

Abstract
Background and aim:
Acute exacerbation (AE) of idiopathic pulmonary fibrosis (IPF) is a fatal condition with no established treatment. Intravenous immunoglobulin (IVIG) is a unique therapy with both anti-inflammatory and anti-infective effects. Therefore, we hypothesized that IVIG may have a positive effect on AE of interstitial pneumonia. This study aimed to determine the effect of IVIG in patients with AE of fibrotic idiopathic interstitial pneumonias (IIPs), including IPF.
Methods:
We retrospectively analyzed consecutive patients who were diagnosed with AE of fibrotic IIPs and treated with pulse corticosteroid therapy (methylprednisolone 500–1000 mg/day for 3 days) between April 2018 and May 2021 at Kagawa Rosai Hospital and KKR Takamatsu Hospital.
Results:
This study included 52 patients with AE of fibrotic IIPs (IPF,41; fibrotic IIPs other than IPF,11). Thirteen patients received IVIG (5 g/day for 3–5 days) concurrently with pulse corticosteroid therapy. The remaining 39 patients were assigned to the control group. The survival rate on day 90 was significantly higher in the IVIG group than that in the control group (76.9% vs. 38.5%, p = 0.02). IVIG administration (odds ratio [OR], 0.11; 95% confidence interval [CI], 0.02–0.69; p = 0.02) and C- reactive protein (OR, 1.19; 95% CI, 1.06–1.33, p < 0.01) were independently associated with 90-day mortality.
Conclusions:
The results indicate that administration of IVIG may improve the survival of patients with AE of fibrotic IIPs. We are now conducting a prospective study to confirm the effect of IVIG on AE of IPF since May 2022 (jRCT1061220010).
Keywords: Intravenous immunoglobulin, Acute exacerbation, Idiopathic pulmonary fibrosis, Idiopathic interstitial pneumonia
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and fatal lung disease (1). Some patients with IPF show acute exacerbation (AE), defined as an acute, clinically significant respiratory deterioration characterized by evidence of new, widespread alveolar abnormality (2). AE is one of the leading causes of death in patients with IPF (3). However, AE is not unique to IPF. Previous studies reported that it also occurs in other types of interstitial pneumonia, such as idiopathic nonspecific interstitial pneumonia, interstitial pneumonia associated with collagen-vascular disease, and chronic hypersensitivity pneumonitis (4, 5). The majority of case series report the mortality rate of AE of IPF to be > 50%, and effective therapies have not yet been established (2). The efficacy of recombinant thrombomodulin and cyclophosphamide for AE of IPF was studied in a phase III trial, but these studies did not show an improvement in prognosis (6, 7). However, the pathophysiology of AE remains unclear. Inflammatory processes are believed to be one of the possible causes because an increased number of white blood cells and neutrophils and elevated serum levels of C-reactive protein (CRP) and inflammatory cytokines are observed in patients with AE (8, 9). Thus, corticosteroids or corticosteroids with immunosuppressive drugs are typically used to treat AEs (10, 11). In addition, some AEs are believed to be associated with infection; therefore, broad-spectrum antibiotics are typically initiated (12).
Intravenous immunoglobulin (IVIG) is a unique therapy with both anti-inflammatory and anti-infective effects (13-15). In Japan, IVIG is approved for the treatment of immune thrombocytopenia, Kawasaki disease, chronic inflammatory demyelinating polyneuropathy, Guillain–Barré syndrome, pemphigus, eosinophilic granulomatosis with polyangiitis, polymyositis/dermatomyositis, myasthenia gravis, multifocal motor neuropathy, optic neuritis, Stevens–Johnson syndrome, toxic epidermal necrolysis, hypogammaglobulinemia, agammaglobulinemia, and severe infections. Considering the possible mechanisms of AE as inflammation and infection, IVIG may provide a positive effect on AE of interstitial pneumonia. At Kagawa Rosai Hospital and KKR Takamatsu Hospital, we administered IVIG to some patients with AE of interstitial pneumonia, expecting these effects.
In this study we aimed to retrospectively analyze the effect of IVIG on patients with AE of fibrotic idiopathic interstitial pneumonias (IIPs).
Materials and methods
Study design and patients
This retrospective, observational study was conducted at Kagawa Rosai Hospital and KKR Takamatsu Hospital. We enrolled consecutive patients who were diagnosed with AE of fibrotic IIPs and treated with pulse corticosteroid therapy (methylprednisolone 500–1000 mg/day for 3 days) between April 2018 and May 2021. Fibrotic IIPs included IPF, idiopathic nonspecific interstitial pneumonia (iNSIP), and unclassifiable fibrotic IIPs. iNSIP and unclassifiable fibrotic IIPs were clinically diagnosed based on medical interviews regarding clinical data, physical examination, and computed tomography images due to inadequate pathological data. The following patients were excluded from the analysis to avoid bias caused by treatment and disease conditions: those who did not receive pulse dose of corticosteroid therapy for the treatment of AE, those with known causes of interstitial pneumonia such as collagen-vascular disease, and those with a history of AE. One patient who died of cardiovascular disease within 90 days of admission was also excluded. We collected the clinical data of the patients from their medical records. The diagnosis of IPF and IIPs other than IPF was based on established criteria (1, 16, 17). We diagnosed AE of interstitial pneumonia based on the International Working Group Report of Acute Exacerbation of Idiopathic Pulmonary Fibrosis (2). This study was approved by the institutional review boards of Kagawa Rosai Hospital (R3-25) and KKR Takamatsu Hospital (E222). The requirement for written informed consent was waived because this study was based on a retrospective analysis.
Statistical analysis
Statistical analyses were performed using the EZR software (Saitama Medical Center, Jichi Medical University, Saitama, Japan) (18). Patient characteristics were compared using the Fisher’s exact test for binary variables and Mann–Whitney U test for continuous variables. The survival rate on day 90 post admission was compared between the IVIG and control groups using Fisher’s exact test. Survival curves were plotted using the Kaplan–Meier method. Univariate and multivariate analyses with logistic regression models were performed to identify risk factors for mortality at 90 days post admission. We selected variables (Krebs-von-Lungen-6 [KL-6], CRP levels, and IVIG administration) for multivariate analysis based on previous reports (8, 19). The PaO2/FiO2 ratio was excluded from the variables owing to the large number of missing values (data were available for 42 of 52 patients). Differences were considered statistically significant at p < 0.05.
Results
Study population
This study included 52 patients with AE of fibrotic IIPs. Of the 52 patients, 41 had IPF and 11 had IIPs other than IPF (iNSIP,8; unclassifiable fibrotic IIPs,3). The median patient age was 79 years (interquartile range [IQR], 73–83 years), and 39 patients (75.0%) were men. Seven patients (13.5%) received antifibrotic drugs (pirfenidone or nintedanib) at the time of AE diagnosis. The median PaO2/FiO2 ratio was 192 (IQR, 108–263). Median KL-6 and CRP values were 1146 U/mL (IQR, 663–1625 U/mL) and 9.1 mg/dL (IQR, 4.5–14.3 mg/dL), respectively. Bronchoscopy was performed in only four patients because of severe respiratory failure. Microbiologic tests of bronchoalveolar lavage samples obtained from these patients were all negative. Sputum culture identified specific pathogen in only four cases (Klebsiella pneumoniae: two cases, methicillin-resistant Staphylococcus aureus: one case, Enterococcus faecalis: one case). There were no cases of acute exacerbation associated with severe acute respiratory syndrome coronavirus 2. Thirteen patients received IVIG (5 g/day for 3–5 days) concurrently with pulse corticosteroid therapy immediately after admission. The remaining 39 patients were assigned to the control group. A comparison of the baseline clinical characteristics between the IVIG and control groups is presented in Table 1.
Table 1.
Patient characteristics of IVIg group and control group on admission.
| Characteristics | IVIG group | Control group |
|---|---|---|
| (n = 13) | (n = 39) | |
| Age, yr — median (IQR) | 78 (72-82) | 79 (74-84) |
| Male sex — no. (%) | 8 (61.5) | 31 (79.5) |
| Body-mass index — median (IQR) | 23.3 (19.7-25.2) | 22.7 (21.5-25.1) |
| Smoking history | ||
| Never — no. (%) | 6 (46.2) | 7 (17.9)* |
| Former or current — no. (%) | 7 (53.8) | 31 (79.5)* |
| Clinical diagnosis | ||
| IPF — no. (%) | 12 (92.3) | 29 (74.4) |
| Other than IPF — no. (%) | 1 (7.7) | 10 (25.6) |
| Use of antifibrotic drugs | ||
| Pirfenidone — no. (%) | 1 (7.7) | 1 (2.5) |
| Nintedanib — no. (%) | 2 (15.4) | 3 (7.7) |
| PaO2/FiO2 ratio** — median (IQR) | 205 (114-298) | 189 (109-248) |
| Leukocytes, ×103/μl — median (IQR) | 11.7 (10.6-18.1) | 10.4 (7.6-14.0) |
| KL-6, U/ml — median (IQR) | 1380 (579-1734) | 1010 (664-1580) |
| LDH, U/l — median (IQR) | 332 (282-453) | 354 (279-385) |
| CRP, mg/dl — median (IQR) | 8.7 (6.2-15.7) | 9.4 (4.0-14.2) |
* Smoking history of one patient was not available.
** Data were available for 11 patients in the IVIG group and 31 in the control group.
Abbreviations: IVIG, Intravenous immunoglobulin; IPF, idiopathic pulmonary fibrosis; KL-6, Krebs-von-Lungen-6; LDH, lactate dehydrogenase; CRP, C-reactive protein
The baseline characteristics, including age, sex, body mass index, smoking history, clinical diagnosis, use of antifibrotic drugs, PaO2/FiO2 ratio, and laboratory data, were similar between the IVIG and control groups. Table 2 shows the therapeutic interventions for AE of fibrotic IIPs. All patients were treated with pulse corticosteroid therapy. Empiric antibiotic therapy was administered to all patients in the IVIG group and to 37 of 39 patients (94.9%) in the control group. The percentages of patients who used immunosuppressants, high-flow nasal cannulas, non-invasive ventilation, and invasive mechanical ventilation were similar between the two groups.
Table 2.
Therapeutic interventions for AE-IIPs.
| Characteristics | IVIG group | Control group |
|---|---|---|
| (n = 13) | (n = 39) | |
| Methylprednisolone pulse therapy — no. (%) | 13 (100) | 39 (100) |
| Empiric antibiotic therapy — no. (%) | 13 (100) | 37 (94.9) |
| Immunosuppressant — no. (%) | 5 (38.5) | 7 (17.9) |
| HFNC — no. (%) | 6 (46.2) | 12 (30.8) |
| NIV — no. (%) | 2 (15.4) | 13 (33.3) |
| IMV — no. (%) | 0 (0) | 2 (5.1) |
Abbreviations: IVIG, Intravenous immunoglobulin; HFNC, high-flow nasal cannulas; NIV, noninvasive ventilation; IMV, invasive mechanical ventilation
Effect of IVIG and predictors of survival
Figure 1 shows the survival curve for each group until day 90 post admission. The survival rate on day 90 was significantly higher in the IVIG group than that in the control group (76.9% vs. 38.5%, p = 0.02; Table 3). We performed univariate and multivariate analyses with logistic regression models to identify the risk factors for death up to 90 days post admission (Table 4). Univariate analysis revealed that IVIG administration (Odds ratio [OR], 0.19; 95% confidence interval [CI], 0.04–0.79; p = 0.02) and CRP (OR, 1.14; 95% CI, 1.03-1.25; p = 0.01) were associated with 90-days mortality. Multivariate analysis confirmed the independent association between IVIG administration (OR 0.11, 95% CI 0.02-0.69, p = 0.02) and CRP (OR 1.19, 95% CI 1.06-1.33, p < 0.01) and 90-days mortality.
Figure 1.
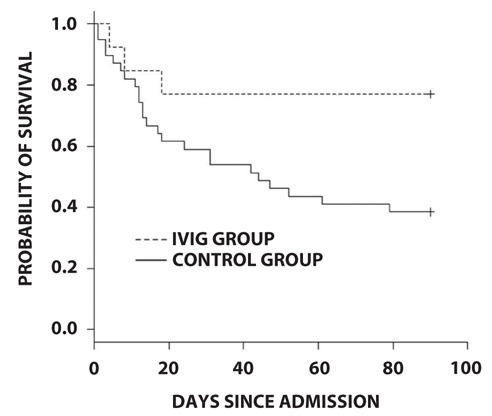
Survival curve for each group until day 90 post admission
Abbreviations: IVIG, Intravenous immunoglobulin.
Table 3.
Survival rate on day 90 post admission.
| Survival rate on day 90 | p value | |
|---|---|---|
| IVIG group | 76.9% (10/13) | 0.02 |
| Control group | 38.5% (15/39) |
Abbreviations: IVIG, Intravenous immunoglobulin
Table 4.
Risk factors of 90-days mortality.
| Variables | OR | 95% CI | p value |
|---|---|---|---|
| Univariate analysis | |||
| Age, yr | 1.05 | 0.98-1.12 | 0.20 |
| Male sex | 0.90 | 0.26-3.18 | 0.87 |
| Body-mass index | 0.92 | 0.81-1.06 | 0.25 |
| Clinical diagnosis IPF | 0.54 | 0.14-2.15 | 0.39 |
| Use of antifibrotic drugs | 1.28 | 0.26-6.36 | 0.77 |
| Leukocytes, ×103/μl | 1.00 | 1.00-1.00 | 0.44 |
| KL-6, U/ml | 1.00 | 1.00-1.00 | 0.26 |
| LDH, U/l | 1.00 | 1.00-1.00 | 0.70 |
| CRP, mg/dl | 1.14 | 1.03-1.25 | 0.01 |
| IVIG administration | 0.19 | 0.04-0.79 | 0.02 |
| Multivariate analysis | |||
| KL-6, U/ml | 1.00 | 1.00-1.00 | 0.89 |
| CRP, mg/dl | 1.19 | 1.06-1.33 | <0.01 |
| IVIG administration | 0.11 | 0.02-0.69 | 0.02 |
Abbreviations: KL-6, Krebs-von-Lungen-6; LDH, lactate dehydrogenase; CRP, C-reactive protein; IVIG, Intravenous immunoglobulin
Adverse Events
No adverse events were considered to be related to IVIG therapy during the study period.
Discussion
This study showed that the survival rate on day 90 post admission in the IVIG group was superior to that in the control group. Although this is the first study on the effect of IVIG on AE-IIPs or fibrotic IIPs, several studies have been conducted in other conditions for interstitial pneumonia. Enomoto et al. reported the efficacy of IVIG for IPF in its non-AE phase (20). They showed that the monthly administration of IVIG reduced the decline in vital capacity. Huapaya et al. suggested that IVIG is a potential salvage therapy in patients with active progressive interstitial lung disease associated with antisynthetase syndrome, who do not respond to the combination of steroids and immunosuppressant drugs (21). The study showed that IVIG increased forced vital capacity by more than 10% in approximately 40% of the patients. In addition, a preclinical study showed the efficacy of IVIG for bleomycin-induced pulmonary fibrosis (22). Although these studies were preliminary with small sample sizes, IVIG is considered to be an attractive candidate for lung fibrosis.
Respiratory infection is considered as one of the possible causes of AE of IPF (2, 8). In a previous study analyzing 52 autopsy cases with AE of IPF, 15 (28.8%) patients had respiratory infections, including fungal, cytomegalovirus, and bacterial infections (23). Another study showed that 14 of 43 patients (33%) with AE of IPF were positive for some viruses in the bronchoalveolar lavage fluid (24). As IVIG is a product prepared from pooled plasma of healthy individuals, it is enriched with a certain amount of immune antibodies against various bacteria, bacterial toxins, and viruses. The anti-infective effect of IVIG on AE of IIPs is one possible mechanism of action of the therapy.
The anti-inflammatory effect of IVIG is another possible mechanism of action of the therapy because excessive inflammation occurs in patients with AE. Increased numbers of white blood cells and neutrophils and elevated serum levels of CRP and inflammatory cytokines have been observed in patients with AE-IPF (8, 9). IVIG is believed to exert its anti-inflammatory effects through the following actions: suppression or neutralization of autoantibodies or cytokines, neutralization of activated complement components, restoration of idiotypic-anti-idiotypic networks, blockade of leukocyte-adhesion-molecule binding, modulation of maturation and function of dendritic cells, blockade of neonatal crystallizable fragment (Fc) receptor, blockade of activating receptor for the Fc portion of IgG (FcγR), upregulation of inhibitory FcγRIIB, and immunomodulation by sialylated immunoglobulin G (13). With regards to neutralization of autoantibodies, Donahoe et al. proposed a regimen of plasma exchange + rituximab + IVIG for AE of IPF, and they reported that improvements in pulmonary gas exchange after treatment were observed in 9 of 11 patients (25). Based on these promising results, a phase II study is currently underway in the United States to confirm the efficacy of this treatment strategy (STRIVE-IPF; NCT03286556). The anti-inflammatory effects of IVIG may improve the prognosis of AE-fibrotic IIPs in our study; however, it should be noted that all our patients received 5 g/day of IVIG in accordance with the approved dose for severe infections, whereas high-dose IVIG (more than 400 mg/kg/day) is usually administered for autoimmune disorders for their anti-inflammatory effects. Therefore, dose escalation may be necessary to maximize the anti-inflammatory effect of AE on fibrotic IIPs.
Regarding the safety of IVIG for IPF, Enomoto et al. reported that adverse events occurred in two of 10 patients with IPF (20). A mild rash and infectious pneumonia developed in one patient, and a mild, non-progressive retinal break was found in another. Although we administered relatively low-dose IVIG (5 g/day) compared to their study (400 mg/kg/day), no adverse events were considered to be related to IVIG during the study period. Considering these results, IVIG is safe and tolerable even in patients with interstitial pneumonia.
Our study has several limitations. First, it was retrospective, non-randomized and had a small sample size, particularly in the IVIG group. Second, the dose of IVIG in this study may not have been optimal because we used a relatively low dose of IVIG (5 g/day) in accordance with the approved dose for severe infections. Third, we do not routinely perform bronchoalveolar lavage at the time of AE diagnosis to exclude infection because of severe respiratory failure; hence, the possibility of infection could not be completely ruled out. Although our study has several limitations, we believe that it is significant in terms of proposing a new promising treatment for AE of fibrotic IIPs, for which there is no effective treatment.
Conclusion
Our study results indicate that the administration of IVIG may improve the survival of patients with AE of fibrotic IIPs. Based on these results, we are now conducting a prospective study to confirm the effect of IVIG on AE of IPF since May 2022 (jRCT1061220010).
Acknowledgements:
We would like to thank Edit age (www.editage.com) for English language editing.
Conflict of Interest:
The authors have no conflicts of interest to declare.
References
- 1.Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198:e44–e68. doi: 10.1164/rccm.201807-1255ST. [DOI] [PubMed] [Google Scholar]
- 2.Collard HR, Ryerson CJ, Corte TJ, et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am J Respir Crit Care Med. 2016;194:265–275. doi: 10.1164/rccm.201604-0801CI. [DOI] [PubMed] [Google Scholar]
- 3.Natsuizaka M, Chiba H, Kuronuma K, et al. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med. 2014;190:773–779. doi: 10.1164/rccm.201403-0566OC. [DOI] [PubMed] [Google Scholar]
- 4.Park IN, Kim DS, Shim TS, et al. Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis. Chest. 2007;132:214–220. doi: 10.1378/chest.07-0323. [DOI] [PubMed] [Google Scholar]
- 5.Miyazaki Y, Tateishi T, Akashi T, Ohtani Y, Inase N, Yoshizawa Y. Clinical predictors and histologic appearance of acute exacerbations in chronic hypersensitivity pneumonitis. Chest. 2008;134:1265–1270. doi: 10.1378/chest.08-0866. [DOI] [PubMed] [Google Scholar]
- 6.Kondoh Y, Azuma A, Inoue Y, et al. Thrombomodulin Alfa for Acute Exacerbation of Idiopathic Pulmonary Fibrosis. A Randomized, Double-Blind Placebo-controlled Trial. American Journal of Respiratory and Critical Care Medicine. 2020;201:1110–1119. doi: 10.1164/rccm.201909-1818OC. [DOI] [PubMed] [Google Scholar]
- 7.Naccache JM, Jouneau S, Didier M, et al. Cyclophosphamide added to glucocorticoids in acute exacerbation of idiopathic pulmonary fibrosis (EXAFIP): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. 2022;10:26–34. doi: 10.1016/S2213-2600(21)00354-4. [DOI] [PubMed] [Google Scholar]
- 8.Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37:356–363. doi: 10.1183/09031936.00159709. [DOI] [PubMed] [Google Scholar]
- 9.Oishi K, Mimura-Kimura Y, Miyasho T, et al. Association between cytokine removal by polymyxin B hemoperfusion and improved pulmonary oxygenation in patients with acute exacerbation of idiopathic pulmonary fibrosis. Cytokine. 2013;61:84–89. doi: 10.1016/j.cyto.2012.08.032. [DOI] [PubMed] [Google Scholar]
- 10.Arai T, Tachibana K, Sugimoto C, et al. High-dose prednisolone after intravenous methylprednisolone improves prognosis of acute exacerbation in idiopathic interstitial pneumonias. Respirology. 2017;22:1363–1370. doi: 10.1111/resp.13065. [DOI] [PubMed] [Google Scholar]
- 11.Homma S, Sakamoto S, Kawabata M, et al. Cyclosporin treatment in steroid-resistant and acutely exacerbated interstitial pneumonia. Intern Med. 2005;44:1144–1150. doi: 10.2169/internalmedicine.44.1144. [DOI] [PubMed] [Google Scholar]
- 12.Ding J, Chen Z, Feng K. Procalcitonin-guided antibiotic use in acute exacerbations of idiopathic pulmonary fibrosis. Int J Med Sci. 2013;10:903–907. doi: 10.7150/ijms.4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2012;367:2015–2025. doi: 10.1056/NEJMra1009433. [DOI] [PubMed] [Google Scholar]
- 14.Alejandria MM, Lansang MA, Dans LF, Mantaring JB., 3rd Intravenous immunoglobulin for treating sepsis, severe sepsis and septic shock. Cochrane Database Syst Rev. 2013:CD001090. doi: 10.1002/14651858.CD001090.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masaoka T. Combination therapy of antibiotics and intravenous immunoglobulin. Nihon Rinsho. 2001;59:781–784. [PubMed] [Google Scholar]
- 16.Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205:e18–e47. doi: 10.1164/rccm.202202-0399ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48:452–458. doi: 10.1038/bmt.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kishaba T, Tamaki H, Shimaoka Y, Fukuyama H, Yamashiro S. Staging of acute exacerbation in patients with idiopathic pulmonary fibrosis. Lung. 2014;192:141–149. doi: 10.1007/s00408-013-9530-0. [DOI] [PubMed] [Google Scholar]
- 20.Enomoto N, Chida K, Suda T, et al. An exploratory trial of intravenous immunoglobulin therapy for idiopathic pulmonary fibrosis: a preliminary multicenter report. Clinical Respiratory Journal. 2016;10:746–755. doi: 10.1111/crj.12281. [DOI] [PubMed] [Google Scholar]
- 21.Huapaya JA, Hallowell R, Silhan L, et al. Long-term treatment with human immunoglobulin for antisynthetase syndrome-associated interstitial lung disease. Respir Med. 2019;154:6–11. doi: 10.1016/j.rmed.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molina V, Haj-Yahia S, Solodeev I, Levy Y, Blank M, Shoenfeld Y. Immunomodulation of experimental pulmonary fibrosis by intravenous immunoglobulin (IVIG) Autoimmunity. 2006;39:711–717. doi: 10.1080/08916930601061272. [DOI] [PubMed] [Google Scholar]
- 23.Oda K, Ishimoto H, Yamada S, et al. Autopsy analyses in acute exacerbation of idiopathic pulmonary fibrosis. Respiratory Research. 2014:15. doi: 10.1186/s12931-014-0109-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wootton SC, Kim DS, Kondoh Y, et al. Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:1698–1702. doi: 10.1164/rccm.201010-1752OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donahoe M, Valentine VG, Chien N, et al. Autoantibody-Targeted Treatments for Acute Exacerbations of Idiopathic Pulmonary Fibrosis. PLoS One. 2015;10:e0127771. doi: 10.1371/journal.pone.0127771. [DOI] [PMC free article] [PubMed] [Google Scholar]