

Abstract
Idiopathic pulmonary fibrosis is a progressive lung disease with limited therapeutic options that is characterized by pathological fibroblast activation and aberrant lung remodeling with scar formation. YAP (Yes-associated protein) is a transcriptional coactivator that mediates mechanical and biochemical signals controlling fibroblast activation. We previously identified HMG-CoA (3-hydroxy-3-methylglutaryl coenzyme A) reductase inhibitors (statins) as YAP inhibitors based on a high-throughput small-molecule screen in primary human lung fibroblasts. Here we report that several Aurora kinase inhibitors were also identified from the top hits of this screen. MK-5108, a highly selective inhibitor for AURKA (Aurora kinase A), induced YAP phosphorylation and cytoplasmic retention and significantly reduced profibrotic gene expression in human lung fibroblasts. The inhibitory effect on YAP nuclear translocation and profibrotic gene expression is specific to inhibition of AURKA, but not Aurora kinase B or C, and is independent of the Hippo pathway kinases LATS1 and LATS2 (Large Tumor Suppressor 1 and 2). Further characterization of the effects of MK-5108 demonstrate that it inhibits YAP nuclear localization indirectly via effects on actin polymerization and TGFβ (Transforming Growth Factor β) signaling. In addition, MK-5108 treatment reduced lung collagen deposition in the bleomycin mouse model of pulmonary fibrosis. Our results reveal a novel role for AURKA in YAP-mediated profibrotic activity in fibroblasts and highlight the potential of small-molecule screens for YAP inhibitors for identification of novel agents with antifibrotic activity.
Keywords: idiopathic pulmonary fibrosis, drug screening, Aurora kinase A inhibitor, YAP, human lung fibroblast
Idiopathic pulmonary fibrosis (IPF) is a debilitating lung disease with an average time to respiratory failure and death of 3–5 years from the time of diagnosis. IPF is characterized by the progressive loss of pulmonary function due to progressive scarring of the lungs (1, 2). In 2014, two medications, nintedanib and pirfenidone, received U.S. Food and Drug Administration approval for patients with IPF after clinical trials demonstrated that these drugs slowed the rate of lung function decline (3, 4). However, there remains a great need for additional therapies that can stop and ultimately reverse established fibrosis.
IPF is believed to result from dysregulated wound repair after lung injury (2). Studies over the last decade have shown that the pathophysiology is highly complex, involving nearly all cell types in the lung, with multiple factors contributing to the development of the disease, including the environment, sex, aging, and genetics (5–8). Central to the process is the activation and recruitment of lung fibroblasts to the alveolar structures, which then secrete extracellular matrix molecules like collagen, forming a scar and stiffening the lung parenchyma. Although this is part of the normal mechanism of wound repair, in IPF lungs this process is persistent, leading to progressive loss of lung function (9). The exact mechanisms of fibroblast activation and persistence in IPF are unclear, and a better understanding of these processes may help identify novel therapeutic targets for IPF.
It is known that fibroblasts sense and respond to the stiffness of the tissue microenvironment, leading to their activation and further extracellular matrix production (10, 11). The transcription coactivators YAP (Yes-associated Protein) and TAZ (Transcription coactivator with PDZ-binding motif) are important effectors of mechanosignaling in fibroblasts. In response to high stiffness, YAP and TAZ translocate to the nucleus, where they help upregulate profibrotic gene expression (12–14). First discovered in Drosophila, YAP and TAZ are regulated by the Hippo pathway kinases LATS1 and LATS2 (Large Tumor Suppressor 1 and 2) through phosphorylation on multiple serine residues (15). When the Hippo pathway is active, YAP and TAZ are phosphorylated and are sequestered in the cytoplasm, where they bind with 14-3-3 proteins and are targeted for degradation (16–18). When the Hippo pathway is inactive, YAP and TAZ are dephosphorylated and can translocate into the nucleus and bind to TEAD (transcriptional enhanced associate domain) to promote target gene expression. The targets of YAP and TAZ include genes promoting cell survival, growth, and proliferation. CTGF (connective tissue growth factor) and CYR61 (cysteine rich angiogenic inducer 61) are both direct target genes for YAP and are also upregulated in TGFβ-induced fibrosis (19–21). Consistent with this, YAP and TAZ are required for increased CTGF, CYR61, ACTA2 (α-smooth muscle actin), and COL1A1 (collagen type I α 1 chain) expression by TGFβ-activated myofibroblasts (22). In addition, previous studies have found that YAP and TAZ are enriched in the nuclei of fibroblasts in fibrotic areas of the IPF lung (23). Furthermore, adoptive transfer of fibroblasts overexpressing nonphosphorylatable forms of YAP and TAZ into mouse lungs confer them with fibrogenic capability and lead to the development of lung fibrosis (23). These data suggest that inhibiting YAP/TAZ activity could be a potential therapeutic target in IPF.
We performed a small-molecule screen to identify compounds that can induce YAP nuclear exclusion, using high-throughput fluorescent imaging. Using this screen, we previously identified HMG-CoA reductase inhibitors (statins) as potent inhibitors of YAP nuclear translocation and demonstrated their antifibrotic effects in primary human lung fibroblasts (HLFs) and in a bleomycin mouse model of pulmonary fibrosis (24).
A number of the compounds in the top hits from the screen have previously been reported to have effects on fibroblasts or in clinical trials on IPF (25–30), validating that our screen design is effective. Here we report that several Aurora kinase inhibitors were identified in the top hits, including MK-5108, a highly selective inhibitor for AURKA (Aurora kinase A). The Aurora kinases are important regulators of mitosis and are overexpressed in a number of tumors (31–42). AURKA and AURKB inhibitors have been developed and tested in clinical trials for solid tumors and hematological malignancies (43, 44), but the therapeutic potential for treating fibrosis by inhibiting Aurora kinases has not been well studied. In this study, we characterize the antifibrotic effect of inhibiting AURKA with MK-5108 through actin cytoskeleton–mediated YAP regulation and report its efficacy in preventing bleomycin-induced lung fibrosis in a mouse model of IPF. Our study also demonstrates the potential of small-molecule screens for YAP inhibitors for identification of novel agents with antifibrotic activity.
Some of the results of these studies have been previously reported in the form of an abstract (45).
Methods
HLFs Culture
Primary HLFs from patients with IPF or healthy control donors were collected through the Massachusetts General Hospital Fibrosis Translational Research program from deidentified discarded excess tissue from clinically indicated surgical lung resections or lung transplant explants. Cells were routinely grown in Dulbecco’s modified Eagle medium (Lonza) supplemented with 10% FBS (Lonza), 2 mM l-glutamine (Lonza), 100 U/ml penicillin, and 100 μg/ml streptomycin (Lonza) in a humidified incubator with 5% CO2 at 37°C. Unless otherwise stated, experiments were conducted using fibroblasts from a healthy control donor with patient ID number 699.
High-Throughput Drug Screening Analysis
A small-molecule screen for YAP inhibitors using primary HLFs was previously conducted at the ICCB-Longwood Screening Facility at Harvard Medical School (24). A total of 13,232 small molecules were screened in duplicate and assigned a score based on their ability to displace YAP from the nucleus into the cytoplasm with minimum toxicity. Here we defined high hits and low hits as compounds with a score higher than 55 and 45, respectively. These cutoffs were determined by visual inspection of multiple random images and qualitatively correlating score values to cell phenotypes. Out of the 13,232 small molecules tested, 386 were considered high hits and 443 were considered low hits. Compounds with the same simplified molecular-input line-entry system (SMILES) were grouped together, resulting in 227 high hits and 328 low hits.
Consolidation of Screening Library
Chemical structures of the screening library have been consolidated using the data science workflow software BIOVIA Pipeline Pilot. Protonation states of the structures have been standardized, and counter ions have been eliminated. We used canonical SMILES as a unique linear textual representation of the chemical structure. This way, the initial 13,232 structures could be mapped onto 7,696 unique canonical SMILES, of which 4,329 are represented by a single well in the library and 3,367 occur in up to 19 wells. Multiple occurrences of individual canonical SMILES could be traced to multiple vendors and/or multiple molar concentrations of the individual probes.
Library Annotation and Target Enrichment Analysis
The condensed library of 7,656 compounds tested in the primary screen was annotated with known targets based on information from ChEMBL, considering a threshold of activity <100 nM. The target signatures of the primary screen hits (high and low) and the nonhits were compared with identify targets that are significantly enriched in the hit population. These data were used to filter the 118 compounds with a good dose–response profile down to 46 compounds with targets enriched in the hit population. Target enrichment determination has been performed in two ways. Given the following contingency table per target x:
| Compound Is Hit in
Assay |
|||
|---|---|---|---|
| Yes | No | ||
| Compound is annotated with Target x | Yes | a | b |
| No | c | d | |
we calculated the enrichment E as the ratio between the hit rate among all compounds annotated with a specific target and the hit rate of the entire dataset given by:
In addition, we used the statistics package R to estimate the statistical significance of the target-specific contingency table by Fisher exact test. P values, derived by Fisher exact test, have been corrected using the Bonferroni-Holm method.
Compound Quality Control
Forty-four compounds with targets enriched in the hit population were obtained from independent sources. Compounds at 10 mM in DMSO solution were analyzed by HPLC/mass spectrometry. Purity was assessed by peak area, and authenticity was determined by mass spectrometry.
Additional Methods
Additional experimental details and methods are provided in the online data supplement.
Results
A High-Throughput Small-Molecule Screen Identifies Aurora Kinase Inhibitors as High-Efficacy YAP Inhibitors
We previously reported initial results using a high-throughput phenotypic screen for YAP inhibitors in primary HLFs (24). In our primary screen, 13,232 compounds were tested in duplicate from the known bioactives collection and the academic collection at the ICCB-Longwood Screening Facility at Harvard Medical School (https://iccb.med.harvard.edu/compound-libraries). Compounds were assigned a score between 0 and 100 based on their ability to displace YAP from the nucleus into the cytoplasm with minimal toxicity (24). Because of library redundancy, some molecules were screened multiple times. We grouped together compounds with the same simplified molecular-input line-entry system, yielding 7,656 unique compounds (Figure 1A). Out of these, 227 compounds were classified as high-potency hits (score >55), and 328 were classified as low-potency hits (scores between 45 and 54) (Figure 1A).
Figure 1.
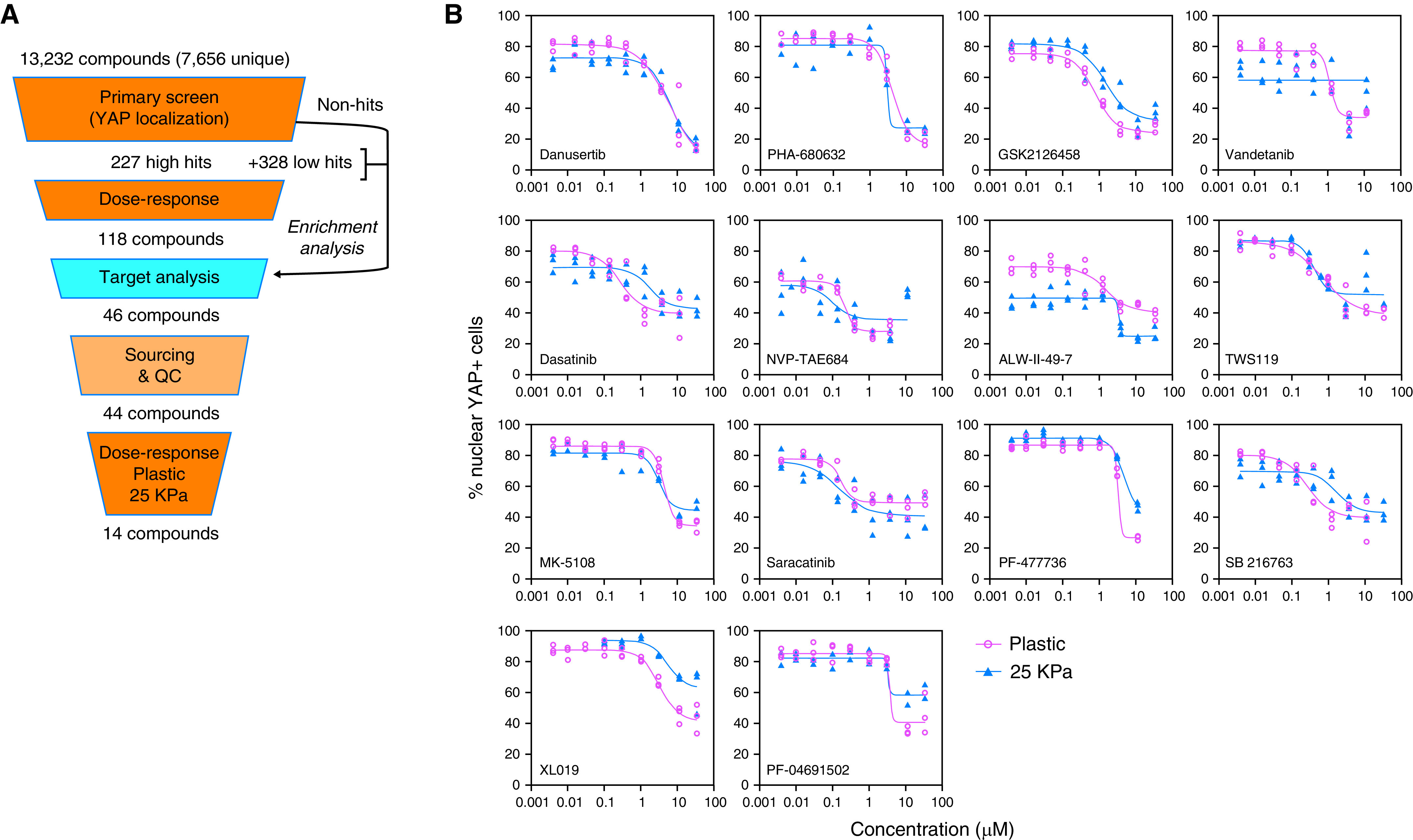
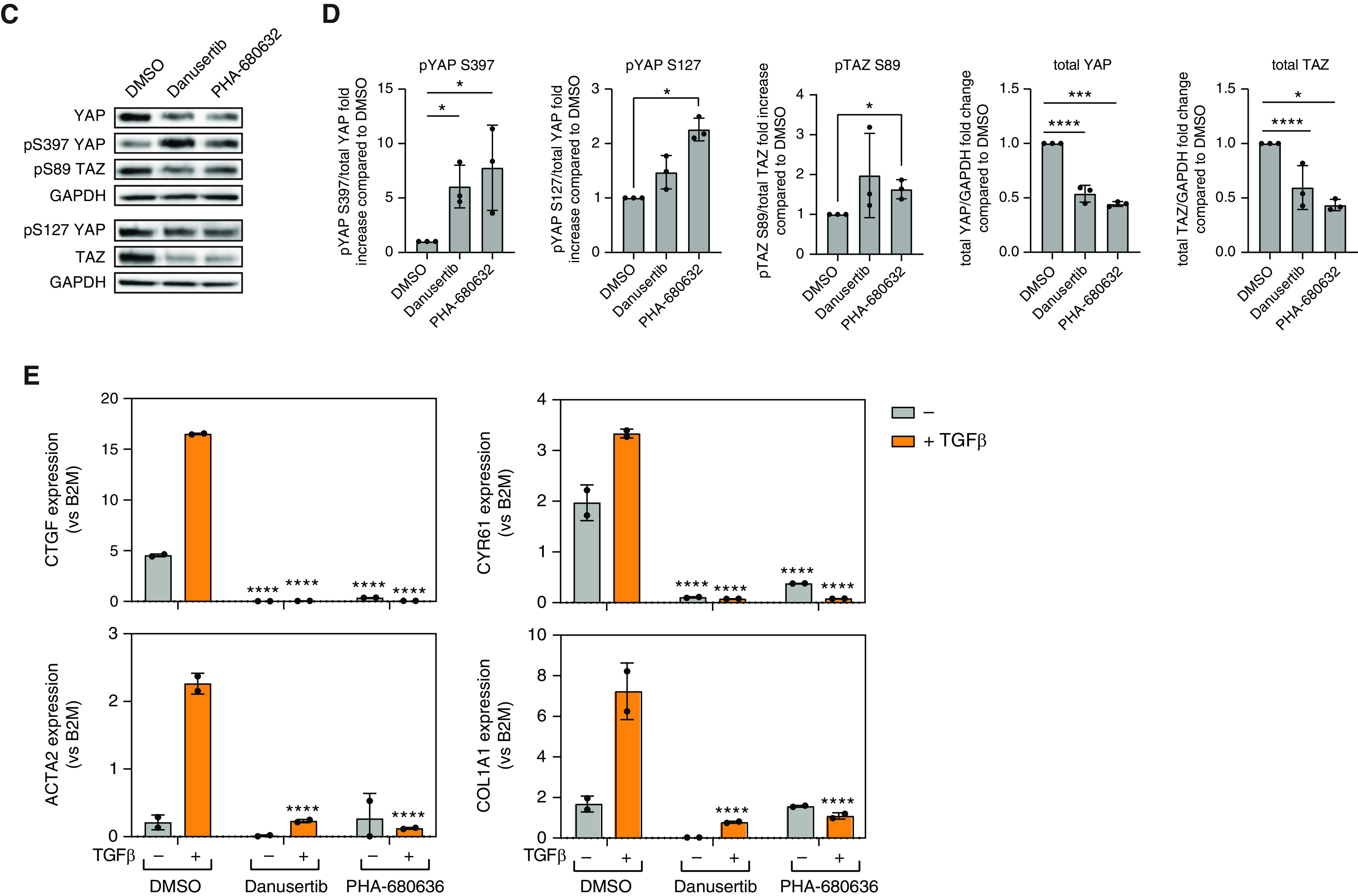
High-throughput small-molecule screen for inhibitors of YAP (Yes-associated protein) nuclear localization identifies Aurora A inhibitors as novel hits. (A) Scheme of small-molecule screen. (B) Dose–response curve of 14 top hits of the screen generated on plastic or 25 kPa Matrigel. (C) Western blots and (D) quantifications showing effects of pan-Aurora inhibitors danusertib and PHA-680632 in YAP/TAZ (Transcription coactivator with PDZ-binding motif) phosphorylation and degradation. Phosphorylated YAP/TAZ and total YAP/TAZ were both normalized to the corresponding GAPDH, and the ratio of phosphorylated YAP/TAZ to total YAP/TAZ was normalized to DMSO control. Data represent mean ± SD, n = 3. *P < 0.05, ***P < 0.001, and ****P < 0.0001, unpaired t test. (E) Quantitative PCR (qPCR) results showing that pan-Aurora inhibitors danusertib and PHA-680632 significantly reduced expression of profibrotic genes CTGF (connective tissue growth factor), CYR61 (cysteine rich angiogenic inducer 61), COL1A1 (collagen type I α 1 chain), and ACTA2 (α-smooth muscle actin). Data represent mean ± SD, n = 2. ****P < 0.0001, two-way ANOVA with Sidak’s multiple comparisons test. QC = quality control.
We performed a dose–response analysis of YAP localization (nuclear/cytoplasmic ratio) and cell number on the 227 high-potency hits. The dose–response curves were rated by four independent investigators as described in the Methods section, and 118 of the compounds were further selected based on the effectiveness of the inhibition of YAP nuclear translocation. We also annotated the condensed library of 7,656 screened compounds with their known targets based on information from public databases. We then compared the target signatures of the hits and the nonhits to identify targets enriched in the hit population. We cross-referenced this list with the selective list of 118 compounds and further selected 46 compounds with enriched targets in the high-potency hit population. Next, we obtained 44 of the 46 compounds and confirmed compound purity. We performed dose–response experiments with these 44 compounds on primary HLFs plated on plastic and on 25 kPa hydrogels coated with collagen (which mimics the conditions of a fibrotic lung). Fourteen top hits were identified according to the following criteria: 1) first effective concentration <4 μM in both plastic and 25 kPa; and 2) reduction of nuclear YAP of ⩾30% compared with DMSO control values in either plastic or 25 kPa (Figure 1B and Table 1). Notably, the two hits with the highest reduction from baseline were danusertib (81% and 78% for plastic and 25 kPa hydrogel, respectively) and PHA-680632 (76% and 68% for plastic and 25 kPa hydrogel, respectively), both pan-Aurora kinase inhibitors. The list of top hits also included MK-5108, a highly selective AURKA inhibitor.
Table 1.
Validated Top Hits from Phenotypic Screen for YAP Inhibitors in Human Lung Fibroblasts
| Compound | Maximum Reduction
(%) |
First Effective Concentration
(μM) |
IC50
(μM) |
Compound Targets | |||
|---|---|---|---|---|---|---|---|
| 25 kPa | Plastic | 25 kPa | Plastic | 25 kPa | Plastic | ||
| Danusertib | 78 | 81 | 1.20 | 1.20 | 6.42 | 6.04 | Aurora A/B/C |
| PHA-680632 | 68 | 76 | 3.67 | 1.20 | ∼3.13 | 4.29 | Aurora A/B/C |
| GSK2126458 | 64 | 70 | 1.20 | 0.40 | 1.49 | 0.78 | PI3K (α/β/δ/γ) and mTORC1/2 |
| Vandetanib | 63 | 60 | 1.20 | 1.20 | N/A | 1.14 | VEGFR2, VEGFR3, and EGFR |
| Dasatinib | 61 | 59 | 0.016 | 0.016 | 1.73 | 0.28 | Abl, Src, and c-Kit |
| NVP-TAE684 | 58 | 63 | 0.14 | 0.14 | 0.10 | 0.21 | ALK |
| ALW-II-49-7 | 56 | 43 | 3.67 | 1.20 | ∼3.36 | 1.41 | EPHB2, DDR1, EPHA2, PDGFRA, EPHA5, EPHA4, EPHA3, EPHA8, KIT, PDGFRB, EPHB4, EPHB3, DDR2 |
| TWS119 | 52 | 51 | 0.40 | 0.40 | 0.41 | 0.74 | GSK-3β |
| MK-5108 | 49 | 59 | 3.67 | 3.67 | 3.08 | 4.23 | Aurora A |
| Saracatinib | 47 | 43 | 0.14 | 0.14 | 0.15 | 0.17 | Src, c-Yes, Fyn, Lyn, Blk, Fgr, and Lck |
| PF-477736 | 46 | 69 | 3.67 | 3.67 | 4.67 | ∼3.24 | Chk1, VEGFR2, Aurora A, FGFR3, Flt3, Fms (CSF1R), Ret, and Yes |
| SB 216763 | 43 | 52 | 0.14 | 0.14 | 1.73 | 0.28 | GSK-3 inhibitor |
| XL019 | 32 | 50 | 3.67 | 1.20 | 5.23 | 3.16 | JAK2 |
| PF-04691502 | 31 | 59 | 3.67 | 3.67 | ∼3.26 | ∼3.60 | PI3K (α/β/δ/γ) and mTOR |
Definition of abbreviations: IC50 = Half Maximal Inhibition Concentration; N/A = Not Available; YAP = Yes-associated protein.
Aurora Kinase Inhibitors Promote YAP Sequestration and Degradation
We further investigated the effects of Aurora kinase inhibitors on YAP. Western blot analysis of HLFs treated 16–20 hours with danusertib or PHA-680632 revealed a decrease in total amount of YAP and an increase in serine 397 (S397) phosphorylation, which promotes YAP proteasomal degradation. There was also an increase in the ratio of serine 127 phosphorylation (S127) to total YAP protein amount, which promotes cytoplasmic sequestration. TAZ, a YAP paralogue, was similarly regulated (Figures 1C and 1D). HLFs treatment with either danusertib or PHA-680632 reduced total TAZ amount and increased the ratio of serine 89 (S89) phosphorylation (equivalent to S127 phosphorylation in YAP).
We next tested whether Aurora kinase inhibition modulates YAP activity and profibrotic HLF activation. Treatment of HLFs with the profibrotic cytokine TGFβ for 48 hours increased the expression of CTGF, CYR61, and other fibrotic markers such as ACTA2 and COL1A1. Expression of these genes was inhibited by either danusertib or PHA-680632 (Figure 1E). Aurora kinase inhibitors also inhibited baseline expression of CTGF and CYR61, indicating effects independent of TGFβ stimulation.
AURKA-Specific Inhibition Decreased YAP Nuclear Localization and TGFβ-induced Fibroblast Activation
Because both pan-Aurora inhibitors and the AURKA-specific inhibitor MK-5108 were among the top hits, we wanted to determine which Aurora kinase is the key to this regulation. HLFs were plated in 96-well plates. After 48 hours, cells were treated with DMSO or specific Aurora kinase inhibitors with or without TGFβ. Barasertib, a specific inhibitor for AURKB, did not alter nuclear YAP localization, whereas 10 μM MK-5108 significantly reduced nuclear YAP localization in both the presence and absence of TGFβ (Figures 2A and 2B). Similar results were confirmed using human primary lung fibroblasts from multiple normal donors or patients with IPF (see Figure E1 in the online supplement). MK-5108 alone or together with barasertib decreased expression of CTGF, CYR61, COL1A1, and ACTA2, but barasertib alone did not (Figure 2C). Given that pharmacological kinase inhibitors can have off-target effects, we wanted to confirm the effects using siRNAs. HLFs were transfected with siRNAs against AURKA, AURKB, AURKC, or a combination of the three. The specificity and efficiency of siRNA knockdown were confirmed using quantitative PCR and Western blot (Figure E2). AURKA knockdown resulted in a slight decrease in AURKB expression and vice versa. Interestingly, knocking down AURKC resulted in a compensatory increase in AURKA and AURKB expression. We also observed an increase in AURKA expression after siAURKB at earlier time points (data not shown). Consistent with the pharmacological compound treatments, siAURKA decreased nuclear YAP localization (Figure E2C) and expression of TGFβ-induced profibrotic genes (Figure 2D). At baseline, siAURKA also resulted in decreased CTGF, CYR61, and COLA1 expression. Interestingly, there were small increases in CTGF, CYR61, COL1A1, and ACTA2 expression when AURKB or AURKC are knocked down, which may be due to compensatory changes in AURKA expression. In summary, these results indicate that AURKA-specific inhibition reduces YAP nuclear localization and TGFβ-induced profibrotic gene expression in HLFs.
Figure 2.
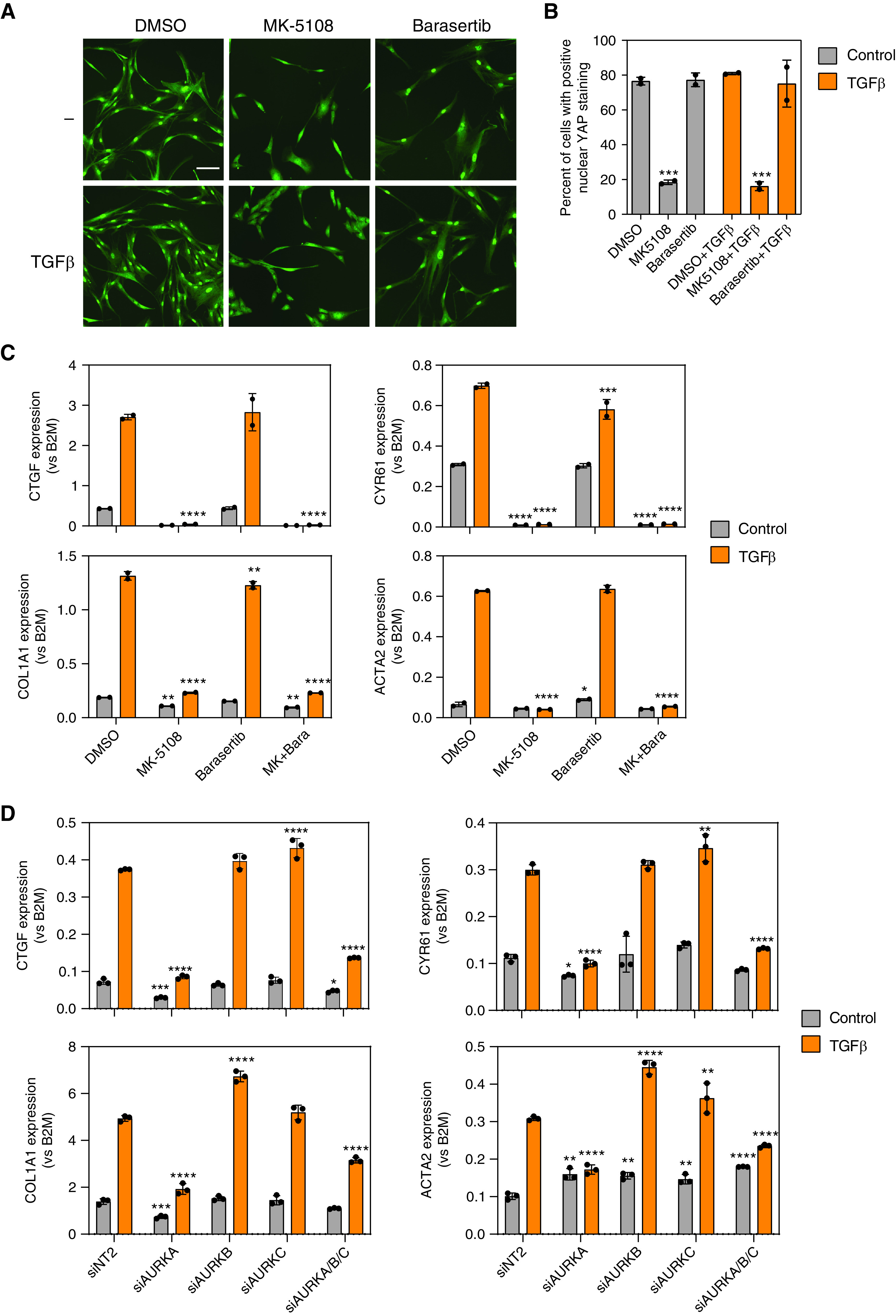
Inhibiting Aurora A, but not Aurora B, significantly decreased YAP nuclear localization and TGFβ-induced fibroblast activation. (A) Representative images and (B) quantification of YAP staining in human lung fibroblasts (HLFs) treated with MK-5108 or barasertib, in the presence or absence of TGFβ. Data represent mean ± SD, n = 3. ***P < 0.001, unpaired t test. Scale bar, 10 μm. (C) qPCR results showing the expression of profibrotic genes in HLFs after MK-5108, barasertib, or both, in the presence or absence of TGFβ. Data represent mean ± SD, n = 2. *P < 0.05, **P < 0.01, and ****P < 0.0001, two-way ANOVA with Sidak’s multiple comparisons test. (D) qPCR results showing that only siRNAs against Aurora A kinase, but not Aurora B or Aurora C, significantly reduced expression of profibrotic genes in HLFs. Data represent mean ± SD, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, two-way ANOVA with Sidak’s multiple comparisons test.
MK-5108 Inhibits AURKA Autophosphorylation on T-288 in HLFs and Induces YAP Inactivation Independent of Effects on Mitosis
To demonstrate target engagement of MK-5108 in HLFs, we examined amount of AURKA with phosphorylation at T-288, which is an autophosphorylation site that reflects its kinase activity (46). Within 1 hour of 10 μM MK-5108 addition, there was a dramatic reduction of p-T288 AURKA–to– total AURKA ratio compared with DMSO control, confirming that MK-5108 efficiently inhibited the kinase activity of AURKA in HLFs (Figure 3A).
Figure 3.
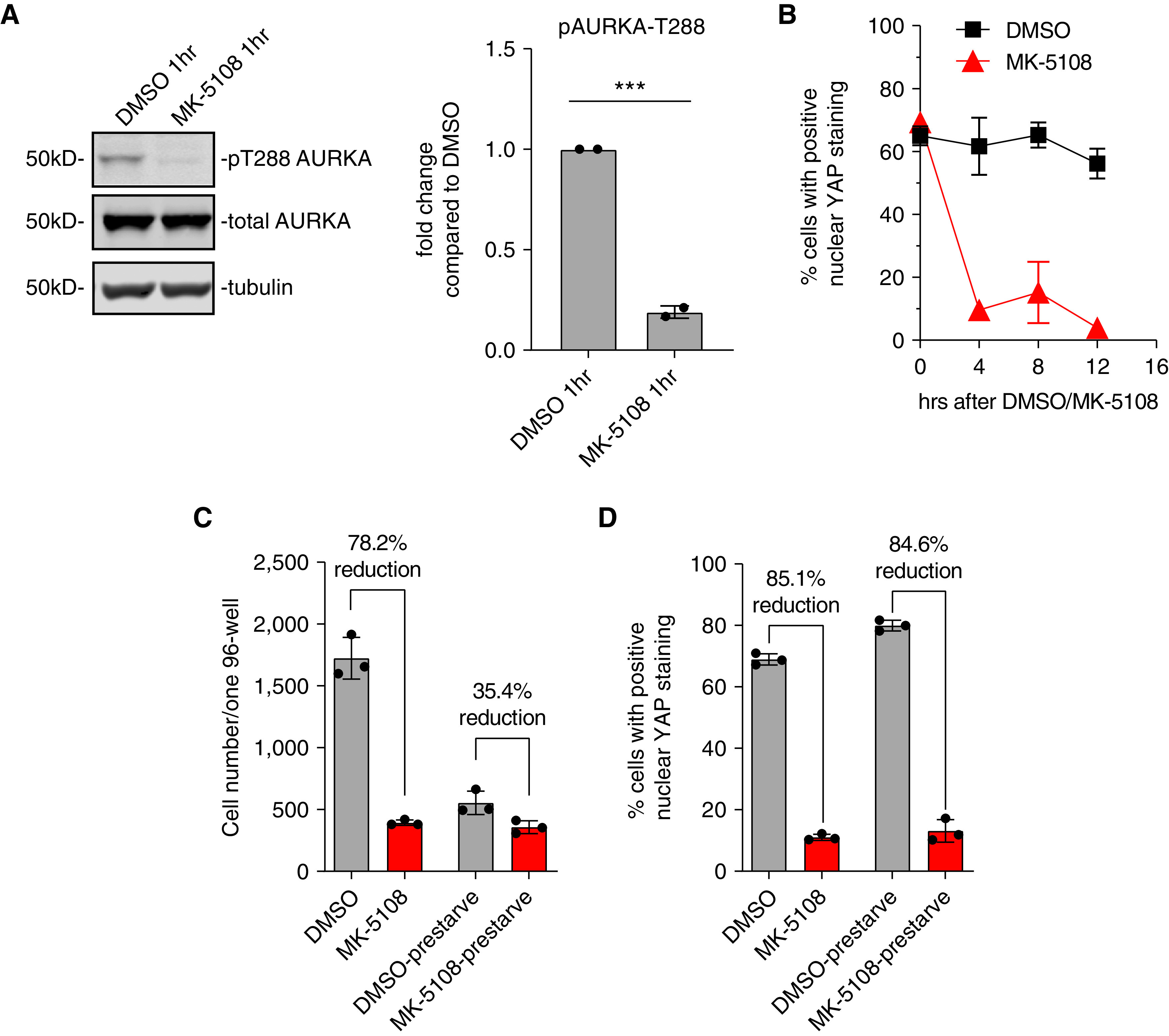
Effect of Aurora A inhibition in YAP inactivation is independent of effects on mitosis. (A) Western blot and quantification showing the decrease in phosphorylated AURKA on T288 1 hour after changing to serum-free media and MK-5108 treatment. ***P < 0.001. (B) Average percentage of cells with positive nuclear YAP localization during a time course of MK-5108 treatment, after overnight prestarvation of the cells in serum-free medium. Data represent mean ± SD. (C) Average cell numbers of three individual wells after immunostaining in 96-well plates, after MK-5108 treatment in HLFs, with or without prestarvation for 16 hours. Data represent mean ± SD. (D) Average percentage of cells with YAP nuclear localization correlation coefficient greater than 0.7, in DMSO- or MK-5108–treated HLFs, with or without prestarvation for 16 hours. Data represent mean ± SD.
AURKA’s role in regulating mitosis has been well studied. Inhibition of AURKA delays mitosis by inhibiting centrosome maturation and causes mitotic arrest because of abnormal spindle formation, which then leads to cellular apoptosis (31). To determine if AURKA regulates YAP through its mitotic function, we synchronized the cells by double thymidine block and release. Samples were collected every 4 hours for 12 hours for immunostaining to determine YAP localization. After thymidine release, YAP was localized in the nucleus. Addition of MK-5108 induced YAP cytoplasmic translocation within 4 hours, when most of the cells had not yet entered mitosis (Figure 3B). Cell numbers decreased in the MK-5108–treated wells consistent with the known induction of apoptosis with AURKA inhibition (47). Moreover, when the HLFs were prestarved in serum-free medium overnight, MK-5108 similarly diminished YAP nuclear localization, while having comparable total cell numbers to the DMSO-control group (Figures 3C and 3D). These results suggest that AURKA inhibition by MK-5108 inactivates YAP independent of its effects on the cell cycle.
AURKA-Specific Inhibition Leads to YAP Translocation Independent of the Canonical Hippo Pathway Kinases LATS1 and LATS2
In canonical Hippo signaling, YAP and TAZ are phosphorylated and inhibited by the Hippo pathway kinases LATS1 and LATS2 (16–18, 48), which are activated by MST1 and MST2 (Mammalian Ste20-like kinases 1 and 2) phosphorylation (49, 50). When phosphorylated, YAP binds with 14-3-3 protein in the cytoplasm and is targeted for degradation (17, 18). In noncanonical Hippo signaling, YAP and TAZ can also be phosphorylated by other kinases, including AURKA (51–55). To determine if MK-5108 inhibits YAP through canonical Hippo pathway regulators, we tested if depleting LATS1/2 or inhibiting MST1/2 can prevent YAP inactivation by MK-5108. siLATS1 and siLATS2 reduced the baseline amount of YAP phosphorylation on S397 and S127, indicating the depletion of LATS1 and LATS2 have the expected effects of reducing YAP phosphorylation at these sites (Figure 4A). When LATS1 and LATS2 were depleted, MK-5108 induced more YAP phosphorylation on S397 than DMSO-treated control HLF, albeit to a lesser degree than a nontargeting control siRNA. MK-5108 also induced YAP phosphorylation on S127 when LATS1 and LATS2 were depleted (Figures 4A and 4B). These results indicate that inhibiting AURKA causes YAP cytoplasm sequestration independent of the canonical Hippo pathway kinases LATS1 and LATS2; however, it may partially depend on LATS1 and LATS2 to induce phosphorylation on YAP S397 (Figures 4A and 4B). Consistent with this, YAP total protein amount was maintained when LATS1 and LATS2 were depleted after adding MK-5108 (Figure 4B), but the amount of YAP cytoplasmic localization was not changed (Figure 4C). As expected, YAP target genes CTGF, CYR61, and COL1A1 were reduced after MK-5108 treatment, even with LATS1 and LATS2 depletion (Figure 4E). The MST1/2 inhibitor XMU-MP-1 also did not restore YAP nuclear localization when combined with MK-5108 treatment (Figure 4D). These results suggest that AURKA-specific inhibition by MK-5108 causes YAP phosphorylation and cytoplasmic localization primarily through a mechanism independent of the canonical Hippo pathway.
Figure 4.
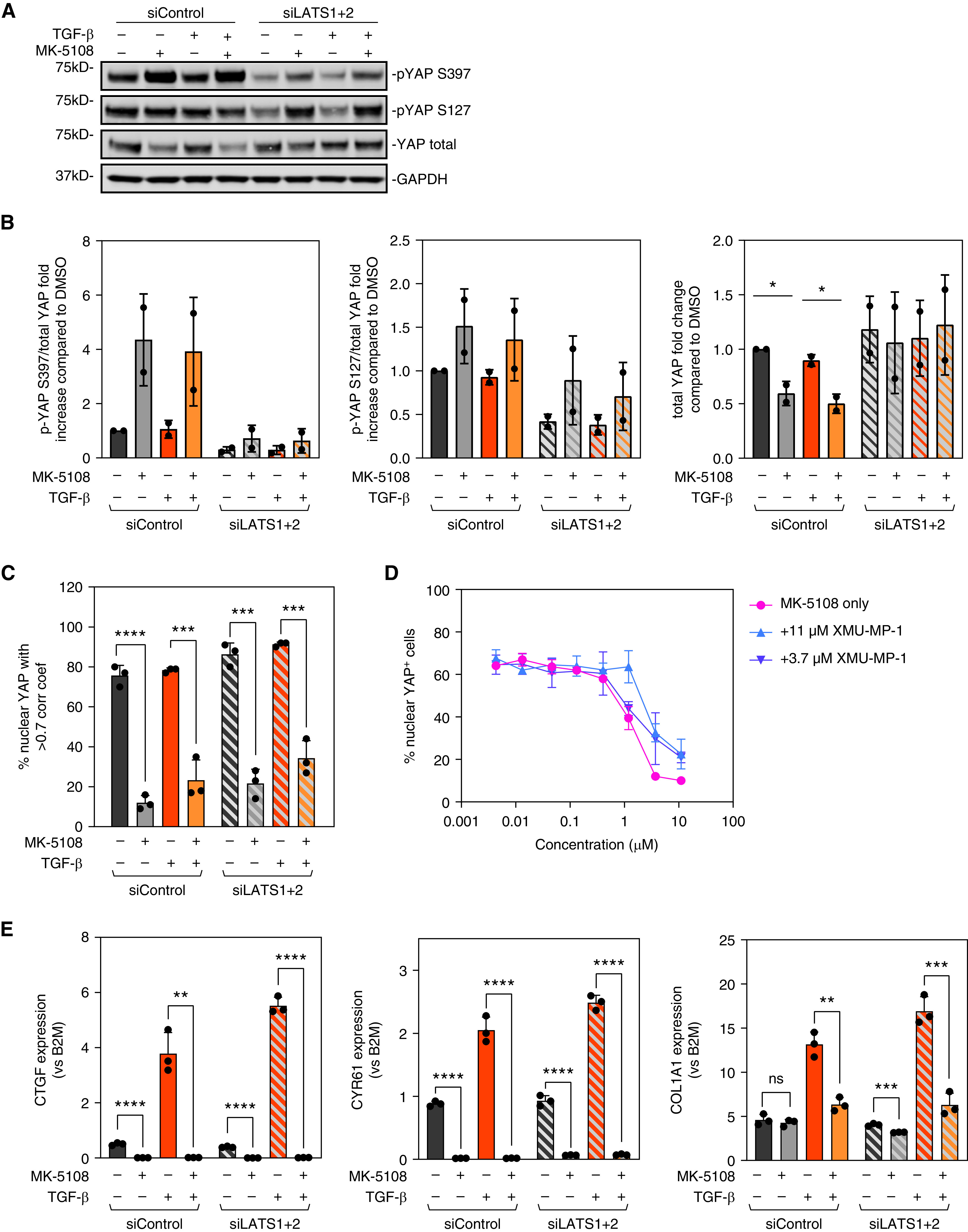
YAP inactivation and antifibrotic effect of Aurora A inhibition is not dependent on canonical Hippo pathway kinases LATS1 and LATS2 (Large Tumor Suppressor 1 and 2). (A) Western blot images and (B) quantification showing the amount of total and phosphorylated YAP. Phosphorylated YAP/TAZ and total YAP/TAZ were both normalized to the corresponding GAPDH, and the ratio of phosphorylated-to-total YAP/TAZ was normalized to DMSO control. Data represent mean ± SD, n = 2. *P < 0.01, unpaired t test. (C) quantification of the YAP staining in HLFs after MK-5108 treatment combined with siNT2 or siLATS1 + 2, in the presence or absence of TGFβ. Data represent mean ± SD, n = 3. ***P < 0.001 and ****P < 0.0001, unpaired t test. (D) Quantification of the YAP staining after treating HLFs with a dose curve of MK-5108 combined with MST (Mammalian Ste20-like kinase) inhibitor XMU-MP-1. (E) qPCR results showing the effect of MK-5108 on CTGF, CYR61, and COL1A1 expression with control and LATS1 and LATS2 siRNA, in the presence or absence of TGFβ. Data represent mean ± SD. ns = non-significant. **P < 0.01, ***P < 0.001, and ****P < 0.0001, unpaired t test.
MK-5108 Displaces Nuclear YAP and Inhibits Fibroblast Activation through Disruption of Actin Polymerization and Focal Adhesion
YAP and TAZ act as mechanosensors and mechanotransducers (56–59). Specifically, F-actin polymerization and stress fiber formation can activate YAP and TAZ, whereas disruption of the actin cytoskeleton sequesters YAP and TAZ in the cytoplasm (14). It has been shown that lysophosphatidic acid (LPA) and sphingosine 1-phosphophate (S1P) can act through G12/13-coupled receptors to inhibit LATS1 and LATS2, therefore activating YAP. LPA and S1P have also been shown to induce YAP nuclear localization and activation through Rho GTPase activation and F-actin polymerization, independent of the canonical Hippo pathway (60). When HLFs were treated with MK-5108, actin fibers were dramatically disrupted, and the cells changed from a spindle shape to a more rounded shape (Figure 5A). Thus, we wanted to determine if modulating actin cytoskeleton dynamics in MK-5108–treated HLFs with Rho activator II, LPA, or S1P, can restore YAP nuclear localization and profibrotic gene expression. Although all these treatments increased actin fiber polymerization in MK-5108–treated HLFs both with and without TGFβ stimulation, only LPA restored nuclear YAP localization at 4 hours to an amount comparable to control cells treated with DMSO (Figures 5A and 5B). Rho activator II also increased stress fiber formation and partially restored YAP nuclear localization, but to a lesser extent than LPA (Figures 5A and 5B). LPA and Rho activator II were not as effective at 16 hours compared with 4 hours but were still able to restore some YAP nuclear localization (Figure 5B). S1P mainly increased F-actin cortical ring staining of the cells and only restored YAP nuclear localization at 4 hours, but not at 16 hours (Figures 5A and 5B). Interestingly, gene expression of CTGF and CYR61 could not be restored to control amounts after adding Rho activator II, LPA, or S1P. LPA was able to partially restore CYR61 expression at 4 hours, but not at 16 hours (Figures 5C and 5D).
Figure 5.
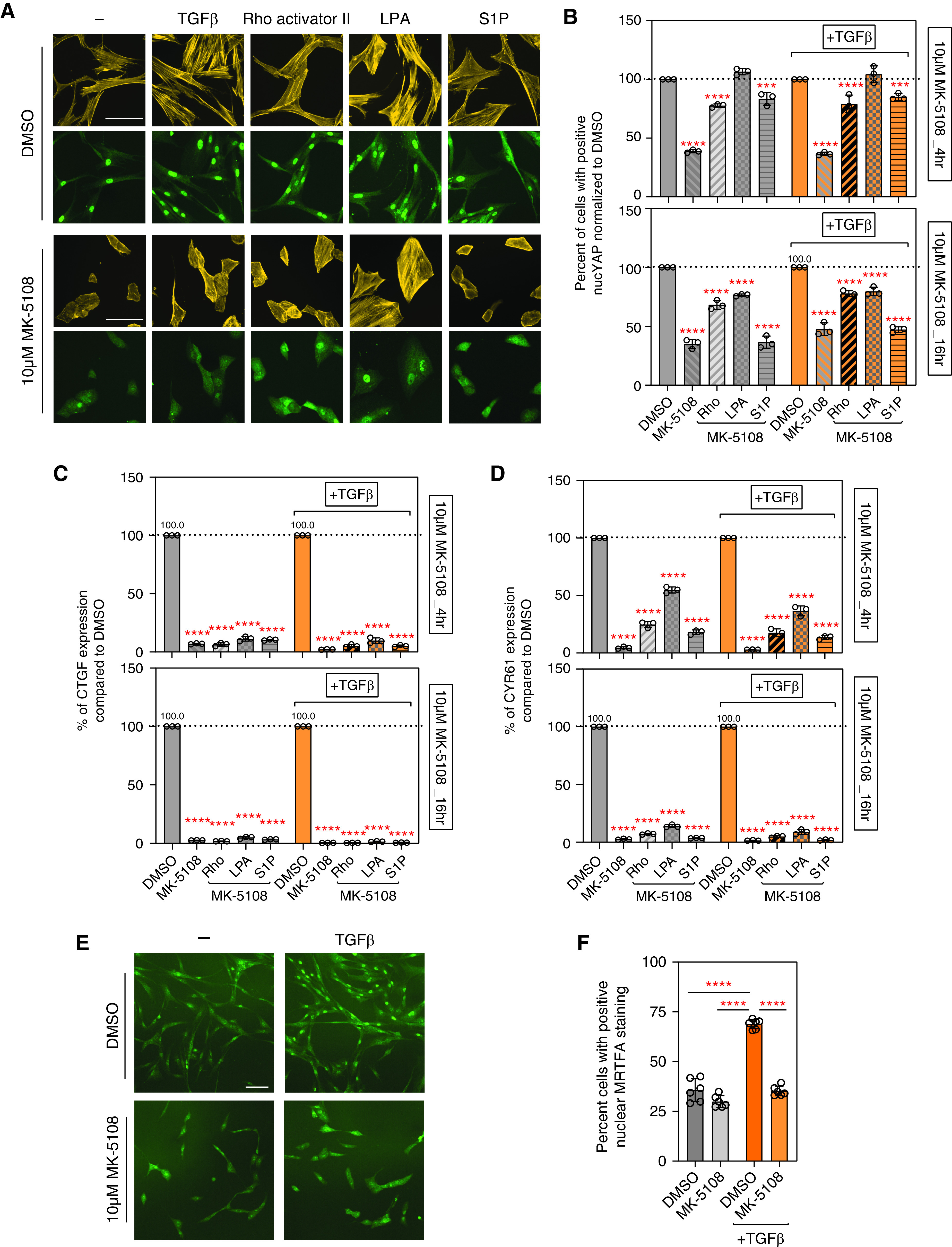
Aurora A inhibition inactivates YAP and decreases fibrotic gene expression partly through regulating actin polymerization. (A) Rhodamine phalloidin (yellow) and YAP (green) fluorescent images showing F-actin fibers and YAP localization after treating HLFs with DMSO or MK-5108, combined with TGFβ (5 ng/ml), Rho activator II (0.5 μg/ml), lysophosphatidic acid (LPA) (10 μM), or sphingosine 1-phosphophate (S1P) (1 μM) treatment. Scale bars, 10 μm. (B) Quantification of YAP nuclear localization in A. Percentage of cells with positive nuclear YAP staining normalized to control are shown as percentage of control value. Data represent mean ± SD, n = 3. ****P < 0.0001 and ***P < 0.001, one-way ANOVA with Sidak’s multiple comparisons test. (C and D) Profibrotic genes CTGF and CYR61 expression at 4 hours or 16 hours normalized to DMSO shown as percentage of control value, after treating HLFs with 10 μM MK-5108 alone or combined with TGFβ and Rho activator II, LPA, or S1P. Data represent mean ± SD, n = 3. ****P < 0.0001, one-way ANOVA with Sidak’s multiple comparisons test. (E) Fluorescent images (scale bar, 10 µm) and (F) quantifications showing MRTFA (Myocardin-Related Transcription Factor A) localization after treating HLFs with DMSO or MK-5108, combined with TGFβ (5 ng/ml). Scale bar, 10 μm. Data represent mean ± SD, n = 6. ****P < 0.0001, unpaired t test.
We reasoned that a high concentration of MK-5108 may have a strong and long-lasting effect that may be difficult to counteract with short-lived mediators such as LPA and S1P. Therefore, we tried using a lower concentration of MK-5108 (3 μM) in these assays. YAP nuclear localization was reduced by 3 μM MK-5108 at 4 hours and 16 hours. Rho activator II, LPA, or S1P all restored nuclear YAP localization to that of DMSO-treated control cells at 4 hours, but only Rho activator II and LPA could maintain nuclear YAP localization at 16 hours (Figure E3A). Furthermore, only LPA increased CTGF and CYR61 expression at 4 hours when added together with MK-5108. At 16 hours, LPA restored higher expression of CTGF and CYR61 than Rho activator II or S1P (Figures E3B and E3C). Interestingly, TGFβ could not increase YAP nuclear localization or profibrotic gene expression in MK-5108–treated HLFs, and MK-5108 treatment leads to lower SMAD2 phosphorylation after TGFβ treatment than control cells (Figure E3F).
It has been reported that AURKA promotes actin polymerization through inhibitory phosphorylation on Cofilin, an actin depolymerization factor (61). Consistent with this, we also observed a decrease in Cofilin phosphorylation in HLFs after MK-5108 treatment. In addition, siRNA knockdown of Cofilin partially increased YAP nuclear localization (Figures E3D and E3E). It is also notable that HLF cell morphology changed dramatically after MK-5108 treatment, which may reflect a change in cell–matrix adhesion. Therefore, we checked FAK (Focal Adhesion Kinase) activity after MK-5108 treatment. FAK is a nonreceptor tyrosine kinase that is activated by cell adhesion via pY397. As expected, MK-5108 treatment of HLFs also decreased the amount of FAK phosphorylated on tyrosine 397 (Figure E3D).
To further examine the effects of AURKA on downstream effects of changes to the actin cytoskeleton, we determined if MRTFA (Myocardin-Related Transcription Factor A) localization is altered after MK-5108 treatment of HLFs. MRTFA is a coactivator of the transcription factor SRF (Serum Response Factor) and binds to monomeric G-actin. Thus, MRTFA is sequestered in the cytoplasm if actin polymerization is reduced (62), thereby reducing the transcription of genes involved in fibroblast functions, including profibrotic activity. MRTFA nuclear localization after TGFβ stimulation of HLFs was reduced with MK-5108 treatment (Figures 5E and 5F). Overall, these data demonstrate that AURKA can modulate YAP nuclear localization through multiple mechanisms that affect actin polymerization and signaling.
MK-5108 Reduces Lung Collagen Deposition in a Bleomycin Mouse Model of Pulmonary Fibrosis
To determine if AURKA-specific inhibition by MK-5108 has antifibrotic effects in vivo, we used the bleomycin mouse model of pulmonary fibrosis (63). We injected mice intratracheally with 1 unit/kg of bleomycin and treated mice with either vehicle or 30 mg/kg MK-5108 twice a day for 2 weeks via oral gavage. At the end of the 2 weeks, the lungs were harvested and analyzed by hydroxyproline assay to measure the total collagen content (Figure 6A). Administering MK-5108 alone did not have adverse effects on body weight or change the baseline amount of hydroxyproline (Figures 6B and 6D). As expected, bleomycin injection induced lung fibrosis and increased hydroxyproline amount in vehicle-treated mice (Figures 6C and 6D). In contrast, mice treated with MK-5108 had lower hydroxyproline amount than mice treated with vehicle (Figures 6C and 6D). This result suggests that inhibiting AURKA in vivo protects mice from bleomycin-induced lung fibrosis.
Figure 6.
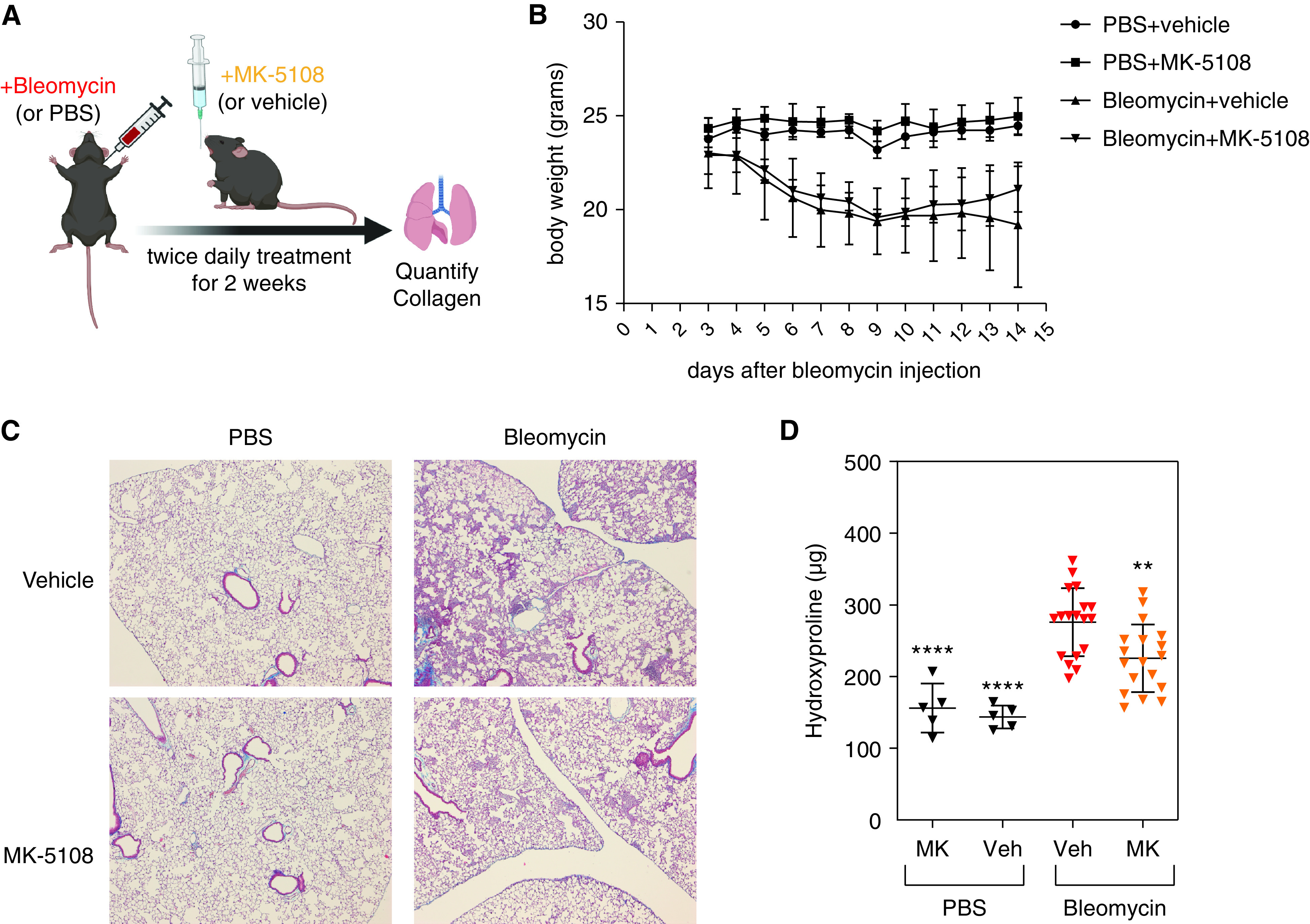
Aurora A–specific inhibition protects mice from bleomycin-induced lung fibrosis. (A) Scheme of administering bleomycin and MK-5108. Mice were given 1 unit/kg of bleomycin intratracheally. Starting the following day, 30 mg/kg MK-5108 was given by oral gavage two times a day for 14 days, before whole lungs were harvested and assayed for hydroxyproline amount. (B) Weight curve of mice treated with vehicle or MK-5108. (C) Masson’s trichrome staining (on 5X lung sections) showing significant reduction of hydroxyproline amount in the lungs of the mice treated with MK-5108. (D) Quantification of hydroxyproline content in the whole lungs of mice treated with vehicle or MK-5108, after injection of PBS or bleomycin. Data represent mean ± SD. **P < 0.01 and ****P < 0.0001, one-way ANOVA with Sidak’s multiple comparisons test.
Discussion
In this study, we used a previously validated high-throughput small-molecule screen (24) for modulators of YAP nuclear localization in HLFs to identify compounds with potential antifibrotic activity. This screen identified several Aurora kinase inhibitors as potential targets, including MK-5108, a highly selective AURKA inhibitor. Further studies show that inhibiting AURKA, but not AURKB or AURKC, regulates YAP activity. These effects were independent of the effects of AURKA on the mitotic cell cycle and did not depend on the canonical Hippo pathway kinases. In vitro studies demonstrate that inhibiting AURKA reduced actin polymerization and FAK activity and inhibited TGFβ signaling, which led to YAP cytoplasm sequestration and decreased profibrotic gene expression. Finally, we show that MK-5108 treatment reduces lung collagen deposition in the bleomycin mouse model of pulmonary fibrosis. To the best of our knowledge, this is the first description of the use of an AURKA inhibitor as a potential therapeutic agent for fibrotic disease.
YAP and TAZ proteins are transcriptional coactivators that act as mechanosensors. In a high-stiffness environment, YAP and TAZ are activated and translocate into the nucleus, where they bind with transcription factors to increase expression of genes promoting proliferation and growth. In IPF, fibroblasts in fibrotic areas of the lung have increased nuclear YAP and TAZ, consistent with a profibrotic phenotype (23). Mechanistically, YAP localization and activity can be regulated by phosphorylation in different amino acid residues. Phosphorylation on serine 127 sequesters YAP in the cytoplasm, and phosphorylation on serine 397 targets YAP for degradation. The Aurora kinases are highly conserved serine/threonine kinases that are important regulators of the cell cycle and cell division. Chang and colleagues showed that AURKA promotes YAP phosphorylation on serine 397, the same residue phosphorylated by the Hippo pathway kinases LATS1 and LATS2 (55). Furthermore, in lung cancer cells, AURKA expression was found to correlate with YAP expression and may modulate YAP activity via inhibition of autophagy (64). These studies suggest that AURKA may modulate YAP expression and activity; however, the mechanisms by which AURKA can control YAP localization are incompletely understood.
In our in vitro studies, we show that MK-5108 inhibits actin polymerization and FAK autophosphorylation, as well as TGFβ-induced SMAD2 phosphorylation. FAK and actin stress fiber formation are well-known factors contributing to mechanotransduction, and SMAD2 phosphorylation is a key mediator of canonical TGFβ signaling. These data suggest that AURKA indirectly promotes YAP nuclear translocation and profibrotic gene expression via a broad range of effects on actin polymerization and TGFβ signaling. Interestingly, there does not seem to be a direct effect of AURKA on YAP, although prior work does suggest that AURKA may phosphorylate YAP in the nucleus (55). It is also possible that some of these changes are due to off-target effects of MK-5108, which can occur with kinase inhibitors. However, we were able to show that siRNA-mediated knockdown of AURKA resulted in decreased YAP nuclear localization and TGFβ-induced profibrotic gene expressions, confirming that AURKA is required for robust YAP nuclear localization and TGFβ activity. Further studies are needed to identify the key targets of AURKA activity that mediate its profibrotic actions.
Given their role in cell proliferation, inhibition of the Aurora kinases has been well studied as a potential therapeutic for cancer (43, 44). However, the therapeutic potential of inhibiting Aurora kinases for treating fibrosis has not been well studied. Recent work has demonstrated that AURKB inhibition attenuates lung fibrosis induced by either TGFα or intradermal bleomycin injection (65). Our work provides a likely antifibrotic mechanism for AURKA inhibitors in IPF and suggests that further studies into the clinical potential of these agents in IPF may be warranted.
In conclusion, we have developed an effective screening platform for YAP inhibitors and validated an AURKA inhibitor as an antifibrotic agent. Our findings provide evidence that AURKA promotes YAP nuclear localization and TGFβ signaling activity in HLFs and suggest that AURKA inhibitors may have therapeutic value in IPF. Further study is needed to fully elucidate the utility of AURKA inhibition for treatment in lung fibrosis and other fibrotic diseases.
Acknowledgments
Acknowledgment
The authors thank the staff at the ICCB Longwood Screening Facility and Image and Data Analysis Core at Harvard Medical School for help with the small-molecule screen and the development of an image analysis custom module, respectively. They also thank the members of the Medoff lab at Massachusetts General Hospital for helpful comments.
Footnotes
Supported by Boehringer Ingelheim grant 228453 as part of a collaboration with the Harvard Fibrosis Network.
Author Contributions: Y.Y. designed and performed experiments, analyzed data, prepared the figures, and wrote the manuscript. D.M.S. designed and performed experiments, analyzed data, prepared figures, and edited the manuscript. L.P. developed the screen analysis and scoring system, analyzed screen data, and generated figures. R.K., E.A., A.L., G.P., and F.G. performed experiments. K.E.B. and J.J.S. collected and banked the human lung fibroblasts. M.B. characterized the screen hit set. C.G. provided medicinal chemistry and compound annotation support. P.N., D.W., P.S., and F.E.H conceived the project and provided intellectual input. A.M.T. conceived the project, provided intellectual input, and supervised the initial screening study. B.D.M. provided intellectual input, supervised the study, and edited the manuscript. All authors reviewed the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0428OC on April 4, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. American Thoracic Society; European Respiratory society; Japanese Respiratory Society; Latin American Thoracic Association An official ATS/ERS/JRS/ALAT Clinical Practice Guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 Clinical Practice Guideline Am J Respir Crit Care Med 2015192e3–e19.. [Published erratum appears in Am J Respir Crit Care Med 192:644.]26177183 [Google Scholar]
- 2. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med . 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 3. King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. ASCEND Study Group A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med . 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 4. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. INPULSIS Trial Investigators Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med . 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 5. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet . 2017;389:1941–1952. doi: 10.1016/S0140-6736(17)30866-8. [DOI] [PubMed] [Google Scholar]
- 6. Spagnolo P, Cottin V. Genetics of idiopathic pulmonary fibrosis: from mechanistic pathways to personalised medicine. J Med Genet . 2017;54:93–99. doi: 10.1136/jmedgenet-2016-103973. [DOI] [PubMed] [Google Scholar]
- 7. Spagnolo P, Kropski JA, Jones MG, Lee JS, Rossi G, Karampitsakos T, et al. Idiopathic pulmonary fibrosis: disease mechanisms and drug development. Pharmacol Ther . 2021;222:107798. doi: 10.1016/j.pharmthera.2020.107798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol 20149157–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis: aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med . 2014;190:867–878. doi: 10.1164/rccm.201403-0509PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parker MW, Rossi D, Peterson M, Smith K, Sikström K, White ES, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest . 2014;124:1622–1635. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med . 2012;186:866–876. doi: 10.1164/rccm.201204-0754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M, et al. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J . 2000;19:6778–6791. doi: 10.1093/emboj/19.24.6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J . 1999;18:2551–2562. doi: 10.1093/emboj/18.9.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature . 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 15. Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev . 2010;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang J, Wu S, Barrera J, Matthews K, Pan D. The hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell . 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 17. Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D, et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFbeta-TrCP E3 ligase. J Biol Chem . 2010;285:37159–37169. doi: 10.1074/jbc.M110.152942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP) Genes Dev . 2010;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev . 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choi HJ, Zhang H, Park H, Choi KS, Lee HW, Agrawal V, et al. Yes-associated protein regulates endothelial cell contact-mediated expression of angiopoietin-2. Nat Commun . 2015;6:6943. doi: 10.1038/ncomms7943. [DOI] [PubMed] [Google Scholar]
- 21. Zhang H, Pasolli HA, Fuchs E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci USA . 2011;108:2270–2275. doi: 10.1073/pnas.1019603108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Szeto SG, Narimatsu M, Lu M, He X, Sidiqi AM, Tolosa MF, et al. YAP/TAZ are mechanoregulators of TGF-β-Smad signaling and renal fibrogenesis. J Am Soc Nephrol . 2016;27:3117–3128. doi: 10.1681/ASN.2015050499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol . 2015;308:L344–L357. doi: 10.1152/ajplung.00300.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santos DM, Pantano L, Pronzati G, Grasberger P, Probst CK, Black KE, et al. Screening for YAP inhibitors identifies statins as modulators of fibrosis. Am J Respir Cell Mol Biol . 2020;62:479–492. doi: 10.1165/rcmb.2019-0296OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu H, Mi S, Li Z, Xiaoxi Lv, Li K, Hua F, et al. SB216763, a selective small molecule inhibitor of glycogen synthase kinase-3, improves bleomycin-induced pulmonary fibrosis via activating autophagy. Acta Pharm Sin B . 2013;3:226–233. [Google Scholar]
- 26. Hu M, Che P, Han X, Cai GQ, Liu G, Antony V, et al. Therapeutic targeting of SRC kinase in myofibroblast differentiation and pulmonary fibrosis. J Pharmacol Exp Ther . 2014;351:87–95. doi: 10.1124/jpet.114.216044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine . 2019;40:554–563. doi: 10.1016/j.ebiom.2018.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lukey PT, Harrison SA, Yang S, Man Y, Holman BF, Rashidnasab A, et al. A randomised, placebo-controlled study of omipalisib (PI3K/mTOR) in idiopathic pulmonary fibrosis. Eur Respir J . 2019;53:1801992. doi: 10.1183/13993003.01992-2018. [DOI] [PubMed] [Google Scholar]
- 29. Hettiarachchi SU, Li YH, Roy J, Zhang F, Puchulu-Campanella E, Lindeman SD, et al. Targeted inhibition of PI3 kinase/mTOR specifically in fibrotic lung fibroblasts suppresses pulmonary fibrosis in experimental models. Sci Transl Med . 2020;12:eaay3724. doi: 10.1126/scitranslmed.aay3724. [DOI] [PubMed] [Google Scholar]
- 30. Verstovsek S. Therapeutic potential of JAK2 inhibitors. Hematology Am Soc Hematol Educ Program . 2009:636–642. doi: 10.1182/asheducation-2009.1.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fu J, Bian M, Jiang Q, Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res . 2007;5:1–10. doi: 10.1158/1541-7786.MCR-06-0208. [DOI] [PubMed] [Google Scholar]
- 32. Andrews PD. Aurora kinases: shining lights on the therapeutic horizon? Oncogene . 2005;24:5005–5015. doi: 10.1038/sj.onc.1208752. [DOI] [PubMed] [Google Scholar]
- 33. Sorrentino R, Libertini S, Pallante PL, Troncone G, Palombini L, Bavetsias V, et al. Aurora B overexpression associates with the thyroid carcinoma undifferentiated phenotype and is required for thyroid carcinoma cell proliferation. J Clin Endocrinol Metab . 2005;90:928–935. doi: 10.1210/jc.2004-1518. [DOI] [PubMed] [Google Scholar]
- 34. Chieffi P, Troncone G, Caleo A, Libertini S, Linardopoulos S, Tramontano D, et al. Aurora B expression in normal testis and seminomas. J Endocrinol . 2004;181:263–270. doi: 10.1677/joe.0.1810263. [DOI] [PubMed] [Google Scholar]
- 35. Araki K, Nozaki K, Ueba T, Tatsuka M, Hashimoto N. High expression of Aurora-B/Aurora and Ipll-like midbody-associated protein (AIM-1) in astrocytomas. J Neurooncol . 2004;67:53–64. doi: 10.1023/b:neon.0000021784.33421.05. [DOI] [PubMed] [Google Scholar]
- 36. D’Assoro AB, Haddad T, Galanis E. Aurora-A kinase as a promising therapeutic target in cancer. Front Oncol . 2015;5:295. doi: 10.3389/fonc.2015.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ohmine S, Salisbury JL, Ingle J, Pettinato G, Haddox CL, Haddad T, et al. Aurora-A overexpression is linked to development of aggressive teratomas derived from human iPS cells. Oncol Rep . 2018;39:1725–1730. doi: 10.3892/or.2018.6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reichardt W, Jung V, Brunner C, Klein A, Wemmert S, Romeike BF, et al. The putative serine/threonine kinase gene STK15 on chromosome 20q13.2 is amplified in human gliomas. Oncol Rep . 2003;10:1275–1279. [PubMed] [Google Scholar]
- 39. Tanaka T, Kimura M, Matsunaga K, Fukada D, Mori H, Okano Y. Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res . 1999;59:2041–2044. [PubMed] [Google Scholar]
- 40. Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, et al. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res . 2003;9:1420–1426. [PubMed] [Google Scholar]
- 41. Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet . 1998;20:189–193. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 42. Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J . 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bavetsias V, Linardopoulos S. Aurora kinase inhibitors: current status and outlook. Front Oncol . 2015;5:278. doi: 10.3389/fonc.2015.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Borisa AC, Bhatt HG. A comprehensive review on Aurora kinase: small molecule inhibitors and clinical trial studies. Eur J Med Chem . 2017;140:1–19. doi: 10.1016/j.ejmech.2017.08.045. [DOI] [PubMed] [Google Scholar]
- 45. Yang Y, Santos DM, Pantano L, Knipe R, Abe E, Logue A, et al. A YAP inhibitor screen identifies the Aurora kinase A inhibitor MK-5108 as a modulator of lung fibrosis [abstract] Am J Respir Crit Care Med . 2021;203:A4443. [Google Scholar]
- 46. Littlepage LE, Wu H, Andresson T, Deanehan JK, Amundadottir LT, Ruderman JV. Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora-A. Proc Natl Acad Sci USA . 2002;99:15440–15445. doi: 10.1073/pnas.202606599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Görgün G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood . 2010;115:5202–5213. doi: 10.1182/blood-2009-12-259523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev . 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou D, Zhang Y, Wu H, Barry E, Yin Y, Lawrence E, et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc Natl Acad Sci USA . 2011;108:E1312–E1320. doi: 10.1073/pnas.1110428108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chan EH, Nousiainen M, Chalamalasetty RB, Schäfer A, Nigg EA, Silljé HH. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene . 2005;24:2076–2086. doi: 10.1038/sj.onc.1208445. [DOI] [PubMed] [Google Scholar]
- 51. Cho YS, Zhu J, Li S, Wang B, Han Y, Jiang J. Regulation of Yki/Yap subcellular localization and Hpo signaling by a nuclear kinase PRP4K. Nat Commun . 2018;9:1657. doi: 10.1038/s41467-018-04090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhao Y, Khanal P, Savage P, She YM, Cyr TD, Yang X. YAP-induced resistance of cancer cells to antitubulin drugs is modulated by a Hippo-independent pathway. Cancer Res . 2014;74:4493–4503. doi: 10.1158/0008-5472.CAN-13-2712. [DOI] [PubMed] [Google Scholar]
- 53. Moon S, Kim W, Kim S, Kim Y, Song Y, Bilousov O, et al. Phosphorylation by NLK inhibits YAP-14-3-3-interactions and induces its nuclear localization. EMBO Rep . 2017;18:61–71. doi: 10.15252/embr.201642683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hong AW, Meng Z, Yuan HX, Plouffe SW, Moon S, Kim W, et al. Osmotic stress-induced phosphorylation by NLK at Ser128 activates YAP. EMBO Rep . 2017;18:72–86. doi: 10.15252/embr.201642681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chang SS, Yamaguchi H, Xia W, Lim SO, Khotskaya Y, Wu Y, et al. Aurora A kinase activates YAP signaling in triple-negative breast cancer. Oncogene . 2017;36:1265–1275. doi: 10.1038/onc.2016.292. [DOI] [PubMed] [Google Scholar]
- 56. Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev . 2012;26:54–68. doi: 10.1101/gad.173435.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sansores-Garcia L, Bossuyt W, Wada K, Yonemura S, Tao C, Sasaki H, et al. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J . 2011;30:2325–2335. doi: 10.1038/emboj.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fernández BG, Gaspar P, Brás-Pereira C, Jezowska B, Rebelo SR, Janody F. Actin-Capping Protein and the Hippo pathway regulate F-actin and tissue growth in Drosophila. Development . 2011;138:2337–2346. doi: 10.1242/dev.063545. [DOI] [PubMed] [Google Scholar]
- 59. Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell . 2013;154:1047–1059. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 60. Miller E, Yang J, DeRan M, Wu C, Su AI, Bonamy GM, et al. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem Biol . 2012;19:955–962. doi: 10.1016/j.chembiol.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 61. Ritchey L, Chakrabarti R. Aurora A kinase modulates actin cytoskeleton through phosphorylation of Cofilin: Implication in the mitotic process. Biochim Biophys Acta . 2014;1843:2719–2729. doi: 10.1016/j.bbamcr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mizuguchi M, Fuju T, Obita T, Ishikawa M, Tsuda M, Tabuchi A. Transient α-helices in the disordered RPEL motifs of the serum response factor coactivator MKL1. Sci Rep . 2014;4:5224. doi: 10.1038/srep05224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jenkins RG, Moore BB, Chambers RC, Eickelberg O, Königshoff M, Kolb M, et al. ATS Assembly on Respiratory Cell and Molecular Biology An official American Thoracic Society Workshop report: use of animal models for the preclinical assessment of potential therapies for pulmonary fibrosis. Am J Respir Cell Mol Biol . 2017;56:667–679. doi: 10.1165/rcmb.2017-0096ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang P, Gong Y, Guo T, Li M, Fang L, Yin S, et al. Activation of Aurora A kinase increases YAP stability via blockage of autophagy. Cell Death Dis . 2019;10:432. doi: 10.1038/s41419-019-1664-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kasam RK, Ghandikota S, Soundararajan D, Reddy GB, Huang SK, Jegga AG, et al. Inhibition of Aurora Kinase B attenuates fibroblast activation and pulmonary fibrosis. EMBO Mol Med . 2020;12:e12131. doi: 10.15252/emmm.202012131. [DOI] [PMC free article] [PubMed] [Google Scholar]