

Abstract

Digital PCR (dPCR) was first conceived for single-molecule quantitation. However, current dPCR systems often require DNA templates to share partitions due to limited partitioning capacities. Here, we introduce UltraPCR, a next-generation dPCR system where DNA counting is performed in a single-molecule regimen through a 6-log dynamic range using a swift and parallelized workflow. Each UltraPCR reaction is divided into >30 million partitions without microfluidics to achieve single template occupancy. Combined with a unique emulsion chemistry, partitions are optically clear, enabling the use of a three-dimensional imaging technique to rapidly detect DNA-positive partitions. Single-molecule occupancy also allows for more straightforward multiplex assay development due to the absence of partition-specific competition. As a proof of concept, we developed a 222-plex UltraPCR assay and demonstrated its potential use as a rapid, low-cost screening assay for noninvasive prenatal testing for as low as 4% trisomy fraction samples with high precision, accuracy, and reproducibility.
One of the most transformative methods for precise DNA quantification is digital PCR (dPCR), which originates from the concept of isolating single DNA molecules into individual compartments for amplification and detection.1−5 In early studies, DNA samples were diluted and quantified using PCR in a limited dilution protocol, which involved a highly laborious partitioning process.5,6 With advances in microfluidics, modern dPCR platforms have emerged that take advantage of various methods for DNA partitioning.7−9 Microfluidics-based partitioning systems not only simplify counting of DNA molecules but also improve the sensitivity of rare molecule detection and PCR signal amplitudes even in the presence of PCR inhibitors.10
Despite these advancements, current dPCR systems offer relatively low partition capacity (≤100 K) per sample due to physical limitations and manufacturing constraints.11,12 In many applications, DNA quantitation with these systems often occurs outside of the single-molecule domain, where >1 target molecules occupy the same partition (Figure 1). In this regime, multiple adaptations are required for a successful dPCR experiment. First, Poisson statistics is used to estimate the original DNA input. This necessitates the determination of both DNA-positive and DNA-negative partitions,11−13 which can increase the workflow time14−16 per sample and/or limit partition imaging capacity. Poisson correction has its limitations especially in high DNA occupancy regimes, as quantitative precision reduces exponentially until saturation of all partitions is achieved.12 Second, when multiple DNA templates are housed in the same partitions, partition-specific competition (PSC) can occur in multiplexing assays, where two or more amplicons compete for limited PCR reagents.12,13 To reduce the effect of PSC, multiplex dPCR assays require a more rigorous assay optimization process to balance multiplex PCR efficiencies.13 Altogether, current dPCR systems have found success in applications where the DNA dynamic range requirement is low, and assays can be standardized, such as in clinical workflows for cancer liquid biopsy and infectious diseases.14
Figure 1.
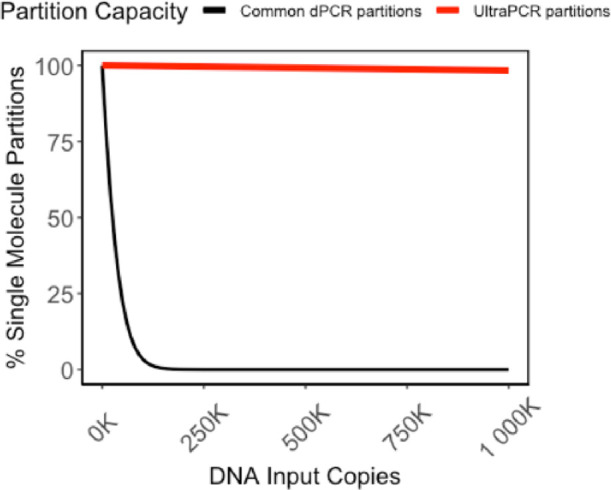
Single-molecule partition vs DNA input. Partitions with a single DNA molecule over all DNA-positive partitions are calculated based on Poisson statistics for a partition capacity of 20 K partitions common across commercially available dPCR platforms11 versus 30 million partitions in UltraPCR’s. Low partition systems fall out of the single-molecule domain rapidly and require Poisson correction to derive quantitative accuracy up to a threshold number of counts.20,21 In contrast, even at 1 million DNA input copies, UltraPCR still maintains the limited dilution regime, allowing for true single-molecule counting across a 6-log dynamic range.
The ideal dPCR system would involve a significantly greater number of partitions to reduce or eliminate partition saturation, thereby increasing the quantitative precision and dynamic range.12 Many high partition dPCR systems have had limited commercial success due to prohibitively high cost, low throughput, and long workflow time.17−19
Here, we have developed UltraPCR, a system for true single-molecule DNA counting with a 6-log dynamic range using a simple and cost-effective workflow without microfluidics. Every 50 μL sample is partitioned into >30 million droplets by centrifugation, enabling single-molecule occupancy via limited dilution, easily extending beyond 1 million DNA molecules (Figure 1). Using a unique formulation of emulsion reagents, UltraPCR emulsions are optically transparent, allowing for the use of a 3D imaging technique to rapidly scan each closed PCR tube for DNA-positive droplets. Furthermore, due to single-molecule partitioning, UltraPCR enables straightforward multiplexing, avoiding PSC and providing leniency in assay design aspects associated with balancing PCR efficiency across targets. As a proof of principle, we demonstrate the use of a 222-plex UltraPCR assay for counting loci of chromosomes 13, 18, and 21 for noninvasive fetal aneuploidy detection, paving the way to replace NGS and the microarray with a low-cost and rapid workflow in clinical labs.22−24
Experimental Section
UltraPCR Workflow
All UltraPCR reaction mixes were prepared using an UltraPCR TaqMan mix (Enumerix) according to the manufacturer’s protocol. For most reactions except for the volume titration experiment, 50 μL of the UltraPCR mix was added to an ultrapartitioning droplet generator (Enumerix), outfitted into PCR strip tubes carrying emulsifying reagents (Enumerix) as a strip assembly. The strip assembly was then fitted into a custom UltraPCR swing bucket (Enumerix) for use in an Eppendorf 5430 centrifuge installed with a swing bucket rotor. For each centrifugation, up to 12 strip assemblies (48 samples) were spun for 20 min at 16,000g to form UltraPCR emulsions. After centrifugation, the droplet generator was discarded, and the PCR tubes containing the emulsions were sealed and placed into a Bio-Rad C1000 thermal cycler for amplification. The same PCR strip tubes were then placed into an UltraPCR 3D Imager (Enumerix) for target-positive droplet scanning, where a laser light sheet was translated across the PCR tube and the illuminated partitions were imaged. For each sample, 500 frames of the emulsions were sampled and compiled to generate a 3D reconstruction of the PCR tube to count target-positive droplets using custom software written in C++ and MATLAB. Unless specified, all UltraPCR count data were normalized to 50 μL input.
Droplet Characterization
The droplet size was measured using bright-field microscopy in a hemocytometer. Briefly, an UltraPCR emulsion was generated by centrifuging a 50 μL PCR emulator mix (without a DNA template and dNTPs) directly into PCR strip tubes with a spin duration of 20 min at 16,000g. Droplets were aspirated and diluted to 1:15 with emulsion oil (Enumerix). Diluted droplets (10 μL) were pipetted into a disposable hemocytometer chip (Incyto DHC-N01) and imaged on a bright-field microscope (AmScope IN200TB). The droplet diameter was quantified with a custom MATLAB script. The packing efficiency of the emulsion was analyzed using 3D light sheet microscopy with Nile Red (Sigma)-stained emulsion oil.
DNA Template Generation
DNA templates for UltraPCR reactions were generated from either a synthesized DNA template for the gene prfA from Listeria monocytogenes or N1 and RPP30 plasmids provided by Integrated DNA Technologies for SARS-CoV-2 detection assay development. For each template generation assay, approximately 100,000 DNA templates were amplified with template generation primers for 16 cycles, using a hot start polymerase (New England Biolabs) for 16 cycles of 95 °C for 20 s, 55 °C for 60 s, and 68 °C for 60 s with a 10 min final extension at 68 °C followed by DNA purification using a DNA Clean & Concentrator-5 (Zymo Research). The DNA template concentration was determined by serially diluting the PCR products and quantified using the UltraPCR platform.
UltraPCR TaqMan Assay Conditions
Sequences of all primers and TaqMan probes, their final concentrations, and thermal cycling conditions used in all UltraPCR experiments in this study are provided in Tables S1 and S2.
UltraPCR Characterizations
For UltraPCR repeat scanning tests, a one-color prfA TaqMan assay was performed and 3D scanned twice immediately after thermal cycling (scans 1–2). The emulsion was then placed into cold storage between 2 and 8 °C overnight before proceeding with four scans (day 2, scans 3–6) and placed into cold storage again. On day 7, the emulsion was scanned again six times (scans 7–12) and placed into cold storage again. On day 209 (approximately 7 months), the emulsion was scanned five times (scans 13–17). prfA counts, droplet signal, and emulsion background values were compared to determine emulsion scanning stability over time.
For UltraPCR emulsion volume tests, a one-color RPP30 TaqMan assay mastermix was made with synthetic RPP30 templates with a target of 100,000 templates per 50 μL reaction, and the same mastermix was loaded into droplet generators at different volumes from 20 to 50 μL with four technical replicates per volume tested. RPP30 counts were normalized to 50 μL for analysis.
For the 6-log dynamic range test, a concentrated stock tube of the RPP30 template with ∼440k counts per μL was used to generate dilutions serially. An UltraPCR mastermix was made with multiplex RPP30 and N1 primers, spiked with ∼50k N1 copies per final PCR reaction to measure potential partition-specific competition effects. This PCR mastermix was aliquoted into 4.4× volumes to add the serially diluted RPP30 template DNA from 0 to ∼1.3 million copies to measure the UltraPCR dynamic range. Four technical replicates of each RPP30 dilution were used to measure platform precision while minimizing pipetting and other technical errors as much as possible.
Benchmarking against Conventional ddPCR
A Bio-Rad QX200 two-color ddPCR system was used according to the manufacturer’s instructions. All dPCR reactions used the ddPCR supermix for probes (Bio-Rad) according to the manufacturer’s instructions using the same DNA template serial dilutions, primers, and probes as were tested in UltraPCR. Annealing temperatures for RPP30 and SARS-CoV-2 N1 assays were optimized by performing using an annealing gradient in a two-step amplification protocol according to the manufacturer’s instructions. For example, in Figure 4, the amplification condition was 95 °C for 10 min followed by 40 cycles of 94 °C for 30 s, 54 °C for 1 min, and 98 °C for 10 min. Data analysis was performed using the manufacturer’s Quantasoft software (Bio-Rad), where droplet amplitude thresholds were manually determined based on droplet amplitudes displayed by nontemplate control (NTC) samples. In contrast, positive droplet counting by UltraPCR software (Enumerix) was performed independently for each sample.
Figure 4.

6-Log dynamic range on the UltraPCR platform. Demonstration of a 6-log dynamic range in serial dilution experiments, measuring DNA-positive partitions vs target RPP30 input copies. For each dilution, four technical replicates are performed, with total error (%CV) represented in the error bars.
Fetal Aneuploidy Assay Designs
The 222-plex NIPT-UltraPCR TaqMan assay panel (available for purchase from Enumerix) targeted 74 regions each on chromosomes 13, 18, and 21. The 74 regions on each chromosome shared a set of common 10-mer probe sequences, and each set of chromosome-specific probes was conjugated with the same fluorophore. The panel was developed based on in silico primer design followed by empirical optimization using UltraPCR. Primers were designed to target regions outside of common CNVs and SNPs and with minimal interactions with probes and other primers in the panel. Each individual primer pair was tested on genomic DNA samples to ensure that it produced the correct chromosomal ratios before combining into the final panel.
Fetal Aneuploidy Testing Sample Preparation
Contrived DNA samples were generated using nucleosomal DNA from GM12878 (Coriell) and Detroit 532 (ATCC). Nucleosomal DNA was isolated from ∼10 million cells using a Zymo Research EZ Nucleosomal DNA Prep kit and Atlantis dsDNase for nucleosomal DNA enrichment and DNA purification. Purified DNA was quantified using Agilent TapeStation D1000 high-sensitivity ScreenTape or cfDNA ScreenTape. The ploidy of the GM12878 and Detroit 532 was confirmed via NGS as follows: sequencing libraries were generated using a NEBNext Ultra II DNA Library Prep kit from Illumina (New England Biolabs), and libraries were sequenced on a MiSeq (Illumina) with 10% PhiX spike-in. NGS data were analyzed by mapping sequencing reads via CNVkit25 to determine aneuploidy status. Various mock trisomy 21 fetal fraction samples were generated by spiking Detroit 532 (trisomy 21) DNA into GM12878 to generate contrived DNA mixtures after quantifying the genome equivalent (GE) per μL using UltraPCR. Each contrived sample is normalized to ∼25 ng of DNA (3788 GEs) per μL, and 11 ng of DNA (1667 GEs) was loaded per sample for the analytical study of the fetal aneuploidy assay using UltraPCR.
For cell-free DNA (cfDNA), 10 mL of blood was collected in cell-free DNA BCT tubes (Streck) from healthy donors. Cell-free plasma was isolated using the Apostle MiniMax kit (Beckman Coulter) according to the manufacturer’s protocol with an elution volume of 60 μL. cfDNA was quantified using Agilent TapeStation with the cell-free DNA ScreenTape assay. For each cfDNA sample, 20 μL of DNA was loaded into each UltraPCR reaction.
Analytical Testing of the Fetal Aneuploidy Assay
The NIPT-UltraPCR assay was applied with contrived samples containing 0, 4, 6, and 10% trisomy 21 DNA using thermal cycling conditions outlined in Table S2. Technical replicates (18–20) were performed per contrived sample. The number of positive partitions for each fluorescent channel was counted, and the ratios of counts of chromosomes 21/13, 21/18, and 13/18 were calculated.
Results and Discussion
Single-Tube UltraPCR Closed Workflow
The UltraPCR workflow uses a single closed tube workflow per sample, as outlined in Figure 2. Partitioning is achieved via the centrifugation of the PCR mix through disposable droplet generator strip columns and directly into PCR strip tubes (Figure 2). UltraPCR also uses a unique formulation of reagents to form optically clear emulsions with near 100% packing efficiency, which contribute to minimal droplet coalescence (Figure 3A and Figure S1A) during physical movement of PCR strip tubes and thermocycling of emulsions. At this packing efficiency, ultrapartitions have no relative motion within the PCR tube and can be directly scanned inside the strip tubes after PCR via 3D light sheet microscopy in under a minute per channel (Figures 2 and 3B).
Figure 2.

UltraPCR workflow. UltraPCR uses disposable droplet generators to rapidly partition a PCR mix via centrifugation directly into PCR strip tubes. The same PCR strip tubes are loaded directly into a standard thermal cycler for target amplification and then transferred to the UltraPCR imager for 3D light sheet scanning of positive partitions.
Figure 3.

Ultrapartition characteristics. (A) Ultrapartitions settle in the bottom of the tube (shown in red brackets) and are optically clear for direct in-tube 3D imaging. (B) Slice of 3D light sheet scan of a sample with ∼500,000 counts of human RPP30 to showcase low DNA occupancy of the system.
UltraPCR samples can also be stored and rescanned repeatedly (Figure S1B). Throughout this single-tube workflow, the UltraPCR mix is contained within a closed system from the point of ultrapartitioning until imaging (Figure 2) with no exchange between samples, thereby significantly reducing the risk of sample-to-sample contamination.
Parallelized Ultrapartitioning in a Flexible Manner
UltraPCR’s ultrapartitions are ∼25 times smaller in volume than another study using centrifugation26 and at least 500 times smaller in volume than current commercially available dPCR systems.11,12,20 The average droplet diameter is 14.1 μm (Figure S2A), equivalent to 1.47 pL in volume. More than 30 million droplets are generated per 50 μL PCR mix from a 20 min spin using a standard benchtop centrifuge. UltraPCR’s droplet generation system is parallelized and has higher sample throughput, currently with up to 48 samples or >1.5 billion partitions per run.
The number of droplets generated is tunable, as the UltraPCR droplet generators also accommodate flexible assay volumes, like qPCR. Up to 50 μL of the PCR mix can be loaded (Figure S2B). For assays with concentrated DNA, a lower reaction volume (i.e., 20 μL for ∼12 million partitions) can be used to minimize cost of reagents. For assays with extremely diluted DNA samples (such as cfDNA), as much as 35 μL of the template in a 50 μL PCR volume has been successfully processed for downstream DNA quantification after cfDNA extraction. This high template–volume feature provides a useful alternative to many other dPCR systems with fixed volumes that are often too low for proper sampling of cfDNA.11,27,28
Ultrapartitioning with a 6-Log Dynamic Range
The partitioning capacity of UltraPCR enables single-molecule counting over a broad dynamic range. Even at 1 million DNA copies (or λ = 0.03), Poisson distribution calculates that 98.34% of the occupied partitions have only 1 DNA copy (Figure 1). As a proof of principle, we performed a serial dilution experiment of the synthetic template of the human gene RPP30 targeting a 6-log dynamic range, multiplexed with fixed synthetic SARS-CoV-2 N1 input, and counted only positive partitions in UltraPCR without the need for Poisson correction. We observed linearity up to 1.3 million positive RPP30, where 1 positive partition translates to 1 DNA copy, with a consistent ∼1% total error throughout this range (Figure 4), while N1 counts remain consistent as expected (Figure S2C). To our knowledge, UltraPCR has the highest dynamic range compared to any commercially available dPCR systems while uniquely operating in a single-molecule domain.
Orthogonal Testing for UltraPCR’s Linearity and Accuracy
Next, we performed orthogonal testing against a Bio-Rad QX-200 droplet dPCR (ddPCR). In this study, a synthetic SARS-CoV-2 N1 template was added to the PCR master mix at ∼2 K counts per sample prior to dividing the master mix into aliquots to add serially diluted synthetic RPP30 templates across a 5-log range from 0 to ∼200 K copies (denoted S1–S10) (Figure 5 and Tables S3 and S4). This multiplex assay was optimized by testing an annealing gradient on both ddPCR and UltraPCR. The N1 amplitude was highly sensitive to the annealing temperature in ddPCR only, with an inverse correlation between the annealing temperature and the N1 amplitude (Figure S3A,B), whereas UltraPCR’s N1 amplitude remained constant (Figure S3C) regardless of the annealing temperature. To achieve the best signal-to-noise separation in ddPCR, an annealing temperature of 54 °C was used for both systems for direct comparisons of RPP30 input count accuracy. Using this condition, both ddPCR and UltraPCR had similar N1 counts throughout the dilution points (Tables S3 and S4).
Figure 5.

Orthogonal testing between UltraPCR and ddPCR. Scatterplots of RPP30 counts where the same serially diluted sets of samples were tested on (A) ddPCR with the Poisson 95% confidence interval (CI) as error bars as indicated by the manufacturer and (B) UltraPCR with positive partitions normalized to 50 μL. Note that “NTC” is defined as 0 expected RPP30 input from serial dilution but contains N1 input.
First, we measured the dead volume for each system. In ddPCR, the median number of accepted droplets was 15,620 (Table S3), as compared to the total number of 23,529 droplets generated,12 equating to ∼33% sample loss. In UltraPCR, via 3D light sheet imaging, we measured an average flowthrough of ∼53 μL from a target 50 μL PCR mix input. This negligible dead volume suggests that centrifugal forces during partitioning allow nearly complete sample utilization. This feature, alongside with up to 35 μL DNA sample input per 50 μL reaction, allows UltraPCR to be particularly useful in applications that require maximal usage of the template volume for detecting rare molecules, such as in liquid biopsies.
Next, we compared the total RPP30 copies detected using comparable metrics. For UltraPCR, we reported the number of positive partitions from the entire emulsion, normalized to 50 μL. For ddPCR, we reported the dead-volume adjusted copies per 20 μL from the manufacturer’s software. In low RPP30 input samples with <50 K counts (S7 to S10), we observed similar RPP30 counts between the two platforms (Figure 5 and Tables S3 and S4), suggesting that the two platforms have comparable accuracy at this range.
At higher RPP30 inputs, such as beyond 100K counts (S1–S6), counts between ddPCR and UltraPCR began to diverge (Figure 5). Almost all droplets in ddPCR are saturated with DNA, harboring increasing Poisson uncertainty within each technical replicate as RPP30 input increases (Table S3 and Figure 5). The Poisson uncertainty percentages between dilution inputs overlapped, and within the same dilution sample, technical errors (such as from pipetting) were masked by Poisson errors12 (Table S3 and Figure 5). To derive quantitative accuracy in ddPCR at this high range, multiple sample wells are required to distribute DNA counts at a lower occupancy. On the other hand, UltraPCR’s single-molecule counting achieves low uncertainty and high resolution in DNA counting by having extremely high partition capacity.14,29 This is exemplified by easily separable RPP30 counts between tight serial dilutions while also maintaining low and consistent %CVs across this range (Table S4 and Figure 5).
The ability to maintain a high dynamic range and a high precision counting profile for UltraPCR has distinct advantages. First, for experiments involving DNA quantitation at a high dynamic range, such as gene expression analysis, users do not need to perform further sample dilutions to achieve the desirable precision range, making experimental design more straightforward and with fewer technical errors. Second, in dPCR systems that take advantage of Poisson correction to stretch the dynamic range,20 each positive partition (or negative partition) represents an increasing number of DNA copies within the sample as input increases and thereby reduces the ability to discern DNA count changes. On the other hand, since UltraPCR operates through a single-molecule regimen through a 6-log range, every DNA copy is represented proportionally by positive partition counting to achieve high-resolution, high-precision DNA quantification.
UltraPCR Eliminates Partition-Specific Competition (PSC)
Multiple DNA templates housed within the same partition may be subjected to PSC. This phenomenon occurs when multiple templates are present in the same partition and compete for PCR resources, such as primers, dNTPs, and polymerases, leading to reduction in fluorescence.12,13 During our orthogonal testing, we observed PSC in ddPCR. As RPP30 input increased, the amplitude of N1 decreased (Figure S4A,B). Analysis of the ddPCR 2D plot showed that RPP30 and N1 dual-positive droplets had a lower N1 amplitude compared to N1-only positive droplets (Figure S4C), likely due to unbalanced PCR efficiencies between the two targets (as demonstrated in the 60 °C annealing condition). The decrease in the N1 amplitude led to a drop-off of DNA counts (Figure S4A), lowering the sensitivity and accuracy of the assay. On the contrary, in UltraPCR, the N1 assay amplitude was unaffected with increasing RPP30 inputs (Figure S4D) in both 54 and 60 °C annealing temperature conditions.
The absence of PSC in UltraPCR is attributed to its large partitioning capacity and low DNA occupancy per partition (∼3% with 1 million occupied partitions), enabling virtually every DNA template to be partitioned, amplified, and detected independently from other targets. The elimination of PSCs in UltraPCR allows more straightforward multiplex assay optimization. Multiplex assay design detecting rare molecules in the backdrop of a highly expressed control gene, such as infectious disease and minimal residual disease applications, can be performed more directly without PSC diminishing rare molecule signals. Moreover, single template separation in UltraPCR can aid high-order multiplexing assay designs to achieve NGS-like precision DNA counting.
UltraPCR NIPT Assay
In addition to the elimination of PSCs, UltraPCR’s partition capacity also enables new applications beyond standard dPCR, such as NIPT. Fetal aneuploidy screening has become a common form of noninvasive prenatal testing for expectant mothers in the past decade.22 The most common method of fetal aneuploidy screening relies on counting chromosomal copies using technologies such as NGS or microarrays.30−33 These tests count copies from different chromosomes in maternal cell-free DNA. Since the fetal fraction in maternal cfDNA is low,14,23,34 technologies that enable high-order DNA counting, such as NGS, are used to achieve statistical confidence to detect aneuploidy in as low as 4% fetal fraction conditions.35−37 At this fetal fraction, these NIPT tests are required to detect chromosomal count differences of 2%. The estimated chromosomal count required to achieve this goal is >220,000 counts for reference chromosomes.24 The more molecules counted, the higher statistical confidence for samples with low fetal fractions. With the flexibility of a 6-log counting range coupled with high precision, we hypothesize that UltraPCR can provide a high-performance and cost-effective alternative to NGS for NIPT.
We first determined the average yield of a standard 10 mL blood draw from healthy donors to be ∼10 ng, which is ∼1500 genome equivalents (GEs). To satisfy the 220,000-count requirement for cfDNA samples, an assay of at least 48-plex per chromosome is required, which is an ∼5× higher requirement than that for published NIPT dPCR studies.38−41 We tackled this problem via rigorous overdesigning of our fetal aneuploidy panel to enable a 74-plex per chromosome, three-color assay for a total of 222-plex. In total, this is over 10× higher in multiplexing capability than any reported NIPT dPCR assays.38−41 In this NIPT-UltraPCR assay, each of the chromosomes Chr13, Chr18, and Chr21 is targeted with a set of optimized chromosome-specific probes, each set having a different fluorophore. The assay achieves a high signal amplitude and low background across various DNA inputs. We validated each of the 222-plex individually to ensure that they produced the expected counts to the expected chromosome with no cross-reaction to another chromosome. The total counts from the 74-plex per chromosome assay also correlated closely to the healthy donor cfDNA input, indicating that the assay is highly efficient (Figure S5A). We have also performed thermal cycler variability testing to ensure reproducible results in chromosomal ratios across three available thermal cyclers (Figure S5B).
We generated contrived DNA samples mimicking different percentages of trisomy 21 (%T21) at 0, 4, 6, and 10% in a background of euploid DNA. We tested them at a conservative target of ∼1667 GE or 11 ng input. A summary of this experiment is provided in Table 1. When considering UltraPCR absolute molecule counts, we observed 2.591 to 3.811% CV between technical replicates, which was likely due to technical errors between pipetting steps. However, when analyzing chromosome ratios Chr21/13, Chr21/18, and Chr13/18, we observed a much lower CV of 0.319 to 0.647% between replicates. Utilizing this three-color assay, we used both Chr13 and Chr18 as reference chromosomes to detect minute Chr21 elevation in the contrived samples. When analyzing Chr21/Chr13 and Chr21/Chr18 ratios, we observed a distinct shift in ratios as %T21 increased (Figure 6A,B), while the Chr13/18 ratio remained similar as expected (Figure 6C).
Table 1. NIPT-UltraPCR Result Summary.
| %trisomy 21 | metric | Chr13 counts | Chr21 counts | Chr18 counts | Chr21/13 ratio | Chr21/18 ratio | Chr13/18 ratio |
|---|---|---|---|---|---|---|---|
| 0 | mean | 272,589 | 284,675 | 270,602 | 1.044 | 1.052 | 1.007 |
| (%CV) | (2.798%) | (2.644%) | (2.696%) | (0.343%) | (0.377%) | (0.319%) | |
| 4 | mean | 276,641 | 293,851 | 273,524 | 1.062 | 1.074 | 1.011 |
| (%CV) | (3.070%) | (3.210%) | (3.089%) | (0.393%) | (0.378%) | (0.397%) | |
| 6 | mean | 262,783 | 281,465 | 259,506 | 1.071 | 1.085 | 1.013 |
| (%CV) | (3.630%) | (3.811%) | (3.777%) | (0.402%) | (0.532%) | (0.647%) | |
| 10 | mean | 266,260 | 289,984 | 263,157 | 1.089 | 1.102 | 1.012 |
| (%CV) | (2.591%) | (2.808%) | (2.692%) | (0.456%) | (0.403%) | (0.359%) |
Figure 6.

UltraPCR NIPT assay analytical study. (A–C) Boxplot of different chromosomal ratios. Chr21/18 and Chr21/13 are expected to increase with more %T21 spiked in, and Chr13/18 is expected to remain constant. (D) ROC curve using all spiked %T21 samples compared to 0% to determine the optimal threshold to separate control versus T21-spiked samples. (E) Accuracy plot vs cutoff where the blue line represents data using Chr13 as a reference chromosome and the orange line represents data using Chr18 as a reference chromosome. Dotted lines represent their respective optimal cutoff to maximize accuracy of the assay.
Next, we performed receiver operating characteristic (ROC) analysis to identify the optimal threshold to different T21-spiked versus 0% T21 contrived samples. Unlike previous NIPT dPCR attempts with only one reference chromosome via a two-color test,6,19,20 we evaluated trisomy cells using two reference chromosomes. Through this analysis, both Chr21/13 and Chr21/18 ratio analysis yielded 100% sensitivity, specificity, and accuracy across all tested %T21 conditions (cutoff values were 1.054 for Chr21/Chr13 and 1.066 for Chr21/Chr18, Figure 6D,E). To our knowledge, this is the first published dataset to distinguish a DNA mixture containing 4% T21 using dPCR without sample enrichment technologies or preamplification steps.
dPCR has long been touted as a method of determining fetal aneuploidy, yet it has not yet been widely adopted clinically due to technical limitations.8,23,39−41 First, no commercially available dPCR system can meet the precision requirement due to low partitioning capacity. Multiple groups have tried splitting a sample into multiple sample wells to add more partitions39 or perform a series of fetal cfDNA pre-enrichment of fetal cfDNA,41 adding cost and complexity to the workflow. Splitting a sample across multiple wells or pre-enrichment of fetal cfDNA can also introduce additional errors that may skew true chromosomal ratios. More recently, a group has added more virtual partitions within the color space using a limited primer approach combined with complex analysis techniques to discern >5% T21 samples.38 In contrast, even in its proof-of-concept phase, UltraPCR already provides the quantitative precision to differentiate chromosomal count differences as low as 2%. This precision extends beyond NIPT and can be applied to discern minute gene expression changes that qPCR and dPCR cannot currently achieve.
Second, no non-NGS or nonmicroarray assay has sufficiently high multiplexing capabilities to interrogate enough genomic loci to achieve the counting requirement using limited cell-free DNA available from a typical blood draw volume (∼10 mL of whole blood). Unlike NGS where bioinformatics pipelines can remove nonspecific sequencing reads, dPCR assays must be developed at a high rigor to have low false-positive and false-negative signals for proper droplet counting. Part of this achievement with our 222-plex assay is due to UltraPCR’s single-molecule amplification regimen with no interference from other templates. In contrast, with low-partitioning systems, as DNA occupancy (or λ) increases, PSC becomes a larger problem, requiring arduous assay development work to obtain enough DNA counts to achieve the precision required for NIPT. This may explain why all published NIPT-dPCR assays utilize designs with low multiplexing capability,38−41 requiring a much higher DNA input, or pre-enriched amplicons18 than what is commonly extracted from peripheral blood. Altogether, UltraPCR’s unique ultrapartitioning method enables unprecedented multiplexing capability with high precision.
Conclusions
UltraPCR is a novel platform that reimagines DNA counting. With >30 million partitions, DNA molecules are partitioned into a single-molecule domain through a 6-log dynamic range (Figure 1), providing high-resolution DNA counting beyond current dPCR capabilities. Every positive partition translates to one DNA copy, enabling straightforward data interpretation and high precision data without error correction. In addition, when single DNA templates are isolated for amplification, there is no PSC, easing the need to perform rigorous assay design to balance PCR efficiency for multiplexing.
In addition to standard dPCR applications, UltraPCR can be extended to research areas where a high dynamic range and/or high precision counting is required. One such example is in RNA expression where the dynamic range is orders of magnitudes larger than DNA and can vary rapidly through transcriptional and post-transcriptional regulation. In these applications, dPCR is not commonly used; instead, users opt for a workflow where RNA markers are identified using NGS and experimentally verified and validated using qPCR. While qPCR is simple with a high dynamic range, it comes with its limitations. qPCR requires a standard curve or housekeeping gene for DNA quantification, and because the signal is quantified after every PCR cycle, the precision is only two-fold. Increasing qPCR resolution is possible and is supported by a study where as many as 18 qPCR replicates are required to discern a 25% difference in DNA counts.42 However, this type of qPCR experimental design is costly, laborious, consuming a lot of the DNA sample, and generally not practical. On the other hand, UltraPCR offers a similar dynamic range in a simple workflow and can discern a DNA count difference as low as 2% as demonstrated in our NIPT analytical study (Figure 6). With such a high resolution and a precision profile that rivals NGS but at a fraction of the cost, UltraPCR can be positioned as a powerful and practical validation partner tool for biomarker research.
Beyond the research setting, UltraPCR provides a swift workflow in high-precision clinical assays. As a proof of concept, we developed an industry-leading 222-plex NIPT assay for fetal aneuploidy. This NIPT assay can likely be further optimized to include additional chromosomes and genes to detect sex aneuploidies and single-gene disorders. While this assay is still at its early stages, the process of UltraPCR is simple and can enable a potential single-day workflow at dPCR-like cost without the need for heavy computational biology support, paving the way for NIPT to be decentralized to hospital labs and beyond first-world countries.14
Acknowledgments
We thank Mr. Ari Chaney, Ms. Kat George, Mr. Ivan Wong, Ms. Camille Barker, and Mr. Billy Chan for their support of this research.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c03649.
Ultrapartitions, UltraPCR emulsion and dynamic range characterization, assay optimization, and additional data (PDF)
Author Contributions
‡ E.Y.S. and J.H.L. contributed equally. E.Y.S., J.H.L., S.L., S.P.A.F., and H.C.F. conceptualized, designed, and developed the UltraPCR technology and research. H.G.L., J.S., V.K.R., and C.K.L. performed the research. E.Y.S., J.H.L., S.L., H.G.L., and J.S. analyzed the data. E.Y.S. wrote the manuscript.
The authors declare the following competing financial interest(s): E.Y.S., J.H.L., S.L., H.G.L., J.S., V.K.R., C.K.L., S.P.A.F., H.C.F. are employees of Enumerix, Inc., a company that commercializes DNA counting technologies. Enumerix, Inc., solely funded this study. In addition, E.Y.S., J.H.L., S.L., S.P.A.F., H.C.F. are inventors on patents and patent applications relating to methods described in this manuscript.
Supplementary Material
References
- Saiki R. K.; Gelfand D. H.; Stoffel S.; Scharf S. J.; Higuchi R.; Horn G. T.; Mullis K. B.; Erlich H. A. Primer-Directed Enzymatic Amplification of DNA with a Thermostable DNA Polymerase. Science 1988, 239, 487–491. 10.1126/SCIENCE.2448875. [DOI] [PubMed] [Google Scholar]
- Jeffreys A. J.; Neumann R.; Wilson V. Repeat Unit Sequence Variation in Minisatellites: A Novel Source of DNA Polymorphism for Studying Variation and Mutation by Single Molecule Analysis. Cell 1990, 60, 473–485. 10.1016/0092-8674(90)90598-9. [DOI] [PubMed] [Google Scholar]
- Ruano G.; Kidd K. K.; Stephens J. C. Haplotype of Multiple Polymorphisms Resolved by Enzymatic Amplification of Single DNA Molecules. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 6296–6300. 10.1073/PNAS.87.16.6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P.; Balfe P.; Peutherer J. F.; Ludlam C. A.; Bishop J. O.; Brown A. J. Human Immunodeficiency Virus-Infected Individuals Contain Provirus in Small Numbers of Peripheral Mononuclear Cells and at Low Copy Numbers. J. Virol. 1990, 64, 864–872. 10.1128/JVI.64.2.864-872.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B.; Kinzler K. W. Digital PCR. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9236–9241. 10.1073/PNAS.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes P. J.; Neoh S. H.; Brisco M. J.; Hughes E.; Condon J.; Morley A. A. Quantitation of Targets for PCR by Use of Limiting Dilution. BioTechniques 1992, 13, 444–449. [PubMed] [Google Scholar]
- Warren L.; Bryder D.; Weissman I. L.; Quake S. R. Transcription Factor Profiling in Individual Hematopoietic Progenitors by Digital RT-PCR. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 17807–17812. 10.1073/PNAS.0608512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H. C.; Quake S. R. Detection of Aneuploidy with Digital Polymerase Chain Reaction. Anal. Chem. 2007, 79, 7576–7579. 10.1021/ac0709394. [DOI] [PubMed] [Google Scholar]
- Ottesen E. A.; Hong J. W.; Quake S. R.; Science J. L. Microfluidic Digital PCR Enables Multigene Analysis of Individual Environmental Bacteria. Science 2006, 314, 1464. 10.1126/science.1131370. [DOI] [PubMed] [Google Scholar]
- Dingle T. C.; Sedlak R. H.; Cook L.; Jerome K. R. Tolerance of Droplet-Digital PCR vs Real-Time Quantitative PCR to Inhibitory Substances. Clin. Chem. 2013, 59, 1670. 10.1373/CLINCHEM.2013.211045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A. S. Digital Assays Part I: Partitioning Statistics and Digital PCR. SLAS Technol. 2017, 22, 369–386. 10.1177/2472630317705680. [DOI] [PubMed] [Google Scholar]
- Whale A. S.; de Spiegelaere W.; Trypsteen W.; Nour A. A.; Bae Y.-K.; Benes V.; Burke D.; Cleveland M.; Corbisier P.; Devonshire A. S.; Dong L.; Drandi D.; Foy C. A.; Garson J. A.; He H.-J.; Hellemans J.; Kubista M.; Lievens A.; Makrigiorgos M. G.; Milavec M.; Mueller R. D.; Nolan T.; O’Sullivan D. M.; Pfaffl M. W.; Rödiger S.; Romsos E. L.; Shipley G. L.; Taly V.; Untergasser A.; Wittwer C. T.; Bustin S. A.; Vandesompele J.; Huggett J. F. The Digital MIQE Guidelines Update: Minimum Information for Publication of Quantitative Digital PCR Experiments for 2020. Clin. Chem. 2020, 66, 1012–1029. 10.1093/CLINCHEM/HVAA125. [DOI] [PubMed] [Google Scholar]
- Whale A. S.; Huggett J. F.; Tzonev S. Fundamentals of Multiplexing with Digital PCR. Biomol. Detect. Quantif. 2016, 10, 15–23. 10.1016/J.BDQ.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nectoux J. Current, Emerging, and Future Applications of Digital PCR in Non-Invasive Prenatal Diagnosis. Mol. Diagn. Ther. 2018, 22, 139–148. 10.1007/S40291-017-0312-X. [DOI] [PubMed] [Google Scholar]
- Huggett J. F.; Cowen S.; Foy C. A. Considerations for Digital PCR as an Accurate Molecular Diagnostic Tool. Clin. Chem. 2015, 61, 79–88. 10.1373/CLINCHEM.2014.221366. [DOI] [PubMed] [Google Scholar]
- Jacobs B. K. M.; Goetghebeur E.; Clement L. Impact of Variance Components on Reliability Ofl Absolute Quantification Using Digital PCR. BMC Bioinf. 2014, 15, 1–13. 10.1186/1471-2105-15-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G. M.; Busby E.; Garson J. A.; Grant P. R.; Nastouli E.; Devonshire A. S.; Whale A. S. Digital PCR Dynamic Range Is Approaching That of Real-Time Quantitative PCR. Biomol. Detect. Quantif. 2016, 10, 31–33. 10.1016/J.BDQ.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyries K. A.; Tropini C.; Vaninsberghe M.; Doolin C.; Petriv O. I.; Singhal A.; Leung K.; Hughesman C. B.; Hansen C. L. Megapixel Digital PCR. Nat. Methods 2011, 8, 649–651. 10.1038/NMETH.1640. [DOI] [PubMed] [Google Scholar]
- Hatch A. C.; Fisher J. S.; Tovar A. R.; Hsieh A. T.; Lin R.; Pentoney S. L.; Yang D. L.; Lee A. P. 1-Million Droplet Array with Wide-Field Fluorescence Imaging for Digital PCR. Lab Chip 2011, 11, 3838–3845. 10.1039/C1LC20561G. [DOI] [PubMed] [Google Scholar]
- Hindson B. J.; Ness K. D.; Masquelier D. A.; Belgrader P.; Heredia N. J.; Makarewicz A. J.; Bright I. J.; Lucero M. Y.; Hiddessen A. L.; Legler T. C.; Kitano T. K.; Hodel M. R.; Petersen J. F.; Wyatt P. W.; Steenblock E. R.; Shah P. H.; Bousse L. J.; Troup C. B.; Mellen J. C.; Wittmann D. K.; Erndt N. G.; Cauley T. H.; Koehler R. T.; So A. P.; Dube S.; Rose K. A.; Montesclaros L.; Wang S.; Stumbo D. P.; Hodges S. P.; Romine S.; Milanovich F. P.; White H. E.; Regan J. F.; Karlin-Neumann G. A.; Hindson C. M.; Saxonov S.; Colston B. W. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzonev S. Fundamentals of Counting Statistics in Digital PCR: I Just Measured Two Target Copies–What Does It Mean?. Methods Mol. Biol. 2018, 1768, 25–43. 10.1007/978-1-4939-7778-9_3. [DOI] [PubMed] [Google Scholar]
- Benn P.; Cuckle H.; Pergament E. Non-Invasive Prenatal Testing for Aneuploidy: Current Status and Future Prospects. Ultrasound Obstet. Gynecol. 2013, 42, 15–33. 10.1002/UOG.12513. [DOI] [PubMed] [Google Scholar]
- el Khattabi L. A.; Rouillac-Le Sciellour C.; le Tessier D.; Luscan A.; Coustier A.; Porcher R.; Bhouri R.; Nectoux J.; Sérazin V.; Quibel T.; Mandelbrot L.; Tsatsaris V.; Vialard F.; Dupont J. M. Could Digital PCR Be an Alternative as a Non-Invasive Prenatal Test for Trisomy 21: A Proof of Concept Study. PLoS One 2016, 11, e0155009 10.1371/JOURNAL.PONE.0155009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M. I.; Wright D. A.; Pergament E.; Cuckle H. S.; Nicolaides K. H. Digital PCR for Noninvasive Detection of Aneuploidy: Power Analysis Equations for Feasibility. Fetal. Diagn. Ther. 2012, 31, 244–247. 10.1159/000337544. [DOI] [PubMed] [Google Scholar]
- Talevich E.; Shain A. H.; Botton T.; Bastian B. C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873 10.1371/JOURNAL.PCBI.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao P.; Jiang M.; Chen Z.; Zhang F.; Sun Y.; Nie J.; Du M.; Wang J.; Fei P.; Huang Y. Three-Dimensional Digital PCR through Light-Sheet Imaging of Optically Cleared Emulsion. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 25628–25633. 10.1073/pnas.2002448117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueck M. E.; Lin R.; Zayac A.; Gallagher S.; Chao A. K.; Jiang L.; Datwani S. S.; Hung P.; Stieglitz E. Precision Cancer Monitoring Using a Novel, Fully Integrated, Microfluidic Array Partitioning Digital PCR Platform. Sci. Rep. 2019, 9, 1–9. 10.1038/s41598-019-55872-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devonshire A. S.; O’Sullivan D. M.; Honeyborne I.; Jones G.; Karczmarczyk M.; Pavšič J.; Gutteridge A.; Milavec M.; Mendoza P.; Schimmel H.; van Heuverswyn F.; Gorton R.; Cirillo D. M.; Borroni E.; Harris K.; Barnard M.; Heydenrych A.; Ndusilo N.; Wallis C. L.; Pillay K.; Barry T.; Reddington K.; Richter E.; Mozioğlu E.; Akyürek S.; Yalçinkaya B.; Akgoz M.; Žel J.; Foy C. A.; McHugh T. D.; Huggett J. F. The Use of Digital PCR to Improve the Application of Quantitative Molecular Diagnostic Methods for Tuberculosis. BMC Infect. Dis. 2016, 16, 336. 10.1186/S12879-016-1696-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube S.; Qin J.; Ramakrishnan R. Mathematical Analysis of Copy Number Variation in a DNA Sample Using Digital PCR on a Nanofluidic Device. PLoS One 2008, 3, e2876 10.1371/JOURNAL.PONE.0002876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi D. W.; Platt L. D.; Goldberg J. D.; Abuhamad A. Z.; Sehnert A. J.; Rava R. P.; Genome-Wide Fetal Aneuploidy Detection by Maternal Plasma DNA Sequencing. Obstet. Gynecol. 2012, 119, 890–901. 10.1097/aog.0b013e31824fb482. [DOI] [PubMed] [Google Scholar]
- Fan H. C.; Blumenfeld Y. J.; Chitkara U.; Hudgins L.; Quake S. R. Noninvasive Diagnosis of Fetal Aneuploidy by Shotgun Sequencing DNA from Maternal Blood. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 16266–16271. 10.1073/PNAS.0808319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu R. W. K.; Akolekar R.; Zheng Y. W. L.; Leung T. Y.; Sun H.; Chan K. C. A.; Lun F. M. F.; Go A. T. J. I.; Lau E. T.; Leung W. C.; Tang R. Y. K.; Au-Yeung S. K. C.; Lam H.; Kung Y. Y.; Zhang X.; van Vugt J. M. G.; Minekawa R.; Tang M. H. Y.; Wang J.; Oudejans C. B. M.; Lau T. K.; Nicolaides K. H.; Lo Y. M. D. Non-Invasive Prenatal Assessment of Trisomy 21 by Multiplexed Maternal Plasma DNA Sequencing: Large Scale Validity Study. Br. Med. J. 2011, 342, c7401. 10.1136/bmj.c7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrich M.; Deciu C.; Zwiefelhofer T.; Tynan J. A.; Cagasan L.; Tim R.; Lu V.; McCullough R.; McCarthy E.; Nygren A. O. H.; Dean J.; Tang L.; Hutchison D.; Lu T.; Wang H.; Angkachatchai V.; Oeth P.; Cantor C. R.; Bombard A.; van den Boom D. Noninvasive Detection of Fetal Trisomy 21 by Sequencing of DNA in Maternal Blood: A Study in a Clinical Setting. Am. J. Obstet. Gynecol. 2011, 204, 205.e1–205.e11. 10.1016/j.ajog.2010.12.060. [DOI] [PubMed] [Google Scholar]
- Lo Y. M. D.; Chan K. C. A.; Sun H.; Chen E. Z.; Jiang P.; Lun F. M. F.; Zheng Y. W.; Leung T. Y.; Lau T. K.; Cantor C. R.; Chiu R. W. K. Maternal Plasma DNA Sequencing Reveals the Genome-Wide Genetic and Mutational Profile of the Fetus. Sci. Transl. Med. 2010, 2, 61ra91. 10.1126/scitranslmed.3001720. [DOI] [PubMed] [Google Scholar]
- Palomaki G. E.; Kloza E. M.; Lambert-Messerlian G. M.; Haddow J. E.; Neveux L. M.; Ehrich M.; van den Boom D.; Bombard A. T.; Deciu C.; Grody W. W.; Nelson S. F.; Canick J. A. DNA Sequencing of Maternal Plasma to Detect Down Syndrome: An International Clinical Validation Study. Genet. Med. 2011, 13, 913–920. 10.1097/gim.0b013e3182368a0e. [DOI] [PubMed] [Google Scholar]
- Ericsson O.; Ahola T.; Dahl F.; Karlsson F.; Persson F.; Karlberg O.; Roos F.; Alftrén I.; Andersson B.; Barkenäs E.; Boghos A.; Brandner B.; Dahlberg J.; Forsgren P. O.; Francois N.; Gousseva A.; Hakamali F.; Janfalk-Carlsson Å.; Johansson H.; Lundgren J.; Mohsenchian A.; Olausson L.; Olofsson S.; Qureshi A.; Skarpås B.; Svahn P.; Sävneby A.; Åström E.; Sahlberg A.; Fianu-Jonasson A.; Gautier J.; Costa J. M.; Jacobsson B.; Nicolaides K. Clinical Validation of a Novel Automated Cell-Free DNA Screening Assay for Trisomies 21, 13, and 18 in Maternal Plasma. Prenatal Diagn. 2019, 39, 1011–1015. 10.1002/PD.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaides K. H.; Syngelaki A.; Gil M.; Atanasova V.; Markova D. Validation of Targeted Sequencing of Single-Nucleotide Polymorphisms for Non-Invasive Prenatal Detection of Aneuploidy of Chromosomes 13, 18, 21, X, and Y. Prenatal Diagn. 2013, 33, 575–579. 10.1002/PD.4103. [DOI] [PubMed] [Google Scholar]
- Jacky L.; Yurk D.; Alvarado J.; Leatham B.; Schwartz J.; Annaloro J.; MacDonald C.; Rajagopal A. Virtual-Partition Digital PCR for High-Precision Chromosomal Counting Applications. Anal. Chem. 2021, 93, 17020–17029. 10.1021/acs.analchem.1c03527. [DOI] [PubMed] [Google Scholar]
- Haidong W.; Zhijie Y.; Picchiassi E.; Tarquini F.; Coata G.; You W.; Youxiang W.; Yu C.; Carlo Di Renzo G.; Chen Y.. Non-Invasive Prenatal Testing of Fetal Aneuploidies Using a New Method Based on Digital Droplet PCR and Cell Free Fetal DNA, 10.1101/2020.12.19.20248553. [DOI]
- Tan C.; Chen X.; Wang F.; Wang D.; Cao Z.; Zhu X.; Lu C.; Yang W.; Gao N.; Gao H.; Guo Y.; Zhu L. A Multiplex Droplet Digital PCR Assay for Non-Invasive Prenatal Testing of Fetal Aneuploidies. Analyst 2019, 144, 2239–2247. 10.1039/C8AN02018C. [DOI] [PubMed] [Google Scholar]
- Dai P.; Yang Y.; Zhao G.; Gu Z.; Ren H.; Hu S.; Liu N.; Jiao W.; Li J.; Kong X. A DPCR-NIPT Assay for Detections of Trisomies 21, 18 and 13 in a Single-Tube Reaction-Could It Replace Serum Biochemical Tests as a Primary Maternal Plasma Screening Tool?. J. Transl. Med. 2022, 20, 1–17. 10.1186/S12967-022-03455-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver S.; Dube S.; Mir A.; Qin J.; Sun G.; Ramakrishnan R.; Jones R. C.; Livak K. J. Taking QPCR to a Higher Level: Analysis of CNV Reveals the Power of High Throughput QPCR to Enhance Quantitative Resolution. Methods 2010, 50, 271–276. 10.1016/J.YMETH.2010.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.