood was collected during surgery in EDTA anticoagulant tubes and mixed with Ca/Mg-free PBS 1:1, then gently slowly layered onto 8 mL Ficoll. After centrifugation, lymphocyte cells remained at the medium layer between plasma and Ficoll and were carefully transferred to a new tube, red blood cell lysis (Solarbio life science, R1010) was performed as appropriated, and then washed twice with PBS. Cell pellet was resuspended with sorting buffer (PBS with 0.5% BSA).
Fresh tumor tissues were cut into approximately 1-mm3 pieces and single-cell suspension was obtained by Tissue Dissociation Kits (Miltenyi, 130,110,201) together with the gentleMACS™ Dissociators and gentleMACS C tubes (Miltenyi, 130,093,237) according to the protocols. Briefly, tissue pieces were mixed using 5 mL enzyme mix (4.7 mL RPMI1640 + 200ul enzyme H+100ul enzyme R+25ul enzyme A) per C tube. After running the gentleMACS program h_tumor_01, incubate sample for 30 min at 37°C with continuous rotation using rotator. And again run the program h_tumor_01 and incubate sample for 30 min at 37°C using rotator. Finally run the program h_tumor_01 and collected the cell pellet to resuspend and filter through a 40 μm cell-strainer until uniform cell suspensions were obtained. Then the pelleted cells were suspended in red blood cell lysis buffer and washed twice, resuspended in sorting buffer.
Single cell sorting and scRNA library construction sequencing by 10x genomics
Based on FACS analysis, T cells (CD45+CD3+, BD Biosciences, 340,943, 555,340) were sorted into tubes containing 0.5% BSA-PBS and stained with0.4% Trypan blue and examined by microscope. When the viability of cells was higher than 80%, use ChromiumTM Controller and ChromiumTM Single Cell 5′ Reagent Version 2 Kit (10x Genomics, Pleasanton, CA) for library construction experiments. In short, GemCode Technology was used to encapsulated sorted cells, reagents and Gel Beads containing barcoded oligonucleotides into nanoliter-sized GEMs. Lysis and barcoded reverse transcription of polyadenylated mRNA from single cells were performed within each GEM. Post RT-GEMs were cleaned up and cDNA were amplified. cDNA was fragmented and repaired at the end of the fragments, and an A-tail was added to the 5′ end. The adaptors were ligated to fragments which were double sided SPRI selected. After sample index PCR, another double sided SPRI selecting was performed. The final library was quality and quantitated using real-time qPCR (TaqMan Probe). The final products were sequenced using the Xten-PE151 platform (BGIShenzhen, China).
TCR V(D)J sequencing
According to the manufacturer’s protocol (10x Genomics), the Chromium Single-Cell V(D)J Enrichment kit was used to enrich the full-length TCR V(D)J segments from amplified cDNA from 5′ libraries via PCR amplification.
Bulk DNA and RNA isolation and sequencing
Genomic DNA of peripheral blood and tissue samples were extracted using the QIAamp DNA Mini Kit (QIAGEN, 51,304) according to the manufacturer’s instruction. Use the Qubit dsDNA HS Kit (Invitrogen, Q32854) to quantify the DNA concentrations and use agarose gel electrophoresis to evaluate the DNA quality. The exon library was constructed using the SureSelectXT target enrichment system for the illumine Double-End Multiplexed Sequencing Library Kit (Agilent). The samples were sequenced on the illumine Hiseq 4000 sequencer, and the paired-end read was 150 bp.
RNA of tumor samples was extracted by Rneasy Mini Kit (QIAGEN, 74,104). The concentration of RNA was quantified by the NanoDrop instrument (Thermo) and the fragment analyzer (AATI) was used to evaluate the quality of RNA. Libraries were constructed using NEBNext Poly(A) mRNA Magnetic Isolation Module Kit (NEB, E7490L) and NEBNext Ultra RNA Library Prep Kit (NEB, E7530L) for illumine Paired-end Multiplexed Sequencing Library. Samples were sequenced on the illumine Hiseq 4000 sequencer with 150 bp paired-end reads.
Bulk TCR sequencing
RNA was extracted as described above and quantity were determined using Nanodrop. HTBI primers and Arm-PCR from iRepertoire were used to construct the libraries including PCR1 and PCR2, inclusively and semi-quantitatively. 5 cycles were used to amplify CDR3 fragments during the first round of PCR1, using the specific primers against each V and J genes. And in the second round, PCR was performed using universal primers.
PCR1
RNA reverse transcription and amplification of the T-cell receptor β CDR3 using the HTBI primers (Huntsville, Alabama, America) was carried out using Qiagen One-Step RT-PCR. The first round of PCR was performed using 200 ng of total RNA mixed with 4 μL random iRepertoire primers, 5 μL 5 × buffer, 1 μL dNTP mix, 0.25 μL Rnasin (40 U/μL), and 1 μL enzyme mix, with nuclease-free water added to reach a total volume of 25 μL. After mixing and centrifugation, the reactions were transferred to a thermal cycler that carried out the following program: one cycle of 50°C for 40 min; one cycle of 95°C for 15 min; 15 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 40 min, and extension for 30 s at 72°C; 10 cycles of denaturation at 94°C for 30 s, annealing and extension at 72°C for 2 min; and a final extension at 72°C for 10 min. The samples were then held at 4°C.
PCR2
A 2 μL sample of the PCR1 product was used as template for a second step of amplification following the addition of 5 μL communal primers, 25 μL Multiplex MM prepared using the Multiplex PCR Kit (Hilden, Nordrhein-Westfalen, Germany), and 18 μL nuclease-free water to reach a total volume of 50 μL. The reactions were then transferred to a thermal cycler that carried out the following program: one cycle of 95°C for 15 min; 40 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s and extension at 72°C for 30 s; and final extension at 72°C for 5 min. The samples were then held at 4°C. Size selection was used to purify 250-bp PCR products on magnetic beads (Agencourt No. A63882, Beckman, Beverly, MA, USA). After gel purification, the PCR product was subjected to HTS using the Hiseq PE151 platform.
Immunohistochemistry
The specimens were collected within 30 min after the tumor resection and fixed in formalin for 48 h. Paraffin-embedded tissues were subsequently cut into 4 μm slides and mounted on glass slides. Tissues were subjected to deparaffinization and then rehydrated in 100%, 90%, 70% alcohol successively. Antigen was retrieved prior to antibody staining, and then endogenous peroxidase was inactivated by incubation in 3% H2O2 for 30 min. After 10% normal goat serum blocking non-specific sites for 1 h, 37°C, slides were stained with primary antibody overnight at 4°C (anti-CD8 antibody, Maxim biotechnologies; anti-CD4 antibody, Maxim biotechnologies; anti-FAP, 1:250, abcam, ab207178; anti-MHC-I, 1:100, abcam, ab52922). Negative controls were treated identically, but with normal serum. After the sections were washed with PBS twice for 5 min, the antigenic binding sites were visualized using HRP conjugated secondary antibody (Beyotime, A0208 and A0216). Staining is visualized using DAB. Slides were scanned fully automatic by Shengqiang Technology slide scanning image system SQS-40P and observed by reading software. Digital images were taken from five different fields and saved as a jpeg file. A 6 × 6 grid was adapted to each image and examined for nuclear keratinocyte counting73 using the digital image processing software ImageJ (v1.8.0). The mean of positive cells was calculated for each case. H-score analysis was performed on FAP and MHC-I IHC described by Melanie.11 The H-score was calculated by adding up the percentage of cells in each scoring category multiplied by the corresponding score using a semiquantitative five category grading system: 0, no staining; 1, 1%–10% staining; 2, 11%–50% staining; 3, 50-75% staining; and 4, >75% staining. Resulting in scores on a scale of 0-400. Staining score was determined separately by two experts under the same conditions, while discordant scores were reevaluated by another expert.
Flow cytometry analysis
Single-cell suspension obtained and stained by following mAbs: Live/dead dye (Fixable Viability stain 620, BD Biosciences, 564,996), anti-CD45 antibody (BD Biosciences, 560,178), anti-CD3 antibody (BD Biosciences, 563,800), anti-CD4 antibody (BD Biosciences, 566,392), anti-CD8 antibody (BD Biosciences, 563,821), anti-CD25 antibody (BD Biosciences, 564,467), anti-CD127 antibody (BD Biosciences, 558,598), anti-CD45RA antibody (BD Biosciences, 563,031), anti-CCR7 antibody (BD Biosciences, 562,555), anti-PD-1 antibody (BD Biosciences, 563,245), anti-LAG3 antibody (BD Biosciences, 565,774), anti-CD39 antibody (BD Biosciences, 561,444). Single cell suspensions were stained with 1 μg/sample fluorochrome-labeled antibodies for specific surface marker at 4°C for 30 min in 100 μL PBS. Stained single cell suspension of tumor tissue were processed to flow cytometry using Cytoflex LX. The data were analysis by using FlowJo V10.6.2 software.
Opal multiplex IHC
Deparaffinization of formalin fixed paraffin embedded (FFPE) sections was done through xylenes. Rehydration was done through decreasing graded alcohol. AR9 buffer (Perkin Elmer, AR900250ML) was used for retrieving antigens in a microwave oven and a hydrophobic pen was used to circle tissue sections. Before primary antibody incubation, tissue sections were blocked with blocking/antibody diluent (Akoya antibody diluent/block, ARD1001) for 30 min at RT. The tissue sections were incubated with primary antibodies for 60 min at RT. Then washed in 1×TBST and incubated with secondary antibody (Perkin Elmer opal polymer HRP Ms + Rb ARH1001EA, 30 min at RT). The HRP-conjugated secondary antibody polymer was detected using fluorescent tyramide signal amplification using Opal dyes 520, 540, 620 and 690 (Perkin Elmer FP1487001KT, FP1494001KT, FP1495001KT, FP1497001KT) for 10 min at RT. The covalent tyramide reaction was followed by heat induced stripping of the primary/secondary antibody complex using Perkin Elmer AR9 buffer (AR900250ML) at 100°C for 15 min preceding the next cycle (each cycle for each marker). After 4 sequential rounds of staining, sections were stained with DAPI (Perkin Elmer, FP1490A) to visualize nuclei. Five color multiplex-stained slides were imaged using the Vectra Multispectral Imaging System version 2 (Perkin Elmer). Scanning was performed at 20×(200× final magnification). Filter cubes used for multispectral imaging were DAPI, FITC, Cy3, Texas Red and Cy5. A spectral library containing the emitted spectral peaks of the fluorophores in this study was created using the Inform analysis software (Perkin Elmer). Using multispectral images from single-stained slides for each marker, the spectral library was used to separate each multispectral cube into individual components allowing for identification of the five marker channels of interest using Inform 2.4 image analysis software. Anti-CD8 (MAB-0021, 6 mL volume, Maxim Biotechnology, 1:5, 60 min, opal540), anti-PD-1 (ab137132, Abcam, 1:1500, 60 min, opal620), anti-GZMB (ab4059, Abcam, 1:1500, 60 min, opal690) and anti-pan-cytokeratin (pan-CK) (RAB-0050, 6 mL volume, Maxim Biotechnology, 1:10, 60 min, opal520) respectively at RT sequentially.
Gene expression quantification
Raw paired-end reads are filtered to remove adapter sequence using pipeline in-house. And then align to reference genome hg19 by STAR54 (v2.7.3) with default patameters. RSEM55 was used to quantify gene expression based on uniquely mapped reads. GENECODE V19 is used for annotation.
Somatic mutation calling
Raw reads are pre-processed to remove adapter sequences and low-quality reads. The processed clean reads are mapped to hg19 using BWA56 with the default parameter. Picard are used to mark duplicates; GATK4 are employed for base quality correction and realignments. Mutect257 are used for somatic SNV/Indel calling. Mutations were filtered with supported reads ≥4 (≤2) and coverage ≥10 in tumor and (normal tissue), whereas indels were filtered with supported reads ≥5 (≤1) and coverage ≥10 in tumor and (normal tissue. Moreover, somatic mutations and indels were annotated by Oncotator58 with database of version “v1_ds_April052016”. TMB is measured as the total number of somatic mutations per megabase within genome, and we used 2800Mb as the effective size for each genome.
Copy number calling and tumor purity estimation
We estimated copy number profiling over 200 bp bins using Patchwork59 with the default parameters, and then calculated the normalized ratio of standardized, average depth between normal tissue and tumor tissue. Fifty bins are further merged into 10 kb windows. Segmentation performed all the 10 kb windows. After that, tumor ploidy and purity were quantified using Patchwork based on the VAF of each somatic SNV and the copy number status of each segment.
BRCA germline variants
Germline indels of BRCA1/2 were called by SvABA60 using default parameters. Germline deleterious SNVs of BRCA1/2 were selected with annotating be pathogenic in the ClinVar database.
Mutational process
We applied the R package deconstructSigs61 to estimate the contributions of 30 mutational signatures documented by the COSMIC62 for each sample with the default parameters. The 30 signatures are annotated as mutagenesis forms based on COMSIC.
Somatic SV detection
We applied SvABA60 to predict somatic SVs and their breakpoints using the suggested parameters. SV with Q value less than ten is filtered. SVs that are marked by TSI-L are omitted.
Chromothripsis
We used four criteria to infer chromothripsis proposed by Campbell74: A). the number four types of SV type (tail-to-tail, head-to-head, head-to-tail, tail-to-head) are comparable. B). the number of segments involved in chromothripsis is more than 5. C). the copy number oscillated between 2 or 3 copy number states. D). there are interspersed LOH within affected regions.
BFB detection
We inferred BFB events by detecting fold-back inversion and telomere loss which is introduced by Campbell.75 Fold-back inversions were detected based on three criteria: 1) the single inversions were without reciprocal support-read clusters, 2) the inversion associated with a copy number change (q < 0.001), and 3) the two ends of the breakpoints had to be separated by 30 kb.
Neoantigen identification
The HLA type for each sample was inferred using HLA-VBSeq v263 by optimizing read alignments to HLA allele sequences and abundance of reads on HLA alleles by variational Bayesian inference. MHC binding affinities are inferred as IC50 values for each peptide sequence and patient HLA type. All mutant peptide sequences considered to be neoantigens meet a standard cut-off: the IC50 of mutant peptide <500 nM and the IC50 of the wild peptide >500 nM. NetMHCpan4.064 predicted Peptide-MHC class I binding affinity, while NetMHCIIpan-3.065 is applied to identify peptides that bind to MHC-II molecules. Neoantigens with at least three RNA-seq reads contained the mutated base was considered to be expressed.
Differentially expressed genes between omen and ovary
We used a rank-sum test (Python package stats) to compare gene expression between samples of omen and ovary groups. P-values are adjusted using FDR, and genes with FDR <0.1 are regarded as significant. GSEA is used for pathway enrichment.
Bulk RNA-seq analyses immune cell infiltration
Twenty-eight immune cell types (Figure 1C) in the tumor microenvironment are quantified by ssGSEA.66 Gene set for each cell type was obtained from a pan-cancer research.76 The ssGSEA score represented the relative abundance of each immune cell type. Unsupervised clustering was performed on the scores for all tumor samples with Ward distance. Feature genes (157 genes) for 6 immune signaling pathway in Figure 1D is from Desbois et al., 2020.11 ssGSEA was used to quantify the scores for each pathway within tumor samples. Samples in each group were sorted based on the sum values of scores of 6 pathways.
Bulk TCR-seq data analysis
Raw sequencing data were processed by the tool IMonitor (v1.4.1).67 Briefly, the raw paired-end (PE) reads were merged to one sequence by the overlapped region. Low-quality sequences were filtered out. The clean sequences were aligned to reference that including V, D and J germline sequences (www.imgt.org). Originated V, D and J genes were determined for sequences and CDR3 regions were identified. Sequencing errors in CDR3 sequences were corrected according to the base sequencing quality and CDR3 frequency. Nucleotide CDR3 sequences were translated to amino acid sequences. Finally, multiple diversity indexes were calculated, and Figs were generated to display the TCR repertoire.
TCR repertoire annotated by disease associated TCR database
Disease-associated TCR sequences consisted by three published databases, including VDJdb,69,68 McPAS-TCR70 and TBAdb.71 The three databases includes 116,875 records of TCR β sequences covering 43 sorts of infectious diseases. Only high-quality records (VDJdb: Score ≥2; McPAS-TCR: Antigen.identification.method ≤2.5; TBAdb: Grade>=4) were selected for further analysis. For each TCR sample, CDR3 amino acid sequences were compared with the CDR3s in three databases by Levinshtein distance. The CDR3 sequence in the sample was supposed to be related to the disease if the Levinshtein distance ≤1 between CDR3 in sample and CDR3 associated with a disease in database. At last, the proportion of disease-related CDR3s, the number of disease-related CDR3s divided by total number of CDR3 in a sample, was calculated. For bulk TCR-seq sample, top 9000 CDR3 sequences ranked by frequency.
Inferring neoantigen associated TCRs
Both MHC I and MHC II restricted neoantigens peptides were predicted from the data of WGS. As the peptide-MHC complex (pMHC) can be specifically recognized by TCRs, neoantigen and associated TCRs are supposed to present at the same regional tissue. Thus, according to the location consistency between them, associated TCRs could be inferred by the distribution of neoantigens. Specifically, for a patient, if the neoantigen was identified in multiple samples, the TCRs that were observed in all the same samples and were not observed in other samples were regarded as the neoantigen associated TCRs. The neoantigens identified in only one sample were excluded. MHC I restricted neoantigens were used to find associated CD8 TCRs while MHC II restricted neoantigens were used for CD4 TCRs. To reduce potential error of TCR, the TCR with at least two cells was regarded as presence in the sample. Additionally, for each patient, the equal number of TCRs as control were selected randomly from all remaining TCRs that didn’t include inferred neoantigen associated TCRs and were from at least two cells.
Pairwise similarity calculation between TCRs
Both α and β CDR3 amino acid sequences were used to calculate the CDR3 sequence was deconstructed into series of contiguous triplet amino acids, started from the first amino acid and with stride 1. For example, the length of CDR3 with 15 amino acids could be deconstructed into 13 triplets. The similarity is equal to the number of shared triplet amino acids divided by total number of triplets from pair TCRs:
where, is the number of shared triplet amino acids from α CDR3, is the number of shared triplet amino acids from β CDR3, is the total number of triplet amino acids from pairwise α CDR3s, is the total number of triplet amino acids from pairwise β CDR3s.
scRNA/TCR-seq data processing
Sequencing reads were aligned to the GRCh38 human reference genome. Seurat v4 (version 4.0.4) R package was used to analyze the scRNA-seq data. Seurat v4 (version 4.0.4) R package was used to analyze the scRNA-seq data. Cells from all samples were merged. Cells with ≤ 200 or ≥ 6,000 transcripts and cells with ≥ 20% of reads mapping to mitochondrial genes were excluded from the dataset. To remove the doublet cells, DoubletFinder (version 2.0.3)77 was used to detect doublet and only singlet cells (∼95% cells in each sample) were retained. Although T cells were sorted in terms of multiple makers by FACS before, there might be a small part of non-T cells. To remove these non-T cells, cells with <3 transcripts of CD3 (CD3D + CD3E + CD3G) were removed. Furthermore, each cell was compared with the cells of 18 clusters from ovarian cancer patients in a previous study.78 The clusters included T cells, B cell, dendritic cell (DC), erythrocyte, fribroblast and cancer cells. For each cell, spearman’s correlation coefficients were calculated between cell’s gene expression and the average gene expression of anyone of the 18 clusters, so each cell obtained 18 correlation coefficients. Then we calculated the maximal ratio for cell ,
where, is spearman’s correlation coefficient, is one of 18 clusters.
If the ratio >1.05, the cell was regarded as non-T cell and were removed from our data. Next, by using Seurat, the data was normalized with scale factor set to 10,000, the top 2000 highly variable genes identified was used for data integration, and the data was scaled using ‘ScaleData’ function. To eliminate potential batch effects (on sample level), we utilized canonical correlation analysis (CCA) by the Seurat function IntegrateData,79 to get a shared low-dimension space (Figure S2B). The Seurat FindNeighbors function (with 30 PCs) was used to calculate the shared nearest neighbor graph based on the calculated Euclidean distance, and FindClusters function with a resolution of 0.2 was used to cluster our data using the Louvain algorithm. The RunUMAP function was used to display the clustered data using the UMAP dimensional reduction.
Signature gene sets
Terminally exhausted CD8+ signature, T effector memory signature, CD39−CD69− signature, and tumor specific CD8+ signature was got from previous studies.26,27,29 These gene sets were used as modules for the AddModuleScore function in Seurat.
Proliferation state definition
The average expression of known proliferation-related genes (ZWINT, E2F1, FEN1, FOXM1, H2AFZ, HMGB2, MCM2, MCM3, MCM4, MCM5, MCM6, MKI67, MYBL2, PCNA, PLK1, CCND1, AURKA, BUB1, TOP2A, TYMS, DEK, CCNB1 and CCNE1) was defined as the proliferation score.80
Identification of signature genes and TCGA data analysis
We identified differentially expressed genes (DE-Gs) based on the FindAllMarkers function of seurat by using wilcox test.79 The DE-Gs with FDR<0.01 and log2(FC) > 1 were selected as the signature genes of CD8_C03 terminal exhausted cluster. The TCGA bulk RNA-seq and clinical data were obtained from UCSC Xena (https://xenabrowser.net/datapages/). The mean value of the expression (log2(tpm+0.001)) of the CD8_C03 signature genes (CD8A, CXCL13, DUSP4, LAG3, GZMB, CCL5, CCL3, CCL4, NKG7) was calculated as the signature score. Kaplan-Meier survival curves were used to show the survival differences between different groups (high group, greater than or equal to the median signature score, vs. low group, less than the median signature score). The R packages survival v3.2-13 and survminer v0.4.9 were used to perform all survival analyses. TIDE (Tumor Immune Dysfunction and Exclusion) was used to predict the immunotherapy responses as described in a previous study.38
TCR analysis
TCR-seq data for each sample were processed using Cell Ranger software (v3.0.2), with the command “cellranger vdj” using the human reference genome GRCh38. To integrate TCR results with the gene expression data, the TCR-based analysis was performed only for cells that were identified as T cells. T cells with TCR information were used to perform the STARTRAC analysis as we previously described.14
Gene set enrichment analysis
Different gene expression between T cells from ovarian (Ov) and T cells from omental (Om) were calculated based on the FindAllMarkers function of seurat by using wilcox test. Sorted (by log fold change) different expression gene list was used to perform the gene set enrichment analysis (GSEA) by using clusterProfiler (version3.18.0) package.
Ligand-receptor expression analysis
To analyze cell-cell interactions between clusters of interest, CellPhoneDB39 (v2.1.1) was used to identify significant ligand-receptor pair in our data. We randomly selected 10% cells per cluster to perform the analysis. Potential ligand-receptor interactions were identified based on specific expression of a ligand by one T cell cluster and the corresponding receptor by another. The ligand-receptor expression analysis of cells from different lesion sites were performed separately.
Trajectory analysis
To compute pseudotime alignment of our transcriptomes, Monocle2 (v2 2.4.0) was used by using the first 30 PCs of the integrated matrix to preform preprocessing and UMAP reduction. DDRTree algorithm was then used to reconstruct the tree embedding.
Software versions
Data were collected using Cell Ranger software (10x Genomics) v3.0.2 and analyzed using R v.4.0.3, and the following packages and versions in R for analysis: Seurat v4.0.4, clustree v0.4.3, and cluster v2.1.2 two-dimensional gene expression maps, were generated using coordinates from the UMAP algorithm using the R package uwot v0.1.10 implementation. Figs were produced using the following packages and versions in R: ggplot v3.3.5, ComplexHeatmap v2.8.0, ggchicket v0.5.2, patchwork v1.1.1, circlize v0.4.13, ggtern v3.3.5, ggpubr v0.4.0, igraph v1.2.7, and RColorBrewer v1.1-2.
Quantification and statistical analysis
Python (v3.6) package sklearn is used to fit Gaussian Mixture Models (GMMs). p values based on two groups are computed using python package stats. Plots are mainly based on matplotlib and seaborn. Paired t test was used to compare differences between two matched groups (Figure 7D). Two-sided Student’s t test or wilcox.test was used to compare differences between two groups of disease stage. If the multiple groups data followed a normal distribution, we used ANOVA test for multiple comparisons. IHC staining data was plotted and multiple compared by Tukey’s test using GraphPad Prism 8.0.2 software. Data are presented as means ± SEM and p < 0.05 was considered significant. Correlation between groups was determined by Pearson correlation test. ANOVA was used to compare differences among multiple groups.
Acknowledgments
We thank the anonymous referees for their useful suggestions and all the enrolled patients for their dedication to science. This study is supported by grants from the National Key R&D Program of China (2022YFC2704200, 2022YFC2704202, and 2016YFC1303100), the Nature and Science Foundation of China (81572569, 81874106, 81402163, and 81974408), and Wuhan Municipal Health Commission (WX18Q16). G.B.M. received a kind gift from the Dr. Miriam and Sheldon Adelson Medical Research Foundation.
Author contributions
C.S. and G.C. conceived of and supervised the project. B.Y. collected the samples and performed IHC staining. Xiong Li, W.Z., and J.F. performed the bioinformatics analyses. B.Y. designed, carried out, and interpreted the wet laboratory experiments. B.Y. wrote the manuscript. B.Y. and X.Y. checked the format of the paper. B.Y., W.L., and J.Y. scanned the IHC figures and calculated the score. E.G., Xi Li, Y.F., S. Liu, R.X., D.H., X.Q., F.L., Z.W., and T.Q. collected the patients’ clinical information and contributed to data processing and analyses. Q.Z., D.M., S. Li., and G.B.M. provided expertise and feedback. G.B.M., C.S., and G.C. contributed to project management and provided valuable critical discussion.
Declaration of interests
G.B.M. has licensed an HRD assay to Myriad Genetics and on Digital Spatial Profiling to Nanostring and is an SAB member/consultant with Amphista, AstraZeneca, Chrysallis Biotechnology, GSK, ImmunoMET, Ionis, Lilly, PDX Pharmaceuticals, Signalchem Lifesciences, Symphogen, Tarveda, Turbine, and Zentalis Pharmaceuticals.
Published: December 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100856.
Contributor Information
Wei Wang, Email: 951102572@qq.com.
Gang Chen, Email: gumpc@126.com.
Chaoyang Sun, Email: suncydoctor@gmail.com.
Supplemental information
Data and code availability
-
•
All data have been deposited at GSA for Human (https://ngdc.cncb.ac.cn/gsa-human/) with accession number HRA002767 and are publicly available as of the date of publication.
-
•
This paper dose not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D., Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Torre L.A., Trabert B., DeSantis C.E., Miller K.D., Samimi G., Runowicz C.D., Gaudet M.M., Jemal A., Siegel R.L. Ovarian cancer statistics, 2018. CA A Cancer J. Clin. 2018;68:284–296. doi: 10.3322/caac.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P., Allison J.P. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 4.González-Martín A., Sánchez-Lorenzo L. Immunotherapy with checkpoint inhibitors in patients with ovarian cancer: still promising? Cancer. 2019;125:4616–4622. doi: 10.1002/cncr.32520. [DOI] [PubMed] [Google Scholar]
- 5.Jayson G.C., Kohn E.C., Kitchener H.C., Ledermann J.A. Ovarian cancer. Lancet. 2014;384:1376–1388. doi: 10.1016/S0140-6736(13)62146-7. [DOI] [PubMed] [Google Scholar]
- 6.Ma X. The omentum, a niche for premetastatic ovarian cancer. J. Exp. Med. 2020;217:e20192312. doi: 10.1084/jem.20192312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McPherson A., Roth A., Laks E., Masud T., Bashashati A., Zhang A.W., Ha G., Biele J., Yap D., Wan A., et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016;48:758–767. doi: 10.1038/ng.3573. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z.C., Birkbak N.J., Culhane A.C., Drapkin R., Fatima A., Tian R., Schwede M., Alsop K., Daniels K.E., Piao H., et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clin. Cancer Res. 2012;18:5806–5815. doi: 10.1158/1078-0432.CCR-12-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ewing A., Meynert A., Churchman M., Grimes G.R., Hollis R.L., Herrington C.S., Rye T., Bartos C., Croy I., Ferguson M.J.C.C.R. Structural variants at the BRCA1/2 loci are a common source of homologous repair deficiency in high-grade serous ovarian carcinoma. Clin. Cancer Res. 2021;27:3201–3214. doi: 10.1158/1078-0432.CCR-20-4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desbois M., Udyavar A.R., Ryner L., Kozlowski C., Guan Y., Dürrbaum M., Lu S., Fortin J.-P., Koeppen H., Ziai J., et al. Integrated digital pathology and transcriptome analysis identifies molecular mediators of T-cell exclusion in ovarian cancer. Nat. Commun. 2020;11:5583–5614. doi: 10.1038/s41467-020-19408-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hornburg M., Desbois M., Lu S., Guan Y., Lo A.A., Kaufman S., Elrod A., Lotstein A., DesRochers T.M., Munoz-Rodriguez J.L., et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell. 2021;39:928–944.e6. doi: 10.1016/j.ccell.2021.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Li H., van der Leun A.M., Yofe I., Lubling Y., Gelbard-Solodkin D., van Akkooi A.C.J., van den Braber M., Rozeman E.A., Haanen J.B.A.G., Blank C.U., et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019;176:775–789.e18. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L., Yu X., Zheng L., Zhang Y., Li Y., Fang Q., Gao R., Kang B., Zhang Q., Huang J.Y., et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–272. doi: 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 15.Bongen E., Vallania F., Utz P.J., Khatri P. KLRD1-expressing natural killer cells predict influenza susceptibility. Genome Med. 2018;10:1–12. doi: 10.1186/s13073-018-0554-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou L., Adrianto I., Wang J., Wu X., Datta I., Mi Q.-S. Single-cell RNA-Seq analysis uncovers distinct functional human NKT cell sub-populations in peripheral blood. Front. Cell Dev. Biol. 2020;8:384. doi: 10.3389/fcell.2020.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pizzolato G., Kaminski H., Tosolini M., Franchini D.-M., Pont F., Martins F., Valle C., Labourdette D., Cadot S., Quillet-Mary A., et al. Single-cell RNA sequencing unveils the shared and the distinct cytotoxic hallmarks of human TCRVδ1 and TCRVδ2 γδ T lymphocytes. Proc. Natl. Acad. Sci. USA. 2019;116:11906–11915. doi: 10.1073/pnas.1818488116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Germano G., Amirouchene-Angelozzi N., Rospo G., Bardelli A. The clinical impact of the genomic landscape of mismatch repair–deficient cancers. Cancer Discov. 2018;8:1518–1528. doi: 10.1158/2159-8290.CD-18-0150. [DOI] [PubMed] [Google Scholar]
- 19.McGranahan N., Furness A.J.S., Rosenthal R., Ramskov S., Lyngaa R., Saini S.K., Jamal-Hanjani M., Wilson G.A., Birkbak N.J., Hiley C.T., et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pellegrino B., Musolino A., Llop-Guevara A., Serra V., De Silva P., Hlavata Z., Sangiolo D., Willard-Gallo K., Solinas C. Homologous recombination repair deficiency and the immune response in breast cancer: a literature review. Transl. Oncol. 2020;13:410–422. doi: 10.1016/j.tranon.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choucair K., Morand S., Stanbery L., Edelman G., Dworkin L., Nemunaitis J. TMB: a promising immune-response biomarker, and potential spearhead in advancing targeted therapy trials. Cancer Gene Ther. 2020;27:841–853. doi: 10.1038/s41417-020-0174-y. [DOI] [PubMed] [Google Scholar]
- 22.Telli M.L., Timms K.M., Reid J., Hennessy B., Mills G.B., Jensen K.C., Szallasi Z., Barry W.T., Winer E.P., Tung N.M., et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast CancerHRD predicts response to platinum therapy in TNBC. Clin. Cancer Res. 2016;22:3764–3773. doi: 10.1158/1078-0432.CCR-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghorani E., Reading J.L., Henry J.Y., Massy M.R.d., Rosenthal R., Turati V., Joshi K., Furness A.J.S., Ben Aissa A., Saini S.K., et al. The T cell differentiation landscape is shaped by tumour mutations in lung cancer. Nat. Can. 2020;1:546–561. doi: 10.1038/s43018-020-0066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu H., Bengsch F., Svoronos N., Rutkowski M.R., Bitler B.G., Allegrezza M.J., Yokoyama Y., Kossenkov A.V., Bradner J.E., Conejo-Garcia J.R., Zhang R. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16:2829–2837. doi: 10.1016/j.celrep.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sade-Feldman M., Yizhak K., Bjorgaard S.L., Ray J.P., de Boer C.G., Jenkins R.W., Lieb D.J., Chen J.H., Frederick D.T., Barzily-Rokni M., et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175:998–1013.e20. doi: 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van der Leun A.M., Thommen D.S., Schumacher T.N. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer. 2020;20:218–232. doi: 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wherry E.J., Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oliveira G., Stromhaug K., Klaeger S., Kula T., Frederick D.T., Le P.M., Forman J., Huang T., Li S., Zhang W., et al. Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature. 2021;596:119–125. doi: 10.1038/s41586-021-03704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simoni Y., Becht E., Fehlings M., Loh C.Y., Koo S.-L., Teng K.W.W., Yeong J.P.S., Nahar R., Zhang T., Kared H., et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 31.Kim T.-S., Shin E.-C. The activation of bystander CD8+ T cells and their roles in viral infection. Exp. Mol. Med. 2019;51:1–9. doi: 10.1038/s12276-019-0316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang A.W., McPherson A., Milne K., Kroeger D.R., Hamilton P.T., Miranda A., Funnell T., Little N., de Souza C.P.E., Laan S., et al. Interfaces of malignant and immunologic clonal dynamics in ovarian cancer. Cell. 2018;173:1755–1769.e22. doi: 10.1016/j.cell.2018.03.073. [DOI] [PubMed] [Google Scholar]
- 33.Joshi K., de Massy M.R., Ismail M., Reading J.L., Uddin I., Woolston A., Hatipoglu E., Oakes T., Rosenthal R., Peacock T., et al. Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat. Med. 2019;25:1549–1559. doi: 10.1038/s41591-019-0592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., III, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones N., Cronin J.G., Dolton G., Panetti S., Schauenburg A.J., Galloway S.A.E., Sewell A.K., Cole D.K., Thornton C.A., Francis N.J. Metabolic adaptation of human CD4+ and CD8+ T-cells to T-cell receptor-mediated stimulation. Front. Immunol. 2017;8:1516. doi: 10.3389/fimmu.2017.01516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J.W., Field S.J., Gore L., Thompson M., Yang H., Fujiwara Y., Cardiff R.D., Greenberg M., Orkin S.H., DeGregori J. E2F1 and E2F2 determine thresholds for antigen-induced T-cell proliferation and suppress tumorigenesis. Mol. Cell Biol. 2001;21:8547–8564. doi: 10.1128/MCB.21.24.8547-8564.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan O., Giles J.R., McDonald S., Manne S., Ngiow S.F., Patel K.P., Werner M.T., Huang A.C., Alexander K.A., Wu J.E., et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature. 2019;571:211–218. doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang P., Gu S., Pan D., Fu J., Sahu A., Hu X., Li Z., Traugh N., Bu X., Li B., et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 40.Borst L., van der Burg S.H., van Hall T. The NKG2A–HLA-E axis as a novel checkpoint in the tumor microenvironment. Clin. Cancer Res. 2020;26:5549–5556. doi: 10.1158/1078-0432.CCR-19-2095. [DOI] [PubMed] [Google Scholar]
- 41.Abraham G., Colonno R.J. Many rhinovirus serotypes share the same cellular receptor. J. Virol. 1984;51:340–345. doi: 10.1128/jvi.51.2.340-345.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Etienne-Manneville S., Chaverot N., Strosberg A.D., Couraud P.-O. ICAM-1-coupled signaling pathways in astrocytes converge to cyclic AMP response element-binding protein phosphorylation and TNF-α secretion. J. Immunol. 1999;163:668–674. [PubMed] [Google Scholar]
- 43.Thommen D.S., Schumacher T.N. T cell dysfunction in cancer. Cancer Cell. 2018;33:547–562. doi: 10.1016/j.ccell.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hervas-Stubbs S., Perez-Gracia J.L., Rouzaut A., Sanmamed M.F., Le Bon A., Melero I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011;17:2619–2627. doi: 10.1158/1078-0432.CCR-10-1114. [DOI] [PubMed] [Google Scholar]
- 45.Zheng C., Zheng L., Yoo J.-K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J.Y., Zhang Q., et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169:1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 46.Dhatchinamoorthy K., Colbert J.D., Rock K.L. Cancer immune evasion through loss of MHC class I antigen presentation. Front. Immunol. 2021;12:636568. doi: 10.3389/fimmu.2021.636568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiu S., Hong R., Zhuang Z., Li Y., Zhu L., Lin X., Zheng Q., Liu S., Zhang K., Huang M. A single-cell immune atlas of triple negative breast cancer reveals novel immune cell subsets. bioRxiv. 2019 doi: 10.1101/566968. Preprint at. [DOI] [Google Scholar]
- 48.Blank C.U., Haining W.N., Held W., Hogan P.G., Kallies A., Lugli E., Lynn R.C., Philip M., Rao A., Restifo N.P., et al. Defining ‘T cell exhaustion. Nat. Rev. Immunol. 2019;19:665–674. doi: 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luca B.A., Steen C.B., Matusiak M., Azizi A., Varma S., Zhu C., Przybyl J., Espín-Pérez A., Diehn M., Alizadeh A.A. Atlas of clinically distinct cell states and ecosystems across human solid tumors. Cell. 2021;184:5482–5496. doi: 10.1016/j.cell.2021.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hornburg M., Desbois M., Lu S., Guan Y., Lo A.A., Kaufman S., Elrod A., Lotstein A., DesRochers T.M., Munoz-Rodriguez J.L., et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell. 2021;39:928–944.e6. doi: 10.1016/j.ccell.2021.04.004. [DOI] [PubMed] [Google Scholar]
- 51.Azizi E., Carr A.J., Plitas G., Cornish A.E., Konopacki C., Prabhakaran S., Nainys J., Wu K., Kiseliovas V., Setty M., et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174:1293–1308.e36. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., Kang B., Liu Z., Jin L., Xing R., et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 53.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramos A.H., Lichtenstein L., Gupta M., Lawrence M.S., Pugh T.J., Saksena G., Meyerson M., Getz G. Oncotator: cancer variant annotation tool. Hum. Mutat. 2015;36:E2423–E2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mayrhofer M., DiLorenzo S., Isaksson A. Patchwork: allele-specific copy number analysis of whole-genome sequenced tumor tissue. Genome Biol. 2013;14:R24. doi: 10.1186/gb-2013-14-3-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wala J.A., Bandopadhayay P., Greenwald N.F., O'Rourke R., Sharpe T., Stewart C., Schumacher S., Li Y., Weischenfeldt J., Yao X., et al. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res. 2018;28:581–591. doi: 10.1101/gr.221028.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenthal R., McGranahan N., Herrero J., Taylor B.S., Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nariai N., Kojima K., Saito S., Mimori T., Sato Y., Kawai Y., Yamaguchi-Kabata Y., Yasuda J., Nagasaki M. Vol. 2. Springer; 2015. pp. 1–6. (HLA-VBSeq: Accurate HLA Typing at Full Resolution from Whole-Genome Sequencing Data). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jurtz V., Paul S., Andreatta M., Marcatili P., Peters B., Nielsen M. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017;199:3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nielsen M., Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8:33–39. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang W., Du Y., Su Z., Wang C., Zeng X., Zhang R., Hong X., Nie C., Wu J., Cao H., et al. IMonitor: a robust pipeline for TCR and BCR repertoire analysis. Genetics. 2015;201:459–472. doi: 10.1534/genetics.115.176735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bagaev D.V., Vroomans R.M.A., Samir J., Stervbo U., Rius C., Dolton G., Greenshields-Watson A., Attaf M., Egorov E.S., Zvyagin I.V., et al. VDJdb in 2019: database extension, new analysis infrastructure and a T-cell receptor motif compendium. Nucleic Acids Res. 2020;48:D1057–D1062. doi: 10.1093/nar/gkz874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shugay M., Bagaev D.V., Zvyagin I.V., Vroomans R.M., Crawford J.C., Dolton G., Komech E.A., Sycheva A.L., Koneva A.E., Egorov E.S., et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 2018;46:D419–D427. doi: 10.1093/nar/gkx760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tickotsky N., Sagiv T., Prilusky J., Shifrut E., Friedman N. McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics. 2017;33:2924–2929. doi: 10.1093/bioinformatics/btx286. [DOI] [PubMed] [Google Scholar]
- 71.Sun C., Guo E., Zhou B., Shan W., Huang J., Weng D., Wu P., Wang C., Wang S., Zhang W., et al. Correction to: a reactive oxygen species scoring system predicts cisplatin sensitivity and prognosis in ovarian cancer patients. BMC Cancer. 2020;20:210. doi: 10.1186/s12885-020-6691-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics: a journal of integrative biology. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bologna-Molina R., Damián-Matsumura P., Molina-Frechero N. An easy cell counting method for immunohistochemistry that does not use an image analysis program. Histopathology. 2011;59:801–803. doi: 10.1111/j.1365-2559.2011.03954.x. [DOI] [PubMed] [Google Scholar]
- 74.Korbel J.O., Campbell P.J. Criteria for inference of chromothripsis in cancer genomes. Cell. 2013;152:1226–1236. doi: 10.1016/j.cell.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 75.Campbell P.J., Yachida S., Mudie L.J., Stephens P.J., Pleasance E.D., Stebbings L.A., Morsberger L.A., Latimer C., McLaren S., Lin M.L., et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., Hackl H., Trajanoski Z. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 77.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Izar B., Tirosh I., Stover E.H., Wakiro I., Cuoco M.S., Alter I., Rodman C., Leeson R., Su M.J., Shah P., et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020;26:1271–1279. doi: 10.1038/s41591-020-0926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., III, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Whitfield M.L., George L.K., Grant G.D., Perou C.M. Common markers of proliferation. Nat. Rev. Cancer. 2006;6:99–106. doi: 10.1038/nrc1802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data have been deposited at GSA for Human (https://ngdc.cncb.ac.cn/gsa-human/) with accession number HRA002767 and are publicly available as of the date of publication.
-
•
This paper dose not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.