

Abstract
Virus-like particles (VLPs) are versatile protein-based platforms that can be used as a vaccine platform mainly in infectiology. In the present work, we compared a previously designed, non-infectious, adenovirus-inspired 60-mer dodecahedric VLP to display short epitopes or a large tumor model antigen. To validate these two kinds of platforms as a potential immuno-stimulating approach, we evaluated their ability to control melanoma B16-ovalbumin (OVA) growth in mice. A set of adjuvants was screened, showing that polyinosinic-polycytidylic acid (poly(I:C)) was well suited to generate a homogeneous cellular and humoral response against the desired epitopes. In a prophylactic setting, vaccination with the VLP displaying these epitopes resulted in total inhibition of tumor growth 1 month after vaccination. A therapeutic vaccination strategy showed a delay in grafted tumor growth or its total rejection. If the “simple” epitope display on the VLP is sufficient to prevent tumor growth, then an improved engineered platform enabling display of a large antigen is a tool to overcome the barrier of immune allele restriction, broadening the immune response, and paving the way for its potential utilization in humans as an off-the-shelf vaccine.
Keywords: virus-like-particles, antigen display, cancer vaccine, immuno-oncology, melanoma, tumor associated antigens, neoantigens
Graphical abstract
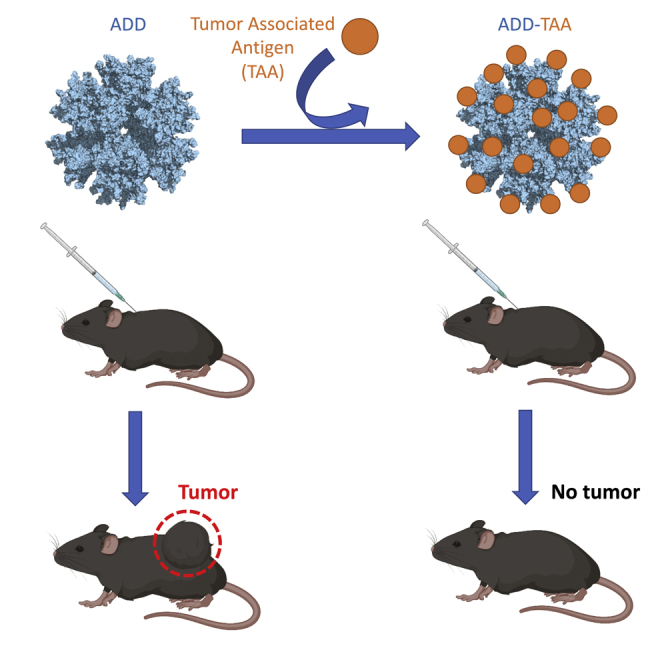
Besson et al. report a non-infectious, adenovirus-inspired, self-assembling particle designed to genetically encode small tumor epitopes or spontaneously and covalently display large tumor antigens. Immunization of mice with these vaccines results in an efficient immune response capable of preventing melanoma tumors in vivo.
Introduction
Despite the genomic vaccine revolution seen during the coronavirus disease 2019 (COVID-19) pandemic,1 protein-based vaccines remain attractive because of their low cost of production, ease of transport and storage, and better social acceptance. Virus-like-particles (VLPs) are non-infectious nanoparticles that assemble spontaneously, mimicking their corresponding virus and, thus, their immunogenicity. Two of the best known examples are the surface antigen of hepatitis B virus (HBV)2 and the structural L1 protein of the human papillomavirus (HPV), which elicit antibodies preventing virus-induced cervical cancer from different HPV types.3 Beyond this, VLPs can be used as a scaffold for display of epitopes derived from other biological targets. The immunogenicity is highly increased through presentation to the immune system in a multimeric manner, and a number of vaccine platforms have been developed to this aim, with various advantages and limitations.4 One advantage is that the small size of VLPs (less than 100 nm) permits efficient drainage to the lymph nodes, which can favor the immune response.5,6 Formulation with an adequate adjuvant, such as inorganic salts (alum), an oil emulsion (MF59), or a lipid A component (monophosphoryl lipid [MPL]) is often used and depends on the platform/antigen tandem and the targeted application.7
From a manufacturing point of view, two main approaches can be considered for presentation of epitopes at the VLP surface. Chemical cross-linking has been reported to induce a good titer of neutralizing antibodies but makes the manufacturing workflow more complex and increases the cost of production. Genetic insertion of the epitope into the VLP enables an easier way of production and purification but may face the risk of epitope misfolding and/or VLP self-assembly issues during production. Because this limitation increases with the size and complexity of the epitope to be inserted, large antigen display cannot be considered using this approach. To overcome this issue, a two-component system called SpyTag/SpyCatcher (ST/SC) has been developed. In this system, derived from Streptococcus pyogenes, an aspartate residue from the 13-amino-acid ST peptide genetically inserted in the VLP can spontaneously create a covalent bond with a lysine residue in the complementary SC module that can be fused to large antigens.8,9,10,11
We have reported previously that a non-infectious VLP derived from the human adenovirus of type 3 and consisting of 60 identical penton base monomers could be exploited to display epitopes of interest on its surface.6,12,13 In this vaccine platform, called adenovirus dodecamer (ADDomer), exposed loops of the penton base protein were engineered to allow insertion of foreign peptides, such as a linear neutralizing epitope from chikungunya virus. However, this design did not permit insertion of structurally complex antigens. To overcome this limitation while keeping the immunogenicity advantage of ADDomer, we redesigned this platform by genetically inserting an ST. Spontaneous display of large and structurally complex antigens with potential post-translational modifications, such as the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) receptor binding domain (RBD) fused to the SC was achieved. This system has proven to be highly effective to elicit potent neutralizing antibodies against SARS-CoV-2.14
In this work, we assess the potential of these two vaccine platforms (direct epitope display and large antigen fusion via SC) in the context of cancer immunotherapy, using the melanoma B16-ovalbumin (OVA) model in mice. An investigation of the cellular immune response underlying tumor growth control is also performed.
Results
MHC class I and class II OVA epitopes can be displayed successfully on the ADDomer surface
The ADDomer is a non-infectious nanoparticle formed of 12 bricks of the homopentameric penton base from the human adenovirus type 3. Two exposed loops can be used for insertion of epitopes: the variable loop (VL) and the RGD (Arg-Gly-Asp) loop (RGD-L). The major histocompatibility complex (MHC) class I epitope (SIINFEKLTEWTSS, called OT-I) and the MHC class II epitope (ISQAVHAAHAEINEAGR, called OT-II) from the OVA were genetically inserted into the RGD-L and VL respectively. These two epitopes have been chosen to initiate a CD8+ T lymphocyte immune response (via the epitope OT-I presented to CD8 by dendritic cells) as well as a CD4+ T lymphocyte immune response (via the epitope OT-II presented to CD4+ T cells by dendritic cells). Because of the spontaneous homo-oligomerization of the 12-pentameric penton bases, 60 copies of each epitope are exposed on the surface of the ADDomer particle, called ADDomer-Duo (ADD-Duo) (Figure 1A). The ADD-Duo was successfully produced in a baculovirus expression system and purified in two steps (Figure 1B). Negative-stain electron microscopy showed that particles harboring these two epitopes were well folded (Figure 1A). An ADDomer displaying only the OT-I epitope was also generated.
Figure 1.

Design and characterization of ADD-Ø, ADD-Duo, ADD-OT-I, and ADD-Blov
(A) Diagram showing the insertion of epitopes (OT-I, blue; OT-II, red) and SpyTag (ST; pink) in the 2 loops of the ADDomer monomer. ST (pink) can make an isopeptidic bond with SpyCatcher (SC; blue) fused to the OVA protein (orange) containing the OT-I and OT-II epitopes. Negative-stain electron micrographs of ADD-Ø, ADD-Duo, ADD-OT-I, and ADD-Blov (bar, 30 nm) are shown on the right. (B) SDS-PAGE profile of reduced and boiled samples of ADD-Ø, ADD-Duo, ADD-OT-I, and ADD-Blov, showing a higher-molecular weight (MW) covalent adduct for the ADD-Blov (ADD-ST/Blov-SC). (C) Structure of the OVA protein with the epitopes OT-I and OT-II highlighted, respectively, in blue and red. The protein structure colored in white was removed and replaced by the SC to give the Blov-SC protein. (D) Sequence of Blov-SC with OVA (orange; OT-I and OT-II epitopes highlighted, respectively, in blue and red), SC (blue), and additional linkers and histidine tag (black).
Immunization with ADD-Duo in combination with the poly(I:C) adjuvant elicits a potent cellular and humoral anti-OVA response compared with the other adjuvants used
We aimed to define the best adjuvant to use in combination with the ADDomer vaccine. To do so, mice were vaccinated with ADD-Duo without or with a different set of adjuvants according to a prime-boost schedule (Figures 2A and 2B). Mice vaccinated with an “empty” ADDomer (ADD-Ø; i.e., an ADDomer that does not display any OVA epitopes) were used as a control. For the adjuvant screening, three different adjuvants were screened separately or in combination (i.e., the three adjuvants together). These different adjuvants aim to target different Toll-like receptors (TLRs) on the three human dendritic cells subtypes (Figure 2B), each of them being representative of one subset. MPLA targets the TLR4 mostly present on cDC2, poly(I:C) (polyinosinic-polycytidylic acid) targets the TLR3 mostly present on cDC1, and ODN 2395 (oligodeoxynucleotide containing an unmethylated CpG motif) targets the TLR9 mostly present on pDC (plasmacytoid Dendritic Cell). To define the best ADD-Duo/adjuvant combination, mice were split into 6 groups (Figure 2B). On day 14, mice were sacrificed, and their immune response was analyzed in their circulating blood, spleen, and injection site-draining lymph nodes (Figure 2A).
Figure 2.

ADD-Ø and ADD-Duo immunogenicity evaluation and adjuvant screening
(A) Immunization schedule, blood sampling, and organ collection for all groups. s.c., subcutaneous. (B) Different groups were evaluated regarding the immunogenicity of ADD-Ø (G1), ADD-Duo (G2), ADD-Duo + 1 adjuvant (G3, G4, G5), and ADD-Duo + 3 adjuvants (G6). (C) Percentage of CD8+ dextramer OT-I+ T lymphocytes in the draining lymph nodes and spleen for all groups (n = 3). (D) Ab response against OT-II peptide on day 14 (n = 3).
The frequency of antigen-specific CD8+ T cells has been determined in the draining lymph nodes and the spleen using a dextramer staining. Poly(I:C) alone was the adjuvant that gave the most homogeneous anti OT-I immune response in the draining lymph nodes (Figure 2C). In the spleen, it was more difficult to determine the best combination because two mice of three in group 5 (poly(I:C)) showed a similar response as the other groups. However, the third mouse of this group showed the highest percentage of specific CD8+ T lymphocytes compared with all other mice of all groups.
Humoral response against the OT-II peptide was also investigated by ELISA (Figure 2D). Again, poly(I:C) gave the highest responses, and the combination of the three adjuvants together did not significantly improve the response compared with poly(I:C) alone (Figure 2D).
The results show that the association of the three adjuvants did not give a better response (cellular and humoral) than poly(I:C) alone. In particular, in the draining lymph nodes, the response obtained with the association of the three adjuvants is lower than the one obtained for poly(I:C) alone. This suggests that the cDC1 subset was probably the main dendritic cell (DC) subset involved in mounting an immune response against the epitopes displayed by the ADDomer. Poly(I:C) is thus a well-suited adjuvant for ADDomer vaccination in vivo. This is in line with other studies using poly(I:C) for therapeutic anti-cancer vaccines.15,16
Immunization with ADDDuo+poly(I:C) protects against B16-OVA tumor challenge
To assess the functionality of antigen-specific T cells induced by ADDomer vaccination, vaccinated mice were challenged with a subcutaneous OVA-expressing tumor inoculation (prophylactic setting). Mice were vaccinated 5 times every 2 or 3 days with ADD-Duo+poly(I:C) (Figure 3A). ADD-Ø was used as a control (Figure 3B). Nearly 4 weeks (25 days) after the last injection, mice were challenged with subcutaneous implantation of the tumor cell line B16-OVA. This cell line expresses the OVA antigen containing the two epitopes OT-I and OT-II, which are displayed on ADD-Duo.
Figure 3.

Tumor challenge of mice vaccinated previously with ADD-Ø and ADD-Duo
(A) Immunization and tumor challenge schedule for all groups. (B) Different groups were evaluated regarding the protective effect of ADD-Ø (G1) and ADD-Duo (G2) against a tumor challenge. Both ADDomers were injected with the poly(I:C) adjuvant, and the tumor challenge was performed using the B16-OVA cell line. (C) Tumor size evolution days after tumor inoculation (n = 3).
Tumor growth was monitored, and mice were sacrificed on day 17, when the first tumor reached the ethical tumor size limit. Mice that received ADD-Ø developed tumors, whereas mice that received ADD-Duo did not have visible tumors on day 17. (Figure 3C). This result shows that the immune response induced by vaccination was specific to the tumor antigens borne by the vehicle and not due to an indirect stimulating response triggered by the vehicle itself. Remarkably, even though a gap of nearly 4 weeks passed between the last vaccination and the challenge, none of the mice showed tumor growth, reflecting that a robust response was mounted during this immunization period.
Large OVA antigen can be successfully displayed on the ADDomer’s surface
To get rid of the allelic restriction and elicit a multi-specific anti-tumor response, display of large antigens rather than small epitopes on the ADDomer surface is desirable. To do so, an ADDomer displaying the ST peptide (ADD-ST) on its surface was used to spontaneously make an isopeptidic bond with an SC fused to the OVA protein (1–355), called binder long OVA (Blov), containing the OT-I and OT-II epitopes (Figures 1A and 1D). The OVA C terminus sequence (356–396, colored white in Figure 1C) buried in the protein was removed and replaced with the SC prolonged with a histidine tag. This OVA C-terminus deletion aimed to make the SC more accessible to the ST to favor Blov-SC assembly to ADD-ST. A melittin signal peptide was added to Blov fused to SC (Blov-SC) to allow protein post-translational modifications (PTMs), such as glycosylations and secretion from insect cells. ADD-ST and Blov-SC were produced separately in insect cells using the baculovirus expression system.
Covalent binding of Blov-SC to ADD-ST was assessed by SDS-PAGE. As expected, an adduct of a higher molecular weight corresponding to Blov-SC bound to the ADD-ST monomer was clearly seen (Figure 1B). This faint band, observed around 180 kDa, reflects weak binding of Blov-SC to ADD-ST (3 Blov-SCs per ADD-ST particle), which is lower than the theoretical ratio used for incubation (ratio of 4:1, meaning 15 Blov-SCs per ADD-ST particle). Electron micrographs showed that particles remained intact after antigen binding (Figure 1A).
Therapeutic vaccination with ADD-Duo, ADD-OT-I, and ADD-Blov drastically decreases tumor growth and improves mouse survival
To assess the efficacy of the ADDomer vaccine in a therapeutic setting, mice were vaccinated 5 times every 2–3 days with ADD-Duo, ADD-OT-I, and ADD-Blov from 1 day after tumor inoculation. ADD-Ø was used as a control, and all ADDomers were injected with poly(I:C) (Figures 4A and 4B). Each group was composed of 12 mice; 6 were euthanized on day 15 to evaluate immunological responses and the others (5 or 6 per group according to the success of tumor implantation) when the maximal ethical tumor size was reached to investigate the long-term clinical response.
Figure 4.

Effect of therapeutic vaccination with ADDomers on B16-OVA tumor growth
(A) Tumor inoculation, immunization, blood sampling, and organ collection for all groups. (B) Different groups were evaluated regarding the therapeutic effect of ADD-Ø (G1), ADD-Duo (G2), ADD-OT-I (G3), and ADD-Blov (G4). ADDomers were injected with the poly(I:C) adjuvant, and tumor inoculation was performed using the B16-OVA cell line. Each group is composed of 12 mice; 6 were sacrificed on day 15 to evaluate the short-term clinical and immunological response, and the remaining mice (n = 5 for ADD-Ø/Duo/OT-I, n = 6 for ADD-Blov) were sacrificed when the endpoint was reached to investigate a long-term response. (C) Tumor size evolution days after tumor inoculation (n = 11 for ADD-Ø/Duo/OT-I, n = 12 for ADD-Blov). (D) Photographs showing the difference in tumor size and aspect on mice vaccinated with ADD-Ø or ADD-OT-I.
On day 15, mice vaccinated with ADD-Duo, ADD-OT-I, and ADD-Blov had a significatively smaller tumor than mice vaccinated with ADD-Ø. For example, on day 15, the mean tumor size for the ADD-Ø group was 81.7 mm2 versus 16.8 mm2 for the ADD-Duo group (Figure 4C). Visual inspection showed a massive difference in tumor size between the ADD-Ø and ADD-OT-I groups (Figure 4D, center panel). Macroscopic observation showed a well-vascularized tumor in mice vaccinated with ADD-Ø, whereas a much smaller and very poorly vascularized tumor was seen in mice vaccinated with ADD-OT-I (Figure 4D, left and right panel, respectively).
Cellular response was investigated (Figures 5 and S2). In the spleen, a higher OVA-specific CD8+ T cell frequency was observed with the ADD-Duo and ADD-OT-I groups compared with the ADD-Ø group. However, the difference between the ADD-Blov and ADD-Ø groups was not significant (Figure 5A, left). In the tumor, tumor-infiltrating OVA-specific CD8+ T cell frequency was higher with the ADD-Duo, ADD-OT-I, and ADD-Blov groups versus the ADD-Ø group (Figures 5A, right, and 5B). This observation could only be made on one tumor for the treated groups (ADD-Duo, ADD-OT-I, and ADD-Blov) because only one tumor was available for analysis as a result of the high efficiency of the vaccination regarding tumor growth. A similar trend was observed in the spleen and in the tumor when looking at the absolute number of OVA-specific CD8+ T lymphocytes (Figure 5C).
Figure 5.

Short-term immunological responses
(A) Percentage of CD8+ dextramer OT-I+ T lymphocytes in the spleen (n = 6) and in the tumor (n = 5 for ADD-Ø, n = 1 for the other groups) for all groups. (B) Dot plot representation of the percentage of CD8+ dextramer OT-I+ T lymphocytes in the tumor of one mouse per group. (C) Absolute number of CD8+ dextramer OT-I+ T lymphocytes in the spleen (n = 6) and in the tumor (n = 5 for ADD-Ø, n = 1 for the other groups) for all groups.
Therapeutic vaccination improves mouse survival in all vaccinated groups and notably for the group of mice vaccinated with ADD-OT-I and ADD-Blov (Figure 6A). Individual tumor growth experiments showed that all tumors were well controlled, even over 60 days of follow up, and only one mouse from two groups escaped from immunological control (Figure 6B, groups ADD-OT-I and ADD-Blov).
Figure 6.

Tumor growth and survival monitoring for therapeutic vaccination
(A) Kaplan-Meier survival analysis showing the percentage of surviving mice. (B) Tumor growth curves for individual mice of each group. The numbers of tumor-free mice are shown in parentheses.
Discussion
In this study, we assessed the potential use of an adenovirus-inspired non-infectious VLP as a potential therapeutic anticancer vaccine. The investigated VLP originally results from spontaneous assembly of 12 homopentameric penton bases from a human adenovirus of type 3 (HAdV-B3).17 Engineering of this 60-mer VLP, called ADDomer, enabled easy and rapid genetic insertion of short epitopes at two different locations of the particle surface. This vaccine platform has been shown recently to trigger a significant neutralizing humoral response against a 19-amino-acid epitope from chikungunya virus.6
OVA is a well-characterized antigen encompassing the immunodominant MHC class I-restricted OVA257–264 (SIINFEKL) and the MHC class II-restricted OVA323–339 (ISQAVHAAHAEINEAGR) epitopes. This OVA system is often used to investigate the immunization potential of novel vaccine platforms or vaccine candidates against different cancers, including melanoma.18,19,20 To bind the MHC class I proteins, class I epitopes must be processed by the proteasome into 8- to 10-amino-acid peptides. Because the cleavage efficiency is influenced by the flanking regions,21,22 special attention must be paid to the directly neighboring epitope. It is known that small amino acid residues, such as glycine or alanine, favor antigen processing by the proteasome.23 Here we genetically inserted the MHC class I OVA epitope flanked by GSGG linkers alone in the RGD-L or in combination with an MHC class II OVA epitope in the VL (Figure 1A). In both cases, the VLPs displaying MHC class I alone or both epitopes at once resulted in nicely folded VLPs, as seen by transmission electron microscopy (Figure 1A).
To obtain an optimized vaccine formulation, it is often necessary to combine an adjuvant to the vaccine platform/antigen tandem. Adjuvants enhance the magnitude, breadth, and durability of the immune response and can orient the balance between the humoral and cellular response.24 Despite identification of TLRs expressed on DCs sensing the different pathogen-associated molecular patterns (PAMPs),25,26 the choice of adjuvant is still empirical. The immune response triggered by ADD-Duo occurred in the absence or presence of a set of adjuvants known to activate different subsets of DCs. ODN 2395, MPLA, and poly(I:C), activating pDC, cCD2, and cCD1, respectively, were tested (Figure 2). Poly(I:C) gave the most homogeneous OVA-specific CD8 response in the draining lymph nodes and spleen as well as a good humoral response against the MHC class II OVA epitope. Poly(I:C) has been reported to interact with TLR327 and is often used in cancer immunotherapy trials28 because it triggers potent Th1-mediated anti-tumor immunity through cDC1 targeting.
In a first attempt, we studied the possibility of preventing tumor growth in vaccinated mice. For this purpose, immunizations were performed with ADD-DUO in the presence of poly(I:C) with a delay of 25 days between the last injection and tumor implantation. Tumor growth was monitored until the ethical endpoint was reached. Contrary to the rapid increase in tumor size seen in the group vaccinated with the empty ADDomer (i.e., vehicle not displaying OVA epitopes), no tumors were seen in mice vaccinated with ADD-Duo (Figure 3C). This result shows that the anti-tumor effect was indeed due to the epitope displayed by the vector and not due to non-specific activation of the immune system. This protection seems to last for a long time because there was a delay of 25 days between the last immunization and tumor implantation, suggesting elicitation of a potent memory CD8+ T cell response.
Genetic insertion of sequences coding short epitopes, such as OT-I and OT-II (14 and 17 residues, respectively), in the two exposed loops of the ADDomer did not affect the overall particle structure (Figure 1A), and the slightly faster migration of OT-I compared to ADDomer-Ø is likely due to its sequence composition29 because mass spectroscopy analyses confirmed the exact molecular weight of these two constructs. In humans, an epitope-based vaccine would represent an important limitation because use is restricted to a particular HLA-bearing population (for instance an HLA-A2-restricted epitope-based vaccine would be suitable only for around 40% of the european population). It would be desirable to overcome this limitation by developing a fully protein-based vaccine, enabling epitope presentation in all individuals (i.e., developing an off-the-shelf vaccine).30 Thus, displaying large antigens rather than short epitopes would be valuable. Unfortunately, genetic insertion of large folded antigens results in subunit misfolding and insolubility.31 Villegas et al.20,32 have shown that the OVA248–376 antigen could be fused to WW domains (containing two conserved tryptophans) to make interactions with the adenovirus PPxY sequences in the adenovirus penton base. However, this non-covalent interaction relied on a relatively low KD (65 nM)33 that did not warrant a homogeneous batch required for clinical trials. To overcome this limitation, insertion of an ST (13 residues) in the exposed loops of the ADDomer has been reported to permit spontaneous and covalent attachment of large and correctly folded antigens, such as the SARS-CoV-2 RBD, to the particle.8,14 Fusion of OVA1–355 (Blov) to the SC successfully resulted in display of this antigen to the vaccine platform (Figures 1A and 1B).
Therapeutic vaccination where tumor implantation occurred prior to vaccination has been performed using an ADDomer with genetic epitope insertion and large OVA antigen display using the ST/SC system. Both systems were efficient at preventing B16-OVA tumor growth (Figure 4C). The different OVA-bearing VLPs (30 nm for the undecorated particle) are within the size range (20–200 nm) of particles readily drained to lymph nodes, potentiating uptake by antigen-presenting cells and cross-presentation.34,35 These immunizations seemed to elicit robust and sustained immune responses capable of rejecting the highly aggressive B16-OVA melanoma in prophylactic and therapeutic strategies (Figures 3 and 4).
Because CD8+ T cells are key players in immuno-oncology, most efforts in vaccine development focus on generation of this response by improving antigen delivery and presentation on the DC surface for the initial T cell encounter.36
In our experiments, immunological characterization showed a significant OVA-specific CD8+ T cell response in the spleen of mice vaccinated with ADD-Duo and ADD-OT-I (Figures 5A and 5C). A similar tendency was also observed in the only residual tumor where this analysis was feasible (Figures 5B and 5C). However, because of the low number of mice presenting a tumor with a size suitable for this analysis, we cannot draw a solid conclusion regarding this observation. Surprisingly, the anti-OT-I CD8+ T cell response in the spleen of mice vaccinated with ADD-Blov was not significant compared with control ADD-Ø. By contrast, this response seemed to be significant compared with the control group within the tumor (4.08% versus 0.28%). Despite the low amount of tumor material that could be analyzed in the ADD-Blov group, this tendency is highly supported by tumor growth control as well as long-term mouse survival (Figures 4C and 6A, respectively). In Blov mice, multi-specific immune responses could have been triggered, improving the tumor control, even though such multi-specific responses were not investigated here (only response against a single epitope was investigated). Such multi-specific responses could be investigated through cytokine production upon ex vivo T cell restimulation with APC loaded with OVA.
Another unexpected feature was the presence of a strong, anti-OVA CD8+ T cell response in the spleen and in measurable tumors of mice vaccinated with ADD-Duo, which was not correlated with the long-term survival curves. We propose two possible hypotheses to explain these data: generation of regulatory T cells and/or tumor escape. It is conceivable that addition of a unique CD4+ T cell epitope, as opposed to ADDomer-OT-I or full OVA protein, triggered a regulatory T cell response which, in turn, impaired efficient anti-tumor immunity despite a high proportion of anti-OVA CD8+ T cells. Another explanation could be tumor escape from anti-OVA CD8+ T cells by MHC class I loss and/or strong immune checkpoint expression. The ADD-Duo vaccine triggered the highest immunization in spleen and tumor (Figure 5). One can imagine that, upon strong immune pressure, tumor cell clones lacking MHC class I expression were selected and/or that OVA-specific T cell-derived interferon γ (IFNɣ) triggered high PD-L1 expression by tumor cells, leading to their inhibition and subsequent tumor escape. To address these hypotheses, fluorescence-activated cell sorting (FACS) analysis of OVA-specific FoxP3 regulatory T cells as well as MHC class I, PD-L1, and PD-1 expression would be required.
Our data show that the ADDomer is well suited to mount an efficient anti-tumoral response. Genetic insertion of epitopes directly in the ADDomer loops is based on single-particle expression but limited to short epitopes, thus restricting its use to personalized medicine or stratified patients. Display of large antigen required expression of ADD-ST and the large cargo fused to the SC. Because of the autocatalytic reaction, simple addition of both components resulted in “ready-to-use” immunotherapy applicable to a large population. With this latter strategy, a mosaic vaccine displaying several different antigens at once can be produced,37,38 paving the way for next-generation products with a broader immunological spectrum for off-the shelf cancer immunotherapy.
Improvement of the technology can be envisioned to combine our platform with other therapeutic strategies, such as oncolytic viruses (OVs). Indeed, a similar approach has already been reported with oncolytic adenovirus encoding OVA or melanoma epitopes directly into the tumor.39 It could be feasible to combine the respective advantages of OVs and particle display by designing an oncolytic adenovirus encoding the ADDomer-ST and the antigen of interest fused to an SC to benefit from tumor lysis and in situ immune stimulation.
Materials and methods
Baculovirus production
The baculovirus expression system was used for production of the different ADDomers (ADD-Ø, ADD OT-I, ADD-Duo, and ADD-ST) and for OVA fused to SC (Blov-SC). Synthetic DNA (Genscript) was cloned in pACEBac1 using the restriction sites BamHI and HindIII. For Blov-SC, the OVA sequence (1–355) was cloned upstream of the SC, and a His6 tag was added to the C-terminus of SC. This fusion protein was secreted using the melittin signal peptide in the vector. Recombinant baculoviruses were made by transposition with an in-house bacmid expressing yellow fluorescent protein, as described previously.6 The baculovirus was amplified on Sf21 cells at a low multiplicity of infection (MOI), and after two amplification cycles, it was used to infect insect cells for 64–72 h at a high MOI. For ADDomer production, the infected cells were pelleted and recovered, whereas for Blov-SC, cells were discarded, and the supernatant was saved.
Protein purification
ADDomer purification
The ADDomers were purified according to a classic protocol.40 Briefly, after lysis of the insect cell pellet by three cycles of freezing-thawing in the presence of Complete protease inhibitor cocktail (Roche) and removal of debris, the lysate was loaded onto a 20%–40% sucrose density gradient. The gradient was centrifuged for 18 h at 4°C at 41,000 rpm on an SW41 rotor in a Beckman XPN-80 ultracentrifuge. The dense collected fractions at the bottom of the tubes were dialyzed against HEPES (10 mM, pH 7.4) and NaCl (150 mM) and then loaded onto a Macroprep Q cartridge (Bio-Rad). After elution by a 150–600 mM linear NaCl gradient in HEPES (10 mM, pH 7.4), ADDomer-containing fractions were checked by SDS-PAGE and concentrated on Amicon (MWCO, 100 kDa) with buffer exchange to HEPES (10 mM, pH 7.4) and NaCl (150 mM).
Blov-SC purification
The insect cell supernatant was centrifuged after thawing for 15 min at 5,000 × g for 30 min. The new supernatant was supplemented with 10 mM imidazole-HCl and Complete protease inhibitor cocktail EDTA free (Roche). The His-tagged Blov-SC protein was then purified by loading the supernatant onto a 1-mL His GraviTrap prepacked column (GE Healthcare), pre-equilibrated with 20 column volumes (CVs) of 500 mM sodium chloride, 10 mM HEPES (pH 7.4), and 10 mM imidazole. Columns were then washed with 20 CVs of the same buffer to remove non-specifically bound material. A second wash was performed at 12.5 mM imidazole in a similar buffer. Proteins were eluted with 10 CVs of buffer containing 200 mM imidazole. Blov-SC-containing fractions were checked by SDS-PAGE and concentrated on Amicon (MWCO, 30 kDa) with buffer exchange to HEPES (10 mM, pH 7.4) and NaCl (150 mM).
ADD-Blov complex formation for characterization and mouse immunization experiments
Covalent complex formation was obtained by incubation of purified ADD-ST with purified Blov protein fused to SC (Blov-SC). Incubation was performed at 25°C under agitation on a Thermomixer at 300 rpm. The Blov-SC ratio per ADD-ST was fixed to 1:4 (1 Blov-SC for 4 monomers) to avoid steric hindrance between the Blov-SC proteins on the ADDomer surface. For immunization experiments, a ratio of 3 copies of Blov-SC per ADD-ST was chosen to leave minimal free Blov-SC. This ratio was calculated on SDS-PAGE using ImageLab software (Bio-Rad). The integrity of ADD-Blov was checked by negative-stain electron microscopy.
B16-OVA culture and preparation for tumor challenge in mice
B16 melanoma cells stably expressing chicken OVA (B16-OVA) were provided by Dr. Dalil Hannani (TIMC Laboratory, Université Grenoble Alpes, France). The cells were tested for OVA expression (Figure S1) and for pathogens (mouse essential panel and PVM (Pneumonia Virus of Mice) PCR, Charles River Laboratories). The cell lines were maintained in RPMI-1640 Glutamax (Gibco) containing 10% fetal bovine serum (Gibco), 5,000 U/100 mL of penicillin-streptomycin (Gibco), and 500 μg/mL Geneticin at 37°C in 5% CO2. Cells were cultivated in T75 flasks, washed with PBS, and passaged with trypsin. For mouse injection, cells were resuspended at 2.106/mL in PBS.
In vivo experiments and vaccination
Animals were housed and bred at the Plateforme de Haute Technologie Animale (PHTA) core facility (Grenoble, France; EU0197, Agreement C38-51610006) under specific pathogen-free conditions in a temperature-controlled environment with a 12-h light/dark cycle and ad libitum access to water and food. Animal housing and procedures were in accordance with the recommendations from the Direction des Services Vétérinaires, Ministry of Agriculture of France, according to European Communities Council Directives 2010/63/EU and according to recommendations for health monitoring from the Federation of European Laboratory Animal Science Associations. Protocols involving animals were reviewed by the local ethics committee (Comité d’Ethique pour l’Expérimentation Animale no. 12, Cometh-Grenoble) and approved by the Ministry of Research (APAFIS 2021032418404959_v3, APAFIS 2018062601109404_v2, and APAFIS 2020112016376739_v1).
Vaccination experiments were performed according to ethics guidelines. Five-week-old female C57Bl6J mice were purchased from Janvier (Le Genest-Saint-Isle, France).
Adjuvant screening
Each mouse (6 groups of 3 mice each) received 100 μL of vaccine: 50 μL of ADDomer (40 μg per mouse) in HEPES (10 mM) and NaCl (150 mM) adjuvanted with an equivalent volume of ODN 2395 (50 μg per mouse), MPLA (20 μg per mouse), or poly(I:C) (50 μg per mouse) (VacciGrade-InvivoGen). Mice were injected subcutaneously in the right flank twice (days 0 and 7) and sacrificed on day 14 for organ harvest (spleen and injection site draining lymph nodes [axillary, brachial, and inguinal lymph nodes on the right flank]). Blood was collected by retro-orbital sampling on day 0 and day 14 under anesthesia using 4% isoflurane.
Prophylactic vaccination
Mice received 5 doses, injected subcutaneously in the left flank, of the poly(I:C) adjuvanted ADDomer vaccine on days 0, 2, 5, 7, and 9. On day 34 (i.e., 25 days after the last vaccination), the mice were challenged with inoculation of B16-OVA cells (2.105 cells/mice in a total volume of 100 μL of PBS, right flank). Tumor growth was monitored for 17 days after the challenge.
Therapeutic vaccination
Mice received the first tumor inoculation (B16-OVA, 2.105 cells, right flank) before receiving 5 doses of the therapeutic poly(I:C)-adjuvanted ADDomer vaccine on days 1, 3, 6, 8, and 10. On day 15, 6 of the 12 animals were sacrificed, and the blood, tumor, spleen, and tumor side draining lymph nodes (axillary, brachial, and inguinal lymph nodes on the right flank) were collected for immune analyses. The 5 or 6 remaining vaccinated and tumor-bearing mice were then monitored for survival until the ethical endpoint was reached.
Flow cytometry analyses of the CD8+ dextramer OT-I+ response
After collection, organs (lymph nodes, spleens, and tumors) were resuspended, washed, and filtered using a 100-μm cell strainer. For spleens and tumors, Red Blood Lysis Buffer (1-min incubation, 1181438900, Merck) and RPMI-1640 Glutamax (Gibco) supplemented with 25 μg/mL of Liberase (20-min incubation, Liberase Research Grade, 5401119001, Merck), respectively, were used during this process. Two million cells were then incubated at room temperature (RT) in the dark for 30 min with the dextramer OT-I (H-2 Kb [SIINFEKL], JD2163-PE, Immudex). CD45, Live/Dead, CD3, CD4, and CD8 antibodies (BD Biosciences) were then added for 20 min at 4°C in the dark. Cells were then washed and analyzed on a flow cytometer (LSRII Analyzer, BD Biosciences).
ELISA
The peptide OT-II (peptide OVA 23–339, InvivoGen) was diluted at 2 μg/mL in PBS, and 50 mL was coated overnight at 4°C in a 96-well plate (Maxisorp Nunc Immunoplate, 442404). Plates were washed using a Thermo Scientific microplate washer (5165040). After three washes with 100 μL of PBS-Tween (0.05%), plates were blocked with PBS-BSA (3%) for 1 h. Mouse serum was serially diluted in PBS and incubated for 1 h (50 mL/well) at RT. After five washes with PBS-Tween (0.05%), a goat anti-mouse immunoglobulin G (IgG) (H + L) secondary antibody linked to horseradish peroxidase (JIR 115-035-062) diluted at 1:2,500 in PBS-Tween (0.05%) was added for 1 h. After five washes, 50 mL of transmembrane domain substrate was distributed per well. The enzymatic reaction was stopped after 70 s by addition of 50 mL of H2S04 (1 M), and plates were read at 450 nm with a Tecan Spark 10M plate reader.
Electron microscopy: Negative staining
Samples of 3.5 mL were adsorbed on the clean side of a carbon film evaporated previously on mica and then stained using 2% (w/v) sodium silicotungstate (pH 7.4) for 30 s. The sample/carbon ensemble was then transferred to a grid and air dried. Images were acquired under low-dose conditions (<30 e−/Å2) on an F20 electron microscope operating at 120 kV using a CETA camera.
Statistical analyses
Because percentages and absolute numbers of CD8+ dextramer OT-I+ T lymphocytes datasets followed a non-normal, heteroscedastic distribution, a non-parametric test was used for comparison of the different groups. Kruskal-Wallis tests were performed, followed by a Dunn’s multiple comparison test for each figure. Differences were considered significant when the p value was below 0.05. Statistics were performed using GraphPad software v.8.
Acknowledgments
We thank Jean Paufique and Brigitte Closs from the Fondation d’Entreprise SILAB for scientific and financial support, which enabled us to perform this work. We are grateful to Daphna Fenel and Guy Schoehn for electron microscopy. We are indebted to Emilie Stermann, Marie-Claire Dagher, Christopher Chevillard, Axelle Amen, Pascal Poignard, and Laurence Chaperot for help and advice. We thank the zootechnicians of the PHTA facility for animal housing and care. This work was supported by Fondation d’Entreprise SILAB - Jean PAUFIQUE. S.B. was the recipient of the SILAB price 2020–2022 dedicated to skin cancer research. S.B was granted by a “bourse doctorale” from the Université Grenoble Alpes. D.H. is supported by GEFLUC Dauphiné-Savoie, Ligue Contre le Cancer Comité Isère, Ligue Contre le Cancer Comité Savoie, Université Grenoble Alpes IDEX Initiatives de Recherche Stratégiques, and Fondation du Souffle-Fonds de Recherche en Santé Respiratoire (FdS-FRSR). This work used the platforms of the Grenoble Instruct-ERIC Centerre (ISBG; UAR 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for Structural Biology (PSB), supported by FRISBI (ANR-10-INBS-05-02) and GRAL, financed within the Université Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003). The electron microscope facility is supported by the Auvergne-Rhône-Alpes region, the Fondation pour la Recherche Médicale (FRM), the Fonds FEDER, and the GIS-Infrastructures en Biologie Sante et Agronomie (IBISA).
Author contributions
Conceptualization, S.B., C.A., D.H., and P.F.; investigation, S.B., E.B., O.M., D.L., C.A., and D.H.; animal experiments, S.B., C.A., D.L., E.B., and D.H.; funding acquisition, S.B. and P.F. S.B. and P.F wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.12.003.
Supplemental information
Data availability
Data are available upon request.
References
- 1.Bok K., Sitar S., Graham B.S., Mascola J.R. Accelerated COVID-19 vaccine development: milestones, lessons, and prospects. Immunity. 2021;54:1636–1651. doi: 10.1016/j.immuni.2021.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ho J.K.-T., Jeevan-Raj B., Netter H.-J. Hepatitis B virus (HBV) subviral particles as protective vaccines and vaccine platforms. Viruses. 2020;12:126. doi: 10.3390/v12020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villa L.L., Costa R.L.R., Petta C.A., Andrade R.P., Ault K.A., Giuliano A.R., Wheeler C.M., Koutsky L.A., Malm C., Lehtinen M., et al. Prophylactic quadrivalent human papillomavirus (types 6, 11, 16, and 18) L1 virus-like particle vaccine in young women: a randomised double-blind placebo-controlled multicentre phase II efficacy trial. Lancet Oncol. 2005;6:271–278. doi: 10.1016/S1470-2045(05)70101-7. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen B., Tolia N.H. Protein-based antigen presentation platforms for nanoparticle vaccines. NPJ Vaccines. 2021;6:70. doi: 10.1038/s41541-021-00330-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy S.T., van der Vlies A.J., Simeoni E., Angeli V., Randolph G.J., O’Neil C.P., Lee L.K., Swartz M.A., Hubbell J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 6.Vragniau C., Bufton J.C., Garzoni F., Stermann E., Rabi F., Terrat C., Guidetti M., Josserand V., Williams M., Woods C.J., et al. Synthetic self-assembling ADDomer platform for highly efficient vaccination by genetically encoded multiepitope display. Sci. Adv. 2019;5 doi: 10.1126/sciadv.aaw2853. eaaw2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caldeira J.C., Perrine M., Pericle F., Cavallo F. Virus-like particles as an immunogenic platform for cancer vaccines. Viruses. 2020;12:488. doi: 10.3390/v12050488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zakeri B., Fierer J.O., Celik E., Chittock E.C., Schwarz-Linek U., Moy V.T., Howarth M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA. 2012;109:E690–E697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brune K.D., Leneghan D.B., Brian I.J., Ishizuka A.S., Bachmann M.F., Draper S.J., Biswas S., Howarth M. Plug-and-Display: decoration of Virus-Like Particles via isopeptide bonds for modular immunization. Sci. Rep. 2016;6:19234. doi: 10.1038/srep19234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L., Fierer J.O., Rapoport T.A., Howarth M. Structural analysis and optimization of the covalent association between SpyCatcher and a peptide tag. J. Mol. Biol. 2014;426:309–317. doi: 10.1016/j.jmb.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keeble A.H., Howarth M. Power to the protein: enhancing and combining activities using the Spy toolbox. Chem. Sci. 2020;11:7281–7291. doi: 10.1039/D0SC01878C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fender P., Ruigrok R.W., Gout E., Buffet S., Chroboczek J. Adenovirus dodecahedron, a new vector for human gene transfer. Nat. Biotechnol. 1997;15:52–56. doi: 10.1038/nbt0197-52. [DOI] [PubMed] [Google Scholar]
- 13.Besson S., Vragniau C., Vassal-Stermann E., Dagher M.C., Fender P. The adenovirus dodecahedron: beyond the platonic story. Viruses. 2020;12:718. doi: 10.3390/v12070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chevillard C., Amen A., Besson S., Hannani D., Bally I., Dettling V., Gout E., Moreau C.J., Buisson M., Gallet S., et al. Elicitation of potent SARS-CoV-2 neutralizing antibody responses through immunization with a versatile adenovirus-inspired multimerization platform. Mol. Ther. 2022;30:1913–1925. doi: 10.1016/j.ymthe.2022.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martins K.A.O., Bavari S., Salazar A.M. Vaccine adjuvant uses of poly-IC and derivatives. Expert Rev. Vaccines. 2015;14:447–459. doi: 10.1586/14760584.2015.966085. [DOI] [PubMed] [Google Scholar]
- 16.Liu C., Chu X., Sun P., Feng X., Huang W., Liu H., Ma Y. Synergy effects of Polyinosinic-polycytidylic acid, CpG oligodeoxynucleotide, and cationic peptides to adjuvant HPV E7 epitope vaccine through preventive and therapeutic immunization in a TC-1 grafted mouse model. Hum. Vaccin. Immunother. 2018;14:931–940. doi: 10.1080/21645515.2017.1420446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fender P., Hall K., Schoehn G., Blair G.E. Impact of human adenovirus type 3 dodecahedron on host cells and its potential role in viral infection. J. Virol. 2012;86:5380–5385. doi: 10.1128/JVI.07127-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellone M., Cantarella D., Castiglioni P., Crosti M.C., Ronchetti A., Moro M., Garancini M.P., Casorati G., Dellabona P. Relevance of the tumor antigen in the validation of three vaccination strategies for melanoma. J. Immunol. 2000;165:2651–2656. doi: 10.4049/jimmunol.165.5.2651. [DOI] [PubMed] [Google Scholar]
- 19.Epaulard O., Toussaint B., Quenee L., Derouazi M., Bosco N., Villiers C., Le Berre R., Guery B., Filopon D., Crombez L., et al. Anti-tumor immunotherapy via antigen delivery from a live attenuated genetically engineered Pseudomonas aeruginosa type III secretion system-based vector. Mol. Ther. 2006;14:656–661. doi: 10.1016/j.ymthe.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 20.Villegas-Mendez A., Garin M.I., Pineda-Molina E., Veratti E., Bueren J.A., Fender P., Lenormand J.-L. In vivo delivery of antigens by adenovirus dodecahedron induces cellular and humoral immune responses to elicit antitumor immunity. Mol. Ther. 2010;18:1046–1053. doi: 10.1038/mt.2010.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Del Val M., Schlicht H.-J., Ruppert T., Reddehase M.J., Koszinowski U.H. Efficient processing of an antigenic sequence for presentation by MHC class I molecules depends on its neighboring residues in the protein. Cell. 1991;66:1145–1153. doi: 10.1016/0092-8674(91)90037-Y. [DOI] [PubMed] [Google Scholar]
- 22.Kloetzel P.-M. Antigen processing by the proteasome: ubiquitin and proteasomes. Nat. Rev. Mol. Cell Biol. 2001;2:179–187. doi: 10.1038/35056572. [DOI] [PubMed] [Google Scholar]
- 23.Nussbaum A.K., Dick T.P., Keilholz W., Schirle M., Stevanović S., Dietz K., Heinemeyer W., Groll M., Wolf D.H., Huber R., et al. Cleavage motifs of the yeast 20S proteasome β subunits deduced from digests of enolase 1. Proc. Natl. Acad. Sci. USA. 1998;95:12504–12509. doi: 10.1073/pnas.95.21.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pulendran B., Arunachalam P.S., O’Hagan D.T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug Discov. 2021;20:454–475. doi: 10.1038/s41573-021-00163-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 26.Kawai T., Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 27.Alexopoulou L., Holt A.C., Medzhitov R., Flavell R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 28.Ammi R., De Waele J., Willemen Y., Van Brussel I., Schrijvers D.M., Lion E., Smits E.L.J. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol. Ther. 2015;146:120–131. doi: 10.1016/j.pharmthera.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 29.Rath A., Glibowicka M., Nadeau V.G., Chen G., Deber C.M. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. USA. 2009;106:1760–1765. doi: 10.1073/pnas.0813167106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao W., Wu J., Chen S., Zhou Z. Shared neoantigens: ideal targets for off-the-shelf cancer immunotherapy. Pharmacogenomics. 2020;21:637–645. doi: 10.2217/pgs-2019-0184. [DOI] [PubMed] [Google Scholar]
- 31.Vragniau C. PhD Thesis. 2018. Modification des dodécaèdres bases de l’adénovirus de sérotype 3: Design et caractérisation d’un nouveau vecteur multi-épitopique polyvalent.https://hal.archives-ouvertes.fr/tel-02110396 [Google Scholar]
- 32.Villegas-Méndez A., Fender P., Garin M.I., Rothe R., Liguori L., Marques B., Lenormand J.-L. Functional characterisation of the WW minimal domain for delivering therapeutic proteins by adenovirus dodecahedron. PLoS One. 2012;7 doi: 10.1371/journal.pone.0045416. e45416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galinier R., Gout E., Lortat-Jacob H., Wood J., Chroboczek J. Adenovirus protein involved in virus internalization recruits Ubiquitin−Protein ligases. Biochemistry. 2002;41:14299–14305. doi: 10.1021/bi020125b. [DOI] [PubMed] [Google Scholar]
- 34.Bachmann M.F., Jennings G.T. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010;10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 35.Zabel F., Kündig T.M., Bachmann M.F. Virus-induced humoral immunity: on how B cell responses are initiated. Curr. Opin. Virol. 2013;3:357–362. doi: 10.1016/j.coviro.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 36.Shibagaki N., Udey M.C. Dendritic cells transduced with protein antigens induce cytotoxic lymphocytes and elicit antitumor immunity. J. Immunol. 2002;168:2393–2401. doi: 10.4049/jimmunol.168.5.2393. [DOI] [PubMed] [Google Scholar]
- 37.Cohen A.A., Gnanapragasam P.N.P., Lee Y.E., Hoffman P.R., Ou S., Kakutani L.M., Keeffe J.R., Wu H.-J., Howarth M., West A.P., et al. Mosaic nanoparticles elicit cross-reactive immune responses to zoonotic coronaviruses in mice. Science. 2021;371:735–741. doi: 10.1126/science.abf6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen J. The dream vaccine. Science. 2021;372:227–231. doi: 10.1126/science.372.6539.227. [DOI] [PubMed] [Google Scholar]
- 39.Feola S., Russo S., Martins B., Lopes A., Vandermeulen G., Fluhler V., De Giorgi C., Fusciello M., Pesonen S., Ylösmäki E., et al. Peptides-coated oncolytic vaccines for cancer personalized medicine. Front. Immunol. 2022;13:826164. doi: 10.3389/fimmu.2022.826164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fender P. Use of dodecahedron “VLPs” as an alternative to the whole adenovirus. Methods Mol. Biol. 2014;1089:61–70. doi: 10.1007/978-1-62703-679-5_4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request.