

Abstract
The clonal dynamics following hematopoietic stem progenitor cell (HSPC) transplantation with busulfan conditioning are of great interest to the development of HSPC gene therapies. Compared with total body irradiation (TBI), busulfan is less toxic and more clinically relevant. We used a genetic barcoded HSPC autologous transplantation model to investigate the impact of busulfan conditioning on hematopoietic reconstitution in rhesus macaques. Two animals received lower busulfan dose and demonstrated lower vector marking levels compared with the third animal given a higher busulfan dose, despite similar busulfan pharmacokinetic analysis. We observed uni-lineage clonal engraftment at 1 month post-transplant, replaced by multilineage clones by 2 to 3 months in all animals. The initial multilineage clones in the first two animals were replaced by a second multilineage wave at 9 months; this clonal pattern disappeared at 13 months in the first animal, though was maintained in the second animal. The third animal maintained stable multilineage clones from 3 months to the most recent time point. In addition, busulfan animals exhibit more rapid HSPC clonal mixing across bone marrow sites and less CD16+ NK-biased clonal expansion compared with TBI animals. Therefore, busulfan conditioning regimens can variably impact the marrow niche, resulting in differences in clonal patterns with implications for HSPC gene therapies.
Keywords: busulfan, rhesus macaque, HSPC transplantation, barcode
Graphical abstract
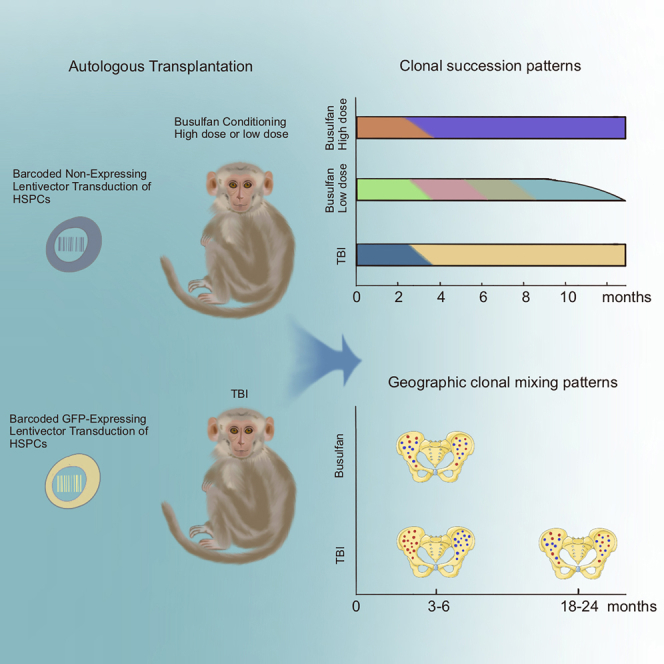
Abraham and colleagues examine the impact of different pre-HSPC transplant conditioning regimens, busulfan and TBI, on the clonal behavior of engrafting HSPCs in rhesus macaques. Longitudinal clonal tracking of HSPCs in busulfan- and TBI-treated animals reveals distinct clonal patterning of hematopoietic reconstitution in busulfan-conditioned animals.
Introduction
Hematopoietic stem cell transplantation (HSCT) and HSC-directed gene therapies are potentially curative treatments for a variety of blood, bone marrow, immune system, and metabolic storage disorders.1,2,3 Namely, HSCT is used to replace defective endogenous hematopoietic stem and progenitor cells (HSPCs) with allogeneic healthy donor or genetically modified autologous HSPCs. Bone marrow conditioning is a necessary component of HSCT, as it depletes endogenous HSPCs, thus clearing bone marrow niches and facilitating competitive engraftment of transplanted therapeutic HSPCs. Furthermore, engraftment of allogeneic HSPCs in addition requires profound immunosuppression to prevent rejection.
Historically, total body irradiation (TBI) has been best studied both experimentally and clinically as the basis of conditioning, due to potent HSPC ablative and immunosuppressive properties.1,4 However, TBI results in significant acute and chronic toxicities, limiting its clinical utilization. Given the large number of patients who could benefit from HSCT, effective conditioning regimens with less toxicity have been investigated.5 The chemotherapy drug and alkylating agent busulfan has been used in combination with immunodepleting agents as a conditioning regimen for patients undergoing allogeneic transplantation for hematological malignancies, initially to avoid the detrimental effects of TBI on brain development in children.6,7,8 Although busulfan-induced myelosuppression is sufficient to deplete endogenous HSCs, busulfan is not potently immunosuppressive, thus the need for additional immunosuppression when used in allogeneic HSCT. In addition, busulfan alone has been by far the most common agent used as conditioning prior to transplantation of virally transduced or gene-edited autologous HSPC in clinical gene therapy protocols targeting inherited marrow disorders.9,10,11,12,13,14,15 Combination of busulfan with additional immunosuppressants may be needed to prevent immune-mediated rejection of transplanted genetically modified HSPCs expressing previously absent proteins that may elicit an immune response.10,16
Conditioning agents may also impact components of the bone marrow niche, with later indirect impact on hematopoietic recovery and post-transplantation long-term hematopoiesis. The niche consists of a complex array of cellular components such as mesenchymal stromal cells, endothelial cells, osteoblasts, and immune cells, along with extracellular matrix proteins such as collagen and laminin. Interactions between components and with HSPCs continue to be intensely studied, but there is increasing evidence that microenvironmental characteristics can have profound impact on steady-state hematopoiesis, engraftment, and long-term hematopoietic recovery from transplantation, as well as malignant HSPC transformation.17
It is challenging to study the impact of conditioning on such a complex microenvironment in vivo, and to date most investigations have used in vitro assays of stromal cell numbers or function, with heterogeneous results. In mice, ablative doses of TBI have been reported to markedly decrease marrow fibroblast colony-forming units as well as stromal layers able to support in vitro hematopoiesis both acutely and long term.18 The impact of busulfan has been less clear, with one study finding no loss of HSPC-supportive stroma in mice,19 but another reporting equivalent loss of human marrow stromal layer formation with busulfan conditioning as compared with TBI.20 Quantitative analyses of hematopoiesis in vivo comparing various conditioning regimens has not yet been performed.
Given the close phylogenetic similarity and HSPC characteristics between non-human primates (NHPs) and humans, rhesus macaques serve as an effective model to develop HSPC gene therapies and investigate fundamental characteristics of hematopoietic reconstitution after HSCT.21,22 We and others have previously used high-diversity lentiviral barcoding of NHP HSPCs in an autologous transplantation model to study clonal dynamics of thousands of HSPCs following TBI conditioning.23,24 In our previous studies, we reported short-term reconstitution from lineage-restricted clones for several months, followed by stable contributions originating from multipotent clones persisting for up to 7 years post-transplantation. We used this model to analyze the geographic distribution of HSPC clones within the bone marrow, which in TBI-conditioned animals somewhat surprisingly was characterized by geographically restricted output from HSPC clones, comparing across different bone marrow (BM) sites, for up to 2 years post-transplantation.25 Finally, we made the novel observation of natural killer (NK)-biased massively expanded mature NK cell clones emerging and waxing and waning over time, suggesting peripheral mature NK self-renewal.26 Investigators have used vector insertion site retrieval from hematopoietic cells following human autologous HSPC gene therapies to study clonal dynamics post-transplantation, notably all using busulfan or other chemotherapy conditioning, not TBI.27,28,29 Overall, the same pattern of transient contributing clones being replaced by long-term stable multilineage clones was observed.
In the current study, we tracked the hematopoietic reconstitution of lentivirally barcoded autologous HSPCs following busulfan conditioning and directly compared clonal dynamics to patterns observed previously in the macaque model with TBI conditioning. In doing so, we asked whether potential differences in the impact of busulfan versus TBI on the marrow microenvironment or clearing of HSPCs impacted the patterns of clonal reconstitution. Our study provides the first direct comparison of the impact of conditioning on clonality and can serve as a platform for further comparisons with novel conditioning approaches such as stem cell-targeted cytotoxic antibodies.
Results
Experimental design
To investigate the impact of busulfan conditioning on hematopoietic reconstitution following autologous HSPC transplantation, we tracked the clonal output from transplanted HSPCs in rhesus macaques using high-diversity lentiviral barcoding, as previously described.23,24 As shown in Figure 1, Figure 3A and 3 animals were transplanted with barcoded HSPCs following busulfan conditioning. Since busulfan is not sufficiently immunosuppressive to prevent rejection of cells expressing foreign proteins such as green fluorescent protein (GFP) in macaques,16 we used two barcoded vectors for the current studies engineered to be less immunogenic. The first vector contains a human truncated nerve growth factor receptor (tNGFR), an engineered signaling-impaired version of the endogenous receptor (Figures S1A and S1B), and was expected to be minimally antigenic in macaques, given greater than 99.3% homology to the rhesus macaque receptor at the amino acid level.30 This marker gene was chosen to facilitate flow cytometric identification and sorting of vector-containing cells, as previously demonstrated in both murine xenografted mice conditioned with busulfan and macaques conditioned with TBI.31,32 CD34+ cells transduced with the tNGFR lentivirus were cultured in vitro for 48 to 96 h to measure the in vitro transduction efficiency by flow cytometric analysis of tNGFR expression (Table 1). The second vector was engineered without any expressed marker gene via excision of the copGFP single-cell DNA (termed NE) from the pCDH-EF1α-MCS∗-T2A-CopGFP backbone (CD526A-1, Systmbio company). This vector was used to ensure engraftment with barcoded cells even if tNGFR-expressing cells were rejected (Figure 1A).31,33
Figure 1.

Experimental design and marking summary
(A) Animal-specific conditioning and transplantation schema. Mobilized peripheral blood (PB) CD34+ cells from each animal were split into two equal aliquots and each fraction was transduced with lentiviral barcode library containing an elongation factor 1-α (EF1-α) promoter, with no expressed transgene marker (NE) or with a truncated nerve growth factor receptor marker gene (tNGFR), and distinguished by a unique library ID. After transduction, both fractions were cryopreserved during busulfan conditioning, then thawed and infused intravenously into the autologous macaque. Animals 11021142 and 10U004 received 5.5 mg/kg of intravenous busulfan for 4 consecutive days, while animal H84D received 6.0 mg/kg for 4 consecutive days. (B) Vector copy number (VCN) of PB granulocytes (Gr) and T cells from the three busulfan animals over time determined by digital droplet PCR (ddPCR) using a probe targeting to the RRE region shared by both vectors, and a probe detecting the housekeeping gene TERT. (C) Summary of GFP marking levels of PB Gr and T lineages from five macaques conditioned with myeloablative TBI and transplanted with CD34+ HSPCs transduced with cop-GFP expressing lentiviral barcode libraries. Solid lines: Gr, dotted lines: T cells, with each color representing an individual animal. (D) Molecular marking level comparisons of PB Gr and T between NE and tNGFR barcode libraries in the three busulfan animals over time. The total VCN determined in 1B (VCN) was multiplied by the fraction of reads retrieved carrying each specific library ID from Illumina sequencing to calculate the VCN attributed to the tNGFR and the NE vectors. “X” in the bar graphs indicates the sample for that time point is unavailable, thus no results are provided.
Figure 3.

Clonal geographic distribution in bone marrow
(A) Heatmaps depicting the contributions of the top 10 clones in purified left (L) and right (R) BM CD34+ cells and concurrently sampled PB Gr over time (m: months post-transplantation) for the three busulfan-conditioned animals and one representative TBI-conditioned animal (ZJ31). The heatmaps were generated as detailed in Figure 2A. (B) Pairwise Pearson correlation coefficients comparing clonal contributions retrieved from left (L) and right (R) BM CD34+ cells and concurrent PB Gr over time (m: months post-transplantation) for each animal. The color scale for r values is shown on the right. (C) Fold bias line graphs showing the fractional bias between left and right BM CD34+ HSPC clones over time for the three busulfan animals and one representative TBI animal (ZJ31) over time. Each time point is shown by a different color line as indicated on the right (in months). The x axis shows the fractional bias comparing left and right BM CD34+ clonal contributions. The y axis shows the cumulative percentage contributions of these clones to overall retrieved barcodes within the two samples, with the maximal possible contribution of 200%; −2 to 2 regions on the x axis indicate nonbiased clones.
Table 1.
Busulfan animal transplantation and engraftment parameters
| Animal ID |
11021142 |
10U004 |
H84D |
|||
|---|---|---|---|---|---|---|
| Vector | pCDH-EF1α-T2A- no marker gene library 15 (without WPRE) | pCDH-EF1α-T2A- tNGFR library 17 (without WPRE) | pCDH-EF1α-T2A- no marker gene library 15 (without WPRE) | pCDH-EF1α-T2A- tNGFR library 17 (without WPRE) | pCDH-EF1α-T2A- no marker gene library 21 (with WPRE) | pCDH-EF1α-T2A- tNGFR library 11 (with WPRE) |
| Date of transplant | 02/01/2019 | 11/29/2019 | 07/31/2020 | |||
| Transduction MOI | 25 | 25 | 25 | 25 | 20 | 20 |
| Transduction condition | FN+ Cytokines + protamine sulfatea | FN+ Cytokines + protamine sulfatea | FN + Cytokines + p407 + PGE2b | |||
| Transduction efficiencyc | N/A | 31.5% | N/A | 31.2% | N/A | 66.0% |
| Number of cells collected (× 106) | 28 | 27.5 | 28 | |||
| Number of cells infused (× 106) | 36.0 | 36.0 | 45.0 | 41.0 | 25.9 | 25.6 |
| Transplantation dose (CD34+ cells × 106/kg) | 15.0 | 16.4 | 10.5 | |||
| Busulfan conditioning dose (mg/kg) × 4 days | 5.5 | 5.5 | 6.0 | |||
| Day until neutrophil recovery to >500/μL | Day 11 | Never Below 500/μL | Never below 500/μL | |||
| Last follow-up (months post- transplant) | 36 | 26 | 18 | |||
| Total number of barcoded clones detected | 2,118 | 2,250 | 18,803 | |||
| Number of barcoded clones detected at the last follow-up, which include all lineages (T, B, Mo, Gr, CD16+, and CD56+NK) | 13 (24m) | 52 (22.5m) | 1,778 (18m) | |||
| d Estimated Minimum frequency long-term CD34+ engraftment (at last follow-up) | 0.00011% | 0.00037% | 0.01% | |||
Fibronectin (FN) coated plate + % HSA + cytokines (Flt-3, SCF, TPO all at 100 ng/mL) + protamine sulfate (4 μg/mL).
Fibronectin (FN) coated plate + cytokines (Flt-3, SCF, TPO all at 100 ng/mL) + P407 (100 μg/mL) + PGE2 (10 μM).
Transduction efficiency is measure by FACS for NGFR.
Estimated Minimum frequency long-term CD34+ engraftment (at last follow-up) is calculated by using the “unique clones number that tracked at the last follow-up” to divide “initial infused transduced CD34+ cells numbers.”
In each animal, half of the CD34+ cells were transduced with the tNGFR barcoded vector containing one 6-base pair (bp) library ID followed by a high-diversity barcode library, and the other half the NE barcoded vector containing a different 6-bp library ID followed by the barcode library (Figure 1A). The libraries were of sufficient diversity to ensure a >95% chance that each unique barcode would be present in no more than one initial HSPC, and the transduction efficiency was optimized to avoid multiple barcode insertions per HSPC.23,34 Cells were cryopreserved at the end of transduction, following 4 days of busulfan conditioning, both aliquots for each animal were thawed, mixed, and infused. Transplantation parameters, including busulfan dose, estimated transduction efficiency, and number of cells infused for each animal are summarized in Table 1. Transplantation parameters for TBI monkeys are summarized in Table S1. Leukocyte, neutrophil, red cell, and platelet counts decreased markedly as expected in the days following busulfan administration, but recovered to near baseline within 2 weeks post-transplantation (Figure S1F).
Overall molecular marking of vector transduced HSPCs varies between animals
To determine the overall engraftment level with transduced HSPCs, we measured the vector copy number (VCN) using digital droplet PCR (ddPCR) targeting a common RRE region in the lentiviral backbones of both the tNGFR vector and NE vector. The overall VCN at all time points was less than 0.04 in blood granulocytes (Gr) and T cells from animals 11021142 and 10U004, respectively (Figure 1B). However, in H84D the overall VCN in Gr reached and stabilized above 0.3–0.4, 10 times higher than the first two animals. The T cell VCN was lower than Gr in all three animals (Figure 1B), as previously reported with both busulfan and TBI conditioning,16 shown for comparable previously reported rhesus macaques receiving autologous HSPCs barcoded with GFP-expressing vectors following ablative TBI, summarized and updated in Figure 1C.24
To determine whether the low level of engraftment in the first animals was due to a lower exposure to busulfan, we performed busulfan pharmacokinetic (PK) analyses (Table S4). We compared the areas under the curve (AUC) of plasma busulfan concentrations in 11021142, an animal given 5.5 mg/kg busulfan for 4 days, previously shown to result in engraftment with genetically modified HSPCs in our rhesus macaque model,16 dose and H84D, the animal given a 10% higher dose of 6 mg/kg for 4 days. Surprisingly, despite the marked differences in engraftment levels with transduced cells, we found that 11021142 had an AUC of 3,675 μM∗min and H84D had only a 3% higher AUC of 3,791 μM∗min, not explaining the marked difference in engraftment of transduced cells between the animals. However, the transduction efficiency in bulk CD34+ cells was 2-fold higher as measured by fluorescence-activated cell sorting (FACS) for animal H84D compared with the other two animals (Table 1), and the transduction enhancers p407 and PGE2 were used, perhaps resulting in more marked improvement in engrafting HSPC transduction and also contributing to the much better engraftment with transduced HSPCs in vivo.
HSPCs transduced with the NE vector demonstrated higher and more stable marking compared with those transduced with the tNGFR vector in vivo
We next compared the engraftment of HSPCs transduced with both vectors in vivo in each animal. The percentage of tNGFR+ cells in various populations can be detected by flow cytometry; however, we observed that T and B cells express some background NGFR in pre-transplantation samples, with less background NGFR expression in Gr (Figure S1C). Monitoring tNGFR expression within hematopoietic cell populations over time post-transplantation by flow cytometry revealed very low levels or levels not clearly over background of tNGFR expressing blood cells in all three animals (Figure S1D). These findings suggested that tNGFR-transduced HSPC engraftment is very low, despite transduction efficiencies of 31% to 66% present in aliquots of CD34+ cells maintained in culture post-transduction (Tables 1 and S1E). This suggests that tNGFR-expressing HSPCs were being rejected prior to or soon after engraftment, and that the limited amino acid differences between human and rhesus macaque NGFR sequences might be immunogenic.
We next investigated the engraftment of cells containing the two vector libraries at the molecular level. The total molecular marking level can be accessed by ddPCR and the contributions of the two different vector barcode libraries can be measured by comparing the number of reads retrieved via sequencing containing the library ID specific to each vector library. In all three animals, the tNGFR vector molecular marking levels were very low, with almost undetectable levels in 10U004 and H84D (Figure 1D), confirming the results from FACS. Therefore, we focused our subsequent clonal analyses solely on the library encoded by the NE vector in each animal, given the neglectable contributions from the tNGFR-transduced cells.
Clonal dynamics following busulfan conditioning
To study HSPC clonal dynamics following busulfan conditioning, we analyzed peripheral blood Gr, monocytes (Mo), T cells, B cells, and NK cells post-transplantation in the three animals. Our previous data on clonal dynamics using barcoded autologous transplantation with myeloablative TBI conditioning in more than 10 animals documented an initial set of uni-lineage clones contributing short term for 1–2 months, subsequently replaced by stable contributions from multilineage long-term HSPC clones persisting for up to 10 years23,24 (and unpublished), as visualized in heatmaps showing the fractional contributions of individual barcodes (clones) to all barcodes retrieved for the top 10 largest contributing clones in each lineage and time point mapped over all time points. In the animals receiving busulfan conditioning, we also observed uni-lineage clones contributing for 1 to 2 months post-transplantation in all three animals (Figure 2A). Multilineage clonal contributions emerged and showed substantial contributions as early as 2 months post-transplantation (Figure 2A). However, in contrast to TBI-conditioned animals, we observed significant reduction in relative contributions from this initial wave of multilineage clones by 9 months post-transplantation in the first two animals (11021142 and 10U004), replaced by contributions from a new set of multilineage clones. Surprisingly, 11021142 then demonstrated a loss of most of this second wave of multilineage clones by 18 months, with the very low-level residual marking (Figures 1B and 1D) emanating from a very small number of multilineage clones (Figure 2A), whereas 10U004 retained contributions from this second wave of multilineage clones (Figure 2A).
Figure 2.

Clonal contributions to hematopoietic lineages in peripheral blood over time
(A) Heatmaps depicting the top contributing clones for various PB lineages: T, B, Monocyte (Mo), Granulocyte (Gr), CD16+ NK, and CD56+NK over time (m: months post-transplantation) for each busulfan-conditioned animal (11021142, 10U004, H84D) and a representative TBI-conditioned animal (ZG66). Each row of the heatmap represents a unique barcoded clone. Each column is purified lineage sample at various time points. The heatmap for each animal was generated by selecting the top 10 most abundant clones contributing in each sample (designated by the asterisks in that sample) and plotting the abundance of these clones across all samples, with a color scale indicating the fractional contribution of each barcode to all barcodes retrieved from that sample. (B) Autocorrelation plots showing the Spearman correlation between all clonal contributions in each PB Gr sample and the previous time point. The y axis represents the autocorrelation R value. Samples with close to identical clonal contributions will have an autocorrelation close to 1 and very dissimilar clonal contributions will have an autocorrelation near 0. Samples from the three busulfan-conditioned animals (colored lines) and five TBI-conditioned animals (ZG66, ZH19, ZJ31, ZH33, ZK22, shown in black lines) were analyzed. (C) The top three graphs show the number of unique barcodes (y axis) in PB lineages over time (x axis) retrieved from each busulfan-conditioned animal (11021142, 10U004, and H84D). The bottom graph compares the number of unique barcodes in PB Gr between busulfan-treated animals (colored lines) and five TBI-conditioned animals (black lines). (D) The top three graphs display the Shannon diversity indices for PB lineages over time in busulfan-conditioned animals. The bottom plot directly compares the Shannon diversity indices for PB Gr between busulfan (colored lines) and five TBI (black lines)-conditioned animals. (E) The top three graphs show the cumulative number of unique barcodes retrieved from PB lineages in busulfan-conditioned animals. The bottom graph directly compares the number of cumulative barcodes retrieved from PB Gr samples between busulfan (colored lines), and five TBI (black lines)-conditioned animals. (F) Normalized unique barcodes retrieved from PB Gr of three busulfan (colored lines) and five TBI (black lines)-conditioned animals over time. Normalization: unique barcode counts divided by the vector marking level (NE vector VCN) (busulfan) or % GFP marking level (TBI) from the same sample. (G) Normalized cumulative barcode number retrieved from the longest time point PB Gr (11021142 at 18 months, 10U004 at 22.5 months, and H84D at 14 months) from busulfan-conditioned animals and equivalent time points from TBI-conditioned animals (ZG66 at 17 months, ZH19 at 15.5 months, ZH33 at 23 months, ZJ31 at 20 months, and ZK22 at 15.5 months). Normalization: cumulative barcode number from the last time point divided by the NE vector VCN or % GFP marking from the last time point sample. Unpaired t test with p < 0.05 is considered as significant.
Pearson correlations can be used to determine the degree of shared clonal contributions between two samples,23,26,35 taking into account all clones, in contrast to the heatmap analyses focused on only the large contributing clones. In the first two animals, we observed two groups of samples correlated to each other due to the two waves of multipotent clones emerging over time. In H84D, the correlation between samples remains high starting from 3 months post-transplantation except the CD16+ NK population is less correlated with other lineages, which is the similar to TBI animal ZG66 (Figure S2). Therefore, the overall clonal patterns confirmed those seen with the largest clones.
Since the granulocyte population is short-lived and depends on active HSPC output, the clonal patterns in Gr reflect the active HSPCs pool. We applied autocorrelation analyses to Gr, calculating Spearman correlations between the clonal contributions of peripheral blood (PB) Gr at one time point to the previous time point for busulfan and TBI animals as another measure of clonal stability over time. In the TBI animals, the autocorrelation values became very stable at 5 to10 months, greater than 0.5 in all TBI animals (Figure 2B). However, in the busulfan animals, 11021042 autocorrelations remained lower than 0.5 until 30 months post-transplantation, and 10U004 until 15 months. Yet, the H84D autocorrelations were similar to the TBI animals by 5 months (Figure 2B).
Both the number of unique contributing clones (barcodes) at each time point in each lineage in animals 11021142 and 10U004 (Figure 2C), and the clonal Shannon diversity index (Figure 2D), which takes into account both clone number and distribution of contributions,35 dropped markedly post-transplantation, suggesting loss or absence of engrafting transduced true long-term repopulating HSPCs. The unique clone counts across all lineages in these first two animals over time were very low compared with the prior TBI animals (Figure 2C), and the total cumulative barcode counts in 11021142 and 10U004 are less than 2,500 clones (Figure 2E). In comparison, H84D, which received a 10% higher dose of busulfan, exhibited stable contributions from multilineage clones from 2 to 3 months to up to 18 months post-transplantation, the last follow-up time point (Figure 2A), and the clonal diversity remained high and stable after 6 months post-transplantation (Figure 2D). The total cumulative barcode counts in H84D reached 18,803 clones at 18 months (Figure 2E) and the unique clone number that could be retrieved at the last follow-up was 1,778 (Figure 2C), very similar to the TBI animals with similar overall levels of marking/VCN.
The vector marking levels of the lineage cells in our monkeys vary across lineages and individual monkeys. For more valid comparisons of clone numbers across different levels of engraftment with transduced cells, we normalized the unique and cumulative clone counts by the sample VCN (NE) in busulfan monkeys and the GFP+ marking level in the TBI monkeys. The normalized unique clone counts of Gr in the busulfan monkeys over time were somewhat lower compared with the TBI monkeys (Figure 2F). We compared the normalized cumulative clone counts of Gr at the last follow-up time point of busulfan monkeys (1102114 at 18 months, 10U004 at 22.5 months, and H84D at 14 months) and five TBI monkeys (ZG66 at 17 months, ZH19 at 15.5 months, ZH33 at 23 months, ZJ31 at 20 months, and ZK22 at 15.5 months), the results showed no statistical significance, indicating the busulfan conditioning overall was able to support a similar number of long-term engrafting HSPCs if similar vector marking levels in vivo were achieved compared with TBI condition animals (Figure 2G).
We did not have a surface marker to check the in vitro transduction efficiency for the NE vector, and the episomal virus and vector plasmid interfere with the integrated vector detection in our system. If we assume the NE lentivirus vector transduction efficiency was similar to the tNGFR in vitro transduction efficiency in vitro, we can estimate the frequency of long-term engrafting clones in H84D as 0.01% of CD34+ cells, calculated by dividing initial infused estimated transduced CD34+ cell number by the number of unique clones retrieved at the last follow-up, which is within the frequency range (0.0056%–0.014%) calculated in our barcoded macaques receiving TBI, matching the overall similar levels of vector-containing cells in H84D and the TBI animals.24 However, the estimated frequency of long-term engrafting clones in 11021142 (0.00011%) and 10U004 (0.00037%) was much lower compared with H84D and the TBI animals24 (Table 1), going along with the very low-level overall engraftment.
Early widespread geographic marrow clonal distribution in busulfan animals
We previously reported asymmetric HSPC distribution in different BM sites for up to 2 years in TBI-conditioned animals.25 For example, as shown in Figure 3, ZJ31 retains segregation of CD34+ HSPC clones between left and right BM sites as late as more than 2 years post-transplantation. We analyzed the clonal geographic distribution of BM HSPCs at several time points post-transplantation in the three busulfan-conditioned animals. There was much more rapid clonal mixing in the busulfan-conditioned animals. In 11021142, both heatmaps and Pearson correlations show mixing of left- and right-sided marrow CD34+ HSPCs clones as early as 3.5 months post-transplantation (Figures 3A and 3B). At 6 months in 11021142, when the wave of multilineage clones was established, CD34+ HSPCs clonal patterns on the left and right BM sites were similar, and also closely matched the PB Gr clonal pattern at the same time point. In 10U004, at 4.5 months, CD34+ HPSCs clonal mixing between right and left BM was incomplete (Figures 3A and 3B) but was fully mixed by 9 months. In H84D there was incomplete clonal segregation at 3.5 months, with more mixing at 7 to 9 months (Figures 3A and 3B). At 19 months, H84D CD34+ clones were fully equilibrated (Figures 3A and 3B). For more quantitative analysis of mixing, we calculated the fraction of CD34+ HSPC clones biased toward contributions to left versus right BM samples as shown in fold bias line plots (Figure 3C). These plots demonstrate highly biased clones persisting long term with very slow gradual mixing and movement toward unbiased clones in ZJ31 and other TBI animals, as previously reported.25 All three busulfan animals showed less biased contributions as early as 3.5 months post-transplantation (Figure 3C). Overall, these results demonstrate more rapid mixing across BM niches in animals conditioned with busulfan compared with the TBI-conditioned animals.
NK cell clonal patterns in busulfan-conditioned animals
In previous studies with TBI-conditioned animals, we observed markedly expanded CD56−CD16+ mature NK cell clones waxing and waning over time, independent of ongoing production from HSPCs, suggesting peripheral mature NK self-renewal (Figure 4A, representative TBI animals ZG66 and ZJ31).23,26,36 In contrast, the CD16+ NK clonal pattern following busulfan conditioning in monkeys 11021142 and 10U004 was more polyclonal and highly correlated with other lineages, including CD56+CD16− NK cells and Gr, once muti-lineage clonal output was established in monkeys 11021142 and 10U004 (Figure 2, Figure 4A, 4A, and 4B). However, in H84D, CD56−CD16+ NK-biased and expanded clones did emerge by 1-year post-transplantation (Figures 4A and 4B).
Figure 4.

Clonal patterns in NK cells
(A) Heatmaps depicting the contributions from the top 10 clones in CD16+ NK, CD56+ NK, and PB Gr samples over time post-transplantation for the three busulfan-conditioned animals and two TBI-conditioned animals (ZG66 and ZJ31). The heatmaps were generated as detailed as in Figure 2A. (B) Pairwise Pearson correlation coefficients between CD16+ NK, CD56+ NK, and PB Gr over time for each animal. The color scale for r values is shown on the right. (C) Total clonal contributions of the ≥10X biased and expanded CD16+ NK clones over time. A 10X biased clone was defined a top 30 contributing clone with a ≥10-fold expansion in fractional contribution to the CD16+ NK lineage compared with all other PB lineages analyzed (T, B, Mo, Gr, and CD56+) at the same time point. The three busulfan-conditioned animals and four TBI-conditioned animals with available samples (ZJ31, ZG66, ZH33, and ZK22) are shown. (D) Total clonal contributions of the 10X biased CD16+ NK clones at each time point post-engraftment comparing busulfan- and TBI-conditioned animals; 10X biased CD16+ NK clones defined as in (C) above. Unpaired t test applied with p < 0.05 considered as significant. (E) Number of 10X biased CD16+ NK clones contributing at each time point post-engraftment comparing busulfan- and TBI-conditioned animals; 10X biased CD16+ NK clones are defined as in (C) above. Unpaired t test applied with p < 0.05 considered as significant. (F) Normalized clonal contribution of each 10X biased CD16+ NK clone from busulfan and TBI-conditioned animals. Samples from different time points of individual busulfan-conditioned animals shown by animal or pooled together for comparison with the samples from four TBI-conditioned animals (ZJ31, ZG66, ZH33, and ZK22); 10X biased CD16+ NK clones are defined as in (C) above. Normalization: total clonal contributions of the 10X biased CD16+ NK clones from a sample divided by the clone numbers of the 10X biased CD16+ NK clones from the same sample. Unpaired t test applied with p < 0.05 considered as significant. (G) Ridge plot showing clonal bias between CD16+ NK and Gr lineages over time for busulfan- and TBI-conditioned animals. Ridges indicate the abundance-weighted density at the value of log-bias on the x axes, and dots indicate individual clones, sized by their overall abundance. Ridge plots stacked along the y axes correspond to the time point of each sample in months post-transplantation for each animal.
We defined biased CD16+ NK clones as large clones ≥10-fold over-represented in CD16+ NK compared with T, B, Mo, Gr, and CD56+ NK cells at the same time point. We plotted the 10X biased CD16+ NK clones’ cumulative clonal contributions as a fraction of the total barcoded CD16+ NK cells at each time point in busulfan and TBI animals. Overall, 10X biased CD16+ NK clonal contributions were lower (11021142 and 10U004) or in the lower range (H84D) compared with TBI monkeys (Figure 4C). Combining all post-engraftment time points, biased expanded clones contributed less to overall CD16+ NK cells in the three busulfan animals as compared with the TBI animals with available data (Figure 4D). The actual number of 10X biased CD16+ NK clones were also less in the busulfan monkeys compared with the TBI monkeys (Figure 4E), perhaps not surprisingly given the lower overall engraftment with barcoded cells in the busulfan animals. However, with normalization by dividing the 10X biased clones’ cumulative contributions from a specific sample by the 10X biased clone numbers retrieved from the same sample, the results showed overall there was no significant difference in each biased clone’s contribution between busulfan and TBI monkeys (Figure 4F). Still, H84D showed the most similar patterns to the TBI animals (Figures 4D and 4F).
To further investigate and visualize CD16+ NK clonal bias, we developed ridge plot visualizations. The log-bias of all clones from each sample was computed, with unbiased clones having a log-bias value near 0. Subsequently, the height of the ridges includes both the density of clones at each value of log-bias (x axis) as well as the abundance of those clones. The ridge height indicates the relative clonal contributions of the clones at the value of log-bias to both samples.35 We plotted CD16+ NK versus other lineages by using ridge plots (Figures 4G and S4). For comparison, we plotted TBI animals, ZG66 and ZJ31, which had exhibited massively expanded CD16+ NK-biased clones.26 We observed that in 10U004 after 2 months post-transplantation, most of the CD16+ NK clones have no bias compared with Gr or other lineages, with a balanced middle peak appearing in the ridge plots. However, in H84D, there was a slight clonal bias shift toward CD16+ NK compared with other lineages (Figures 4G and S4). Of note, the CD16+ NK clonal bias degree in H84D was much less than the degree of CD16+ NK clonal bias degree in the TBI animals ZJ31 and ZG66. We hypothesize that busulfan depletion of endogenous lymphocytes was much less marked than after TBI, producing a lower initial drive for NK clonal expansion, with more bias appearing in the animal receiving the higher busulfan dose.
We have previously linked CD16+ NK clonal bias and expansion to rhesus cytomegalovirus (RhCMV) infection or reactivation.36 All busulfan and TBI animals included in our comparisons were RhCMV seropositive. In contrast to the clear RhCMV reactivation post-TBI, we did not detect RhCMV DNA in serum or saliva via qPCR in 11021142 and 10U004 over time. Fewer than 200 copies per milliliter of RhCMV DNA were detected in saliva from H84D at two time points immediately before and 27 days post-transplantation only in saliva, and never in serum. Further investigation will be needed to assess the contribution of RhCMV infection in these animals.
Discussion
Busulfan conditioning offers a potentially less toxic alternative to TBI in preparing the BM niche for engraftment of genetically modified HSPCs. Several clinical studies have shown that busulfan is better tolerated in patients and results in improved survival outcomes in allogeneic HSPC transplantation,6,7,8 and busulfan or treosulfan have been used in almost all human HSPC autologous gene therapy clinical trials using conditioning prior to HSPC infusion to date.29,37
As a conditioning regimen, busulfan at high doses is primarily myeloablative but not profoundly lymphodepleting or immunosuppressive. A recent rhesus macaque report found that conditioning with busulfan alone permitted stable engraftment of HSPCs expressing a nonimmunogenic antigen, but did not facilitate engraftment of HSPCs expressing a highly immunogenic antigen such as GFP.16 Our previous studies of clonal dynamics in macaques following TBI conditioning included CopGFP in the vector as a marker gene.23,24,25,26,36 Due to the lack of immunosuppression afforded by busulfan, we investigated the use of an alternative surface protein, human tNGFR, which lacks an intracellular signaling domain and differs from endogenous rhesus macaque NGFR by only four amino acids.31,33,38,39 Taking the possibility of immune-mediated rejection of tNGFR-transduced cells into account, we also used a second vector containing no expressed (NE) marker gene. We did not observe efficient or persistent long-term engraftment of tNGFR-transduced HSPCs despite efficient transduction as assessed in vitro. In addition, we detected endogenous expression of NGFR in sorted lineage cells from PB sampled before HSPC transduction. Based on likely immune rejection of tNGFR-expressing cells, and the complications of using tNGFR as a marker gene given some background expression of endogenous NGFR, we focused our further analyses on the NE vector. Our results do raise concerns regarding use of busulfan alone for conditioning in gene therapy trials where patients have complete loss of the therapeutic gene product, and thus may reject cells expressing the protein following gene therapy.
In human clinical trials with myeloablative conditioning, busulfan resulted in polyclonal profiles of unique integration sites without detection of dominant clones through at least 12 months post-transplantation in almost all patients.14,15 Furthermore, Biasco and colleagues conducted a study investigating clonal dynamics in human Wiskott-Aldrich syndrome patients conditioned with busulfan (plus immunosuppressive fludarabine and Rituxan) via quantitative analysis of vector integration sites, finding multiple repopulating waves at the clonal leve.28 Following the exhaustion of short-term HSPC producing an initial wave of contributions from uni-lineage clones, they observed progressive appearance of long-term HSCs in several waves beginning around 6 months post-transplantation and then stabilizing and persisting for at least 3 years.
We have previously carried out detailed quantitative clonal mapping of hematopoiesis following TBI conditioning in the rhesus macaque model, and now compare busulfan conditioning with TBI in this model. In the current study, all three animals conditioned with busulfan also exhibited initial engraftment from short-term uni-lineage progenitors. In contrast to our prior experience with TBI conditioning, busulfan animals 11021142 and 10U004 both exhibited a wave of contributions from intermediate multilineage, but non-persisting HSPC clones appear between 2 and 9 months following transplantation, then replaced by a later wave of multilineage clones. This final wave of multilineage clones in 11021142 largely disappeared completely by 18 months but persisted in 10U004 to up to 26 months post-transplantation. This pattern is similar to that reported in a thalassemia gene therapy trial using moderate-intensity busulfan conditioning resulting in low overall engraftment.40 H84D, with much higher levels of engraftment with marked cells, exhibited a stable and persistent pattern of multilineage clones, similar to our previous studies in animals conditioned with TBI. The busulfan exposure was only 3% higher in this animal based on AUC, suggesting that the marked difference in engraftment in this animal resulted from other factors, including higher transduction efficiency and better maintenance of HSCs with transduction enhancers. In addition, there is limited prior data on busulfan dosing and sensitivity in macaques. In humans and macaques, other underlying genetic loci may affect busulfan sensitivity, in addition to differences in busulfan metabolism.
Our observations and human clinical trials to date suggest that lower dose intensity of busulfan may not clear sufficient niches able to support true LT-HSCs, but that higher intensity busulfan can create niches equivalent to TBI, resulting in similar engraftment patterns at a clonal level. Yet, it is important to note that we only had capacity to transplant one animal with the higher dose and given the heterogeneity between animals, more studies using a range of busulfan doses could be beneficial, as well as carefully comparing available data from clinical trials using different intensities of busulfan conditioning.
In contrast to previous studies with TBI-conditioned animals,25 the clonal geographic distribution of engrafted HSPCs in bone marrow was largely symmetrical early following transplantation, indicating rapid mixing of clones between left and right BM sites during hematopoietic reconstitution in animals conditioned with busulfan. With TBI conditioning, the clonal geographic distribution was segregated with little overlap between clones in left versus right BM sites up to years following transplantation, suggesting that HSPCs preferred to spread locally rather than traveling to different marrow niches via the circulation.41 In contrast, clonal contributions were correlated between both left and right BM sites and with circulating Gr as early as 3.5 months post-transplantation in all three animals following busulfan conditioning. These discrepancies are likely attributed to the more limited impact of busulfan on the marrow niche compared with irradiation, which exerts its potent nonspecific effects on the surrounding heterogeneous BM microenvironment.42,43 Further investigation into the unique effects of busulfan conditioning in the marrow niche should provide greater insight into hematopoietic recovery with implications to gene therapies with busulfan conditioning.
We previously reported evidence of long-term clonal expansion, persistence, and self-renewal of mature NK cells independently of progenitor cells in animals conditioned with TBI.23,24,26 Surprisingly, we observe less frequent and large oligoclonal expansions of NK cells in animals receiving busulfan. Instead, NK clonal contributions correlated with other lineages and demonstrated minimal evidence of NK-biased clones at later time points post-transplantation, reflecting a canonical model of NK hematopoiesis from BM precursors and CD56bright NK cells rather than peripheral expansion and persistence.44,45 It is possible that busulfan conditioning failed to achieve sufficient lymphodepletion to enable the competitive engraftment of newly derived mature NK cell population capable of peripheral expansion and maintenance; or due to the lower engraftment, low numbers of HSPC clones engrafted, thus it is possible that the expanded CD16+ NK clones were present but not marked. Supporting this hypothesis, in the third monkey H84D with higher barcoded HSPC engraftment, the results were more similar to TBI monkeys in terms of both the CD16+ NK-biased clone number and the cumulative contributions of biased clones. We recently reported clonal expansion of mature CD16+ NK cells driven by RhCMV infection.36 Animals that were RhCMV negative displayed less dominant and oligoclonal CD16+ NK-biased clones. In the current study, we did not detect significant levels of RhCMV DNA in serum or saliva samples with busulfan conditioning, in contrast to TBI conditioning, where RhCMV was universally reactivated. Future studies with additional lymphodepletion will be needed to determine whether these NK clonal patterns are a true feature of busulfan conditioning or related to responses toward environmental microbes.
In conclusion, clonal tracking of genetically barcoded HSPCs in rhesus macaques reveals distinct clonal patterns of hematopoietic reconstitution and variably impacts the marrow niche in animals conditioned with busulfan. In contrast to previous studies with TBI conditioning, we observed inefficient engraftment of true long-term repopulating HSPCs in two of three animals, a polyclonal NK clonal pattern correlating with other lineages, and a symmetrical clonal geographic distribution of BM HSPCs. These findings provide better understanding of clonal dynamics following busulfan conditioning, which is of great relevance to autologous HSPC gene therapies.
Materials and methods
Animal model
Animal studies were approved by the National Heart, Lung, and Blood Institute (NHLBI) Animal Care and Use Committee. PB CD34+ HSPCs from rhesus macaques were mobilized with G-CSF and plerixafor, collected via apheresis, and transduced with diverse barcoded lentiviral libraries as previously described23,24,26 (Figure 1A). For each rhesus macaque, purified CD34+ HSPCs were split into equal fractions, and each half transduced respectively with tNGFR (CD271, Figures S1A and S1B) or NE lentiviral vectors carrying high-diversity oligonucleotide barcode libraries, then cryopreserved following removal of aliquots for analysis of transduction efficiency. The in vitro transduction conditions are detailed in Table 1. Following conditioning with intravenous busulfan 5.5 mg/kg/d for 4 consecutive days (days −4 to −1) in animals 11021142 and 10U004, and 6.0 mg/kg/d for 4 consecutive days in H84D,16 each animal’s transduced aliquots of CD34+ cells were thawed, mixed together, and reinfused intravenously.
Busulfan pharmacokinetics
Plasma samples from rhesus macaques 11021142 and H84D were collected immediately before the first dose of busulfan administration, immediately following the 1 h busulfan intravenous infusion, and then serially at 0, 15, and 30 min, then 4, 6 ,and 24 h following the initial busulfan dose. Plasma samples were frozen and busulfan concentrations were performed by the Pharmacokinetics Laboratory at the Seattle Cancer Care Alliance. The maximum concentration (C max), the total AUC, drug half-life (t 1/2), and drug clearance parameters were calculated and are summarized in Table S4.
Cell lineage purifications
PB and BM cells were separated on a density gradient (Lymphocyte Separation Medium, GE Healthcare, cat# 17144002) to obtain a PB mononuclear cell (PBMC) layer and a granulocyte pellet, followed by red blood cell lysis with ACK lysis buffer (Quality Biological, cat# 118156101). PBMCs were stained for FACS to high purity on a BD FACSAria II instrument (antibodies listed in Table S2; gating strategy is detailed in Figure S3). CD34+ HSPCs were isolated from BM mononuclear cells using MACS MicroBeads (Miltenyi Biotec, cat# 130-042-201).
Barcode retrieval
Genomic DNA from purified hematopoietic cells was extracted with the DNeasy Kit (Qiagen). DNA (200–500 ng) underwent 28-cycle PCR using Phusion High-Fidelity DNA Polymerase (ThermoFisher Scientific) with forward and reverse primers. A universal reverse primer and a unique forward primer were used to multiplex samples for sequencing. After gel purification, 24 to 40 multiplexed samples were pooled for sequencing on an Illumina HiSeq 2500 or NovaSeq SP. PCR primers are listed in Table S3.
VCN determination
Quantitation of VCN was performed via ddPCR (Bio-Rad, CA, USA). Primers and probes were designed by Bio-Rad and the unique assay numbers were dCNS219891929 (rhesus TERT gene as internal control, Fluorophore: HEX) and dCNS749431138 (HIV RRE gene found in both vectors, Fluorophore: FAM). Droplets were generated via the QX100 Droplet Generator (Bio-Rad) and assayed on the QX200 droplet reader (Bio-Rad) via a standard protocol.46 Data were analyzed by QuantaSoft software 1.7 (Bio-Rad). RhCMV DNA copy numbers in plasma and saliva were determined via real-time qPCR as previously described.36
Data processing and analysis
Sequencing output files were processed using custom Python code to identify barcoded clones contributing above sequencing error and sampling thresholds.24,35 Data analysis, Pearson correlations, and plot generation were performed using R (Foundation for Statistical Computing) and Prism (GraphPad Software). Custom R code is available on GitHub at https://github.com/dunbarlabNIH/barcodetrackR35 and https://github.com/dunbarlabNIH/Busulfan_paper and the various analytical approaches and visualizations used were described in detail previously.35
Acknowledgments
The authors thank Nathaniel Linde, Theresa Engels, and Justin Golomb from the NHLBI Non-human Primate facility for excellent animal care. The authors also acknowledge the contributions from the NHLBI Flow Cytometry and DNA Sequencing and Genomics cores. All work was supported by the intramural research program of the National Heart, Lung, and Blood Institute.
Author contributions
Conceptualization: C.E.D., C.W., J.F.T. Methodology: D.M.A., R.J.L., X.G., J.A.L., R.D.M., D.A.E., X.F., J.F.T., C.W., C.E.D. Investigation: D.M.A., R.J.L., X.G., J.A.L., R.D.M., D.A.E., X.F., A.B., A.K., C.W., C.E.D. Formal analysis: C.E.D., C.W., D.M.A., J.A.L., R.J.L., K.S. Visualization: C.W., D.M.A., J.A.L. Supervision: C.E.D., C.W. Writing – original draft: R.J.L., C.W., D.M.A., C.E.D. Writing – review & editing: C.W., D.M.A., C.E.D.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.12.001.
Contributor Information
Chuanfeng Wu, Email: wuc3@mail.nih.gov.
Cynthia E. Dunbar, Email: dunbarc@nhlbi.nih.gov.
Supplemental information
Data availability statement
Barcode sequencing data used in this study are publicly available at https://github.com/dunbarlabNIH/Busulfan_paper.
References
- 1.Copelan E.A. Hematopoietic stem-cell transplantation. N. Engl. J. Med. 2006;354:1813–1826. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- 2.Shizuru J.A., Negrin R.S., Weissman I.L. Hematopoietic stem and progenitor cells: clinical and preclinical regeneration of the hematolymphoid system. Annu. Rev. Med. 2005;56:509–538. doi: 10.1146/annurev.med.54.101601.152334. [DOI] [PubMed] [Google Scholar]
- 3.Dunbar C.E., High K.A., Joung J.K., Kohn D.B., Ozawa K., Sadelain M. Gene therapy comes of age. Science. 2018;359:eaan4672. doi: 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- 4.Deeg H.J. Acute and delayed toxicities of total body irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1983;9:1933–1939. doi: 10.1016/0360-3016(83)90365-6. [DOI] [PubMed] [Google Scholar]
- 5.Garcia-Perez L., van Roon L., Schilham M.W., Lankester A.C., Pike-Overzet K., Staal F.J.T. Combining mobilizing agents with busulfan to reduce chemotherapy-based conditioning for hematopoietic stem cell transplantation. Cells. 2021;10:1077. doi: 10.3390/cells10051077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santos G.W., Tutschka P.J., Brookmeyer R., Saral R., Beschorner W.E., Bias W.B., Braine H.G., Burns W.H., Elfenbein G.J., Kaizer H., et al. Marrow transplantation for acute nonlymphocytic leukemia after treatment with busulfan and cyclophosphamide. N. Engl. J. Med. 1983;309:1347–1353. doi: 10.1056/NEJM198312013092202. [DOI] [PubMed] [Google Scholar]
- 7.Clift R.A., Buckner C.D., Thomas E.D., Bensinger W.I., Bowden R., Bryant E., Deeg H.J., Doney K.C., Fisher L.D., Hansen J.A., et al. Marrow transplantation for chronic myeloid leukemia: a randomized study comparing cyclophosphamide and total body irradiation with busulfan and cyclophosphamide. Blood. 1994;84:2036–2043. doi: 10.1182/blood.V84.6.2036.2036. [DOI] [PubMed] [Google Scholar]
- 8.Tang B., Zhu X., Zheng C., Liu H., Hao S., Huang D., Lin D., Li N., Gao S., Liang X., et al. Retrospective cohort study comparing the outcomes of intravenous busulfan vs. total-body irradiation after single cord blood transplantation. Bone Marrow Transplant. 2019;54:1614–1624. doi: 10.1038/s41409-019-0441-4. [DOI] [PubMed] [Google Scholar]
- 9.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.-S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 10.Drysdale C.M., Tisdale J.F., Uchida N. Immunoresponse to gene-modified hematopoietic stem cells. Mol. Ther. Methods Clin. Dev. 2020;16:42–49. doi: 10.1016/j.omtm.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aiuti A., Slavin S., Aker M., Ficara F., Deola S., Mortellaro A., Morecki S., Andolfi G., Tabucchi A., Carlucci F., et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 12.Aiuti A., Cattaneo F., Galimberti S., Benninghoff U., Cassani B., Callegaro L., Scaramuzza S., Andolfi G., Mirolo M., Brigida I., et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 13.Boztug K., Schmidt M., Schwarzer A., Banerjee P.P., Díez I.A., Dewey R.A., Böhm M., Nowrouzi A., Ball C.R., Glimm H., et al. Stem-cell gene therapy for the wiskott–aldrich syndrome. N. Engl. J. Med. 2010;363:1918–1927. doi: 10.1056/NEJMoa1003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ribeil J.-A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., El Nemer W., et al. Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 15.Thompson A.A., Walters M.C., Kwiatkowski J., Rasko J.E.J., Ribeil J.-A., Hongeng S., Magrin E., Schiller G.J., Payen E., Semeraro M., et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N. Engl. J. Med. 2018;378:1479–1493. doi: 10.1056/NEJMoa1705342. [DOI] [PubMed] [Google Scholar]
- 16.Uchida N., Nassehi T., Drysdale C.M., Gamer J., Yapundich M., Bonifacino A.C., Krouse A.E., Linde N., Hsieh M.M., Donahue R.E., et al. Busulfan combined with immunosuppression allows efficient engraftment of gene-modified cells in a rhesus macaque model. Mol. Ther. 2019;27:1586–1596. doi: 10.1016/j.ymthe.2019.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoggatt J., Kfoury Y., Scadden D.T. Hematopoietic stem cell niche in health and disease. Annu. Rev. Pathol. 2016;11:555–581. doi: 10.1146/annurev-pathol-012615-044414. [DOI] [PubMed] [Google Scholar]
- 18.Abbuehl J.P., Tatarova Z., Held W., Huelsken J. Long-term engraftment of primary bone marrow stromal cells repairs niche damage and improves hematopoietic stem cell transplantation. Cell Stem Cell. 2017;21:241–255.e6. doi: 10.1016/j.stem.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Halka K.G., Caro J., Erslev A.J. Long-term marrow cultures from mice with busulfan-induced chronic latent aplasia. J. Lab. Clin. Med. 1987;109:698–705. [PubMed] [Google Scholar]
- 20.Spyridonidis A., Küttler T., Wäsch R., Samek E., Waterhouse M., Behringer D., Bertz H., Finke J. Reduced intensity conditioning compared to standard conditioning preserves the in vitro growth capacity of bone marrow stroma, which remains of host origin. Stem Cells Dev. 2005;14:213–222. doi: 10.1089/scd.2005.14.213. [DOI] [PubMed] [Google Scholar]
- 21.Donahue R.E., Dunbar C.E. Update on the use of nonhuman primate models for preclinical testing of gene therapy approaches targeting hematopoietic cells. Hum. Gene Ther. 2001;12:607–617. doi: 10.1089/104303401300057289. [DOI] [PubMed] [Google Scholar]
- 22.Shepherd B.E., Kiem H.-P., Lansdorp P.M., Dunbar C.E., Aubert G., LaRochelle A., Seggewiss R., Guttorp P., Abkowitz J.L. Hematopoietic stem-cell behavior in nonhuman primates. Blood. 2007;110:1806–1813. doi: 10.1182/blood-2007-02-075382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu C., Li B., Lu R., Koelle S.J., Yang Y., Jares A., Krouse A.E., Metzger M., Liang F., Loré K., et al. Clonal tracking of rhesus macaque hematopoiesis highlights a distinct lineage origin for natural killer cells. Cell Stem Cell. 2014;14:486–499. doi: 10.1016/j.stem.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koelle S.J., Espinoza D.A., Wu C., Xu J., Lu R., Li B., Donahue R.E., Dunbar C.E. Quantitative stability of hematopoietic stem and progenitor cell clonal output in rhesus macaques receiving transplants. Blood. 2017;129:1448–1457. doi: 10.1182/blood-2016-07-728691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu C., Espinoza D.A., Koelle S.J., Potter E.L., Lu R., Li B., Yang D., Fan X., Donahue R.E., Roederer M., Dunbar C.E. Geographic clonal tracking in macaques provides insights into HSPC migration and differentiation. J. Exp. Med. 2018;215:217–232. doi: 10.1084/jem.20171341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu C., Espinoza D.A., Koelle S.J., Yang D., Truitt L., Schlums H., Lafont B.A., Davidson-Moncada J.K., Lu R., Kaur A., et al. Clonal expansion and compartmentalized maintenance of rhesus macaque NK cell subsets. Sci. Immunol. 2018;3:eaat9781. doi: 10.1126/sciimmunol.aat9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scala S., Leonardelli L., Biasco L. Current approaches and future perspectives for in vivo clonal tracking of hematopoietic cells. Curr. Gene Ther. 2016;16:184–193. doi: 10.2174/1566523216666160428104456. [DOI] [PubMed] [Google Scholar]
- 28.Biasco L., Pellin D., Scala S., Dionisio F., Basso-Ricci L., Leonardelli L., Scaramuzza S., Baricordi C., Ferrua F., Cicalese M.P., et al. In vivo tracking of human hematopoiesis reveals patterns of clonal dynamics during early and steady-state reconstitution phases. Cell Stem Cell. 2016;19:107–119. doi: 10.1016/j.stem.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Six E., Guilloux A., Denis A., Lecoules A., Magnani A., Vilette R., Male F., Cagnard N., Delville M., Magrin E., et al. Clonal tracking in gene therapy patients reveals a diversity of human hematopoietic differentiation programs. Blood. 2020;135:1219–1231. doi: 10.1182/blood.2019002350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonini C., Grez M., Traversari C., Ciceri F., Marktel S., Ferrari G., Dinauer M., Sadat M., Aiuti A., Deola S., et al. Safety of retroviral gene marking with a truncated NGF receptor. Nat. Med. 2003;9:367–369. doi: 10.1038/nm0403-367. [DOI] [PubMed] [Google Scholar]
- 31.Barese C.N., Krouse A.E., Metzger M.E., King C.A., Traversari C., Marini F.C., Donahue R.E., Dunbar C.E. Thymidine kinase suicide gene-mediated ganciclovir ablation of autologous gene-modified rhesus hematopoiesis. Mol. Ther. 2012;20:1932–1943. doi: 10.1038/mt.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poletto E., Colella P., Pimentel Vera L.N., Khan S., Tomatsu S., Baldo G., Gomez-Ospina N. Improved engraftment and therapeutic efficacy by human genome-edited hematopoietic stem cells with Busulfan-based myeloablation. Mol. Ther. Methods Clin. Dev. 2022;25:392–409. doi: 10.1016/j.omtm.2022.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deola S., Scaramuzza S., Birolo R.S., Carballido-Perrig N., Ficara F., Mocchetti C., Dando J., Carballido J.M., Bordignon C., Roncarolo M.G., et al. Mobilized blood CD34+ cells transduced and selected with a clinically applicable protocol reconstitute lymphopoiesis in SCID-hu mice. Hum. Gene Ther. 2004;15:305–311. doi: 10.1089/104303404322886156. [DOI] [PubMed] [Google Scholar]
- 34.Lu R., Neff N.F., Quake S.R., Weissman I.L. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat. Biotechnol. 2011;29:928–933. doi: 10.1038/nbt.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Espinoza D.A., Mortlock R.D., Koelle S.J., Wu C., Dunbar C.E. Interrogation of clonal tracking data using barcodetrackR. Nat. Comput. Sci. 2021;1:280–289. doi: 10.1038/s43588-021-00057-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Truitt L.L., Yang D., Espinoza D.A., Fan X., Ram D.R., Moström M.J., Tran D., Sprehe L.M., Reeves R.K., Donahue R.E., et al. Impact of CMV infection on natural killer cell clonal repertoire in CMV-naïve rhesus macaques. Front. Immunol. 2019;10:2381. doi: 10.3389/fimmu.2019.02381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cowan M.J., Dvorak C.C., Long-Boyle J. Opening marrow niches in patients undergoing autologous hematopoietic stem cell gene therapy. Hematol. Oncol. Clin. North Am. 2017;31:809–822. doi: 10.1016/j.hoc.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 38.Reddy U.R., Venkatakrishnan G., Maul G.G., Roy A.K., Ross A.H. Transient expression of full-length and truncated forms of the human nerve growth factor receptor. Brain Res. Mol. Brain Res. 1990;8:137–141. doi: 10.1016/0169-328X(90)90058-L. [DOI] [PubMed] [Google Scholar]
- 39.Johnson D., Lanahan A., Buck C.R., Sehgal A., Morgan C., Mercer E., Bothwell M., Chao M. Expression and structure of the human NGF receptor. Cell. 1986;47:545–554. doi: 10.1016/0092-8674(86)90619-7. [DOI] [PubMed] [Google Scholar]
- 40.Boulad F., Maggio A., Wang X., Moi P., Acuto S., Kogel F., Takpradit C., Prockop S., Mansilla-Soto J., Cabriolu A., et al. Lentiviral globin gene therapy with reduced-intensity conditioning in adults with beta-thalassemia: a phase 1 trial. Nat. Med. 2022;28:63–70. doi: 10.1038/s41591-021-01554-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Körbling M., Freireich E.J. Twenty-five years of peripheral blood stem cell transplantation. Blood. 2011;117:6411–6416. doi: 10.1182/blood-2010-12-322214. [DOI] [PubMed] [Google Scholar]
- 42.Morrison S.J., Scadden D.T. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–334. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilke C., Holtan S.G., Sharkey L., DeFor T., Arora M., Premakanthan P., Yohe S., Vagge S., Zhou D., Holter Chakrabarty J.L., et al. Marrow damage and hematopoietic recovery following allogeneic bone marrow transplantation for acute leukemias: effect of radiation dose and conditioning regimen. Radiother. Oncol. 2016;118:65–71. doi: 10.1016/j.radonc.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freud A.G., Becknell B., Roychowdhury S., Mao H.C., Ferketich A.K., Nuovo G.J., Hughes T.L., Marburger T.B., Sung J., Baiocchi R.A., et al. A human CD34(+) subset resides in lymph nodes and differentiates into CD56brightNatural killer cells. Immunity. 2005;22:295–304. doi: 10.1016/j.immuni.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 45.Freud A.G., Yokohama A., Becknell B., Lee M.T., Mao H.C., Ferketich A.K., Caligiuri M.A. Evidence for discrete stages of human natural killer cell differentiation in vivo. J. Exp. Med. 2006;203:1033–1043. doi: 10.1084/jem.20052507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hindson B.J., Ness K.D., Masquelier D.A., Belgrader P., Heredia N.J., Makarewicz A.J., Bright I.J., Lucero M.Y., Hiddessen A.L., Legler T.C., et al. High-Throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Barcode sequencing data used in this study are publicly available at https://github.com/dunbarlabNIH/Busulfan_paper.