

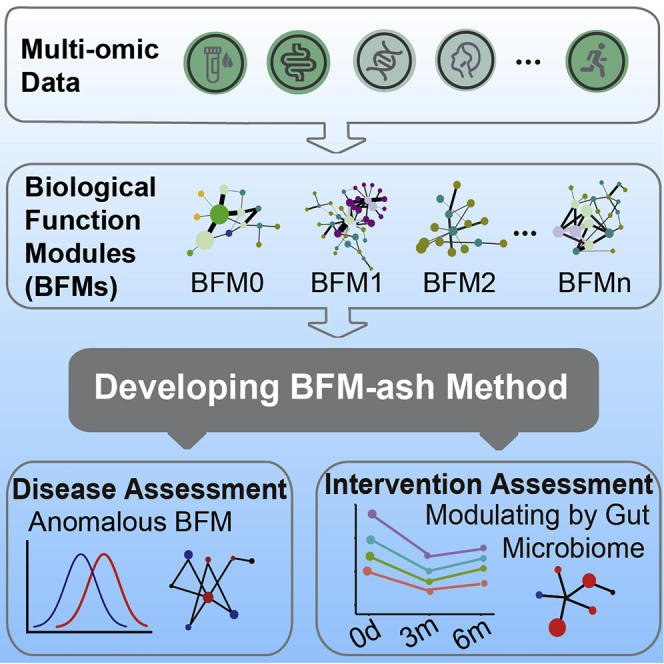
Summary
Recent technological advances in multi-omics and bioinformatics provide an opportunity to develop precision health assessments, which require big data and relevant bioinformatic methods. Here we collect multi-omics data from 4,277 individuals. We calculate the correlations between pairwise features from cross-sectional data and then generate 11 biological functional modules (BFMs) in males and 12 BFMs in females using a community detection algorithm. Using the features in the BFM associated with cardiometabolic health, carotid plaques can be predicted accurately in an independent dataset. We developed a model by comparing individual data with the health baseline in BFMs to assess health status (BFM-ash). Then we apply the model to chronic patients and modify the BFM-ash model to assess the effects of consuming grape seed extract as a dietary supplement. Finally, anomalous BFMs are identified for each subject. Our BFMs and BFM-ash model have huge prospects for application in precision health assessment.
Keywords: multi-omics, correlations, biological functional modules, BFM-ash method, personal health assessment, deitary intervention assessment
Graphical abstract
Highlights
-
•
Mass pairwise feature correlations and 23 BFMs are created from multi-omics data
-
•
BFM-ash method is developed to assess individual health status based on BFMs
-
•
Anomalous BFMs are accurately identified for chronic patients by BFM-ash method
-
•
GSE intervention improves participants’ health status by modulating gut microbiome
Based on the large sample size of multi-omics data, Zhang et al. generate mass correlations and create 23 BFMs. A BFM-ash model is developed to assess individual health status. Using the model, anomalous areas of health are identified for chronic patients, and the effects of dietary intervention for health are assessed.
Introduction
Systems medicine is a global and holistic approach to understanding the basis of human health and disease.1 To achieve this understanding, it is thought that multi-omics profiling should be combined with clinical measurements in an interdisciplinary approach to integrate the data. Currently, medicine is developing toward predictive, preventive, personalized, and participatory approaches because of the development of systems biology and digital technology.1 Unlike reactive healthcare, preventive medicine could reduce healthcare costs and improve health.2 In recent years, advanced technologies, such as high-throughput sequencing and mass spectrometry, have drastically decreased the costs of measuring biological data. Accordingly, multiple studies have begun to utilize multi-omics profiling to investigate health and disease issues.3,4,5,6,7 Compared with traditional studies, investigations based on multi-omics data could be used to unravel biological problems such as genetic and environmental determinants, which could improve health assessments and promote mechanistic discoveries.8,9,10,11
In recent years, multi-omics profiling has been increasingly used in disease research. Multi-dimensional data have been extensively used to understand basic biological principles and processes and uncover causative factors.3,12,13 Two studies of type 2 diabetes (T2D) used longitudinal multi-omics data; one focused on prediabetes and identified that the measurements changed over time and in response to perturbations,4 and the other focused on individuals at risk of T2D and found more than 67 clinically actionable health discoveries.5 A study systematically explored functional dysbiosis of the gut microbiome during active inflammatory bowel disease by following 132 subjects using multi-omics of the gut microbial ecosystem.14 However, most studies have so far focused on diseases, and few have leveraged omics technologies to research healthy people or those in very early stages of disease. Price et al.6 identified many correlation networks between multi-omics features based on 108 subjects and then constructed a community structure using the Girvan and Newman (GN) algorithm, one of the community detection methods.15,16 Using the same strategy, Shomorony et al.17 assessed data for 1,253 individuals to construct correlation modules with the Louvain algorithm, another community detection method18 and reported potential biomarkers for cardiometabolic health and gut microbiome health. These types of investigations are critical for developing preventive medicine; however, larger sample sizes and more multi-omics data are required to precisely evaluate the interconnections among features. The methodology required to harness the findings associated with multi-omics data to assess individual health status is lacking and urgently needed.
In this study, we collected multi-omics profiling data. We investigated the correlations of pairwise features from inter-omics data and identified dozens of biological functional modules (BFMs) by the community detection method. We developed a method to assess individual health status using functional modules. The effects of dietary intervention were precisely assessed by these modules. The correlations and modules provide a theoretical basis for future studies to improve our understanding of health and disease.
Results
Project design and summary of multi-omics data
This study aimed to develop methods for analyzing and harnessing multi-omics data to evaluate the health status of individuals. The study was carried out in five stages. Multi-omics data were first collected from a large cohort of participants. Then pairwise inter-omics correlations were calculated, and BFMs were identified with a community detection algorithm. A method utilizing BFMs to assess health status (BFM-ash) was developed, and two applications were presented, using the BFMs and the BFM-ash method (Figure 1A).
Figure 1.

Project design, sex differences, and inter-omics correlations of pairwise features from two sections of a large cohort
(A) Project design: HE dataset and healthy examination dataset.
(B) Schematic of the data collected in the study. Fourteen sections of data were collected for each sample.
(C) Feature comparison between males and females for each section. A random sampling of 1,000 individuals was used to compare each feature (analysis of covariance [ANCOVA] test, age as the covariate). padj, adjusted p value; ns, non-significant. ∗padj < 0.05, ∗∗padj < 0.01, ∗∗∗padj < 0.001.
(D) Top 1,000 correlations of pairwise features from two sections. A random sampling of 1,000 individuals was used to calculate the correlations for each pairwise feature by linear or logistic regression with age as a covariate. This resampling and calculation step was repeated 501 times, and the median value was taken as the final result. The orange line represents a positive correlation significant at padj < 0.001, and the blue line represents a negative correlation.
(E) The number of significant correlations (padj < 0.001) between data sections. The percentages shown are the proportion of significant correlations of all possible pairwise correlations between data sections.
Our study included 4,277 participants (age range, 20–70 years; 51% males, 49% females) and 1,240 measurements derived from multi-omics data (Figure 1B; Table S1). Briefly, for each participant, blood was collected and used for whole-genome sequencing (WGS), immune repertoire sequencing (IR-seq), and targeted metabolomics profiling (including amino acids, microelements, vitamins, and hormones). Urine and blood samples were used for clinical laboratory tests (labs). Stool samples were collected and used for gut metagenomics sequencing (metagenomics). Facial skin measurement (FSM), electrocardiography (ECG), physical fitness assessment (PFA), and body composition analysis (BCA) were conducted, and psychological questionnaires and lifestyle questionnaires were completed. The three sequencing datasets, including WGS, IR-seq, and metagenomics, were processed by relevant tools to generate features. Single-nucleotide polymorphisms (SNPs) were identified from WGS. Then, polygenic risk scores (PRSs) for 405 diseases and quantitative traits were calculated based on SNPs reported in previous studies (Table S1) that were selected from the National Human Genome Research Institute (NHGRI) catalog of genome-wide association studies (GWASs).19 For IR-seq, which captures the large T cell receptor β (TRB) repertoire, raw data were analyzed by IMonitor20 to calculate four diversity indices, including clone and gene levels, and then 34 disease scores (for which the frequencies of disease-associated TRB clones were found in the sample) were calculated according to three manually curated databases: VDJdb,21 TBAdb,22 and McPAS-TCR.23 78 species and 518 gut microbial modules (GMMs) were identified from the metagenomics data. More details are presented in the STAR Methods.
Mass correlations in males and females were generated from multi-omics data
We found that many features significantly differed between male and female participants (Figure S1). At least 25% of the features were significantly different (padj < 0.001), except in the PRS (Figure 1C). More than 90% of features in the BCA were significantly different, but no gender difference was found for 99% of the PRS features (Figure 1C). Thus, the results highlight the importance of stratification by sex for further analysis. Next, for each dataset stratified by sex, we investigated the inter-omics correlations of pairwise features from two data sections. For each pairwise feature, the data were normalized and processed together, as shown in our detailed workflow (Figure S2A; STAR Methods). The original data types of features, including continuous variables and three discrete variables, could be changed based on the count of zero or categories. According to the variable types, linear regression or one of the different types of logistic regression was used to calculate age-adjusted correlations of pairwise features based on 1,000 resampled samples with 501 iterations (Figure S2B; STAR Methods).
To improve accuracy, we only used highly significant correlations (padj < 0.001) for further analyses. The correlations created an inter-omics network (Figures 1D, S3, and S4A). The male network comprised 1,189 nodes and 4,905 edges, demonstrating a fairly strong connection between sections (Figure S3A). There were strong associations between BCA and the other nine sections, with the proportion of significant correlations ranging from 7.8%–35% (Figure 1E). A large number of significant correlations (n = 2,632, 0.8%) was found between the PRS and metagenomics and between the PRS and psychology (n = 381, 6.3%) (Figure 1E). It has been reported that some gut bacterial species are enriched in many diseases,4,14,24,25 and some diseases have psychological symptoms.26 The female correlation network showed dense connections between sections (Figures S3B and S4A). Like the male network, there were strong associations between BCA and other sections, and the largest number of significant correlations (n = 1945, 0.59%) was found between the PRS and metagenomics (Figure S4B). However, there were some differences. For example, more correlations (105, 39%) were found between BCA and FSM, and no correlation was observed between BCA and hormones in the female network (Figure S4B).
Some of these correlations have been documented previously in the literature. For example, vitamin E is positively correlated with total cholesterol, low-density lipoprotein (LDL), and triglycerides (Figure S5A; Tables S2 and S3), which have been reported to be associated with the metabolism of vitamin E.27 Taurine was positively correlated with platelet distribution width in the blood (Figure S5A), consistent with reports that taurine is related to the aggregation and stability of platelets.28,29 Smoking conditions and the frequency of smoking were positively correlated with blood cadmium levels in males (Figure S5A), consistent with reports of cadmium accumulation in the bodies of smokers.30 Some features were measured in labs and also existed in quantitative PRS, and we found positive correlations between them (Figure S5B), such as platelet distribution width and alkaline phosphatase. Other significant correlations (with small p values) reported in the literature are shown in Tables S2 and S3. We also found so far undocumented correlations (Figure S5C); for instance, phosphoethanolamine in plasma positively correlated with the platelet count. Multiple correlations between genetic traits and gut microbiota were found in the star networks, including known and novel correlations (Figure S5D). To validate the correlations found in our study, an independent dataset from a health examination (HE) project (STAR Methods) involving older people (mean age, 58) was used. The dataset contained data from 86 men, and 121 measurements overlapped with the features in our study. The correlations between any pairwise features of 121 measurements were calculated by the same method (Figure S2) and were compared with those in our cohort (Figure S7B). The measurements covered five of the abovementioned correlations (Figures S5A–S5C), yielding consistent results (Figure S5E). We also examined the top 20 correlations ranked by the padj values in our study, and 19 were significantly correlated (p < 0.05) in the HE dataset (Table S4). These results demonstrated that the correlations in our study were reliable. Given that the HE dataset involved elderly participants, we found specific correlations (Figure S7B; Table S5), such as progesterone and zinc, that were not found in younger populations.
Multi-omics functional module identification from the correlation networks
In the large correlation network that consisted of features, the densely connected features in the network may be related with similar biological function and were considered BFMs. To identify the BFMs, the Louvain community detection algorithm,18 well recognized for its good performance,31,32,33 was used to cluster the nodes in the first step. Then the node initially assigned to a BFM was assigned to another BFM when the node was strongly connected with nodes in another BFM. More details are provided in the STAR Methods. Finally, 11 BFMs in males and 12 BFMs in females with at least four vertices were identified from the cross-sectional inter-omics networks, with 13 nodes in males and 10 nodes in females appearing simultaneously in multiple BFMs (Figures 2A and S6A; Tables S2, S3, S6, and S7). Eigenvector centrality, a measure that reflects a node’s influence in the network, was calculated for every node in the BFMs. The BFMs were dominated by features from multiple sections with different centralities (Figures 2B and S6B). The BFMs in males comprised 109 nodes and 279 edges on average; the largest BFM contained 201 nodes and 522 edges, and the smallest BFM contained 15 nodes and 21 edges (Tables S2 and S6). A comparison of the BFMs between males and females indicated distinct compositions of nodes and edges in most BFMs, which implied significant difference between males and females (Table S8). To check the accuracy of the BFMs, another community detection algorithm, GN,15,16 was used to identify BFMs. Most BFMs identified by the two methods showed a relatively high similarity (Table S9).
Figure 2.

Networks of all BFMs in males
(A) All nodes and edges of BFMs in males. BFMs were constructed by the Louvain method, and overlapping nodes were added. The network in the circle is a BFM, and the top three features ranked by node centrality are listed below the network. The size of the node represents the centrality. The black line represents a positive correlation between paired features, and the gray line represents a negative correlation.
(B) Statistics in each section for each BFM. The boxplot shows the centralities of nodes (left y axis), and the red line shows the number of nodes (right y axis) in each section.
(C) Network and annotation for BFM 7 in males.
BFM annotation of biological functions
Four different annotation methods were used to explore the functions of the BFMs: interpretation of the top 10% of nodes ranked by centrality, GMMs annotated by Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis, bacterial species annotated by MicrobiomeAnalyst,34 and metabolic pathway analysis by MetaboAnalyst.35 Based on our findings, the BFMs were inferred to have different biological functions (Tables S10 and S11). For example, in males, BFM 0 was characterized by cardiometabolic health features, such as triglycerides, high-density lipoprotein (HDL) cholesterol, and LDL; BFM 1 contained multiple features related to hemoglobin; BFM 3 comprised mainly psychological features and disease traits; BFM 4 was associated with platelets; and BFM 6 contained multiple features associated with inflammation. Most features in BFM 7 were associated with immunity, including immune cells and disease scores calculated from the IR-seq data (Figure 2C). The node with the highest centrality in BFM 7 was the percentage of lymphocytes from laboratory measurements (Table S6). Features associated with immune cells comprised three sections of data: laboratory data (the percentage of lymphocytes and neutrophils), IR-seq data (the summary of immune clone number), and WGS data (the PRSs of myeloid white cell counts, lymphocyte percentage of leukocytes, neutrophil percentage of leukocytes, and granulocyte counts). These features were strongly correlated with each other, creating the framework of the network (Figure 2C). Eight infectious disease scores calculated from IR-seq, such as cytomegalovirus, influenza, hepatitis C virus, and tuberculosis, and total pathogen scores existed in BFM 7 (Figure 2C), which reflects the history of infection or the ability to resist pathogens by T cell receptors (TCRs). Analysis using MicrobiomeAnalyst34 on bacterial species revealed a significant enrichment in function associated with granulocytes and leukocytes (Figure 2C). Thus, we concluded that BFM 7 in males was associated with immunity. the BFMs in females were annotated using the same strategy (Figure S6; Table S11).
BFM 0 was interpreted as cardiometabolic health and validated with an independent dataset
Two recent multi-omics studies reported markers of cardiometabolic disease,6,17 and 10 of these were identified in our data. We found that all were included in BFM 0 in males, 8 were enriched in BFM 0 in females (Figures 3A and S7A), and both BFMs were interpreted as cardiometabolic health in our study. Within the BFMs, the features most connected to previously reported cardiometabolic markers36,37,38,39,40,41 were identified, including 10 features in males and 9 in females (Figures 3A and S7A), except for the number of pores on the side face (PSF). The clustering of cardiometabolic markers in BFM 0 indicates that the biological network has inherent structures that community detection algorithms can identify. Next, to validate the interpreted function of BFM 0 in males, we used an independent HE dataset to evaluate it. This dataset contained data from 41 men with carotid plaques (CPs) that reflect the risk of cardiovascular diseases (CVDs)42,43 and data from 45 men without CPs. A total of 118 measurements overlapped with features in our data, including 60 features in BFM 0 in males that we interpreted as cardiometabolic health and 58 features in other BFMs. Compared with the 58 features in other BFMs, more features in BFM 0 (n = 60) were found to be different between groups with and without CPs (Figure 3B), indicating that the features in BFM 0 are associated with cardiometabolic health. The top 5 significantly different features were monocyte count, serine, threonine, number of neutrophils, and isoleucine (Figure 3C). Then we used the 60 features found in BFM 0 to classify the status of CPs using the random forest model. The area under the receiver operating characteristic (ROC) curve (AUC) of 10-fold cross-validation was 76.4% (Figure 3D). As a control, the other 58 features that were excluded in BFM 0 were used to classify the status of CPs by the same method, and the AUC was 62.4% (Figure 3E). The AUC of CP prediction could be increased if more features in BFM 0 were involved. Overall, our results demonstrate that it is plausible to interpret BFM 0 in males as a cardiometabolic health-associated module.
Figure 3.

CP prediction and measurement prediction using the features from the BFMs
(A) Sub-network in BFM 0 in males. The blue nodes are markers associated with cardiometabolic disease from two published studies, and the red nodes are the features closely connected with the markers (each red node connected at least six blue nodes).
(B and C) A total of 118 features were compared between the CP group and the control group (two-sided t test). Sixty features were in BFM 0 of males, and other features were distributed in other BFMs. The counts of features with significant differences between the two groups in BFM 0 (B) and the five discrepant features (with p < 0.01) are shown.
(D) ROC curve showing the classification performance of a classifier for the CP and control groups by 5-repeated 10-fold cross-validation using 60 features included in BFM 0 of males.
(E) ROC curve showing the classification performance of a classifier for the CP and control groups by 5-repeated 10-fold cross-validation using 58 features excluded in BFM 0 of males. The AUC and 95% confidence interval (CI) are listed.
BFM-ash method development to precisely assess health status
It is widely acknowledged that the health status of body systems, such as immunity and cardiovascular function, differs for each person. To precisely evaluate the body systems, we developed a method called BFM-ash, based on BFMs (Figure 4A). The large network of BFM (109 nodes on average) was divided into multiple smaller, tightly connected networks, called sub-BFMs, by the Louvain method. First, we defined two healthy baselines: a benchmark group (the absolute healthy baseline) and peer controls (which represent the relatively healthy baseline). The former was defined by selecting young and healthy individuals from our cohort based on strict criteria (STAR Methods), including 450 males and 507 females. The latter was defined by selecting 20 healthy individuals from our cohort (outside the benchmark group), and the individuals had the same age and sex with the sample required for assessment. The healthy reference range differed for different ages, especially the elderly. Accordingly, age- and sex-matched controls were required. The principle of this method was to compare two distributions of similarity distances: one is between the sample required for assessment and the benchmark group, which shows the distance from the absolute healthy baseline; the other is between the controls and the benchmark group. The two distributions can be compared to determine whether BFM is anomalous. BFM-ash mainly includes three steps (Figure 4A). The first step involves identification of anomalous BFMs. Specifically, for each BFM, Euclidean distances weighted by feature centrality were calculated based on the features between the test sample and each sex-matched sample in the benchmark group as well as between each healthy control and each sex-matched sample in the benchmark group; then, for each BFM, the risk score and p value were calculated in terms of the similarity distances. Finally, an anomalous BFM was identified when the distributions of the two types of similarity distances were significantly different. In the second step, for the identified anomalous BFM, we looked into the sub-BFMs and identified the anomalous sub-BFMs according to the analysis procedure conducted in the first step. For the third step, a feature score that reflects the degree of normality (≤0) or anomaly (>0) was calculated for each feature in the anomalous sub-BFM and was then displayed in the network. More details are provided in the STAR Methods. Theoretically, the composition of the benchmark group did not affect the anomalous BFM identification in BFM-ash. To test this, we randomly selected individuals as the benchmark group and then identified the consistent anomalous BFMs for two samples (Figures 4B and S8).
Figure 4.

Assessment of gastroenteritis and tuberculosis by the BFM-ash method
(A) The pipeline of the BFM-ash method. The similarity distance was calculated by Euclidean distance weighted by feature centrality between the test or controls and the samples of the benchmark group. A feature score for each feature was calculated for the anomalous BFMs/sub-BFMs.
(B) Comparison of the similarity distance in each BFM for the two patients with gastroenteritis (one-tailed t test).
(C) The network of anomalous sub-BFM 1 in BFM 4 for two patients with gastroenteritis. The left panel shows the distance comparison for sub-BFM 1. The right panel shows the annotation summary of the GMMs from the KEGG database. The nodes in the network were classified into four groups: feature score > 0 in both patients (red), feature score > 0 in one patient (pink), feature score ≤ 0 in both patients (blue), and no data (gray). Node size represents the average feature score in the two patients. The line width represents the regression coefficient. The solid line represents positive correlation. A dashed line represents negative correlation.
(D) The distribution of feature scores for all features in the two patients with gastroenteritis.
(E) The number of features in BFMs and sub-BFMs. The left number of “/” is the number of features with a feature score of more than 0.8, and the right number of “/” is the number of all features in the sample. The text marked in red represents anomalous BFMs or sub-BFMs.
(F) Comparison of the similarity distance in each BFM for the two patients with tuberculosis (one-tailed t test).
BFM application for assessing the health status of patients
In our cohort, eight women with complete data available had a previous history of gastroenteritis (n = 2), gastritis (n = 4), and tuberculosis (n = 2). We used the BFM-ash method to assess these patients. For the two patients with gastroenteritis, three anomalous BFMs, including BFM 0, 4, and 7, were found; the similarity distances between the patients and benchmark group were significantly larger than between the controls and benchmark group (Figure 4B), which implies that the biological functions of the three BFMs became worse than the controls. After comparing the similarity distances of the sub-BFMs in the three BFMs, one anomalous sub-BFM in BFM 0 and multiple anomalous sub-BFMs in BFM 4 and 7 were identified (Figure S9). Given that the scores of most nodes in these anomalous sub-BFMs were larger than 0 (Figures 4C, S10, and S11A), these nodes were regarded as anomalous features. For BFM 4, 22 of 26 GMMs in sub-BFM 1 were regarded as anomalous features in at least one patient, and 13 GMMs were anomalous in both patients (Figure 4C). According to KEGG database analysis, nine of the 13 GMMs were enriched in saccharide, polyol, and lipid transport system (Figure 4C). For BFM 7, all 13 GMMs in sub-BFM 0 were anomalous in at least one patient, and the function of all GMMs was associated with environmental information processing (Figure S11B). Overall, the anomalous sub-BFMs identified in gastroenteritis mainly contained gut microbiota, consistent with previous reports showing that the intestinal microbiome is altered in gastroenteritis.44,45 On the other hand, the feature score was independent of BFMs/sub-BFMs, and only represented the degree of normality or anomaly for a single feature. Thus, it could be used for evaluation of the BFM-ash method. In the two patients with gastroenteritis, 18% of features had a feature score larger than 0.8 (Figure 4D), which showed these features’ high degree of anomaly. We then found that most features were enriched in anomalous BFMs/sub-BFMs (Figure 4E). However, some BFMs/sub-BFMs, such as BFM 9 in S1, sub-BFM 2/sub-BFM 4 of BFM 4, and sub-BFM 6 of BFM 7 in S2 (Figure 4E), were detected as anomalies by the BFM-ash method even though there were few or even none of these features (Figure 4E), which suggest the superiority of combined features in BFMs/sub-BFMs compared with a single feature for disease evaluation.
Next, the four patients with gastritis were analyzed using the same method. BFM 0 and BFM 4 were also anomalous in two of the four patients (Figure S12), similar to the results for patients with gastroenteritis, which may be due to the strong correlation between the two diseases. Although BFM 9 was anomalous in three patients, there was no significant difference in all sub-BFMs (Figure S13). For patients with tuberculosis, BFM 3 and sub-BFM 0 of BFM 3 were identified as anomalous (Figures 5F and S14A). In sub-BFM 0, two features related to platelets and lymphocyte count were identified as anomalous features (feature score > 0) in both patients (Figure S14B). Platelets have been reported to regulate inflammation and destruction in tuberculosis.46 Our results demonstrate that the BFM-ash method can be used to evaluate individualized health status and identify common characteristics between multiple patients.
Figure 5.

Systematic assessment of GSE intervention using BFMs
(A) Schematic of GSE intervention.
(B and C) The similarity distance between each participant and each sample of the benchmark group at three time points in three BFMs for the case group (B) and control group (C). The arrow indicates the direction of distance change compared with T0. The dot represents the median similarity distance. Each line represents a participant, and the p value was obtained by paired Mann-Whitney test. ∗∗∗p < 1e−3.
(D) Comparison between the case and control groups by change of similarity distance in four BFMs. For each participant (each boxplot), the change of similarity distance was defined as the distance at T1 or T2 minus that at T0. The p value was obtained by t test based on each participant’s median value.
(E) The networks of sub-BFM 3 and 5 in BFM 7 at T1. Node size represents the absolute value of the intervention score; the nodes were classified into three groups: intervention score < 0 (red), intervention score > 0 (blue), and no data (gray). The line width represents the regression coefficient. The solid line represents positive correlation. The dashed line represents negative correlation.
(F) Summary of 21 GMM (intervention score < 0) annotations from the KEGG database.
BFM application for assessment of dietary intervention
It remains unclear how to precisely evaluate the effects of interventions such as sports, vaccines, and diet. Our BFM-ash method based on multi-omics data could be used to evaluate the effects on human bodies. We designed a dietary intervention project (Figure 5A) where four participants (age, 36–50) in the case group took grape seed extract (GSE) (95% polyphenols) as a daily supplement, and three participants (age, 42–45) in the control group took starch as a placebo for 3 months. Blood, urine, and stool samples were collected at three different time points, including pre-intervention (T0), 3 months after intervention (T1), and 3 months after termination of intervention (T2). A total of 737 multi-omics features were measured for each individual. The data were processed by the modified BFM-ash method (STAR Methods). Compared with T0, the similarity distances of all samples in the case group decreased significantly at T1 in three BFMs, including BFM 6, 7, and 10. At T2, although the distances in most samples in the three BFMs recovered, a significant decrease was found in 66.7% of the samples compared with T0 (Figures 5B and S15). In contrast, in the control group, the similarity distances of most samples increased at T1 and T2 compared with T0 in most BFMs (Figures 5C and S16). The distances increased in some BFMs might be induced by climate change across different seasons. That's because the bacteria associated with seasonal change and the vitamin as one of seasonally fluctuating factors to modulate gut microbiome47 have been found in these BFMs of control group (Figure S17). Next, by comparing the change in similarity distances between the case and control groups, we found that BFM 6, 7, 9, and 10 were significantly different at T1, and the differences remained in BFM 6, 7, and 9 at T2 (Figures 5D and S18). Given that decreased distances in BFM imply a health status closer to the benchmark group, our results illustrated that body functions related to anomalous BFMs were improved after 3 months of diet intervention, and the effect was still observed in most samples 3 months after intervention termination. Then, sub-BFMs in the four BFMs were compared between the case and control groups, and five anomalous sub-BFMs were identified (Figure S19). Instead of the feature score, we defined an intervention score for each feature to quantify the degree of change by the intervention (STAR Methods). The intervention positively affected the feature when the intervention score was less than zero. The intervention scores of most nodes in the four BFMs were less than zero, especially for the anomalous sub-BFMs, and most features were from the metagenomics section of our data (Figures S20, S21, S22, and S23). We then examined anomalous sub-BFM 3 and 5 in BFM 7, and 23 of 27 features had intervention scores less than zero, including 5-methyltetrahydrofolate and 21 GMMs (Figure 5E). Three of the 21 GMMs with high scores were related to phosphate and amino acid transport systems, and the function of 16 GMMs was attributed to environmental information processing (Figure 5F). At T2, 16 of the 23 features had scores less than zero (Figure S24D). In the anomalous sub-BFM 0 and 5 in BFM 6, the intervention scores were less than zero in 10 of 11 features at T1, three of them were related to aromatic degradation, and the feature with the highest score was related to the two-component regulatory system of environmental information processing (Figure S24AC). At T2, nine of the 10 features in both sub-BFMs had intervention scores less than zero (Figure S24B). The effect of GSE intervention on the human body is primarily mediated by the gut microbiome, which enables the body to maintain a better and healthier status. The GSE proanthocyanidin has been reported to affect metabolic health by modulating the gut microbiota in rats.48
Discussion
In this study, we used 4,277 samples with multi-omics data to construct dozens of BFMs. We sought to annotate the biological functions of the modules and finally used the BFMs to assess individual health status. First, our data generated significant cross-sectional correlations between males and females separately because many features were different between sexes. The large sample size enabled us to divide the data into males and females. Age was another confounding factor affecting the features and was used as a covariate for correlation analysis. Because all participants came from one city and had the same ancestry, ancestral origin was not considered. In two recent multi-omics studies, sex, age, and/or ancestry were considered for correlation calculation.6,17 Given the intricacy and heterogeneity of the multi-omics data, we developed a detailed workflow to process the data, and multiple regression methods were used, such as linear regression, logistic regression, ordinal logistic regression, and multinomial logistic regression. To compare the results, the same sample size (n = 1,000) was used, and 501 iterations were conducted for regression analysis. These steps ensured that robust and reliable correlations were generated. We identified some correlations that have not been reported in the literature. We used an independent dataset from the HE dataset to validate partial correlations. Although the dataset was small and contained limited measurements, most significantly different correlations were validated in this dataset. The correlations identified in this study could provide novel insights for future studies.
We used community detection algorithms to construct the BFMs, given that biological networks are similar to social networks. We used the Louvain18 algorithm to construct the initial modules, and then the nodes were allowed to belong to multiple BFMs. Except for biological annotation of the BFMs by several tools, the independent dataset from the HE dataset also proved that BFM 0 of males was related to cardiometabolic health because the features in BFM 0 could predict the participants with or without CP, a marker for CVDs.42,43 Thus, the BFMs created by this method were reliable and meaningful. We also used the GN15,16 algorithm to reconstruct the BFMs and compared the results. The average similarity of edges in the BFMs was approximately 37%, suggesting that many features were included in discrepant BFMs. The discrepancy may be attributed to the different algorithms used. It is widely acknowledged that the Louvain algorithm uses a heuristic method for maximizing modularity by dividing a node or network into any number of communities, whereas the GN algorithm first uses the betweenness centrality to construct networks and then uses the modularity to correct the networks. The two methods detect disjoint communities in which every node belongs to one community; however, biological features may be involved in multiple biological functions. Thus, algorithms that detect overlapping communities are more suitable for biological multi-omics data. Based on previous studies,31,49 we selected the top three most commonly used tools that detect overlapping communities, CFinder,50 Svinet,51 and SLPA,52 to validate our multi-omics data. However, the modules detected by the three methods exhibited significant heterogeneity (data not shown). In brief, the sizes of the BFMs detected by CFinder depended on the parameter k, and the BFM corresponding to a large k representing more reliable results contained few features. For the results by Svinet, one-third of the features could not be assigned to any BFMs, and more than half of the features in the BFMs were overlapping nodes. The BFMs detected by SLPA contained few overlapping nodes, and the sizes of the BFMs were largely uneven. The test results demonstrated that current overlapping community detection methods are not suitable for multi-omics biological data, possibly because of the complexity of overlapping functions in biological data and the difference between biological data and human activity data. In the present study, we did not choose currently available algorithms to detect overlapping communities. Instead, we only identified the overlapping nodes between the BFMs according to the connections between nodes. However, biological data should be processed by a suitable algorithm that considers the biological characteristics. Related algorithms are likely to be developed in the next few years with increasing availability of multi-omics data.
We developed the BFM-ash method based on BFMs to assess the detailed health status of individuals. In this respect, BFMs representing certain biological functions were used as a basic unit for evaluation. The BFMs were derived from multi-omics data integration, which provided more information than traditional single-omics data. Although no single feature was abnormal based on clinical criteria, the combined features in BFM could identify the anomaly. Thus, the BFM-ash method has more advantages than traditional methods. For the BFM-ash method, it was necessary to construct a benchmark group consisting of young and healthy participants as the absolute healthy baseline and select healthy peer controls as the relative healthy baseline. For sample assessment, two baseline references were used for accuracy, given that the healthy reference range for features was different at different ages. The two baseline references were crucial in BFM-ash, and several issues should be noted. First, to assess samples from different races or ancestry, a new benchmark group and peer controls from the same background would be better. Second, because some features, such as the gut microbiota, might be affected by the seasons,53,54,55 a benchmark group consisting of samples from different seasons would be better for accurate assessment. Third, for the BFM-ash method, it was assumed that a shorter distance to the health baseline indicated a better health status. That is reasonable for the general population because the benchmark group consisted of young and healthy individuals. However, very few people, such as athletes, have an exceptional health status, but some features may be outside of the normal reference range of the benchmark group, generating a longer distance to the baseline and inaccurate assessment. These applications using the BFM-ash method depend on whether all BFMs have been fully annotated to biological functions. However, because of the limitations of the current study, some BFMs or sub-BFMs were not fully annotated. It is highly conceivable that all BFMs will be annotated comprehensively in future studies.
The GSE intervention substantiated the performance of the modified BFM-ash method. Accordingly, BFM-ash could be used to evaluate other interventions, such as sports and drugs. During GSE intervention, we tried to evaluate the benefits of intervention by comparing the similarity distances at different time points. Decreased distance in several BFMs and sub-BFMs suggested that improvements in health status were associated with modulations in the gut microbiome. In the control group, the distances of most samples gradually increased at T1 and T2 compared with T0. Bacterial species diversity was comparable at T0, T1, and T2 in the control group (data not shown). These findings may be attributed to the fact that some gut bacteria and GMMs are affected by climate change in different seasons. T0 was in June (summer), and T2 was in December (winter), so the humidity and temperature are different during these two seasons. Multiple studies have reported that gut microbial community compositions exhibit significant differences in different seasons,53,54,55 and some bacteria were found in our BFMs. Vitamins also contributed to the change in distance in the control group, and one study reported that it is one of the seasonally fluctuating factors to modulate the gut microbiome.47 The control group was necessary in this case to obtain more reliable findings. However, we concede that the limited samples in the control group increased the uncertainty, and so more samples would be much better.
In systems medicine, multi-omics data are necessary for comprehensively evaluating individual health status, which could unravel biological problems.11 Thus, more dimensional data, such as genome, transcriptome, epi-genome, proteome, metagenome, immunome, metabolome, and phenotype date, are required. However, because our cohort had a large sample size, factors such as sample storage/transportation and cost had to be considered. To facilitate future applications, data that are easily detectable and easily obtained, such as routine HEs, were analyzed in the present study, whereas transcriptomics, epi-genomics, and proteomics data were excluded. In this study, we included TCR repertoire sequencing and gut metagenomic sequencing. It is widely acknowledged that the TCR repertoire forms a dynamic adaptive immune system to protect the body, whereas the dynamic gut microbiota regulates the health of the human body, which are the core data for health assessment. Diverse datasets are indispensable because cohorts vary in age, sex, race, environment, lifestyle, dietary habits, etc. With more dimensional and diverse datasets, it will be possible to create an overall reference of the health state. Last, individual longitudinal data for large cohorts are valuable for detecting disease early. As more datasets become available and studies are performed, more systematic methods will be developed to precisely monitor individual health status and promote the development of precision medicine.
Limitations of the study
The limitations of this study are as follows. First, although our cohort included multi-omics data, some important data, such as transcriptomics, epi-genomics, and proteomics data, are absent. More dimensional data could create an overall reference of the health state. For the disease assessment and GSE intervention, the sample size was too small, which restricted us to finding more common results and could increase the uncertainty of the results. Second, although an overlapping community detection algorithm for identifying BFMs is more suitable for biological data because each biological feature may be involved in multiple biological functions, we found that the currently available algorithms to detect overlapping communities generate very confusing results. Instead, we only identified overlapping nodes according to the connections between nodes. We believe that suitable algorithms for biological data are likely to be developed with increasing availability of multi-omics data in the future. Third, although we developed 23 BFMs, currently some BFMs have not been fully annotated to biological functions because of the limitations of associated published studies, which may restrict us from a good biological interpretation of anomalous BFMs identified by the BFM-ash method. It is highly conceivable that all BFMs will be annotated to the biological functions in future studies. Fourth, with the BFM-ash method, we assumed that a shorter distance to the healthy baseline indicated a better health status. However, a few people, such as athletes, may have an exceptional health status but a long distance to the health baseline, which may result in inaccurate assessment.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| EDTA-plasma and PBMCs prepared from peripheral blood | This paper | N/A |
| Fresh stool samples | This paper | N/A |
| Critical commercial assays | ||
| HiPure Blood DNA Mini Kit 553 | Magen | Cat#D3111 |
| MGIEasy Kit | MGI | N/A |
| Deposited data | ||
| Individual-level data for our cohort | This paper | CNGB: CNP0003420 |
| The data and summary statistics of HE datasets | This paper | OMIX: OMIX002261, OMIX002286 |
| The data of dietary intervention study | This paper | OMIX: OMIX002266 |
| Summary statistics of correlations between pairwise features of cross-sections in our cohort | This paper | Supplementary file |
| Features and correlations in each BFM | This paper | Supplementary file |
| Software and algorithms | ||
| BWA (v0.7.15) | Li et al., 2009 | https://github.com/lh3/bwa |
| IMonitor(v1.4.0) | Zhang et al., 2015 | https://www.github.com/zhangwei2015/IMonitor |
| GATK(v3.8) | McKenna et al.,2010 | https://newreleases.io/project/github/broadinstitute/gatk/release/ |
| PLINK (v1.07) | Purcell et al. 2007 | http://pngu.mgh.harvard.edu/purcell/plink/ |
| SOAP (v2.22) | Li et al. 2009 | http://soap.genomics.org.cn/ |
| MetaPhlAn2 | Truong et al. 2015 | http://segatalab.cibio.unitn.it/tools/metaphlan2 |
| python-louvain(v0.13) | Blondel et al. 2008 | https://pypi.org/project/python-louvain/0.13/ |
| NetworkX((v2.4)) | Hagberg et al.2008 | https://pypi.org/project/networkx/2.4/ |
| Cytoscape | Shannon et al. 2003 | https://cytoscape.org/ |
| Calculating pairwise feature correlation | This paper | https://github.com/zhangwei2015/Multi-omics/tree/main/Correlations |
| BFM-ash/Modified BFM-ash | This paper | https://github.com/zhangwei2015/Multi-omics/tree/main/BFM-ash |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Chao Nie (niechao@genomics.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
All Chinese volunteers were recruited for the multi-omic study during a health examination from March to May in 2017. Blood, urine and stool samples were collected from the participants. Body composition analysis, an electrocardiogram test and a facial skin assessment were performed for each individual. The participants underwent National Fitness Assessment testing for the physical fitness assessment and completed psychological questionnaires and lifestyle questionnaires with inhouse apps on smart phones. The blood samples were used for whole-genome sequencing (WGS), immune repertoire sequencing (IR-Seq) and targeted metabolomic profiling (including amino acids, microelements, vitamins and hormones). Both the blood and urine samples were used for clinical laboratory tests. The stool samples were used for gut metagenomic sequencing. Finally, we collected data for 4,277 individuals (2085 male; 2,192 female) in this study, and the data were classified into 14 sections. Partial individuals were missing data in one or several sections. The study was approved by the Institutional Review Boards (IRBs) at BGI-Shenzhen, and all participants provided written informed consent at enrollment.
Method details
Whole-genome sequencing and polygenic risk score prediction
The buffy coat was isolated from the whole blood sample, and then DNA was extracted with the HiPure Blood DNA Mini Kit 553 (Magen, Cat. no. D3111) following the manufacturer’s protocol. Two hundred nanograms of DNA for each sample was used for library preparation for WGS and was then processed for single-end 100 bp sequencing using the BGISEQ-500 platform.56 Each sample was sequenced to an average of 30x for the whole genome. We removed the reads with low-quality bases (base quality<5) and adapter sequences. The clean reads were aligned to the human genome reference GRCh38/hg38 by BWA (v0.7.15)57 with default parameters. PCR duplicates were marked with Picardtools (v1.62), and variants were called with the Genome Analysis Toolkit (GATK, v3.8),58 including BaseRecalibrator and HaplotypeCaller. The variants were required to meet the following criteria: (i) genotyping rate >99%; (ii) Hardy-Weinberg equilibrium (HWE) p > 10−5; and (iii) minor allele frequencies (MAFs) > 1%. The samples were required to meet the following criteria: (i) variant calling rate >98%; (ii) heterozygosity < three standard deviations; and (ii) exclusion of related individuals by pairwise identity by descent (IBD, Pi-hat < 0.125) calculated by PLINK (v1.07).59
The National Human Genome Research Institute (NHGRI) Catalog of genome-wide association studies (GWAS) provides a curated collection of human GWAS comprising thousands of genetic traits.19 To calculate the polygenic risk score (PRS) more accurately, we applied several screening procedures to extract the GWAS used in this study: (i) we kept studies with a sample size of at least 5,000 individuals; (ii) provided that multiple studies investigated the same trait with different descriptions, we grouped studies by experimental factor ontology (EFO) IDs and then used the EFO ID as a surrogate for each trait; (iii) in the event that the variant was examined in multiancestry populations for the same trait, we preferentially kept odds ratios/beta coefficients from Asian ancestry studies; (iv) for quantitative traits, to avoid errors related to different studies using inconsistent units for beta coefficients, we selected the publication with the largest sample size; and (v) we used only single nucleotide polymorphisms (SNPs) for all traits, and we excluded the traits associated with three or fewer SNPs. Finally, we retained 405 genetic traits for further analysis, including 144 disease traits and 261 quantitative traits (Table S1). After that, we calculated the PRS for each trait. One SNP was retained in the linkage disequilibrium block. We added the number of risk alleles, weighted by respective log odds ratios or beta-coefficients, across each individual, which is listed as follows:
Where a given trait includes n loci, ci represents the copies of the risk allele and birepresents the beta coefficient or odds ratio in the ith locus.
Gut metagenomic sequencing
Fecal samples were collected with the MGIEasy Kit and processed to extract DNA using the MetaHIT protocol.24 Five hundred nanograms of DNA were used for library preparation and then sequenced for single-end 100 bp using the BGISEQ-500 platform.60 After removing low-quality reads,60 all other high-quality reads were aligned against hg38 with SOAP (v2.22, identity>=0.9)61 to delete the human host DNA. Then, the retained reads were aligned to the integrated gene catalog (IGC)62 using SOAP (v2.22, identity>=0.95). The relative abundance of genes for each sample was calculated24 as follows:
where is the relative abundance of gene i, is the number of mapped reads in gene i, and is the gene length.
The relative abundance of microbial species was calculated using MetaPhlAn263 for all samples. Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG) orthology (KO) profiles were obtained according to KO-associated genes with relative abundance.62 The KO module, which we termed the gut metabolic module (GMM), was calculated by the sum of the relative abundances of associated KOs according to the KEGG database (release 84.0, genes from animals or plants were excluded). Finally, the profile of species frequencies and the profile of GMM frequencies that presented more than 500 samples were used for further multi-omic analysis.
Immune repertoire sequencing
The gDNA was extracted from peripheral blood (PB). For each sample, 1.2 μg gDNA was used to capture the TCR β repertoire using the multi-plex PCR method that we previously described.64,65,66 The PCR products were sequenced for single-end 100 bp using the BGISEQ-500 platform. The raw reads were processed by our developed tool IMonitor (v1.4.0)20 with default parameters. In brief, low-quality reads were filtered out; the variable (V), diversity (D) and joining (J) genes were assigned to clean reads according to the alignment between the reads and reference sequences from the IMGT database; after correcting sequencing errors, complementarity-determining region 3 (CDR3) was identified, and putative amino acid sequences of CDR3 were translated. Then, the sequences derived from cross-sample pollution were filtered out using our previously described method.65 Finally, 1 million productive TCR β sequences were selected at random from each sample for further analysis. Four indices were calculated, including VJ gene usage diversity (VJ gene pairing profile calculated by Shannon index), immune clone number (the unique number of amino acid CDR3 sequences), immune clone diversity (frequencies of amino acid CDR3 sequences calculated by Shannon index) and immune clone evenness (frequencies of amino acid CDR3 sequences calculated by Pielou’s evenness index).
Most diseases can induce an immune response, and disease-associated memory T cells might exist in the body for a very long time. Accordingly, the immune repertoire profile encodes the disease history. Currently, three manually curated databases collect the disease-associated TCRs derived from published studies: VDJdb,21 McPAS-TCR23 and TBAdb.22 Therefore, we used these databases to annotate our immune repertoire data. We obtained 108 diseases, including infectious diseases, autoimmune diseases, cancers, and allergies, from the three databases. We developed an in-house program to calculate the disease score for each sample using the three databases. The disease score was defined as the proportion of disease-associated CDR3s from the databases found in the sample. One Levenshtein distance between the CDR3s in the sample and the CDR3s in the databases was allowed. For each sample, only the top 5000 CDR3s ranked by frequency were used to calculate the disease score.
Quantitative measurement of blood metabolites
The methods for measuring blood metabolites, including amino acids, hormones, vitamins, microelements and heavy metals, were described in detail by Jie et al.67 Briefly, we detected the amino acids from 40 μL plasma using ultra-high pressure liquid chromatography (UHPLC) coupled to an AB Sciex Qtrap 5,500 mass spectrometer (MS) (AB Sciex, US) with an electrospray ionization (ESI) source; to measure hormones, 250 μL plasma was used and detected via UHPLC-MS (AB Sciex Qtrap 5,500) with an atmospheric pressure chemical ionization (APCI) source; water-soluble vitamins were detected from 200 μL plasma via UPLC-MS (Waters Xevo TQ-S Triple Quad, Waters, US) with an ESI source; and fat-soluble vitamins were detected from 250 μL plasma via UPLC-MS (AB Sciex Qtrap 4,500, AB Sciex, US) with an APCI source; we detected the microelements and heavy metals in 200 μL whole blood via an Agilent 7,700x ICP-MS (Agilent Technologies, Japan) equipped with an octupole reaction system (ORS). UPLC-MS was used in positive ion mode. All measured items are shown in Table S1.
Clinical laboratory tests
The blood and urine samples underwent clinical laboratory tests at a licensed physical examination center. The test included basic blood tests, such as the proportion of all types of cells, and blood biochemistry tests associated with lipids, liver function, renal function, fasting blood glucose, etc. Additionally, the participants underwent the 14C-urea breath test to measure Helicobacter pylori infection. Finally, 44 measurements were included (Table S1).
Body composition analysis with inbody
Quantitative measurement of body composition was performed with Inbody (TANITA MC-980MA, China), and a total of 17 items were measured for each individual (Table S1).
Physical fitness assessment
Physical fitness was assessed using the revised 2003 version of the National Physical Fitness Standards for Adults, including vital capacity, step index, grip strength test by hand, timed push-up test (male), timed sit-up test (female), vertical jump test, sit-and-reach test, reaction time and eye-closed and single-legged standing. All fitness indicators are derived from raw measurements following the guidelines.
Facial skin assessment with VISIA
Facial skin features were assessed by the VISIA Complexion Analysis System (Canfield Imaging Systems, Fairfield, NJ, USA). The volunteer’s face without makeup was placed in fixed support, and the eyes were closed during the photographing process. Images were taken in two different views (front and left lateral 37°) to obtain the skin characteristic indicators from both the subjects' cheeks and forehead skin. The indicators included spots, pores, wrinkles, texture, UV spots, porphyrins, brown spots and red areas.
Lifestyle questionnaire
The collection of lifestyle data mainly depended on the subjects filling out the questionnaire. The content of the questionnaire reflected the subjects' lifestyle habits. The participants completed a self-administered questionnaire about personal habits (including 17 multiple-choice questions, Table S1) through an in-house app on their mobile phone.
Psychological questionnaire
The participants were required to complete two questionnaires to assess psychological symptoms and resilience, including the Symptom Checklist 90 (SCL-90)68 and the Connor-Davidson Resilience Scale (CD-RISC).69 The SCL-90 consists of a series of 90 items of symptoms, and each item is rated with regard to severity ranging from 0 (none) to 4 (extreme). The SCL-90 includes ten symptom dimensions, including somatization, obsessive-compulsive, interpersonal sensitivity, depression, anxiety, hostility, phobic anxiety, paranoid ideation, psychoticism and others such as sleep and diet. Each dimension generated a score, and then the sum of all the scores created a total score of psychological symptoms (Table S1). For the CD-RISC, 25 items are included, and each item is rated on a 5-point scale ranging from 0 to 4, with a higher score reflecting greater resilience. There are three dimensions, which are related to optimism, strength and toughness. There are scores in each dimension and a total score of psychological resilience (Table S1).
Samples detected for carotid plaques
Another healthy examination (HE) dataset was obtained consisting of older people (mean age: 58) that underwent B-mode ultrasound imaging and magnetic resonance imaging (MRI) to detect whether they have carotid plaques (CPs). In this study, the participants with CP detected by one of the two methods were used as the case group, and the participants without CP were used as the control group. We used the data of 41 men in the case group and 45 men in the control group, as these participants also performed other tests, including clinical laboratory tests, blood metabolites (amino acids, microelements, vitamins and hormones), body composition analysis and electrocardiogram tests.
Dietary intervention samples
Seven Chinese volunteers in the city of Shenzhen took part in the dietary intervention project in 2017. All participants were relatively healthy (no disease was diagnosed). Four volunteers (females, who were 36, 43, 45 and 50 years old, respectively) in the case group took grape seed extract (GSE) (95% polyphenols, GNC HERBAL PLUS) as a daily supplement, while three volunteers (females, who were 42, 43 and 45 years old, respectively) in the control group took starch as a placebo for three months. Each volunteer took 8.34 mg orally once a day. Blood, urine and stool samples were collected from three timepoints, including pre-intervention (T0, June 2017), three months post-intervention (T1, September 2017) and three months after intervention termination (T2, December 2017). The seven volunteers were required to complete the physical fitness assessment at the three time points. The collected samples were used for clinical laboratory tests, blood metabolites (amino acids, microelements, vitamins and hormones), IR-Seq and metagenomics. Finally, 737 features from multi-omics were measured for each individual.
Data pre-processing and correlation network construction
The multi-omic data were manually checked for errors by data type and normal ranges. The data were separated by gender, and all data analyses in this study were conducted separately for males and females. Next, to calculate the correlations of pairwise inter-omic features from two data sections, paired features were processed together to redefine variable types (Figure S2A). First, each variable was assigned to an initial variable type according to its source. The types included continuous variables, binary discrete variables, multiple ordered discrete variables and multiple unordered discrete variables. Then, for continuous variables, we removed outliers outside three standard deviations away from the mean; we deleted the paired features if the values with zero were in more than 50% of subjects or if the number of subjects was less than 150; then, if the values with zero were in less than 20% of subjects, the values were converted to a normal distribution using inverse normal rank transformation, or the variable type was assigned to binary or multiple categorical fields. For discrete variables, we deleted the paired features if the number of subjects was less than 150; categories with fewer than 20 subjects were deleted; then, the new variable type could be determined according to the number of categories. Finally, a feature initially assigned to a continuous variable could be reassigned to a discrete variable, and multiple ordered or unordered variables could be reassigned to a binary discrete variable.
To detect the associations of pairwise inter-omic features from two data sections, multiple regression models were used according to the redefined variable types (Figure S2B). Specifically, if there was a continuous variable (as an independent variable), linear regression (R package: stats-3.4.1, lm) was prior to choosing; then if there was a binary discrete variable (as independent variable), logistic regression (R package: stats-3.4.1, glm) was used; then if one of two multiple discrete variables were the ordered type (as an independent variable), ordinal logistic regression (OLR, R package: MASS-7.3.51.3, polr) was used; last, if both variables were multiple unordered discrete types, multinomial logistic regression (MLR, R package: nnet-7.3.12, multinom) was used. For both OLR and MLR, we performed the analyses twice with the independent and dependent variables exchanged each time, and we used the median beta coefficients as the final result from multiple beta coefficients and p values. Age as a covariate for all types of regression. All participants were Chinese people from the same ancestry and country, so ancestry was not used as a covariate. Lifestyle was an independent source of data in our study. To make it more comparable between the associations, we randomly resampled 1,000 individuals with replacements for each paired variable to calculate the association and conducted 501 iterations. The median beta coefficient of 501 results and the corresponding p value were used as the final result. All p values were then corrected with the false discovery rate (FDR).
Generating biological function modules
To identify densely connected modules, we performed community detection with the Louvain algorithm (python-louvain, v0.13) through greedy and heuristic optimization of modularity locally.18 The associations of pairwise inter-omic features with padj < 0.001 were used as input and the negative logarithm with base 10 of padj values () was used as the weight. Briefly, in the beginning, a single node was regarded as a community; then, nodes were iteratively merged to increase the gain in modularity. Finally, if the modularity did not increase, the clusters were regarded as the final communities. However, nodes are placed in only one community using the Louvain method, which is inconsistent with the fact that a feature could be involved in multiple biological functions. Thus, we assigned the nodes to a new community if the node degree was more than 30% of the mean degree of the top 3 central nodes in the new community. The node degree was defined as the number of associations with the node. We called the final community the BFM. Then, we computed nodes' weighted eigenvector centrality using NetworkX (v2.4).70 The higher centrality reflects that it is more likely to be the hub of the BFM. All BFM networks in the study were visualized in Cytoscape.71
Additionally, the BFMs were generated by another community detection method called the Girvan and Newman (GN) algorithm.15,16 We used the same data as Louvain used as input, including the associations (padj < 0.001) and the weight (. The codes of the GN method used are provided in a previously published paper.6
BFM annotation
To interpret the biological functions of each BFM, we performed enrichment analysis on microbiome KO modules using a hypergeometric distribution test, and bacterial species were annotated by Taxon Set Enrichment Analysis (TSEA) of the online MicrobiomeAnalyst.34 Features of metabolites were annotated by metabolic pathway analysis by online MetaboAnalyst (v4.0).35 Additionally, the top 10% of nodes (features) ranked by centrality were focused on functions. Finally, we tried to summarize the function of BFM.
Carotid plaque classification
Features were divided into cardiometabolic sets (in BFM 0) and non-cardiometabolic sets (in other BFM 0). Each feature was scaled by Z-score and then transformed to a normal distribution by Box-Cox. A random forest model (R3.4.1, randomForest 4.6–12 package) was used to classify individuals with CP or without CP. Age was added as one feature. Then, we performed 5 repeated 10-fold cross-validation to evaluate the classification accuracy for each set. The performance of this classifier was evaluated by AUC (R 3.4.1, pROC package).
Health status precise assessment by BFM-ash method
First, two steps had to be completed before using the BFM-ash method, listed as follows:
-
(a)
Constructing sub-BFMs: Given that each BFM contains too many features, the BFMs were divided into multiple sub-BFMs by the Louvain method (python-louvain, v0.13) for a more precise assessment of health status. For each BFM, the significant associations and negative logarithm of padj (, as weight) were used as input.
-
(b)
Constructing the benchmark group: A young and healthy group was selected from the cohort of 4,277 subjects. The subjects had to meet the following criteria: (i) no self-reported disease; (ii) more than 80% of all features in clinical laboratory tests and blood metabolites were in the normal reference range; and (iii) ages from 20 to 30 years old. Finally, we selected 450 men and 507 women as the benchmark group.
Next, we can use the BFM method to assess health status. It includes seven steps listed as follows:
-
1)
Selecting peer controls: For a test sample, we first selected several (such as 10) gender- and age-matched and healthy samples as the controls from the cohort of 4,277 subjects.
-
2)
Scaling data: For each feature, the data were scaled with Z-scores. However, for features of metagenomic data, the logarithm of the value was used.
-
3)
Calculating BFM similarity distances: In each BFM, the distance between one sample i from test or control samples and one sample j from the benchmark group was calculated using the average weighted Euclidean distance listed in Equation 1.
| (Equation 1) |
Where m is the feature count in BFM; is the value of feature k in sample i (test or control); is the value of feature k in sample j of benchmark group; and is the centrality of feature (node) k in BFM.
-
4)
Comparing BFM similarity distances and calculating the risk score: We set n samples in the benchmark group and q samples in the control. The test sample obtained n distances, while the control samples obtained q∗n distances. The two distance distributions from the test and control samples were tested with a one-tailed Mann-Whitney test, and we identified significantly different BFMs. The risk score was calculated for each BFM and was defined as the mean of similarity distances.
-
5)
Calculating sub-BFM similarity distances: For the significantly different BFMs, the sub-BFM distance was calculated according to the features in the sub-BFM and Equation 1.
-
6)
Comparing sub-BFM distances and calculating the risk score: As in step 4), we identified the significantly different sub-BFMs.
-
7)
Calculating node feature score: For the significantly different sub-BFMs, we calculated the feature score for each node using the following Equation 2:
| (Equation 2) |
Where q is the number of control samples; is the value of feature k in test sample i; is the value of feature k in sample j of the control.
-
8)
Network visualization: The network was visualized in Cytoscape.71
Dietary intervention assessment by the modified BFM-ash method
For the GSE intervention in our study, one subject had three timepoint samples and a control group, and we mainly paid attention to the change after the intervention. We modified the BFM-ash method in two aspects:
-
1)
We did not compare the similarity distances directly; instead, we focused on the change in similarity distances. It was defined as the similarity distance in T1 or T2 minus that in T0.
| (Equation 3) |
where is the above Equation 1; ( ={T1,T2}) are the time points and T0 for individual ; sample j is from the benchmark group.
-
2)
We did not calculate the feature score for each node; instead, we defined an intervention score for each node and listed the formula (4).
| (Equation 4) |
Where N1 is the number of samples in the case group; N2 is the number of samples in the control group; is the value of feature k in sample i (at T1 or T2) in the case group; is the value of feature k in sample j (at T1 or T2) of the control; and and are the samples at T0 in the case and control groups, respectively.
Quantification and statistical analysis
To compare the features between males and females, for every feature, we randomly sampled 1,000 individuals with replacement from each gender, and performed a one-way ANCOVA with age as the covariate. Gender was as the independent variable and each feature was as the dependent variable. For analysis of HE dataset, we performed a two-sided t-test to compare each feature between case and control. A two-sided t-test was used in Figure 5D. In BFM-ash method, the two distribution of similarity distances were compared with one-sided t-test. Other analyses in the study were done by Mann-Whitney U-test. The p values were corrected for multiple testing using Benjamini & Hochberg. All statistical analysis was performed using R packages. The statistical tests and report significant difference p values were showed in the figure legends.
Acknowledgments
This project was mainly funded by BGI-Shenzhen. We are grateful for the support provided by the China National GeneBank. J.M. was funded by the National Key Research and Development Program of China (2018YFC2000400 and 2018YFA0507801) and the National Natural Science Foundation of China (31970689). C.N. was funded by the National Key Research and Development Program of China (2020YFC2002902). We thank all volunteers for supporting the project.
Author contributions
C.N. and Wei Zhang conceived the study. Wenwei Zhang, H.Y., J.W., X.J., T.L., S.Y., and X.X. contributed to organization of the cohort sample collection. H.Z., M.F., Y. Zong, R.W., P.C., J.M., Yi Z., and Y.Y. were involved in sample collection. Y. Sun, J.D., and Z. Wang contributed to multi-omics data generation. R.L. and Z. Wan processed data. Wei Zhang, Z. Wan, and X.L. developed the code and generated the figures. Z. Wan, X.L., and Wei Zhang performed data analyses. L.L., Y.M., Z.S., Z.L., S.W., Y. Shan, Y.L., and B.C. helped with data analyses. Wei Zhang and Z. Wan wrote the manuscript. All authors approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: December 8, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100847.
Supplemental information
Data and code availability
-
•
All summary statistics that support the findings of this study, including pairwise features’ correlations and BFMs, are available as supplementary files. The data and summary statistics of HE datasets have been deposited at Open Archive for Miscellaneous Data (OMIX) in National Genomics Data Center (NGDC) database (OMIX: OMIX002261, OMIX002286). The data of dietary intervention has been deposited at OMIX (OMIX: OMIX002266). The individual-level data including host genetics, microbiome profile, metabolites, laboratory tests, immune repertoire and other phenotypes have been deposited at China National GeneBank DataBase (CNGBdb: CNP0003420). Access to individual-level data has to be approved by corresponding authors (niechao@genomics.cn), and is subject to the policies and approvals from the Human Genetic Resource Administration, Ministry of Science and Technology of the People’s Republic of China.
-
•
All original codes have been deposited at GitHub: the codes for calculating correlation at Github: https://github.com/zhangwei2015/Multi-omics/tree/main/Correlations; the codes for the BFM-ash pipeline at Github: https://github.com/zhangwei2015/Multi-omics/tree/main/BFM-ash.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Hood L., Flores M. A personal view on systems medicine and the emergence of proactive P4 medicine: predictive, preventive, personalized and participatory. N. Biotechnol. 2012;29:613–624. doi: 10.1016/j.nbt.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Murray C.J.L., Frenk J. Ranking 37th--measuring the performance of the U.S. health care system. N. Engl. J. Med. 2010;362:98–99. doi: 10.1056/NEJMp0910064. [DOI] [PubMed] [Google Scholar]
- 3.Paczkowska M., Barenboim J., Sintupisut N., Fox N.S., Zhu H., Abd-Rabbo D., Mee M.W., Boutros P.C., Functional Interpretation Working. Reimand J. Integrative pathway enrichment analysis of multivariate omics data. Nat. Commun. 2020;11:735. doi: 10.1038/s41467-019-13983-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou W., Sailani M.R., Contrepois K., Zhou Y., Ahadi S., Leopold S.R., Zhang M.J., Rao V., Avina M., Mishra T., et al. Longitudinal multi-omics of host-microbe dynamics in prediabetes. Nature. 2019;569:663–671. doi: 10.1038/s41586-019-1236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schüssler-Fiorenza Rose S.M., Contrepois K., Moneghetti K.J., Zhou W., Mishra T., Mataraso S., Dagan-Rosenfeld O., Ganz A.B., Dunn J., Hornburg D., et al. A longitudinal big data approach for precision health. Nat. Med. 2019;25:792–804. doi: 10.1038/s41591-019-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Price N.D., Magis A.T., Earls J.C., Glusman G., Levy R., Lausted C., McDonald D.T., Kusebauch U., Moss C.L., Zhou Y., et al. A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat. Biotechnol. 2017;35:747–756. doi: 10.1038/nbt.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen R., Mias G.I., Li-Pook-Than J., Jiang L., Lam H.Y.K., Chen R., Miriami E., Karczewski K.J., Hariharan M., Dewey F.E., et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toward Precision Medicine: Building A Knowledge Network For Biomedical Research And A New Taxonomy of Disease . 2012. National Research Council (US) Committee on a Framework for Developing a New Taxonomy of Disease. [PubMed] [Google Scholar]
- 9.Li X., Dunn J., Salins D., Zhou G., Zhou W., Schüssler-Fiorenza Rose S.M., Perelman D., Colbert E., Runge R., Rego S., et al. Digital health: tracking physiomes and activity using wearable biosensors reveals useful health-related information. PLoS Biol. 2017;15:e2001402. doi: 10.1371/journal.pbio.2001402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perkins B.A., Caskey C.T., Brar P., Dec E., Karow D.S., Kahn A.M., Hou Y.C.C., Shah N., Boeldt D., Coughlin E., et al. Precision medicine screening using whole-genome sequencing and advanced imaging to identify disease risk in adults. Proc. Natl. Acad. Sci. USA. 2018;115:3686–3691. doi: 10.1073/pnas.1706096114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karczewski K.J., Snyder M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018;19:299–310. doi: 10.1038/nrg.2018.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mertins P., Mani D.R., Ruggles K.V., Gillette M.A., Clauser K.R., Wang P., Wang X., Qiao J.W., Cao S., Petralia F., et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas Research Network. Electronic address, w.b.e., and Cancer Genome Atlas Research, N Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169:1327–1341.e23. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lloyd-Price J., Arze C., Ananthakrishnan A.N., Schirmer M., Avila-Pacheco J., Poon T.W., Andrews E., Ajami N.J., Bonham K.S., Brislawn C.J., et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–662. doi: 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Girvan M., Newman M.E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA. 2002;99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newman M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA. 2006;103:8577–8582. doi: 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shomorony I., Cirulli E.T., Huang L., Napier L.A., Heister R.R., Hicks M., Cohen I.V., Yu H.C., Swisher C.L., Schenker-Ahmed N.M., et al. An unsupervised learning approach to identify novel signatures of health and disease from multimodal data. Genome Med. 2020;12:7. doi: 10.1186/s13073-019-0705-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blondel V.D., Guillaume J.L., Lambiotte R., Lefebvre E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008;2008:P10008. doi: 10.1088/1742-5468/2008/10/P10008. [DOI] [Google Scholar]
- 19.Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E., et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang W., Du Y., Su Z., Wang C., Zeng X., Zhang R., Hong X., Nie C., Wu J., Cao H., et al. IMonitor: a robust pipeline for TCR and bcr repertoire analysis. Genetics. 2015;201:459–472. doi: 10.1534/genetics.115.176735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shugay M., Bagaev D.V., Zvyagin I.V., Vroomans R.M., Crawford J.C., Dolton G., Komech E.A., Sycheva A.L., Koneva A.E., Egorov E.S., et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 2018;46:D419–D427. doi: 10.1093/nar/gkx760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang W., Wang L., Liu K., Wei X., Yang K., Du W., Wang S., Guo N., Ma C., Luo L., et al. PIRD: Pan immune repertoire database. Bioinformatics. 2020;36:897–903. doi: 10.1093/bioinformatics/btz614. [DOI] [PubMed] [Google Scholar]
- 23.Tickotsky N., Sagiv T., Prilusky J., Shifrut E., Friedman N. McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics. 2017;33:2924–2929. doi: 10.1093/bioinformatics/btx286. [DOI] [PubMed] [Google Scholar]
- 24.Qin J., Li Y., Cai Z., Li S., Zhu J., Zhang F., Liang S., Zhang W., Guan Y., Shen D., et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 25.Feng Q., Liang S., Jia H., Stadlmayr A., Tang L., Lan Z., Zhang D., Xia H., Xu X., Jie Z., et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015;6:6528. doi: 10.1038/ncomms7528. [DOI] [PubMed] [Google Scholar]
- 26.Cohen S., Janicki-Deverts D., Miller G.E. Psychological stress and disease. JAMA. 2007;298:1685–1687. doi: 10.1001/jama.298.14.1685. [DOI] [PubMed] [Google Scholar]
- 27.Schmölz L., Birringer M., Lorkowski S., Wallert M. Complexity of vitamin E metabolism. World J. Biol. Chem. 2016;7:14–43. doi: 10.4331/wjbc.v7.i1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayes K.C., Pronczuk A., Addesa A.E., Stephan Z.F. Taurine modulates platelet aggregation in cats and humans. Am. J. Clin. Nutr. 1989;49:1211–1216. doi: 10.1093/ajcn/49.6.1211. [DOI] [PubMed] [Google Scholar]
- 29.Ahtee L., Boullin D.J., Paasonen M.K. Transport of taurine by normal human blood platelets. Br. J. Pharmacol. 1974;52:245–251. doi: 10.1111/j.1476-5381.1974.tb09707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis G.P., Coughlin L.L., Jusko W.J., Hartz S. Contribution of cigarette smoking to cadmium accumulation in man. Lancet. 1972;1:291–292. doi: 10.1016/s0140-6736(72)90294-2. [DOI] [PubMed] [Google Scholar]
- 31.Newman M.E.J. Second edition. Oxford University Press; 2018. Networks. [Google Scholar]
- 32.Amir E.a.D., Davis K.L., Tadmor M.D., Simonds E.F., Levine J.H., Bendall S.C., Shenfeld D.K., Krishnaswamy S., Nolan G.P., Pe'er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takemura S.Y., Bharioke A., Lu Z., Nern A., Vitaladevuni S., Rivlin P.K., Katz W.T., Olbris D.J., Plaza S.M., Winston P., et al. A visual motion detection circuit suggested by Drosophila connectomics. Nature. 2013;500:175–181. doi: 10.1038/nature12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chong J., Liu P., Zhou G., Xia J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020;15:799–821. doi: 10.1038/s41596-019-0264-1. [DOI] [PubMed] [Google Scholar]
- 35.Chong J., Soufan O., Li C., Caraus I., Li S., Bourque G., Wishart D.S., Xia J. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018;46:W486–W494. doi: 10.1093/nar/gky310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Libert D.M., Nowacki A.S., Natowicz M.R. Metabolomic analysis of obesity, metabolic syndrome, and type 2 diabetes: amino acid and acylcarnitine levels change along a spectrum of metabolic wellness. PeerJ. 2018;6:e5410. doi: 10.7717/peerj.5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Webb C.M., Collins P. Role of testosterone in the treatment of cardiovascular disease. Eur. Cardiol. 2017;12:83–87. doi: 10.15420/ecr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murr C., Grammer T.B., Meinitzer A., Kleber M.E., März W., Fuchs D. Immune activation and inflammation in patients with cardiovascular disease are associated with higher phenylalanine to tyrosine ratios: the ludwigshafen risk and cardiovascular health study. J. Amino Acids. 2014;2014:783730. doi: 10.1155/2014/783730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barbagallo M., Dominguez L.J., Galioto A., Ferlisi A., Cani C., Malfa L., Pineo A., Busardo A., Paolisso G. Role of magnesium in insulin action, diabetes and cardio-metabolic syndrome X. Mol. Aspects Med. 2003;24:39–52. doi: 10.1016/s0098-2997(02)00090-0. [DOI] [PubMed] [Google Scholar]
- 40.Larsson S.C., Burgess S., Michaëlsson K. Serum magnesium levels and risk of coronary artery disease: mendelian randomisation study. BMC Med. 2018;16:68. doi: 10.1186/s12916-018-1065-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mangge H., Zelzer S., Prüller F., Schnedl W.J., Weghuber D., Enko D., Bergsten P., Haybaeck J., Meinitzer A. Branched-chain amino acids are associated with cardiometabolic risk profiles found already in lean, overweight and obese young. J. Nutr. Biochem. 2016;32:123–127. doi: 10.1016/j.jnutbio.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Martinsson A., Östling G., Persson M., Sundquist K., Andersson C., Melander O., Engström G., Hedblad B., Smith J.G. Carotid plaque, intima-media thickness, and incident aortic stenosis: a prospective cohort study. Arterioscler. Thromb. Vasc. Biol. 2014;34:2343–2348. doi: 10.1161/ATVBAHA.114.304015. [DOI] [PubMed] [Google Scholar]
- 43.Inaba Y., Chen J.A., Bergmann S.R. Carotid plaque, compared with carotid intima-media thickness, more accurately predicts coronary artery disease events: a meta-analysis. Atherosclerosis. 2012;220:128–133. doi: 10.1016/j.atherosclerosis.2011.06.044. [DOI] [PubMed] [Google Scholar]
- 44.Jalanka-Tuovinen J., Salojärvi J., Salonen A., Immonen O., Garsed K., Kelly F.M., Zaitoun A., Palva A., Spiller R.C., de Vos W.M. Faecal microbiota composition and host-microbe cross-talk following gastroenteritis and in postinfectious irritable bowel syndrome. Gut. 2014;63:1737–1745. doi: 10.1136/gutjnl-2013-305994. [DOI] [PubMed] [Google Scholar]
- 45.Chen S.Y., Tsai C.N., Lee Y.S., Lin C.Y., Huang K.Y., Chao H.C., Lai M.W., Chiu C.H. Intestinal microbiome in children with severe and complicated acute viral gastroenteritis. Sci. Rep. 2017;7:46130. doi: 10.1038/srep46130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cox D.J., Keane J. Platelets and tuberculosis: small cells, not so innocent bystanders. Am. J. Respir. Crit. Care Med. 2018;198:153–154. doi: 10.1164/rccm.201802-0279ED. [DOI] [PubMed] [Google Scholar]
- 47.Waterhouse M., Hope B., Krause L., Morrison M., Protani M.M., Zakrzewski M., Neale R.E. Vitamin D and the gut microbiome: a systematic review of in vivo studies. Eur. J. Nutr. 2019;58:2895–2910. doi: 10.1007/s00394-018-1842-7. [DOI] [PubMed] [Google Scholar]
- 48.Casanova-Martí À., Serrano J., Portune K.J., Sanz Y., Blay M.T., Terra X., Ardévol A., Pinent M. Grape seed proanthocyanidins influence gut microbiota and enteroendocrine secretions in female rats. Food Funct. 2018;9:1672–1682. doi: 10.1039/c7fo02028g. [DOI] [PubMed] [Google Scholar]
- 49.Harenberg S., Bello G., Gjeltema L., Ranshous S., Harlalka J., Seay R., Padmanabhan K., Samatova N. Community detection in large-scale networks: a survey and empirical evaluation. WIREs Comp. Stat. 2014;6:426–439. doi: 10.1002/wics.1319. [DOI] [Google Scholar]
- 50.Palla G., Derényi I., Farkas I., Vicsek T. Uncovering the overlapping community structure of complex networks in nature and society. Nature. 2005;435:814–818. doi: 10.1038/nature03607. [DOI] [PubMed] [Google Scholar]
- 51.Gopalan P.K., Blei D.M. Efficient discovery of overlapping communities in massive networks. Proc. Natl. Acad. Sci. USA. 2013;110:14534–14539. doi: 10.1073/pnas.1221839110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie J., Szymanski B.K., Liu X. SLPA: uncovering overlapping communities in social networks via a speaker-listener interaction dynamic process. IEEE. 2011 doi: 10.1109/ICDMW.2011.154. [DOI] [Google Scholar]
- 53.Hisada T., Endoh K., Kuriki K. Inter- and intra-individual variations in seasonal and daily stabilities of the human gut microbiota in Japanese. Arch. Microbiol. 2015;197:919–934. doi: 10.1007/s00203-015-1125-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J., Guo Z., Lim A.A.Q., Zheng Y., Koh E.Y., Ho D., Qiao J., Huo D., Hou Q., Huang W., et al. Mongolians core gut microbiota and its correlation with seasonal dietary changes. Sci. Rep. 2014;4:5001. doi: 10.1038/srep05001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smits S.A., Leach J., Sonnenburg E.D., Gonzalez C.G., Lichtman J.S., Reid G., Knight R., Manjurano A., Changalucha J., Elias J.E., et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science. 2017;357:802–806. doi: 10.1126/science.aan4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang J., Liang X., Xuan Y., Geng C., Li Y., Lu H., Qu S., Mei X., Chen H., Yu T., et al. A reference human genome dataset of the BGISEQ-500 sequencer. GigaScience. 2017;6:1–9. doi: 10.1093/gigascience/gix024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fang C., Zhong H., Lin Y., Chen B., Han M., Ren H., Lu H., Luber J.M., Xia M., Li W., et al. Assessment of the cPAS-based BGISEQ-500 platform for metagenomic sequencing. GigaScience. 2018;7:1–8. doi: 10.1093/gigascience/gix133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li R., Yu C., Li Y., Lam T.W., Yiu S.M., Kristiansen K., Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 62.Li J., Jia H., Cai X., Zhong H., Feng Q., Sunagawa S., Arumugam M., Kultima J.R., Prifti E., Nielsen T., et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014;32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 63.Truong D.T., Franzosa E.A., Tickle T.L., Scholz M., Weingart G., Pasolli E., Tett A., Huttenhower C., Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods. 2015;12:902–903. doi: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- 64.Liu X., Zhang W., Zeng X., Zhang R., Du Y., Hong X., Cao H., Su Z., Wang C., Wu J., et al. Systematic comparative evaluation of methods for investigating the TCRbeta repertoire. PLoS One. 2016;11:e0152464. doi: 10.1371/journal.pone.0152464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu X., Zhang W., Zhao M., Fu L., Liu L., Wu J., Luo S., Wang L., Wang Z., Lin L., et al. T cell receptor beta repertoires as novel diagnostic markers for systemic lupus erythematosus and rheumatoid arthritis. Ann. Rheum. Dis. 2019;78:1070–1078. doi: 10.1136/annrheumdis-2019-215442. [DOI] [PubMed] [Google Scholar]
- 66.Cao K., Wu J., Li X., Xie H., Tang C., Zhao X., Wang S., Chen L., Zhang W., An Y., et al. T-cell receptor repertoire data provides new evidence for hygiene hypothesis of allergic diseases. Allergy. 2020;75:681–683. doi: 10.1111/all.14014. [DOI] [PubMed] [Google Scholar]
- 67.Jie Z., Liang S., Ding Q., Tang S., Li F., Wang D., Lin Y., Chen P., Cai K., Rao W. A multi-omic cohort as a reference point for promoting a healthy human gut microbiome. bioRxiv. 2019:585893. doi: 10.1101/585893. Preprint at. [DOI] [Google Scholar]
- 68.Derogatis L.R., Lipman R.S., Covi L. SCL-90: an outpatient psychiatric rating scale--preliminary report. Psychopharmacol. Bull. 1973;9:13–28. [PubMed] [Google Scholar]
- 69.Connor K.M., Davidson J.R.T. Development of a new resilience scale: the connor-davidson resilience scale (CD-RISC) Depress. Anxiety. 2003;18:76–82. doi: 10.1002/da.10113. [DOI] [PubMed] [Google Scholar]
- 70.Hagberg A., Swart P., S Chult D. Los Alamos National Lab.(LANL); 2008. Exploring Network Structure, Dynamics, and Function Using NetworkX. [Google Scholar]
- 71.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All summary statistics that support the findings of this study, including pairwise features’ correlations and BFMs, are available as supplementary files. The data and summary statistics of HE datasets have been deposited at Open Archive for Miscellaneous Data (OMIX) in National Genomics Data Center (NGDC) database (OMIX: OMIX002261, OMIX002286). The data of dietary intervention has been deposited at OMIX (OMIX: OMIX002266). The individual-level data including host genetics, microbiome profile, metabolites, laboratory tests, immune repertoire and other phenotypes have been deposited at China National GeneBank DataBase (CNGBdb: CNP0003420). Access to individual-level data has to be approved by corresponding authors (niechao@genomics.cn), and is subject to the policies and approvals from the Human Genetic Resource Administration, Ministry of Science and Technology of the People’s Republic of China.
-
•
All original codes have been deposited at GitHub: the codes for calculating correlation at Github: https://github.com/zhangwei2015/Multi-omics/tree/main/Correlations; the codes for the BFM-ash pipeline at Github: https://github.com/zhangwei2015/Multi-omics/tree/main/BFM-ash.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.