

Abstract
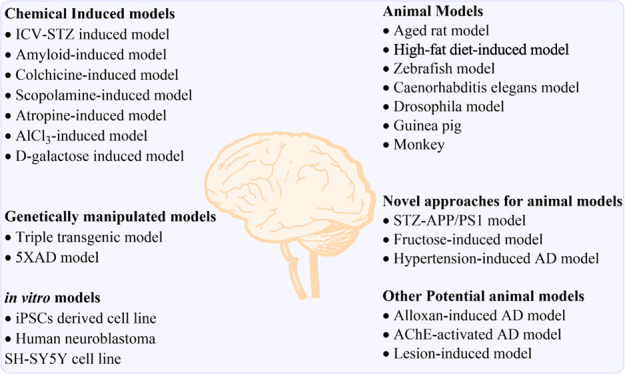
A robust preclinical disease model is a primary requirement to understand the underlying mechanisms, signaling pathways, and drug screening for human diseases. Although various preclinical models are available for several diseases, clinical models for Alzheimer’s disease (AD) remain underdeveloped and inaccurate. The pathophysiology of AD mainly includes the presence of amyloid plaques and neurofibrillary tangles (NFT). Furthermore, neuroinflammation and free radical generation also contribute to AD. Currently, there is a wide gap in scientific approaches to preventing AD progression. Most of the available drugs are limited to symptomatic relief and improve deteriorating cognitive functions. To mimic the pathogenesis of human AD, animal models like 3XTg-AD and 5XFAD are the primarily used mice models in AD therapeutics. Animal models for AD include intracerebroventricular-streptozotocin (ICV-STZ), amyloid beta-induced, colchicine-induced, etc., focusing on parameters such as cognitive decline and dementia. Unfortunately, the translational rate of the potential drug candidates in clinical trials is poor due to limitations in imitating human AD pathology in animal models. Therefore, the available preclinical models possess a gap in AD modeling. This paper presents an outline that critically assesses the applicability and limitations of the current approaches in disease modeling for AD. Also, we attempted to provide key suggestions for the best-fit model to evaluate potential therapies, which might improve therapy translation from preclinical studies to patients with AD.
1. Introduction
Alzheimer’s is a progressive neurodegenerative disease characterized by the deposition of amyloid plaques and neurofibrillary tangles (NFT) in the brain regions of the cerebral cortex and hippocampus, which are the major pathological hallmarks of the disease. The common symptoms of the disease include memory loss, impairment in learning, and retarded intellectual and thinking ability. However, the number of patients with AD-related dementia is expected to reach 152 million globally by the year 2050, with the highest growth anticipated in low-to-middle-income nations.1 According to 2020 statistics, AD patients (≤65 years) in the United States may increase dramatically from 5.8 million to 13.8 million by 2050.2 Further population studies conducted in Japan and China showed a marked increase in AD prevalence in the past several years.3
Additionally, various risk factors contribute to AD progression and symptoms, as shown in Figure 1. Therefore, a proper diagnosis is crucial for patients with cognitive impairment. Significantly, amyloid precursor protein (APP)-mediated generation of amyloid-β (Aβ) and hyperphosphorylated tau protein-generated neurofibrillary tangles are the major pathological hallmarks of AD. Hence, Aβ and tau are the indicative biomarkers of AD. Still, healthy individuals with these biomarkers at normal or above-normal levels often do not show signs and symptoms of AD,4 making it challenging to get a presymptomatic diagnosis. Future obstacles would include the discovery of less invasive and more sensitive biomarkers or procedures that can also be employed for early detection and diagnosis. In any case, future research must investigate evidence-based preventative techniques consistent with the apparent relationship between modifiable risk variables and late-onset AD.
Figure 1.
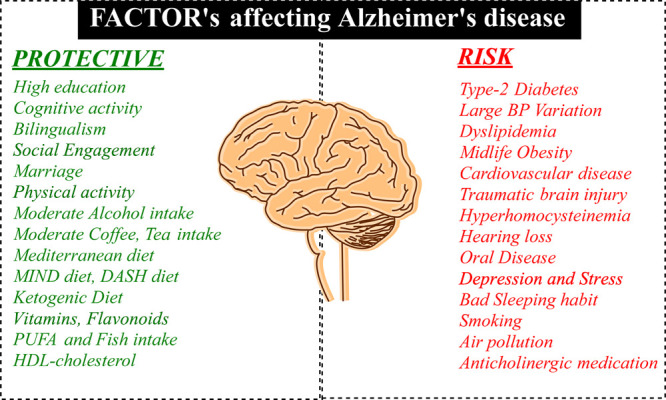
Potential risk factors for AD (PUFA = polyunsaturated fatty acid; HDL-cholesterol = high-density lipoprotein cholesterol).6
According to data from prospective population-based research, the majority of risk variables were pre-existing diseases, poor lifestyles, and environmental exposures that significantly impact the likelihood of dementia in old age.5 Still, psychological circumstances and healthy lifestyles may protect against AD. In addition, many elements appeared to represent both AD risk factors and symptoms, presumably due to reverse causality; these factors are emphasized in Figure 1.6 Late-onset AD is a complex genetic disorder with a 60–80% hereditary rate. APOE genotype is the most significant risk factor for late-onset AD.7
Extracellular amyloid plaques formed by amyloid precursor protein (APP) and intracellular neurofibrillary tangles resulting from hyperphosphorylated tau protein in the cortical and hippocampal regions are the key pathological hallmarks of AD.8 The catalytic proteases involved in APP proteolysis include α-, β-, and γ-secretases. This process forms the basis of the amyloidogenic pathway with β- and γ-secretase and the nonamyloidogenic pathway with α-secretase, generating insoluble and soluble neurotic amyloid plaques, respectively (Figure 2).9
Figure 2.

Amyloidogenic and nonamyloidogenic pathways.
Tau contains 441 amino acids, where 85 potential amino acids (serine, threonine, and tyrosine) are available for phosphorylation. Typically, two to three locations of tau are reported to be phosphorylated in normal physiological conditions. However, tau hyperphosphorylation is a pathological condition having phosphorylation at almost six positions (as shown in Figure 3).10
Figure 3.
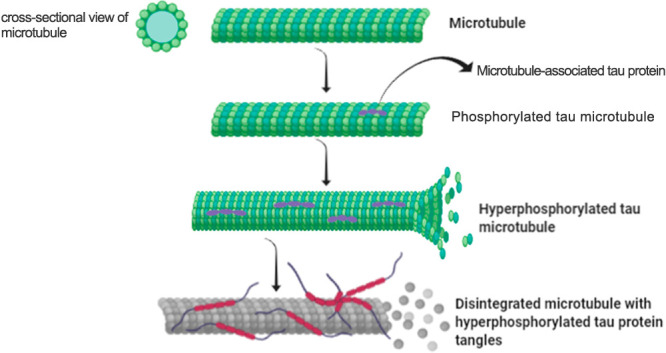
Hyperphosphorylated tau protein-mediated tangles.
Notably, AD has been demonstrated to depict underlying mechanisms through numerous cascades, including reactive oxidative species or oxidative stress, mitochondrial dysfunction, cholinergic dysfunction, neuro-inflammation, glucose metabolism impairment, and, most importantly, insulin signaling pathway dysregulation or insulin resistance11 (as shown in Figure 4). Therefore, targeting these pathways through an inhibitory way can be advantageous and beneficial to minimize AD-related deterioration and symptoms and also retard the neurodegenerative process and disease progression in an advanced case. Animal models to carry over these harmful mechanisms (oxidative stress, mitochondrial dysfunction, cholinergic dysfunction, neuroinflammation, and insulin resistance) will be suitable for finding newer therapeutic interventions and pharmacological treatments against AD.
Figure 4.

Various pathophysiological pathways of Alzheimer’s disease.
The currently approved drugs are either acetylcholinesterase inhibitors (such as rivastigmine, donepezil, and galantamine) or NMDA receptor antagonists (like memantine). However, these drugs only have limited symptomatic relief but fail to prevent AD progression. Several newer molecules that have been identified for AD in the last four decades act by preventing amyloid deposition in the brain and removing existing amyloid plaques along with other recognized mechanisms associated with the disease.12 Although these compounds demonstrate promising data from preclinical AD models, the clinical failure rate in AD treatment is almost 100%. Several factors can be alleged for the failure, but the primary concern remains with the preclinical models used at the preliminary stages of drug development. This indicates the gap between the preclinical data curation and their translational value. The flawed and incomplete preclinical evaluation of candidate drugs for AD may be the cause of the molecules’ blip into clinical trials. Hence, in the context of the significant shortcomings of preclinical models, the reasons for poor translation to clinical outcome could be poor hypothesis or target selection, model selection, validation, and pharmacodynamic/pharmacokinetic (PD/PK) characterization of novel drugs.
Moreover, on the basis of amyloid theory and genetics of AD, a rising dependency on transgenic AD models possessing the targets of amyloid plaque and tau protein has been witnessed.13 Additionally, the mouse brain produces amyloid peptides distinct from the human brain, and the mouse model, even with amyloid deposition, often fails to show a substantial neuronal loss.14 Moreover, comorbidities associated with human AD are not well-mimicked in animal models. Despite numerous limitations associated with the preclinical AD model, these models can somewhat predict the pathophysiology and therapeutic targets. However, selecting a suitable model system for the specific target/pathway with scientific rationalization can reduce the chances of failure in drug screening. This review aims to provide insight into the rationalization and selection of animal models for AD. The objective is to discuss the current and future animal models for AD and their role in exploring pathophysiology/drug development. Despite decent face validity, transgenic models usually lack complete content and predictive validity. In this review, we have discussed the current preclinical AD models used in drug screening for AD. This review should be helpful in guiding researchers toward the rational selection of preclinical models for AD.
2. Chemically Induced Central Administration
2.1. ICV-STZ-Induced Model
Sporadic Alzheimer’s disease (SAD) is a multifactorial disease caused by genetic, epigenetic, environmental, and metabolic factors. Among metabolic factors, impaired glucose metabolism and energy utilization are observed in the initial stages of disease progression.15 In this regard, the animal models have made considerable progress in unveiling the molecular pathways involved in AD’s pathogenesis; thus, it has led to the development of potential therapeutic approaches for AD. One such animal model that is widely used is to produce insulin signaling impairment through intracerebroventricular (ICV) streptozotocin (STZ) administration.
There is an abundance of insulin receptors in brain regions like the cortex, hippocampus, hypothalamus, olfactory bulb, etc.; clinical reports showed downregulation of insulin, insulin receptors, and insulin receptor substrates under the influence of ICV-STZ-mediated insulin resistance.11,16−18 Other AD phenomena associated with the ICV-STZ model include oxidative stress, mitochondrial dysfunction, cholinergic dysfunction, and neuroinflammation. These factors are considered to be the triggering points of neurodegeneration and are adjacently interlinked with insulin resistance.11,16−18 Hence, these characteristics associated with the neuropathology of the ICV-STZ model have the potential to lead to cognitive deficit and memory impairment, signifying a strong reason to validate the sporadic AD model. Various studies have been carried out in the past and recently to confirm and validate the ICV-STZ-induced SAD model and its potential to generate brain insulin resistance, as seen in SAD patients.11,16,17 Hence, it aims to provide insights into the molecular mechanisms involved in causing brain IR-induced AD by ICV-STZ, and further, its advantages and limitations are elaborated. Recent reports of the ICV-STZ model have depicted the downregulation of the α7-nicotinic acetylcholine receptor (α7AchR). This report has shown it to attenuate disease pathology with the insulin-sensitizing agent metformin signifying the role of the cholinergic and insulin signaling pathway in the ICV-STZ model of AD.19
2.2. Amyloid-Induced Model
As a replacement to ICV-STZ, the amyloid-β-42, amyloid-β-40, and amyloid-β-25–30 can be administered via the intracerebroventricular or intrahippocampal route. These amyloids have been subcategorized based on the number of amino acids possessed by them. Moreover, they have different degrees of pathogenesis in AD, where amyloid-β-42 is the most pathogenic one. The ICV-Aβ injection has been proved to drive the neurodegenerative process and impairment in learning and memory. This outcome could happen through normalizing oxidative and nitrosative parameters.20 There has been an excessive generation of reactive oxygen species (ROS) due to amyloid-β.21 Moreover, APP processing is enhanced in an Aβ-42 model of AD, creating more deposits of senile plaques.22 There is a downregulation of nicotinic acetylcholine receptors in an amyloid-induced model. This downregulation leads to cholinergic dysfunction.23 Amyloid also has a role in transgenic mouse models.24 Further, the central administration of Aβ also leads to the formation of tau-related tangles. Mitochondrial dysfunction through oxidative stress pathways can also arise.25 Amyloid is one of the core pathological hallmarks of AD, has been made to be targeted through a monoclonal antibody aducanumab in the latest development for the therapy of AD, and, hence, has been granted accelerated approval by the U.S. Food and Drug Administration (FDA). This approval is significant versus other conventional treatments of AD as this antibody acts as an antiamyloid-β factor and thereby can halt the progression of the disease pathology and neurodegeneration.26
3. Chemically Induced Oral Administration
3.1. Colchicine-Induced Model
Colchicine has been clinically used for gout treatment and is naturally obtained from a plant species (Colchicum autumnale). It has a unique capacity to bind to microtubule-associated tubulin protein. It causes the destabilization of microtubules and is generally administered through the oral route for treatment.27 However, a dose of 15 μg in a 5 μL vehicle, like distilled water in rats, produces cognitive impairment. The cognitive impairment produced is quite comparable to sporadic AD upon intracerebroventricular administration. Colchicine destroys oxidative balance and cholinergic pathways and aggravates the neuroinflammatory pathways responsible for synaptic dysfunction and neurodegeneration.28 Cycloxygenase-2 (COX-2), prostaglandinE2 (PGE2), interleukin-β (IL-1β), and tumor necrosis factor-α (TNF-α) might be responsible for inflammatory action in the colchicine-induced model. Furthermore, microtubules, the central building part of the axonal and neuronal cytoskeleton, cause significant deterioration, thereby paving the way to neuronal death.29
4. Chemically Induced Intraperitoneal Administration
4.1. Scopolamine-Induced Model
Scopolamine is a tropane alkaloid, also known as hyoscine. It is a potent anticholinergic drug obtained from Hyoscyamus niger. It is generally used before traveling to prevent motion sickness and after surgery to check nausea and vomiting. It is a competitive inhibitor of muscarinic receptors and is useful in many cholinergic-related discomforts and side effects such as increased bowel movements, salivation, lacrimation, sweating, etc.30 Acetylcholine is one of the most crucial neurotransmitters in memory processing by strengthening synaptic connections; hence, scopolamine-mediated blockage of cholinergic nervation is widely used as an animal model of AD. Scopolamine acts by the increased acetylcholinesterase (AchE) activity, enhancing the breakdown of acetylcholine. The dose of scopolamine is ∼2 mg/kg intraperitoneally for an AD model. In this process, scopolamine disrupts several brain regions’ connectivities like spatial memory mapping and functional network.31 The advantage of the scopolamine-induced model is the avoidance of complex surgical procedures like in an ICV model. Furthermore, cholinergic drugs like donepezil and rivastigmine and antioxidants like melatonin have been demonstrated to reverse scopolamine-induced memory impairment, proving the additional involvement of the oxidative stress pathway.32,33 Therefore, this model is mainly preferred for creating preventive options in AD treatment.34
4.2. Atropine-Induced Model
Atropine is also an alkaloidal origin drug obtained from Atropa belladonna and has been used as an anticholinergic drug to treat low heart rate and myopia. Atropine, similar to scopolamine, invades the cholinergic pathway, reducing the muscarinic Ach receptor’s hypofunction. It also blocks the nicotinic one up to a minor extent.35,36 Atropine in a dose of 5 mg/kg intraperitoneally (ip) for 21 days generated amyloid plaques, a pathological hallmark of AD. This process could result from an interlink between the cholinergic pathway and amyloidogenesis.35,37 Furthermore, the reduced release of acetylcholine inflicted by Aβ and vice versa was observed.38
4.3. Aluminum Chloride-Induced Model
Aluminum is an element that in excess causes numerous toxicities. An AD model can be established in rats or mice by ip injection of 4 mg/kg or 40 mL/kg per day of aluminum chloride (AlCl3) for nearly 40 days.39 The weighty triggers in an AlCl3 model are oxidative stress and mitochondrial dysfunction, which are reported to appear by inhibiting the NADH dehydrogenase enzyme of the electron transport chain.40 These phenomena were precisely reported in memory centers of the cortex and hippocampus. Further, neuroinflammatory mediators, including iNOS, NF-κB, COX-2, and proinflammatory cytokines, are altered in an AlCl3 model, leading to neurodegeneration. Additionally, any aluminum salt in doses of 100 mg in 1 day or 20 mg in 5 days has also been found to induce AD-associated neurotoxicity.41 Further, Al salts also cause cholinergic dysfunction and oxidative stress, leading to the apoptotic process.42 This model has also been preferred for prophylaxis treatment of AD. In this way, the therapeutic agents can be made available as a preventive measure rather than a protective one by utilizing this model.43
5. Chemically Induced Subcutaneous Administration
5.1. d-Galactose-Induced Model
d-Galactose is a monosaccharide in dairy products, avocados, sugar beets, etc. (e.g., milk contains 7.12 mg of galactose per 100 g; 100 g of avocado contains 0.66 g of sugar, which includes glucose, fructose, sucrose, and galactose; and sugar beet has 0.65% galactose). The metabolism of d-galactose produces ROS.44d-Galactose in doses of 50, 100, and 200 mg/kg through the subcutaneous route increased escape latency in the Morris water maze (MWM) and lowered the discrimination index in the novel object recognition (NOR) test in a dose-dependent way. This increment shows impaired spatial and recognition memory. Further, it increases oxidative stress in the hippocampus.45,46 There are reports of the attenuated immune system upon d-galactose treatment mimicking an aging brain.45,47 It also halts neurogenesis in the hippocampus and dentate gyrus regions of the brain48 and disrupts calcium homeostasis in the cortex and hippocampus, creating excitotoxicity conditions similar to those found in dementia cases.49 This model can be used in insulin resistance-associated AD cases because d-galactose is a sugar and produces an insulin-resistance-like state.
6. Genetically Manipulated Model
6.1. Triple Transgenic Model
The triple transgenic model is a model of an inherited familial form of AD involving mutations on three genes, such as APP on chromosome 21, presenilin 1 on chromosome 14, and p-tau in mice, hence named the triple transgenic model. The mutations in these genes might lead to AD pathogenesis because AAP and tau are linked to amyloid plaques and NFT, respectively, whereas presenilin 1 is the proteolytic subunit of γ-secretase (involved in APP cleavage). For the development of the model, transgenes encoding the mentioned ones are microinjected into mice. The mutations can happen by knocking in APP-Swe, PS1-M146 V, and tau-P301L. Crossing the mutant mice can also result in a familial AD (FAD) model.50,51 In addition, the development of both amyloid oligomers and paired helical filaments of tau was studied.52
Further, this transgenic model demonstrates brain atrophy, synaptic disruption, and neuronal death and cannot regenerate neurons in the areas of the prefrontal cortex, hippocampus, and dentate gyrus, leading to cognitive decline and memory impairment.53 Both spatial and recognition memory were found to be impaired in transgenic mice. Other than cognitive impairment, phenotypic alterations were also reported due to mutated mice.54 A transgenic mouse with mutations at APP and presenilin 2 on chromosome 1 or only at APP can also be created with closely related features to FAD.55 Even though the mouse models of APPG-F for BACE1 inhibitors (BACE1 is beta-site APP-cleaving enzyme 1 responsible for breaking down APP) and APPP11JL for immunotherapies have been discovered, these are not suitable for the more prevalent sporadic AD.56
6.2. 5XAD Model
5XAD is another transgenic mouse model of FAD, denoting mutations in five genes. 5XAD expresses APP695 with S-K670N, S-M671L, F-I716 V, and L-V717I mutant genes.57 These genes are the types of APP mutations expressed in mice. The mutations in these genes lead to AD pathology. These mutations result in the excessive production of senile plaques from APP. Moreover, this model also represents gliosis, synaptic disruption, and neuronal death.58 The model depicts the features of AD earlier than in other transgenic models; however, phosphorylated tau pathology is less prevalent than amyloid plaques in this model.52 Further, proinflammatory cytokines and immune markers through microglial and caspase-3 activation in the brain regions of the cortex and hippocampus have been reported, indicating neuroinflammation and subsequent apoptosis-generated59 neurodegeneration.60
7. Animal Models without Chemical Induction or Genetic Modification
7.1. Aged Rat Model
Compared to younger ones, aged rats show damage triggered naturally in the hippocampus, temporal lobe, and neocortex, which subsequently causes impairment in learning and memory. This model is preferred over other chemical-induced models due to its noninvasive influence and mimicking late-onset/aged sporadic AD pathological symptoms. The age range of rats used for this model could be taken between 15 and 20 months old. This model is more relevant considering the disease’s clinical aspects.61 Further, aging-induced dementia has depicted neuroinflammatory cytokines, oxidative stress, insulin resistance, and mitochondrial dysfunction resulting from an old age-related phenomena like glucose and energy metabolism, obesity, physical inactivity, etc. This model also produces the condition of amyloidogenesis and tau pathology comparable to other models of AD. Exercise, intermittent fasting, and several other antiaging measures have been proven to reverse these detrimental features of AD, leading to improved synaptic plasticity and memory formation.62
7.2. High-Fat Diet-Induced Model
A high-fat diet is widely used to create a model for insulin resistance, obesity, and diabetes mellitus. However, in several recent research reports, it has also been recommended to be designated as a cognitive dysfunction model. In addition to the peripheral distortion of insulin sensitivity, providing fat-loaded diets for almost 10–14 weeks to the rats or mice instead of a regular diet also potentially induces central insulin resistance up to some extent.63,64 The fatty diet comprises 25% fat, 20% protein, and 50% carbohydrate.64 Dementia and AD have long been characterized to possess distorted brain insulin signaling. This model has a core feature of insulin resistance that is relevant to evaluating memory and improving therapeutic interventions.
Moreover, high-fat diet-induced obesity hampers proper blood flow to the brain regions, reducing oxygen and glucose supply and resulting in vascular dementia. Besides, hypertension and diabetes-induced cognitive decline has also been reflected in situations of a high amount of dietary fat intake.65 Fat-associated cholesterol has a role in the generation of senile plaques by upregulating APP, which is accountable for neuronal loss.66 The other mechanisms involved in memory loss could also be an imbalance in lipid profile and glucose-transport interference. The high-fat diet AD model also exacerbates oxidative stress and neuroinflammation through decreased antioxidant enzymes and increased proinflammatory cytokines.67
8. Animal Models Other than Rat/Mouse Species
8.1. Zebrafish Model
Zebrafish are a freshwater fish found in tropical areas. It has been considered a comfortable and conspicuous model for cellular, molecular, and genetic studies as they have a conspicuous molecular structure and the cellular network is not complex. Mutant genes of APP and presenilin orthologues have been discovered in zebrafish embryos, making it a relevant FAD model.68 This model has numerous advantages over rodent models, such as the optically transparent embryo structure. Also, fewer neurons form a clear picture of the neuronal network, rapid neurogenesis, neuronal development, significant reproductive behavior, and swift manipulations of genetic makeup.69 Smaller size, simpler tissue organization, and high fertility rate make it even more appropriate for high-throughput screening of novel drugs.70 However, limitations include a higher mortality rate, difficulty in maintenance, and lesser resemblance to human physiology than rodents. Apart from natural mutants in zebrafish, artificially created mutations have also been revealed.71 Several genome editing tools in zebrafish include zinc finger nuclease, transcription activator-like effector nuclease, CRISPR, etc. Both knock-out and knock-in methods can be used for genetic manipulations in zebrafish that are suitable for studies on neurodegenerative diseases, specifically AD.72
8.2. Caenorhabditis elegans Model
C. elegans belong to nematodes, and in the last few decades, it has been extensively used as a model for studying human diseases, specifically neurodegenerative diseases. Concerning its anatomy, it is transparent and quite less complicated as compared to rodents and humans. It has almost 40% ortholog genes of APP and tau and has a pivotal role in AD pathogenesis,73 making it appropriate for the revelations of AD genomic-level research. The model’s other advantages are its high breeding power, lower food requirement, and visible neurons under a microscope. However, the model’s disadvantages are a short life span and a smaller size to handle. This is a reliable model for evaluating spatial memory and exploratory behavior.74
Furthermore, the synapses of C. elegans are flexible to modify. Hence, agents that are potentially investigated to target synaptic functions and behavioral parameters find a space here and determine synaptic plasticity and memory processing.75 A transgenic C. elegans model has also been established for studying amyloidogenesis linked to FAD.76
8.3. Drosophila Model
The fruit fly Drosophila melanogaster is another model for cellular and molecular findings of neurodenerative diseases and, thus, is helpful as an AD model. This model better mimics symptoms of sporadic or late-onset AD. The model primarily involves the expression of APP, BACE-1, presenilin, and tau orthologs resulting in amyloid aggregation in the model brain, leading to neurodegeneration and memory loss.77,78 Here, the tau ortholog is the homologous tau genes of Drosophila with humans. Further, presenilin (proteolytic subunit of γ-secretase) regulates the cleavage of other proteins like APP, the mutation of which can generate AD pathology. Additionally, it is also applicable in several other neurodegenerative diseases’ biochemical studies due to its short life span and rapid generation ability.79 The neuronal network in Drosophila has made it a robust model for understanding memory acquisition and consolidation mechanisms. The orthologs found in the Drosophila genome are AAPl and dBACE, where APPl has nearly 30% similarity to human APP, providing profound insights into amyloid-related toxins’ research.80 Additionally, another amyloidogenic enzyme γ-secretase has also been detected in Drosophila. On a further note, genetic-based transgenic Drosophila has also been essential in FAD-related dementia, which overexpresses Aβ in the central nervous system (CNS).78,81
8.4. Guinea Pig
Unlike rats and mice, the guinea pig (Cavia porcellus), a nontransgenic animal model, has a human-like Aβ peptide sequence. High-cholesterol diets enhance BACE1 (β-secretase) transcription and decrease ADAM10 (α-secretase) transcription, which should increase Aβ release from APP.82
Guinea pigs have AD-related isoforms not observed in mice or rats. The guinea pig tau gene, MAPT, encodes isoforms with three and four microtubule-binding domains, like humans but unlike mice. Cholesterol affects the ratio of these isoforms. Guinea pigs are a good model for studying how dietary variables like cholesterol affect AD-related genes. Their AD-related genes are more human-like than rats or mice.82
Guinea pigs are the only small animal model where PS2V generation has been discovered. Human neuroblastoma cells exposed to hypoxia-induced oxidative stress and the brains of people with sporadic, late-onset Alzheimer’s disease had previously been shown to express the PS2V transcript.83
8.5. Monkey
Nonhuman primates have the potential to serve as valuable models of sporadic age-related brain-amyloid deposition as well as the pathologic alterations associated with AD. Some nonhuman primates can develop signs of AD during the aging process that are strikingly comparable to those of people with the disease (Table 1). These symptoms include neuropathy and changes in cognitive and behavioral patterns. Aging animals, on the other hand, are not models of Alzheimer’s disease; instead, they are good models of normal aging and naturally occurring Aβ deposition, and some display cognitive impairment. Deposition of amyloid in the brain parenchyma has been seen in the vast majority of nonhuman primates up to this point, which include rhesus monkeys,84 chimpanzees,85 vervet monkeys,86 marmosets,87 and cynomolgus monkeys.88
Table 1. Monkey Models of AD.
| animal species | rhesus macaques | stump-tailed macaques | mouse lemurs | common marmoset | cynomolgus monkeys |
|---|---|---|---|---|---|
| scientific name | Macaca mulatta | Macaca arctoides | Microcebus murinus | Callithrix jacchus | Macaca fascicularis |
| body length | 45–64 cm | 45–70 cm | 12–13 cm | 10–12 cm | 40–65 cm |
| weight | 5–12 kg | 7–12 kg | 50–100 g | 80–100 g | 9 g |
| lifespan | 34–40 years | >30 years | 14 years | 7–11 years | 35 years |
| age when considered old | 20 years | 24 years | 5 years | 7 years | 20 years |
9. In Vitro Models
In vitro models can also reproduce the disease model at cellular and molecular levels. However, the in vitro models’ robustness is not as profound as compared to in vivo models.
9.1. Neuroblastoma Cell Line
The neuroblastoma cell lines, also known as SH-SY5Y cell lines, can potentially develop neuronal cells functioning as neurons upon treatment with various agents. SH-SY5Y cell lines are obtained from neuroblastoma with the subcloning technique. Neuroblastoma is constituted of Schwann cells and neuroblasts. Therefore, these cell lines can be utilized to develop potential therapeutic agents for treating AD.89
9.2. iPSC-Derived Cell Lines
The generation of induced pluripotent stem cells (iPSC) from AD patients and differentiating them into neuronal cells has been regarded as a well-known model for AD-related studies. Conventional models do not recapitulate the complex form of SAD; hence, human-induced PSC can rejuvenate this field by filling the vacuum. This in vitro model can potentially create a brain-like microenvironment mimicking AD patients.90 The pathological hallmarks like amyloid plaques and NFTs centered at iPSC can be the basis of the model. This model can help study AD pathology and find potential therapeutic drugs for the disease. Familial and sporadic AD can be the model’s basis depending on the patient’s source.91
10. Novel Approaches for AD Animal Model
10.1. STZ-APP/PS1 Model
This STZ-APP/PS1 dual model is a newer approach to treating memory impairment in mice. The strategy combines previously used STZ-induced dementia and a genetically modified transgenic model.92 Hence, it can be speculated that this model will generate a higher amount and resemblance of AD features and can be synergistic compared to individual STZ or APP/PS1 models. Moreover, the characteristics of sporadic and familial AD will probably be overlapped in this newly created animal model. The STZ will be introduced intracerebroventricularly in the same way described previously for mice already having mutated genes in APP and presenilin 1.
10.2. Fructose-Induced Model
Like galactose, fructose is a monosaccharide and structural isomer of glucose. It can interfere with the metabolic process by creating insulin resistance and cause imapairment in glucose metabolism. It can stimulate the release of glucocorticoid hormone, thereby controlling food intake. Fructose, if given through diet in an excess amount, has consequences in terms of neuronal death. The brain regions related to food intake and hunger, such as the hypothalamus and hippocampus, can be areas of concern. Finally, it can affect memory impairment and cognitive dysfunction.93 Many reports give rise to evidence that AD is primarily a metabolic disease, which can also be characterized by hyperglycemia, hyperinsulinemia, glucose intolerance, and brain insulin resistance.11,16,17
10.3. Hypertension-Induced AD Model
Increased blood pressure is the excessive pressure exerted on the blood vessels. This is the case when blood gets affected in different body organs, including the brain. Blood usually carries oxygen and other nutrients that are essential for cellular survival. If hypertension persists chronically, it can hinder blood flow to the brain, thereby restricting the brain from obtaining the required nutrients and oxygen. The peripheral hypertensive state can accompany cerebral blood flow hindrance. Mainly, animal hypertension can be induced through drugs or changing lifestyles like minimizing physical movement, high-salt diet, or cholesterol-rich food. Several recent studies have reported hypertension-enhancing cognitive impairment in rats.94 Even though the type of dementia incurred through the hypertensive pathway is frequently labeled as vascular dementia, repetitive induction of high blood pressure might also lead to AD-like features.94 Again, this is not an AD model but can be combined with the models mentioned earlier to create a more robust model of AD.
11. Potential Animal Models in Future Perspective
Even though the models mentioned earlier represent the AD disease model to some extent, there is still no complete resemblance and imitation of the clinical AD manifestations. Therefore, possible alternatives can give rise to more profound AD-related pathological conditions. Some of the strategies can be as follows. Even though the following models have not yet been investigated, these potential options could be investigated in the future for the generation of AD models, which at present have limitations, as mentioned earlier. Furthermore, these potential future models could be the therapeutic targets of anti-AD drugs.
11.1. Alloxan-Induced AD Model
Alloxan (160 mg/kg body weight) is a toxic chemical generally having the property of attacking insulin-producing beta cells of the pancreas. Alloxan has already been investigated in the case of the diabetic model; because AD shares some of the features of diabetes, like insulin resistance, alloxan can be examined for inducing brain insulin resistance if administered directly into the brain rather than peripherally. Besides, alloxan also has the potential to generate reactive oxygen species (ROS),95 which can further lead to mitochondrial dysfunction, which altogether can be the reason for neurodegeneration. The advantage associated with alloxan is that it is less expensive and readily available as compared to streptozotocin.95 Just like STZ, alloxan is also a diabetogenic compound. The STZ-induced AD model is already established. However, the alloxan-induced model has not yet been tried. Therefore, even though alloxan induction causes diabetes, it will still be investigated in the future for a potential AD model.
11.2. Acetylcholinesterase-Activated AD Model
Acetylcholinesterase (AchE) is an enzyme ubiquitously located in brain regions and is accountable for the breakdown of acetylcholine, a neurotransmitter involved in synaptic plasticity and memory formation. AchE, its analogs or its activators, the oximes like pralidoxime and obidoxime, can directly be administered in brain regions responsible for cognition and memory regulation. Pralidoxime and obidoxime are generally used for organophosphate poisoning96 and treating nerve gas toxicity.
11.3. Lesion-Induced Model
Lesions are defined as injuries, damage, or wound infliction in certain areas. Hence, the specific brain regions like the cerebral cortex and hippocampus regulating learning and memory can be made to go through lesion-mediated destruction so that cognitive dysfunction will appear as a symptom of dementia. A bilateral transaction can be made in the hippocampal region to create a learning deficiency. In addition, radiofrequency lesions have also been reported to cause injury.96 The neuronal injuries produced by the lesions can finally lead to neurodegeneration. The neurodegeneration in those cognition-controlling areas can be the secondary cause of AD-related pathology. Likewise, the region-specific damages in these brain areas precisely linked to special kinds of memories can be achieved. Therefore, spatial and recognition memory can be observed to be distorted accordingly. However, these super invasive methods can pose severe ethical concerns and the chance of permanent brain damage, and in extreme cases, animal mortality can result. Various advantages and disadvantages of the earlier-mentioned models are tabulated in Table 2.
Table 2. Advantages and Disadvantages of the Preclinical Models.
| models/species | advantages | disadvantages |
|---|---|---|
| mammalian transgenic models | ||
| transgenic mice97 | comparable brain anatomy to humans | crossbreeding/microinjection of transgenic lines expensive and time-consuming |
| Aβ plaques and NFT reproducible | strain difference between transgenic lines | |
| learning and memory performance assessable using behavioral tests | ||
| therapeutic benefits through examination of histopathology and behavioral tests | ||
| triple transgenic model50 | age- and region-dependent plaques and tangles development in the 3XTg-AD mice model similar to human AD | high cost for procurement and maintenance |
| 5XAD model60 | earlier representation of AD features than other transgenic models | phosphorylated tau pathology is less prevalent than amyloid plaques in this model |
| PDAPP98 | model shows Alzheimer-like neuropathology; disease progression similar to human; amyloid burden and memory impairment increase with aging | formation of paired helical filament does not accompany neurodegenerative alterations; no global neuronal loss in the cortex region observed through 18 months of age |
| mammalian nontransgenic models | ||
| nonhuman primates99 | APP shares same cellular localization, similar structural, biochemical, and age-related changes to human AD | scarcity of brain specimens from aged primates; cost and maintenance is very expensive compared to rodents |
| dogs100,101 | homologous similarity in several APP processing genes, ApoE, and presenilin between dogs and humans | dogs do not form dense neuritic plaques and neurofibrillary tangles |
| Aβ plaques do not occur in all aged dogs | ||
| rabbit100 | same Aβ peptide sequence to humans | do not develop AD pathology spontaneously |
| ICV-STZ induced model102,103 | behavioral similarities to human AD | involved disease pathophysiology is neuro-inflammation but not accumulation of hyperphosphorylated tau and Aβ |
| easy disease induction | strong surgical skills and precision in administration required | |
| single administration required (quick induction) | ||
| amyloid-induced model104,105 | allows better control on concentration of Aβ, thus better and precise induction of disease | because induction is sudden, progression of disease is not similar to human AD |
| single administration required (quick induction) | strong surgical skills and precision in administration required | |
| colchicine-induced model106 | symptoms of sporadic AD | intracerebroventricular (ICV) administration of colchicine required |
| Alzheimer’s like; time-dependent disease progression observed similar to human subjects | strong surgical skills and precision in administration required | |
| scopolamine or atropine-induced model107,108 | impairs learning and memory | involves degeneration of cholinergic neurons, with little impact on accumulation of hyperphosphorylated tau and Aβ |
| can be given orally as well as ICV | ||
| aluminum chloride-induced model109 | induces memory and cognitive impairment along with Aβ | disease progression nonidentical to human AD |
| induces endoplasmic reticulum (ER) stress and oxidative stress | ||
| easy availability of AlCl3 | ||
| aged rat model110 | natural models of memory deficits and dementia | time-consuming |
| noninvasive and without any neurochemical manipulations | probability of getting AD phenotypes may vary | |
| high-fat diet-induced model107 | gives the correlation between diet and AD | time-consuming model |
| mimics some features of AD | ||
| guinea pig110,111 | close similarity to human Aβ sequence | no typical senile plaques and neurofibrillary tangles in the diseased brain |
| higher activity of the β-secretase pathway | time-consuming experimental manipulations, low reproduction kinetics | |
| unavailability of good behavioral study tools | ||
| nonmammalian models | ||
| zebrafish112 | simulates the pathology of Alzheimer’s disease (AD) and tauopathy | amyloid-beta protein shows neurogenesis in the young zebrafish, which can be confusing |
| simple nervous system compared to rodents | confirmation of results with higher vertebrate models required | |
| high-throughput screening | ||
| ease of genetic manipulation | ||
| Caenorhabditis elegans model113 | admirable molecular genetic model to explore pathways of AD and tauopathies | low translational value |
| easy and promising genetic manipulation approaches | multiple pathways cannot be targeted simultaneously | |
| quick and cheap whole-animal high-throughput screening | far away from mimicking phenotypes of human AD | |
| no BACE present | ||
| Drosophila model113 | 70% of human disease-related genes are conserved in Drosophila | a few of the critical features of the pathological signs of AD are not as obviously conserved |
| easy and promising genetic manipulation approaches | ||
| short generation time and short life span | ||
| in vitro models | ||
| chicken embryo primary culture | chick APP gene is identical to humans | no evidence for age-related changes |
| helpful to investigate the pathways regulating amyloidosis | ||
| brain slices/brain culture110,114 | shows the AD mechanism at the molecular and cellular level; controllable environment | no neurological changes, plaques, or neurofibrillary tangles |
| induced pluripotent stem cells (iPSC) | similarity to human genetic background | no pathological changes |
| neuroblastoma cell lines110 | quick and easy to procure | no pathological changes |
| set protocols and assay parameters | low translational value | |
| 3D human neural cell culture microfluidic model115 | recapitulate several critical aspects of BBB dysfunction observed in AD patients | |
12. Conclusion
In a nutshell, all of the past and existing AD models represent pathological features of human AD to some extent but not as a whole. Despite that, the various preclinical AD models with their characteristics of mimicking clinical AD pathology have led to some research opportunities and therapeutic options for clinical AD. Still, newer approaches could be more exploratory for better imitating the disease and reaching a concrete place to understand AD pathology and its subsequent discovery of potential treatments (Figure 5).
Figure 5.

Illustration of various preclinical models for AD.
Acknowledgments
We gratefully acknowledge the Department of Pharmaceutical Sciences, School of Health Science and Technology, UPES, Dehradun, for providing the facility.
Author Contributions
# A.A. and S.M.G. contributed equally.
This manuscript did not receive any specific grant from public, commercial, or not-for-profit funding agencies.
The authors declare no competing financial interest.
References
- Patterson C.World Alzheimer report 2018; Alzheimer’s Disease International: London, 2018.
- 2019 Alzheimer’s disease facts and figures. Alzheimer's Dementia 2019, 15 (3), 321–387. 10.1016/j.jalz.2019.01.010. [DOI] [Google Scholar]
- Ohara T.; Hata J.; Yoshida D.; Mukai N.; Nagata M.; Iwaki T.; Kitazono T.; Kanba S.; Kiyohara Y.; Ninomiya T. Trends in dementia prevalence, incidence, and survival rate in a Japanese community. Neurology. 2017, 88 (20), 1925–1932. 10.1212/WNL.0000000000003932. [DOI] [PubMed] [Google Scholar]; Chan K. Y.; Wang W.; Wu J. J.; Liu L.; Theodoratou E.; Car J.; Middleton L.; Russ T. C.; Deary I. J.; Campbell H.; et al. Epidemiology of Alzheimer’s disease and other forms of dementia in China, 1990–2010: a systematic review and analysis. Lancet 2013, 381 (9882), 2016–2023. 10.1016/S0140-6736(13)60221-4. [DOI] [PubMed] [Google Scholar]
- Livingston G.; Huntley J.; Sommerlad A.; Ames D.; Ballard C.; Banerjee S.; Brayne C.; Burns A.; Cohen-Mansfield J.; Cooper C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396 (10248), 413–446. 10.1016/S0140-6736(20)30367-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhana K.; Evans D. A.; Rajan K. B.; Bennett D. A.; Morris M. C. Healthy lifestyle and the risk of Alzheimer dementia: Findings from 2 longitudinal studies. Neurology 2020, 95 (4), e374–e383. 10.1212/WNL.0000000000009816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.-X.; Tian Y.; Wang Z.-T.; Ma Y.-H.; Tan L.; Yu J.-T. The epidemiology of Alzheimer’s disease modifiable risk factors and prevention. J. Prev. Alzheimer's Dis. 2021, 8 (3), 313–321. 10.14283/jpad.2021.15. [DOI] [PubMed] [Google Scholar]
- Rabinovici G. D. Late-onset Alzheimer disease. Continuum (Minneap Minn). 2019, 25 (1), 14. 10.1212/CON.0000000000000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M.; Shepardson N.; Yang T.; Chen G.; Walsh D.; Selkoe D. J. Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (14), 5819–5824. 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]; Huang H.-C.; Jiang Z.-F. Accumulated amyloid-β peptide and hyperphosphorylated tau protein: relationship and links in Alzheimer’s disease. J. Alzheimers Dis. 2009, 16 (1), 15–27. 10.3233/JAD-2009-0960. [DOI] [PubMed] [Google Scholar]
- De Strooper B.; Vassar R.; Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6 (2), 99–107. 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustinack J. C.; Schneider A.; Mandelkow E.-M.; Hyman B. T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002, 103 (1), 26–35. 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Akhtar A.; Dhaliwal J.; Saroj P.; Uniyal A.; Bishnoi M.; Sah S. P. Chromium picolinate attenuates cognitive deficit in ICV-STZ rat paradigm of sporadic Alzheimer’s-like dementia via targeting neuroinflammatory and IRS-1/PI3K/AKT/GSK-3β pathway. Inflammopharmacology. 2020, 28 (2), 385–400. 10.1007/s10787-019-00681-7. [DOI] [PubMed] [Google Scholar]
- Oumata N.; Lu K.; Teng Y.; Cavé C.; Peng Y.; Galons H.; Roques B. P. Molecular mechanisms in Alzheimer’s disease and related potential treatments such as structural target convergence of antibodies and simple organic molecules. Eur. J. Med. Chem. 2022, 240, 114578. 10.1016/j.ejmech.2022.114578. [DOI] [PubMed] [Google Scholar]; Srivastava S.; Ahmad R.; Khare S. K. Alzheimer’s disease and its treatment by different approaches: A review. Eur. J. Med. Chem. 2021, 216, 113320. 10.1016/j.ejmech.2021.113320. [DOI] [PubMed] [Google Scholar]; Sang Z.; Wang K.; Dong J.; Tang L. Alzheimer’s disease: Updated multi-targets therapeutics are in clinical and in progress. Eur. J. Med. Chem. 2022, 238, 114464. 10.1016/j.ejmech.2022.114464. [DOI] [PubMed] [Google Scholar]
- Durairajan S. S.; Selvarasu K.; Bera M. R.; Rajaram K.; Iyaswamy A.; Li M. Alzheimer’s disease and other tauopathies: exploring efficacy of medicinal plant-derived compounds in alleviating tau-mediated neurodegeneration. Curr. Mol. Pharmacol. 2022, 15 (2), 361–379. 10.2174/1874467214666210906125318. [DOI] [PubMed] [Google Scholar]; Yang C.; Su C.; Iyaswamy A.; Krishnamoorthi S. K.; Zhu Z.; Yang S.; Tong B. C.; Liu J.; Sreenivasmurthy S. G.; Guan X.; et al. Celastrol enhances transcription factor EB (TFEB)-mediated autophagy and mitigates Tau pathology: Implications for Alzheimer’s disease therapy. Acta Pharm. Sin B 2022, 12 (4), 1707–1722. 10.1016/j.apsb.2022.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullane K.; Williams M. Preclinical models of Alzheimer’s disease: relevance and translational validity. Curr. Protoc Pharmacol. 2019, 84 (1), e57 10.1002/cpph.57. [DOI] [PubMed] [Google Scholar]
- Gong C.-X.; Liu F.; Grundke-Iqbal I.; Iqbal K. Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation. J. Alzheimers Dis. 2006, 9 (1), 1–12. 10.3233/JAD-2006-9101. [DOI] [PubMed] [Google Scholar]
- Akhtar A.; Bishnoi M.; Sah S. P. Sodium orthovanadate improves learning and memory in intracerebroventricular-streptozotocin rat model of Alzheimer’s disease through modulation of brain insulin resistance induced tau pathology. Brain Res. Bull. 2020, 164, 83–97. 10.1016/j.brainresbull.2020.08.001. [DOI] [PubMed] [Google Scholar]
- Akhtar A.; Dhaliwal J.; Sah S. P. 7, 8-Dihydroxyflavone improves cognitive functions in ICV-STZ rat model of sporadic Alzheimer’s disease by reversing oxidative stress, mitochondrial dysfunction, and insulin resistance. J. Psychopharmacol. 2021, 238 (7), 1991–2009. 10.1007/s00213-021-05826-7. [DOI] [PubMed] [Google Scholar]
- Akhtar A.; Sah S. P. Insulin signaling pathway and related molecules: role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020, 135, 104707. 10.1016/j.neuint.2020.104707. [DOI] [PubMed] [Google Scholar]
- Yamini P.; Ray R.; Yadav S.; Dhaliwal J.; Yadav M.; Kondepudi K. K.; Chopra K. α7nAChR activation protects against oxidative stress, neuroinflammation and central insulin resistance in ICV-STZ induced sporadic Alzheimer’s disease. Pharmacol., Biochem. Behav. 2022, 217, 173402. 10.1016/j.pbb.2022.173402. [DOI] [PubMed] [Google Scholar]; Kazkayasi I.; Telli G.; Nemutlu E.; Uma S. Intranasal metformin treatment ameliorates cognitive functions via insulin signaling pathway in ICV-STZ-induced mice model of Alzheimer’s disease. Life Sci. 2022, 299, 120538. 10.1016/j.lfs.2022.120538. [DOI] [PubMed] [Google Scholar]
- Shekarian M.; Komaki A.; Shahidi S.; Sarihi A.; Salehi I.; Raoufi S. The protective and therapeutic effects of vinpocetine, a PDE1 inhibitor, on oxidative stress and learning and memory impairment induced by an intracerebroventricular (ICV) injection of amyloid beta (aβ) peptide. Behav Brain Res. 2020, 383, 112512. 10.1016/j.bbr.2020.112512. [DOI] [PubMed] [Google Scholar]
- Cheignon C.; Tomas M.; Bonnefont-Rousselot D.; Faller P.; Hureau C.; Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. Y.; Yan H.; Tang X. C. Huperzine A enhances the level of secretory amyloid precursor protein and protein kinase C-α in intracerebroventricular β-amyloid-(1–40) infused rats and human embryonic kidney 293 Swedish mutant cells. Neurosci. Lett. 2004, 360 (1–2), 21–24. 10.1016/j.neulet.2004.01.055. [DOI] [PubMed] [Google Scholar]
- Wang D.; Noda Y.; Zhou Y.; Mouri A.; Mizoguchi H.; Nitta A.; Chen W.; Nabeshima T. The allosteric potentiation of nicotinic acetylcholine receptors by galantamine ameliorates the cognitive dysfunction in beta amyloid 25–35 icv-injected mice: involvement of dopaminergic systems. Neuropsychopharmacology. 2007, 32 (6), 1261–1271. 10.1038/sj.npp.1301256. [DOI] [PubMed] [Google Scholar]
- Thakker D. R.; Weatherspoon M. R.; Harrison J.; Keene T. E.; Lane D. S.; Kaemmerer W. F.; Stewart G. R.; Shafer L. L. Intracerebroventricular amyloid-β antibodies reduce cerebral amyloid angiopathy and associated micro-hemorrhages in aged Tg2576 mice. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (11), 4501–4506. 10.1073/pnas.0813404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragicevic N.; Smith A.; Lin X.; Yuan F.; Copes N.; Delic V.; Tan J.; Cao C.; Shytle R. D.; Bradshaw P. C. Green tea epigallocatechin-3-gallate (EGCG) and other flavonoids reduce Alzheimer’s amyloid-induced mitochondrial dysfunction. J. Alzheimers Dis. 2011, 26 (3), 507–521. 10.3233/JAD-2011-101629. [DOI] [PubMed] [Google Scholar]; Alvarez G.; Muñoz-Montaño J. R.; Satrústegui J.; Avila J.; Bogónez E.; Díaz-Nido J. Regulation of tau phosphorylation and protection against β-amyloid-induced neurodegeneration by lithium. Possible implications for Alzheimer’s disease. Bipolar Disord. 2002, 4 (3), 153–165. 10.1034/j.1399-5618.2002.01150.x. [DOI] [PubMed] [Google Scholar]
- Karran E.; De Strooper B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat. Rev. Drug Discovery 2022, 21 (4), 306–318. 10.1038/s41573-022-00391-w. [DOI] [PubMed] [Google Scholar]
- Niel E.; Scherrmann J.-M. Colchicine today. Joint Bone Spine. 2006, 73 (6), 672–678. 10.1016/j.jbspin.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Dogra S.; Prakash A. Protective effect of naringin, a citrus flavonoid, against colchicine-induced cognitive dysfunction and oxidative damage in rats. J. Med. Food. 2010, 13 (4), 976–984. 10.1089/jmf.2009.1251. [DOI] [PubMed] [Google Scholar]; Sil S.; Ghosh T. Role of cox-2 mediated neuroinflammation on the neurodegeneration and cognitive impairments in colchicine induced rat model of Alzheimer’s disease. J. Neuroimmunol. 2016, 291, 115–124. 10.1016/j.jneuroim.2015.12.003. [DOI] [PubMed] [Google Scholar]; Sil S.; Ghosh T.; Gupta P.; Ghosh R.; Kabir S. N.; Roy A. Dual role of vitamin C on the neuroinflammation mediated neurodegeneration and memory impairments in colchicine induced rat model of Alzheimer disease. J. Mol. Neurosci. 2016, 60 (4), 421–435. 10.1007/s12031-016-0817-5. [DOI] [PubMed] [Google Scholar]
- Nakayama T.; Sawada T. Involvement of microtubule integrity in memory impairment caused by colchicine. Pharmacol., Biochem. Behav. 2002, 71 (1–2), 119–138. 10.1016/S0091-3057(01)00634-7. [DOI] [PubMed] [Google Scholar]
- Jakabová S.; Vincze L.; Farkas Á.; Kilár F.; Boros B.; Felinger A. Determination of tropane alkaloids atropine and scopolamine by liquid chromatography–mass spectrometry in plant organs of Datura species. J. Chromatogr A 2012, 1232, 295–301. 10.1016/j.chroma.2012.02.036. [DOI] [PubMed] [Google Scholar]; Xia K.; Liu X.; Zhang Q.; Qiang W.; Guo J.; Lan X.; Chen M.; Liao Z. Promoting scopolamine biosynthesis in transgenic Atropa belladonna plants with pmt and h6h overexpression under field conditions. Plant Physiol Biochem. 2016, 106, 46–53. 10.1016/j.plaphy.2016.04.034. [DOI] [PubMed] [Google Scholar]
- Hosseini M.; Mohammadpour T.; Karami R.; Rajaei Z.; Reza Sadeghnia H.; Soukhtanloo M. Effects of the hydro-alcoholic extract of Nigella sativa on scopolamine-induced spatial memory impairment in rats and its possible mechanism. Chin. J. Integr. Med. 2015, 21 (6), 438–444. 10.1007/s11655-014-1742-5. [DOI] [PubMed] [Google Scholar]; Bajo R.; Pusil S.; Lopez M.; Canuet L.; Pereda E.; Osipova D.; Maestú F.; Pekkonen E. Scopolamine effects on functional brain connectivity: a pharmacological model of Alzheimer’s disease. Sci. Rep. 2015, 5 (1), 9748. 10.1038/srep09748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goverdhan P.; Sravanthi A.; Mamatha T. Neuroprotective effects of meloxicam and selegiline in scopolamine-induced cognitive impairment and oxidative stress. Int. J. Alzheimer's Dis. 2012, 2012, 974013. 10.1155/2012/974013. [DOI] [PMC free article] [PubMed] [Google Scholar]; Muhammad T.; Ali T.; Ikram M.; Khan A.; Alam S. I.; Kim M. O. Melatonin rescue oxidative stress-mediated neuroinflammation/neurodegeneration and memory impairment in scopolamine-induced amnesia mice model. J. Neuroimmune Pharmacol. 2019, 14 (2), 278–294. 10.1007/s11481-018-9824-3. [DOI] [PubMed] [Google Scholar]; Yanev P. G.; Dimitrova D. S.; Getova-Spassova D. P.. Effects of rivastigmine and memantine alone and in combination on learning and memory in rats with scopolamine-induced amnesia. Open Med. 2014, 10 ( (1), ), 10.1515/med-2015-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Gupta S. K.; Ganeshpurkar A.; Gutti G.; Krishnamurthy S.; Modi G.; Singh S. K. Development of Piperazinediones as dual inhibitor for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 150, 87–101. 10.1016/j.ejmech.2018.02.078. [DOI] [PubMed] [Google Scholar]
- Anoush M.; Pourmansouri Z.; Javadi R.; GhorbanPour B.; Sharafi A.; Mohamadpour H.; jafari anarkooli I.; Andalib S. Clavulanic Acid: A Novel Potential Agent in Prevention and Treatment of Scopolamine-Induced Alzheimer’s Disease. ACS omega. 2022, 7 (16), 13861–13869. 10.1021/acsomega.2c00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnicella S.; Pain L.; Oberling P. Cholinergic effects on fear conditioning II: nicotinic and muscarinic modulations of atropine-induced disruption of the degraded contingency effect. Psychopharmacology (Berl). 2005, 178 (4), 533–541. 10.1007/s00213-004-2101-6. [DOI] [PubMed] [Google Scholar]
- Barathi V. A.; Beuerman R. W. Molecular mechanisms of muscarinic receptors in mouse scleral fibroblasts: prior to and after induction of experimental myopia with atropine treatment. Mol. Vis. 2011, 17, 680–692. [PMC free article] [PubMed] [Google Scholar]
- Ionov I. D.; Pushinskaya I. I. Amyloid-β production in aged guinea pigs: atropine-induced enhancement is reversed by naloxone. Neuroscience letters 2010, 480 (1), 83–86. 10.1016/j.neulet.2010.06.010. [DOI] [PubMed] [Google Scholar]; Pákáski M.; Kálmán J. Interactions between the amyloid and cholinergic mechanisms in Alzheimer’s disease. Neurochem. Int. 2008, 53 (5), 103–111. 10.1016/j.neuint.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Melo J. B.; Agostinho P.; Oliveira C. R. Amyloid beta-peptide 25–35 reduces [3H] acetylcholine release in retinal neurons. Involvement of metabolic dysfunction. Amyloid 2002, 9 (4), 221–228. 10.3109/13506120209114097. [DOI] [PubMed] [Google Scholar]
- Maya S.; Prakash T.; Goli D. Evaluation of neuroprotective effects of wedelolactone and gallic acid on aluminium-induced neurodegeneration: relevance to sporadic amyotrophic lateral sclerosis. Eur. J. Pharmacol. 2018, 835, 41–51. 10.1016/j.ejphar.2018.07.058. [DOI] [PubMed] [Google Scholar]
- Martinez C. S.; Piagette J. T.; Escobar A. G.; Martín Á.; Palacios R.; Peçanha F. M.; Vassallo D. V.; Exley C.; Alonso M. J.; Miguel M.; et al. Aluminum exposure at human dietary levels promotes vascular dysfunction and increases blood pressure in rats: A concerted action of NAD (P) H oxidase and COX-2. Toxicology 2017, 390, 10–21. 10.1016/j.tox.2017.08.004. [DOI] [PubMed] [Google Scholar]
- Exley C. A molecular mechanism of aluminium-induced Alzheimer’s disease?. J. Inorg. Biochem. 1999, 76 (2), 133–140. 10.1016/S0162-0134(99)00125-7. [DOI] [PubMed] [Google Scholar]
- Yin S.; Ran Q.; Yang J.; Zhao Y.; Li C. Nootropic effect of neferine on aluminium chloride–induced Alzheimer’s disease in experimental models. J. Biochem. Mol. Toxicol. 2020, 34 (2), e22429 10.1002/jbt.22429. [DOI] [PubMed] [Google Scholar]; Amador F. C.; Santos M. S.; Oliveira C. R. Lipid peroxidation and aluminium effects on the cholinergic system in nerve terminals. Neurotox Res. 2001, 3 (3), 223–233. 10.1007/BF03033261. [DOI] [PubMed] [Google Scholar]
- Karami M.; Geravand S.; Rahimpour M. Protective Effect of L-Arginine in an Animal Model of Alzheimer’s Disease Induced by Intra-Hippocampal Injection of AlCl3. Neurol. India 2022, 70 (2), 548–553. 10.4103/0028-3886.344672. [DOI] [PubMed] [Google Scholar]
- Hao L.; Huang H.; Gao J.; Marshall C.; Chen Y.; Xiao M. The influence of gender, age and treatment time on brain oxidative stress and memory impairment induced by D-galactose in mice. Neurosci. Lett. 2014, 571, 45–49. 10.1016/j.neulet.2014.04.038. [DOI] [PubMed] [Google Scholar]
- Wei H.; Li L.; Song Q.; Ai H.; Chu J.; Li W. Behavioural study of the D-galactose induced aging model in C57BL/6J mice. Behav Brain Res. 2005, 157 (2), 245–251. 10.1016/j.bbr.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Pourmemar E.; Majdi A.; Haramshahi M.; Talebi M.; Karimi P.; Sadigh-Eteghad S. Intranasal cerebrolysin attenuates learning and memory impairments in D-galactose-induced senescence in mice. Exp Gerontol. 2017, 87, 16–22. 10.1016/j.exger.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Cui X.; Li W.; Zhang B. Studies on cell senescence induced by d-galactose in cultured neurons and fibroblasts. Chin. J. Appl. Physiol. 1997, 13 (2), 131–133. [PubMed] [Google Scholar]
- Zhang Q.; Li X.; Cui X.; Zuo P. D-galactose injured neurogenesis in the hippocampus of adult mice. Neurol Res. 2005, 27 (5), 552–556. 10.1179/016164105X25126. [DOI] [PubMed] [Google Scholar]
- Wang X.; Zhu G.; Yang S.; Wang X.; Cheng H.; Wang F.; Li X.; Li Q. Paeonol prevents excitotoxicity in rat pheochromocytoma PC12 cells via downregulation of ERK activation and inhibition of apoptosis. Planta medica 2011, 77 (15), 1695–1701. 10.1055/s-0030-1271033. [DOI] [PubMed] [Google Scholar]; Lu J.; Zheng Y.-l.; Luo L.; Wu D.-m.; Sun D.-x.; Feng Y.-j. Quercetin reverses D-galactose induced neurotoxicity in mouse brain. Behav Brain Res. 2006, 171 (2), 251–260. 10.1016/j.bbr.2006.03.043. [DOI] [PubMed] [Google Scholar]
- Oddo S.; Caccamo A.; Shepherd J. D.; Murphy M. P.; Golde T. E.; Kayed R.; Metherate R.; Mattson M. P.; Akbari Y.; LaFerla F. M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 2003, 39 (3), 409–421. 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S.; Caccamo A.; Kitazawa M.; Tseng B. P.; LaFerla F. M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging. 2003, 24 (8), 1063–1070. 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Iyaswamy A.; Wang X.; Krishnamoorthi S.; Kaliamoorthy V.; Sreenivasmurthy S. G.; Kumar Durairajan S. S.; Song J.-X.; Tong B. C.-k.; Zhu Z.; Su C.-F.; et al. Theranostic F-SLOH mitigates Alzheimer’s disease pathology involving TFEB and ameliorates cognitive functions in Alzheimer’s disease models. Redox Biol. 2022, 51, 102280. 10.1016/j.redox.2022.102280. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang X.; Iyaswamy A.; Xu D.; Krishnamoorthi S.; Sreenivasmurthy S. G.; Yang Y.; Li Y.; Chen C.; Li M.; Li H.-W.; et al. Real-Time Detection and Visualization of Amyloid-β Aggregates Induced by Hydrogen Peroxide in Cell and Mouse Models of Alzheimer’s Disease. ACS Appl. Mater. Interfaces 2022, 10.1021/acsami.2c07859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez J. J.; Jones V. C.; Tabuchi M.; Allan S. M.; Knight E. M.; LaFerla F. M.; Oddo S.; Verkhratsky A. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PloS one 2008, 3 (8), e2935 10.1371/journal.pone.0002935. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kulijewicz-Nawrot M.; Verkhratsky A.; Chvatal A.; Sykova E.; Rodríguez J. J. Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer’s disease. J. Anat. 2012, 221 (3), 252–262. 10.1111/j.1469-7580.2012.01536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietropaolo S.; Feldon J.; Yee B. K. Age-dependent phenotypic characteristics of a triple transgenic mouse model of Alzheimer disease. Behav Neurosci. 2008, 122 (4), 733. 10.1037/a0012520. [DOI] [PubMed] [Google Scholar]; Rodriguez J. J.; Noristani H. N.; Olabarria M.; Fletcher J.; Somerville T. D. D.; Yeh C. Y.; Verkhratsky A. Voluntary running and environmental enrichment restores impaired hippocampal neurogenesis in a triple transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8 (7), 707–717. 10.2174/156720511797633214. [DOI] [PubMed] [Google Scholar]
- Leparulo A.; Mahmud M.; Scremin E.; Pozzan T.; Vassanelli S.; Fasolato C. Dampened Slow Oscillation Connectivity Anticipates Amyloid Deposition in the PS2APP Mouse Model of Alzheimer’s Disease. Cells 2020, 9 (1), 54. 10.3390/cells9010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watamura N.; Sato K.; Saido T. C. Mouse models of Alzheimer’s disease for preclinical research. Neurochem. Int. 2022, 158, 105361. 10.1016/j.neuint.2022.105361. [DOI] [PubMed] [Google Scholar]
- Neuner S. M.; Wilmott L. A.; Hoffmann B. R.; Mozhui K.; Kaczorowski C. C. Hippocampal proteomics defines pathways associated with memory decline and resilience in normal aging and Alzheimer’s disease mouse models. Behav Brain Res. 2017, 322, 288–298. 10.1016/j.bbr.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; Oakley H.; Cole S. L.; Logan S.; Maus E.; Shao P.; Craft J.; Guillozet-Bongaarts A.; Ohno M.; Disterhoft J.; Van Eldik L.; et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 2006, 26 (40), 10129–10140. 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y.; Zhang J.; Zhu Y.; Zhang J.; Shen H.; Lu J.; Pan X.; Lin N.; Dai X.; Zhou M.; et al. Tripchlorolide improves cognitive deficits by reducing amyloid β and upregulating synapse-related proteins in a transgenic model of Alzheimer’s Disease. J. Neurochem. 2015, 133 (1), 38–52. 10.1111/jnc.13056. [DOI] [PubMed] [Google Scholar]
- Fung T. Y.; Iyaswamy A.; Sreenivasmurthy S. G.; Krishnamoorthi S.; Guan X.-J.; Zhu Z.; Su C.-F.; Liu J.; Kan Y.; Zhang Y.; et al. Klotho an Autophagy Stimulator as a Potential Therapeutic Target for Alzheimer’s Disease: A Review. Biomedicines 2022, 10 (3), 705. 10.3390/biomedicines10030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landel V.; Baranger K.; Virard I.; Loriod B.; Khrestchatisky M.; Rivera S.; Benech P.; Féron F. Temporal gene profiling of the 5XFAD transgenic mouse model highlights the importance of microglial activation in Alzheimer’s disease. Mol. Neurodegener. 2014, 9 (1), 33. 10.1186/1750-1326-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]; Eimer W. A.; Vassar R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ 42 accumulation and caspase-3 activation. Mol. Neurodegener. 2013, 8 (1), 2. 10.1186/1750-1326-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurucz A.; Bombicz M.; Kiss R.; Priksz D.; Varga B.; Hortobágyi T.; Trencsényi G.; Szabó R.; Pósa A.; Gesztelyi R. Heme oxygenase-1 activity as a correlate to exercise-mediated amelioration of cognitive decline and neuropathological alterations in an aging rat model of dementia. Biomed Res. Int. 2018, 2018, 7212861. 10.1155/2018/7212861. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gocmez S. S.; Gacar N.; Utkan T.; Gacar G.; Scarpace P. J.; Tumer N. Protective effects of resveratrol on aging-induced cognitive impairment in rats. Neurobiol Learn Mem. 2016, 131, 131–136. 10.1016/j.nlm.2016.03.022. [DOI] [PubMed] [Google Scholar]
- Kincaid B.; Bossy-Wetzel E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front Aging Neurosci. 2013, 5, 48. 10.3389/fnagi.2013.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]; Koltai E.; Zhao Z.; Lacza Z.; Cselenyak A.; Vacz G.; Nyakas C.; Boldogh I.; Ichinoseki-Sekine N.; Radak Z. Combined exercise and insulin-like growth factor-1 supplementation induces neurogenesis in old rats, but do not attenuate age-associated DNA damage. Rejuvenation Res. 2011, 14 (6), 585–596. 10.1089/rej.2011.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lanza I. R.; Short D. K.; Short K. R.; Raghavakaimal S.; Basu R.; Joyner M. J.; McConnell J. P.; Nair K. S. Endurance exercise as a countermeasure for aging. Diabetes 2008, 57 (11), 2933–2942. 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]; Di Loreto S.; Falone S.; D’Alessandro A.; Santini S. Jr; Sebastiani P.; Cacchio M.; Amicarelli F. Regular and moderate exercise initiated in middle age prevents age-related amyloidogenesis and preserves synaptic and neuroprotective signaling in mouse brain cortex. Exp Gerontol. 2014, 57, 57–65. 10.1016/j.exger.2014.05.006. [DOI] [PubMed] [Google Scholar]
- Kothari V.; Luo Y.; Tornabene T.; O’Neill A. M.; Greene M. W.; Geetha T.; Babu J. R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim Biophys Acta Mol. Basis Dis. 2017, 1863 (2), 499–508. 10.1016/j.bbadis.2016.10.006. [DOI] [PubMed] [Google Scholar]; Komaki A.; Karimi S. A.; Salehi I.; Sarihi A.; Shahidi S.; Zarei M. The treatment combination of vitamins E and C and astaxanthin prevents high-fat diet induced memory deficits in rats. Pharmacol., Biochem. Behav. 2015, 131, 98–103. 10.1016/j.pbb.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Petrov D.; Pedrós I.; Artiach G.; Sureda F. X.; Barroso E.; Pallàs M.; Casadesús G.; Beas-Zarate C.; Carro E.; Ferrer I.; et al. High-fat diet-induced deregulation of hippocampal insulin signaling and mitochondrial homeostasis deficiences contribute to Alzheimer disease pathology in rodents. Biochim Biophys Acta Mol. Basis Dis. 2015, 1852 (9), 1687–1699. 10.1016/j.bbadis.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Knopman D.; Boland L.; Mosley T.; Howard G.; Liao D.; Szklo M.; McGovern P.; Folsom A. Cardiovascular risk factors and cognitive decline in middle-aged adults. Neurology 2001, 56 (1), 42–48. 10.1212/WNL.56.1.42. [DOI] [PubMed] [Google Scholar]
- Shie F.-S.; Jin L.-W.; Cook D. G.; Leverenz J. B.; LeBoeuf R. C. Diet-induced hypercholesterolemia enhances brain Aβ accumulation in transgenic mice. Neuroreport 2002, 13 (4), 455–459. 10.1097/00001756-200203250-00019. [DOI] [PubMed] [Google Scholar]
- Kaur J.; Sodhi R. K.; Madan J.; Chahal S. K.; Kumar R. Forskolin convalesces memory in high fat diet-induced dementia in wistar rats—Plausible role of pregnane x receptors. Pharmacol Rep. 2018, 70 (1), 161–171. 10.1016/j.pharep.2017.07.009. [DOI] [PubMed] [Google Scholar]
- Musa A.; Lehrach H.; Russo V. E. Distinct expression patterns of two zebrafish homologues of the human APP gene during embryonic development. Dev Genes Evol. 2001, 211 (11), 563–567. 10.1007/s00427-001-0189-9. [DOI] [PubMed] [Google Scholar]
- Newman M.; Nornes S.; Martins R. N.; Lardelli M. T. Robust homeostasis of Presenilin1 protein levels by transcript regulation. Neurosci. Lett. 2012, 519 (1), 14–19. 10.1016/j.neulet.2012.04.064. [DOI] [PubMed] [Google Scholar]
- Saleem S.; Kannan R. R.. Zebrafish: A Potential Preclinical Model for Neurological Research in Modern Biology. In Zebrafish Model for Biomedical Research; Springer: 2022; pp 321–345. [Google Scholar]
- Lee J.-A.; Cole G. J. Generation of transgenic zebrafish expressing green fluorescent protein under control of zebrafish amyloid precursor protein gene regulatory elements. Zebrafish 2007, 4 (4), 277–286. 10.1089/zeb.2007.0516. [DOI] [PubMed] [Google Scholar]
- Schmid B.; Haass C. Genomic editing opens new avenues for zebrafish as a model for neurodegeneration. J. Neurochem. 2013, 127 (4), 461–470. 10.1111/jnc.12460. [DOI] [PubMed] [Google Scholar]; Hwang W. Y.; Fu Y.; Reyon D.; Maeder M. L.; Tsai S. Q.; Sander J. D.; Peterson R. T.; Yeh J. J.; Joung J. K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31 (3), 227–229. 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaye D. D.; Greenwald I. OrthoList: a compendium of C. elegans genes with human orthologs. PloS one 2011, 6 (5), e20085 10.1371/journal.pone.0020085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. M.; Hill J. J.; Bargmann C. I. A circuit for navigation in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (9), 3184–3191. 10.1073/pnas.0409009101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra S. J. III; Jin Y. Advances in synapse formation: forging connections in the worm. Wiley Interdiscip Rev. Dev Biol. 2015, 4 (2), 85–97. 10.1002/wdev.165. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ardiel E. L.; Giles A. C.; Yu A. J.; Lindsay T. H.; Lockery S. R.; Rankin C. H. Dopamine receptor DOP-4 modulates habituation to repetitive photoactivation of a C. elegans polymodal nociceptor. Learn Mem. 2016, 23 (10), 495–503. 10.1101/lm.041830.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link C. D. C. elegans models of age-associated neurodegenerative diseases: lessons from transgenic worm models of Alzheimer’s disease. Exp Gerontol. 2006, 41 (10), 1007–1013. 10.1016/j.exger.2006.06.059. [DOI] [PubMed] [Google Scholar]; Link C. D.; Taft A.; Kapulkin V.; Duke K.; Kim S.; Fei Q.; Wood D. E.; Sahagan B. G. Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer’s disease model. Neurobiol Aging. 2003, 24 (3), 397–413. 10.1016/S0197-4580(02)00224-5. [DOI] [PubMed] [Google Scholar]
- Mhatre S. D.; Michelson S. J.; Gomes J.; Tabb L. P.; Saunders A. J.; Marenda D. R. Development and characterization of an aged onset model of Alzheimer’s disease in Drosophila melanogaster. Exp. Neurol. 2014, 261, 772–781. 10.1016/j.expneurol.2014.08.021. [DOI] [PubMed] [Google Scholar]
- Crowther D.; Kinghorn K.; Miranda E.; Page R.; Curry J.; Duthie F.; Gubb D.; Lomas D. Intraneuronal Aβ, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 2005, 132 (1), 123–135. 10.1016/j.neuroscience.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Lenz S.; Karsten P.; Schulz J. B.; Voigt A. Drosophila as a screening tool to study human neurodegenerative diseases. J. Neurochem. 2013, 127 (4), 453–460. 10.1111/jnc.12446. [DOI] [PubMed] [Google Scholar]
- Carmine-Simmen K.; Proctor T.; Tschäpe J.; Poeck B.; Triphan T.; Strauss R.; Kretzschmar D. Neurotoxic effects induced by the Drosophila amyloid-β peptide suggest a conserved toxic function. Neurobiol Dis. 2009, 33 (2), 274–281. 10.1016/j.nbd.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finelli A.; Kelkar A.; Song H.-J.; Yang H.; Konsolaki M. A model for studying Alzheimer’s Aβ42-induced toxicity in Drosophila melanogaster. Mol. Cell Neurosci. 2004, 26 (3), 365–375. 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]; McGuire S. E.; Deshazer M.; Davis R. L. Thirty years of olfactory learning and memory research in Drosophila melanogaster. Prog. Neurobiol. 2005, 76 (5), 328–347. 10.1016/j.pneurobio.2005.09.003. [DOI] [PubMed] [Google Scholar]; Greeve I.; Kretzschmar D.; Tschäpe J.-A.; Beyn A.; Brellinger C.; Schweizer M.; Nitsch R. M.; Reifegerste R. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J. Neurosci. 2004, 24 (16), 3899–3906. 10.1523/JNEUROSCI.0283-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharman M. J.; Moussavi Nik S. H.; Chen M. M.; Ong D.; Wijaya L.; Laws S. M.; Taddei K.; Newman M.; Lardelli M.; Martins R. N.; et al. The guinea pig as a model for sporadic Alzheimer’s disease (AD): the impact of cholesterol intake on expression of AD-related genes. PLoS one 2013, 8 (6), e66235 10.1371/journal.pone.0066235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N.; Imaizumi K.; Manabe T.; Taniguchi M.; Hitomi J.; Katayama T.; Yoneda T.; Morihara T.; Yasuda Y.; Takagi T.; et al. Increased production of β-amyloid and vulnerability to endoplasmic reticulum stress by an aberrant spliced form of presenilin 2. J. Biol. Chem. 2001, 276 (3), 2108–2114. 10.1074/jbc.M006886200. [DOI] [PubMed] [Google Scholar]; Smith M. J.; Sharples R. A.; Evin G.; McLean C. A.; Dean B.; Pavey G.; Fantino E.; Cotton R. G.; Imaizumi K.; Masters C. L.; et al. Expression of truncated presenilin 2 splice variant in Alzheimer’s disease, bipolar disorder, and schizophrenia brain cortex. Mol. Brain Res. 2004, 127 (1–2), 128–135. 10.1016/j.molbrainres.2004.05.019. [DOI] [PubMed] [Google Scholar]; Higashide S.; Morikawa K.; Okumura M.; Kondo S.; Ogata M.; Murakami T.; Yamashita A.; Kanemoto S.; Manabe T.; Imaizumi K. Identification of regulatory cis-acting elements for alternative splicing of presenilin 2 exon 5 under hypoxic stress conditions. J. Neurochem. 2004, 91 (5), 1191–1198. 10.1111/j.1471-4159.2004.02798.x. [DOI] [PubMed] [Google Scholar]
- Sani S.; Traul D.; Klink A.; Niaraki N.; Gonzalo-Ruiz A.; Wu C.-K.; Geula C. Distribution, progression and chemical composition of cortical amyloid-β deposits in aged rhesus monkeys: similarities to the human. Acta Neuropathol. 2003, 105 (2), 145–156. 10.1007/s00401-002-0626-5. [DOI] [PubMed] [Google Scholar]
- Edler M. K.; Sherwood C. C.; Meindl R. S.; Hopkins W. D.; Ely J. J.; Erwin J. M.; Mufson E. J.; Hof P. R.; Raghanti M. A. Aged chimpanzees exhibit pathologic hallmarks of Alzheimer’s disease. Neurobiol Aging. 2017, 59, 107–120. 10.1016/j.neurobiolaging.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere C. A.; Beierschmitt A.; Iglesias M.; Spooner E. T.; Bloom J. K.; Leverone J. F.; Zheng J. B.; Seabrook T. J.; Louard D.; Li D.; et al. Alzheimer’s disease Aβ vaccine reduces central nervous system Aβ levels in a non-human primate, the Caribbean vervet. Am. J. Pathol. 2004, 165 (1), 283–297. 10.1016/S0002-9440(10)63296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez M.; Ridley R.; Baker H.; Maclean C.; Honer W.; Francis P. Chronic elevation of amyloid precursor protein in the neocortex or hippocampus of marmosets with selective cholinergic lesions. J. Neural Transm. 2001, 108 (7), 809–826. 10.1007/s007020170031. [DOI] [PubMed] [Google Scholar]
- Kimura N.; Tanemura K.; Nakamura S.-i.; Takashima A.; Ono F.; Sakakibara I.; Ishii Y.; Kyuwa S.; Yoshikawa Y. Age-related changes of Alzheimer’s disease-associated proteins in cynomolgus monkey brains. Biochem. Biophys. Res. Commun. 2003, 310 (2), 303–311. 10.1016/j.bbrc.2003.09.012. [DOI] [PubMed] [Google Scholar]
- de Medeiros L. M.; De Bastiani M. A.; Rico E. P.; Schonhofen P.; Pfaffenseller B.; Wollenhaupt-Aguiar B.; Grun L.; Barbé-Tuana F.; Zimmer E. R.; Castro M. A.; et al. Cholinergic differentiation of human neuroblastoma SH-SY5Y cell line and its potential use as an in vitro model for Alzheimer’s disease studies. Mol. Neurobiol. 2019, 56 (11), 7355–7367. 10.1007/s12035-019-1605-3. [DOI] [PubMed] [Google Scholar]
- Hasan M. F.; Trushina E. Advances in Recapitulating Alzheimer’s Disease Phenotypes Using Human Induced Pluripotent Stem Cell-Based In Vitro Models. Brain Sci. 2022, 12 (5), 552. 10.3390/brainsci12050552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T.; Asai M.; Tsukita K.; Kutoku Y.; Ohsawa Y.; Sunada Y.; Imamura K.; Egawa N.; Yahata N.; Okita K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Ap and differential drug responsiveness. Cell Stem Cell. 2013, 12 (4), 487–496. 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Kelliny S.; Lin L.; Deng I.; Xiong J.; Zhou F.; Al-Hawwas M.; Bobrovskaya L.; Zhou X.-F. A new approach to model sporadic Alzheimer’s disease by intracerebroventricular streptozotocin injection in APP/PS1 mice. Mol. Neurobiol. 2021, 58 (8), 3692–3711. 10.1007/s12035-021-02338-5. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F.; Cipolat R. P.; Royes L. F. F. Dietary fructose as a model to explore the influence of peripheral metabolism on brain function and plasticity. Biochim Biophys Acta Mol. Basis Dis. 2021, 1867 (5), 166036. 10.1016/j.bbadis.2020.166036. [DOI] [PubMed] [Google Scholar]
- Coatl-Cuaya H.; Tendilla-Beltrán H.; de Jesús-Vásquez L. M.; Garcés-Ramírez L.; de Jesús Gómez-Villalobos M.; Flores G. Losartan enhances cognitive and structural neuroplasticity impairments in spontaneously hypertensive rats. J. Chem. Neuroanat. 2022, 120, 102061. 10.1016/j.jchemneu.2021.102061. [DOI] [PubMed] [Google Scholar]
- Ighodaro O. M.; Adeosun A. M.; Akinloye O. A. Alloxan-induced diabetes, a common model for evaluating the glycemic-control potential of therapeutic compounds and plants extracts in experimental studies. Medicina 2017, 53 (6), 365–374. 10.1016/j.medici.2018.02.001. [DOI] [PubMed] [Google Scholar]
- Lorke D. E.; Petroianu G. A. Treatment of organophosphate poisoning with experimental oximes: A review. Curr. Org. Chem. 2019, 23 (5), 628–639. 10.2174/1385272823666190408114001. [DOI] [Google Scholar]
- Sarasa M.; Pesini P. Natural non-trasgenic animal models for research in Alzheimer’s disease. Curr. Alzheimer Res. 2009, 6 (2), 171–178. 10.2174/156720509787602834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires T. L.; Hyman B. T. Transgenic models of Alzheimer’s disease: learning from animals. NeuroRx 2005, 2 (3), 423–437. 10.1602/neurorx.2.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N.; Munoz P.; Palacios A. G.; Castellano-Gonzalez G.; Inestrosa N. C.; Chung R. S.; Sachdev P.; Guillemin G. J. Recent rodent models for Alzheimer’s disease: clinical implications and basic research. J. Neural Transm. 2012, 119 (2), 173–195. 10.1007/s00702-011-0731-5. [DOI] [PubMed] [Google Scholar]
- Johnstone E.; Chaney M.; Norris F.; Pascual R.; Little S. Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Brain Res. Mol. Brain Res. 1991, 10 (4), 299–305. 10.1016/0169-328X(91)90088-F. [DOI] [PubMed] [Google Scholar]
- Pugliese M.; Mascort J.; Mahy N.; Ferrer I. Diffuse beta-amyloid plaques and hyperphosphorylated tau are unrelated processes in aged dogs with behavioral deficits. Acta Neuropathol. 2006, 112 (2), 175–183. 10.1007/s00401-006-0087-3. [DOI] [PubMed] [Google Scholar]; Russell M. J.; White R.; Patel E.; Markesbery W. R.; Watson C. R.; Geddes J. W. Familial influence on plaque formation in the beagle brain. Neuroreport 1992, 3 (12), 1093–1096. 10.1097/00001756-199212000-00015. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Liang Z.; Blanchard J.; Dai C.-L.; Sun S.; Lee M. H.; Grundke-Iqbal I.; Iqbal K.; Liu F.; Gong C.-X. A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol. Neurobiol. 2013, 47 (2), 711–725. 10.1007/s12035-012-8375-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutti G.; Kakarla R.; Kumar D.; Beohar M.; Ganeshpurkar A.; Kumar A.; Krishnamurthy S.; Singh S. K. Discovery of novel series of 2-substituted benzo [d] oxazol-5-amine derivatives as multi-target directed ligands for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 182, 111613. 10.1016/j.ejmech.2019.111613. [DOI] [PubMed] [Google Scholar]
- Kim H. Y.; Lee D. K.; Chung B.-R.; Kim H. V.; Kim Y. Intracerebroventricular injection of amyloid-β peptides in normal mice to acutely induce Alzheimer-like cognitive deficits. J. Vis Exp. 2016, (109), e53308 10.3791/53308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swetha R.; Kumar D.; Gupta S. K.; Ganeshpurkar A.; Singh R.; Gutti G.; Kumar D.; Jana S.; Krishnamurthy S.; Singh S. K. Multifunctional hybrid sulfonamides as novel therapeutic agents for Alzheimer’s disease. Future Med. Chem. 2019, 11 (24), 3161–3178. 10.4155/fmc-2019-0106. [DOI] [PubMed] [Google Scholar]
- Saini N.; Singh D.; Sandhir R. Neuroprotective effects of Bacopa monnieri in experimental model of dementia. Neurochem. Res. 2012, 37 (9), 1928–1937. 10.1007/s11064-012-0811-4. [DOI] [PubMed] [Google Scholar]
- Neha; Sodhi R. K.; Jaggi A. S.; Singh N. Animal models of dementia and cognitive dysfunction. Life Sciences 2014, 109 (2), 73–86. 10.1016/j.lfs.2014.05.017. [DOI] [PubMed] [Google Scholar]
- Chen W. N.; Yeong K. Y. Scopolamine, a toxin-induced experimental model, used for research in Alzheimer’s disease. Curr. Drug Targets CNS Neurol Disord. 2020, 19 (2), 85–93. 10.2174/1871527319666200214104331. [DOI] [PubMed] [Google Scholar]
- Savory J.; Herman M. M.; Ghribi O. Mechanisms of aluminum-induced neurodegeneration in animals: Implications for Alzheimer’s disease. J. Alzheimers Dis. 2006, 10 (2–3), 135–144. 10.3233/JAD-2006-102-302. [DOI] [PubMed] [Google Scholar]; Promyo K.; Iqbal F.; Chaidee N.; Chetsawang B. Aluminum chloride-induced amyloid β accumulation and endoplasmic reticulum stress in rat brain are averted by melatonin. Food Chem. Toxicol. 2020, 146, 111829. 10.1016/j.fct.2020.111829. [DOI] [PubMed] [Google Scholar]
- Li X.; Bao X.; Wang R. Experimental models of Alzheimer’s disease for deciphering the pathogenesis and therapeutic screening. Int. J. Mol. Med. 2016, 37 (2), 271–283. 10.3892/ijmm.2015.2428. [DOI] [PubMed] [Google Scholar]
- Beck M.; Bigl V.; Roßner S. Guinea pigs as a nontransgenic model for APP processing in vitro and in vivo. Neurochem. Res. 2003, 28 (3), 637–644. 10.1023/A:1022850113083. [DOI] [PubMed] [Google Scholar]
- Thawkar B. S.; Kaur G. Zebrafish as a promising tool for modeling neurotoxin-induced Alzheimer’s disease. Neurotox Res. 2021, 39 (3), 949–965. 10.1007/s12640-021-00343-z. [DOI] [PubMed] [Google Scholar]; Saleem S.; Kannan R. R. Zebrafish: an emerging real-time model system to study Alzheimer’s disease and neurospecific drug discovery. Cell Death Discovery 2018, 4 (1), 45. 10.1038/s41420-018-0109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraceno C.; Musardo S.; Marcello E.; Pelucchi S.; Di Luca M. Modeling Alzheimer’s disease: from past to future. Front Pharmacol. 2013, 4, 77. 10.3389/fphar.2013.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C.-X.; Lidsky T.; Wegiel J.; Grundke-Iqbal I.; Iqbal K. Metabolically active rat brain slices as a model to study the regulation of protein phosphorylation in mammalian brain. Brain Res. 2001, 6 (3), 134–140. 10.1016/S1385-299X(00)00046-5. [DOI] [PubMed] [Google Scholar]
- Shin Y.; Choi S. H.; Kim E.; Bylykbashi E.; Kim J. A.; Chung S.; Kim D. Y.; Kamm R. D.; Tanzi R. E. Blood–brain barrier dysfunction in a 3D in vitro model of Alzheimer’s disease. Adv. Sci. (Weinh). 2019, 6 (20), 1900962. 10.1002/advs.201900962. [DOI] [PMC free article] [PubMed] [Google Scholar]