

Abstract
Hepatic encephalopathy describes an array of neurological complications that arise due to liver insufficiency. The pathogenesis of hepatic encephalopathy shares a longstanding association with hyperammonemia and inflammation, and recently, aberrant bile acid signaling has been implicated in the development of key features of hepatic encephalopathy. These key features include neuronal dysfunction, neuroinflammation and blood–brain barrier permeability. This review summarizes the findings of recent studies demonstrating a role for bile acids in the pathogenesis of hepatic encephalopathy via one of three main bile acid receptors and speculates on the possible downstream consequences of aberrant bile acid signaling.
Keywords: FXR, TGR5, S1P2R, neuroinflammation, cholesterol homeostasis, neurosteroids
1. INTRODUCTION
Hepatic Encephalopathy (HE) is the decline in neurological and cognitive function resulting from advanced liver failure. Unlike other liver diseases, HE poses a substantial burden on the families and caregivers of the afflicted patient, resulting in the highest utilization of heath care resources among the liver disorders1. HE is more prevalent in our ageing populations, and is potentially due to viral hepatitis, drug-induced liver injury, or the development of cirrhosis2. It has been reported that these patients require two times the amount of care when compared to non-HE patients2. While the complete pathophysiology of HE is unclear, there is a general consensus that there are at least 3 different subtypes with a possible 4th; Type A, Type B, Type C, and Type D. Type A HE develops from acute liver failure; Type B HE is associated with a portosystemic shunting without intrinsic liver disease, and Type C is caused by chronic liver disease3. Type D is an acute-on-chronic liver failure, an example being a cirrhotic patient overdosing on acetaminophen4. For the purpose of this review, only Types A and C will be discussed.
The classical, and best-characterized, hypothesis in the propagation of HE is the ammonia hypothesis5. The buildup of ammonia, hyperammonemia, due to liver disease or injury results in elevated blood ammonia levels and allows crossing of the blood-brain barrier4. Once the ammonia enters the brain, it is metabolized by astrocytes into glutamine, causing an increase in glutamine levels and results in astrocyte swelling that propagates inflammation4. Due to conflicting studies about the correlation of ammonia levels and development of degreed HE, researchers have proposed that increased ammonia works synergistically with inflammation to develop HE6. While inflammation plays a critical role in HE, oxidative stress, brain energy metabolism, neurotransmitter dysfunction, bile acids, and blood-brain barrier permeability have also been observed impacting the propagation of HE5 (PMID: 28017739).
Additionally, altered neurosteroid homeostasis and excess neuroinhibition, mediated by GABA, have been demonstrated in HE7, 8, although the mechanism by which these factors become dysregulated is unknown. However, an increase of circulating bile acids due to liver failure is another key feature of HE and could be a potential link between neurotransmitter dysfunction and the development of HE. This review will highlight recent studies regarding bile acids and their receptors, and speculate on possible downstream effects of aberrant bile acid signaling in the brain.
2. HEPATIC ENCEPHALOPATHY OVERVIEW
Type A HE results from Acute Liver Failure (ALF): a highly specific and rare syndrome characterized by an acute abnormality of liver blood test in an individual without underlying chronic liver disease9. ALF is typically resultant form an acute to severe liver injury observed from drug related hepatotoxicity, but can also be attributed to viral infection and other causes9, 10. Acetaminophen overdose is one of the most common causes of ALF in the developed world, and accounts for over 50% of ALF cases in the United States10. Current standard of care after acetaminophen overdose is treatment of patients with N-acetylcysteine, which can attenuate the toxic effects, but must be used within the first 8hrs of overdose10. Current clinical treatments for ALF include liver transplant, but this does not treat the neurological deficits associated with HE, and more research into the pathophysiology of HE can help elucidate viable treatment options10.
Type C HE is found in patients with liver cirrhosis. These patients can display either covert (euphoria or anxiety, shortened attention span, and a lack of awareness) or overt HE (advanced confusion, lethargy, obvious personality change and hepatic coma)11. The neuropsychological and neurophysiological symptoms have a broad range in these patients, oftentimes making it hard to diagnose- leading clinicians to exclude other possible brain dysfunctional diseases first11. The patients experiencing altered mental states, and to what degree, is varied and can be episodic12. These episodes are typically caused by infection, constipation, dehydration as well as a multitude of other non-homeostatic disorders. Clinicians have found that treating the stressor helps to end the episode12. While the observable symptoms of HE have been documented, the cellular mechanisms remain elusive. Currently the accepted pathophysiology of chronic liver disease involve the presence of Alzheimer type-2 astrocytosis, activation of microglia, and less commonly observed, neuronal cell death8 (pmid20303348). Activation of microglia indicates a pro-inflammatory response, while Alzheimer type-2 astrocytosis is the manifestation of nuclear pallor, swelling and margination of the chromatin pattern8. Partnered together these cytotoxic effects lead to brain edema and the eventual hepatic coma.
2.1. Hyperpermeability of the blood brain barrier
An increase in blood-brain barrier permeability is just one of the clinical features of both Types A and C HE, and is believed not to arise from a breakdown of the blood-brain barrier, but rather an increase in permeability13, 14. Permeability seems to be dependent on pro-inflammatory factors15, 16 and is not observed until the onset of neurological symptoms in many rodent models16–18.
2.2. Neuroinflammation
Type A and Type C HE are both characterized by microglial activation and the increase of neuroinflammation. In physiological conditions microglia are dampened but their activation is typically maintained by a balance of pro- and anti-inflammatory modulatory signals19. Crosstalk between neurons and astrocytes can play a role in the activation of microglia or these signals may be intrinsic. In a Type A HE mouse model, the pro-inflammatory chemokine ligand 2 (CCL2)20 was upregulated in neurons while the inverse was observed for fractalkine, an anti-inflammatory chemokine19, resulting in the activation of microglia via a dysregulation in the balance between pro- and anti-inflammatory signals.
3. BILE ACID METABOLISM OVERVIEW
Metabolized from cholesterol, bile acids are predominantly produced in the liver, and are then secreted into the small intestine to help in the emulsification and digestion of lipids. The primary bile acids, cholic acid and chenodeoxycholic acid, are altered in the lumen of the small intestine by bacteria to produce the secondary bile acids deoxycholic acid and lithocholic acid. Further modification of these bile acids by glucuronidation or sulfination occurs in the liver and intestine, as well as, the conjugation of the bile acids with glycine or taurine to reduce toxicity21. Due to the various metabolic transformations that bile acids can undergo, there are a wide variety of characteristics in the bile acid pool (i.e. lipophilicity, hydrophilicity or receptor affinity). Along with this wide variation in the pharmacokinetic properties of bile acids, there are also a wide variety of receptors and transporters for bile acids exerting a variety of effects. A summary of these can be found in Table 1.
Table 1: A Brief Overview of Bile Acid Receptors and Transporters-.
Summarized in this table are the most relevant bile acid receptors and transporters, their abbreviations, location in the cell, and the bile acids they interact with.
| Name | Abbreviation | Location | Binding |
|---|---|---|---|
| Farnesoid X Receptor | FXR | Nuclear | Cholic Acid, Chenodeoxycholic acid, Lithocholic Acid, Deoxycholic Acid |
| Pregnane X Receptor | PXR | Nuclear | Lithocholic Acid |
| Vitamin D Receptor | VDR | Nuclear | Lithocholic Acid |
| Glucocorticoid Receptor | GR | Nuclear | Ursodeoxycholic Acid, Taurocholic Acid, Glycochenodeoxycholic Acid |
| Takeda G-Protein Coupled Receptor 5 (G-Protein Couple Bile Acid Receptor 1) | TGR5(GPBAR1) | Membrane | Cholic Acid, Chenodeoxycholic acid, Lithocholic Acid, Deoxycholic Acid |
| Sphingosine 1-Phosphate Receptor 2 | S1P2R | Membrane | Taurocholic Acid, Glycocholic Acid, Taurodeoxycholic Acid, Glycodeoxycholic Acid, Tauroursodeoxychilic Acid |
| Apical Sodium-Dependent Bile Acid Transporter | ASBT | Membrane | Glyco- and Tauro Conjugated bile acids |
| Organic Anion-Transporting Polypeptide | OATP | Membrane | Cholic Acid, Chenodeoxycholic acid, Lithocholic Acid, Deoxycholic Acid |
| Sodium Taurocholate Cotransporting Polypeptide | NTCP | Membrane | Taurocholic Acid, Glycocholic Acid, Taurodeoxycholic Acid, Glycodeoxycholic Acid, Taurochenodeoxycholic Acid, Glycochenodeoxycholic Acid |
Bile acid transporters help to keep the bile acids compartmentalized to the bile acid recycling pathways. The bile acid pool is very tightly regulated and predominantly recycled with de novo synthesis of bile acids accounting for roughly 5%22. Once bile acid metabolism, and the emulsification of lipids, is complete the bile acids are transported back to the liver via the enterohepatic circulation system22, detailed in Figure 1. This highly controlled system keeps increased bile acids from entering systemic circulation and causing off target effects22.
Figure 1:
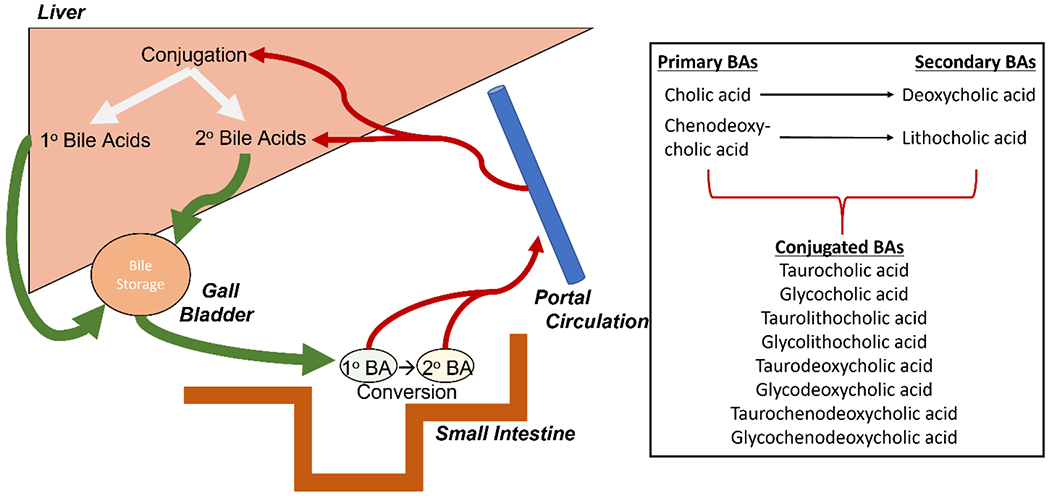
Enterohepatic circulation system. Adapted from Bile Acids in Hepatic Encephalopathy23. Primary bile acids are synthesized in the liver from hepatocytes and then stored in the gall bladder until needed for digestion. Once bile acids are in the small intestine, they are converted from primary to secondary bile acids by the gut microbiome. Bile acids are then taken into portal circulation and returned to the liver where they can be conjugated with taurine and glycine. Accompanied by a list of bile acids and their classifications.
4. SYSTEMIC BILE ACID CONTENT IS INCREASED IN LIVER DAMAGE
Serum bile acids are known to increase in conditions where the liver is damaged which has been observed in acute and chronic liver disorders at varying degrees24, 25. Speculation as to the cause of this increase lies in the potential release of bile acids from damaged hepatocytes or even the impaired recycling of bile acids via damage to the enterohepatic circulation system. It has been suggested that the increase of serum bile acids can be predictive for the onset of acute decompensation and acute-on-chronic liver failure in cirrhotic patients25. Typically, these patients will also develop HE, but as discussed previously, the development of HE is multifactorial. Furthermore, alterations in the plasma bile acid profile has been proposed as a predictor of hepatic decompensation (defined to include the development of HE) in primary sclerosing cholangitis26 and future liver-related events (also defined to include HE) in nonalcoholic fatty liver disease27. Furthermore, treatment with a non-absorbable ASBT inhibitor to specifically inhibit bile acid uptake from the intestine and reduce bile acid content in the blood mitigated the liver damage and symptoms of HE in rodent models of acute and chronic liver disease28. However, without the cofounding effects of hyperammonemia or systemic inflammation, bile acids are not the most likely cause of HE in these patients but rather contribute to the progression of HE by regulating various key features of HE. Further research into the interplay between hyperammonemia, inflammation and bile acid signaling in the context of HE pathogenesis is warranted.
Determining the link between HE and the total bile acid pool is important, but it may prove more beneficial to observe the link between HE and the individual bile acids, as there are different receptor affinities and properties for each bile acid species. Recently observed, in the serum of early and advanced cirrhosis patients, was an increase in total bile acid content along with increased levels of conjugated bile acids that correlated to the severity of the disease29. After analysis of these patients’ stool, there was an observed increase in the prevalence of the primary bile acids over the secondary bile acids correlating to changes in the gut microbiome, or dysbiosis29. The individual bile acid species and their percentage change in the stool or serum of the early and advanced cirrhosis patients compared to controls can be found in Table 2. For absolute values of these bile acids, please see Kakiyama G, et al29.
Table 2: A Brief Comparison of Bile Acid Species in Stool and Serum of Cirrhotic Patients-.
Adapted from Kakiyama G, et al29., this table summarizes the change in individual bile acid species from either the serum or stool of cirrhotic patients as compared to control patients.
| dry stool (μg/100mg) | Early Cirrhosis (n=24) | Advanced Cirrhosis (n=24) |
|---|---|---|
| Total Bile Acids | ↓ | ↓↓ |
| CA | ↑↑ | ↓↓ |
| CDCA | ↑↑↑ | ↑↑↑ |
| LCA | ↓ | ↓↓ |
| DCA | ↓ | ↓↓ |
| serum (μg/L) | ||
| CDCA | ↑ | ↑↑ |
| CA | Not detected | ↑ |
| DCA | ↓↓ | ↓↓ |
| LCA | Not detected | Not detected |
| GUDCA | ↑ | ↑↑ |
| TUDCA | Not detected | ↑ |
| GCA | ↑↑↑↑ | ↑↑↑↑ |
| TCA | ↑↑ | ↑↑↑ |
| GCDCA | ↑↑↑↑ | ↑↑↑↑↑ |
| TCDCA | ↑↑↑ | ↑↑↑ |
| GDCA | ↓↓ | ↓↓↓ |
| TCDCA3S | ↑ | ↑↑↑ |
| GCDCA3S | ↑↑ | ↑↑↑ |
↑= 0-50% ↑↑=51-100% ↑↑↑= 101-1000% ↑↑↑↑= 1001-10,000% ↑↑↑↑↑=10,000+
The observed dysbiosis in the gut microbiome, from those capable of primary to secondary bile acid conversion, is thought to be central to the pathophysiology of chronic liver disease and the development of complications30. Understanding the link between HE and bile acids also requires understanding the link between of HE and the gut microbiome. Bile acids are capable of, and potentially responsible for, changing the gut microbiome31, which then leads to alterations in the composition of the total bile acid pool32. In clinical trials of cirrhotic patients with HE, treatment with the probiotic VSL#3 has dampened the neurological and cognitive deficits33–35, suggesting that targeting the gut microbiome in these patients has therapeutic value. In animal studies, VSL#3 has also been observed to alter the composition of the bile acid pool36 and improve the metabolic profile of rats with HE37. Taken together these observations suggest that there is an observable link between the development of HE and the gut microbiome, as well as individual bile acids in the circulation.
5. EFFECT OF BILE ACIDS IN HEPATIC ENCEPHALOPATHY
5.1. Increased bile acid content in the brain
For bile acids to play a role in the cellular mechanisms of HE, changes in bile acid content in the brain must occur. Early studies have demonstrated that bile acids can be found in the brain in physiological conditions as well as in many neurological disorders and are capable of altering neural function38. Whether bile acids are entering the brain from the periphery through a hyperpermeable blood brain barrier, or via specific bile acid transporters located in the choroid plexus, or are the result of de novo bile acid synthesis in the brain via cholesterol oxidation is unknown39 40. Elevation of total bile acid levels in the cerebrospinal fluid have been observed in fulminant hepatic failure patients41 although it was concluded that the concentration reached in patients was too low to be inhibit brain respiration41. However, with our current understanding of bile acids as ligands and signaling molecules with biological effects at levels that are orders of magnitude lower than those shown to affect brain respiration, it warranted further investigation into the effect of aberrant bile acid signaling in the pathogenesis of HE. Similar to Type A HE, increases specifically in taurocholic acid and glycocholic acid were detected in the cerebrospinal fluid of Type C HE patients as part of a larger metabolic screen42, although whether bile acids in the cerebrospinal fluid can alter neurological function remains to be determined.
Intriguingly, we have demonstrated increased total bile acid content in brain tissue in the azoxymethane mouse model of Type A HE43,44, and specifically changes in isomers of the bile acid taurocholic acid were observed44, 45. However, similar increases in total bile acid content in brain tissue have not been observed in the thioacetamide rodent model of Type A HE46, 47. Similarly, increased total bile acid content in brain tissue has been detected in a model of chronic liver damage prior to HE onset48, and that this may reflect an accumulation of the conjugated and unconjugated forms of the more toxic bile acid lithocholic acid49 or isomers of taurocholic acid45 giving support that bile acids may play a role in HE pathogenesis regardless of the underlying liver pathology.
5.2. Bile acid effects on Blood-Brain Barrier Permeability
As mentioned above, blood brain barrier hyperpermeability is a feature of HE. To date, the mechanisms that bile acids use to enter the cerebrospinal fluid and brain remain unknown, but evidence has suggested that certain bile acids can act on the blood-brain barrier and increase permeability themselves 50. It is believed that these bile acids act on the tight junction protein occludin through Rac1-dependent phosphorylation, and activation of this mechanism was determined to be independent of the bile acid receptors FXR and TGR550, although involvement of other bile acid receptors is unknown.
5.3. Neuronal Dysfunction
In a mouse model of Type A HE, a role of bile acids in neurological dysfunction has been demonstrated43. Specifically, mice were fed a cholestyramine diet to sequester and eliminate bile acids and an observable reduction in serum and brain bile acid content was found43. This reduction in bile acid concentrations alleviated reflex deficits and ataxia, the hallmark neurological impairments associated with HE43. Furthermore, feeding mice a diet enriched with cholic acid and deoxycholic acid altered the bile acid pool and worsened the neurological decline observed in HE43. A further look into the mechanisms behind neuronal dysfunctions caused by bile acids and their receptors is discussed in later sections.
6. BILE ACID RECEPTOR ACTIVITY IN HEPATIC ENCEPHALOPATHY
6.1. Farnesoid X Receptor (FXR)
Farnesoid X Receptor is the quintessential bile acid receptor, and is currently the most researched bile acid receptor in regards to its involvement in HE pathogenesis. FXR, a nuclear receptor, has been found in neurons throughout the brain and many other organ systems43, 51, 52. Expression of FXR has been found to be modulated in different disease states, and in acute liver failure, FXR and its cofactor small heterodimer partner (SHP) have increased expression in the frontal cortex43. Direct infusion of an FXR-specific vivo morpholino into the frontal cortex proved protective against the neurological complications associated with Type A HE43. While this observation is promising for potential HE therapeutics, downstream effects of FXR signaling in HE remain unknown. Figure 2 shows an overview of the array of effects of FXR signaling in the brain.
Figure 2:
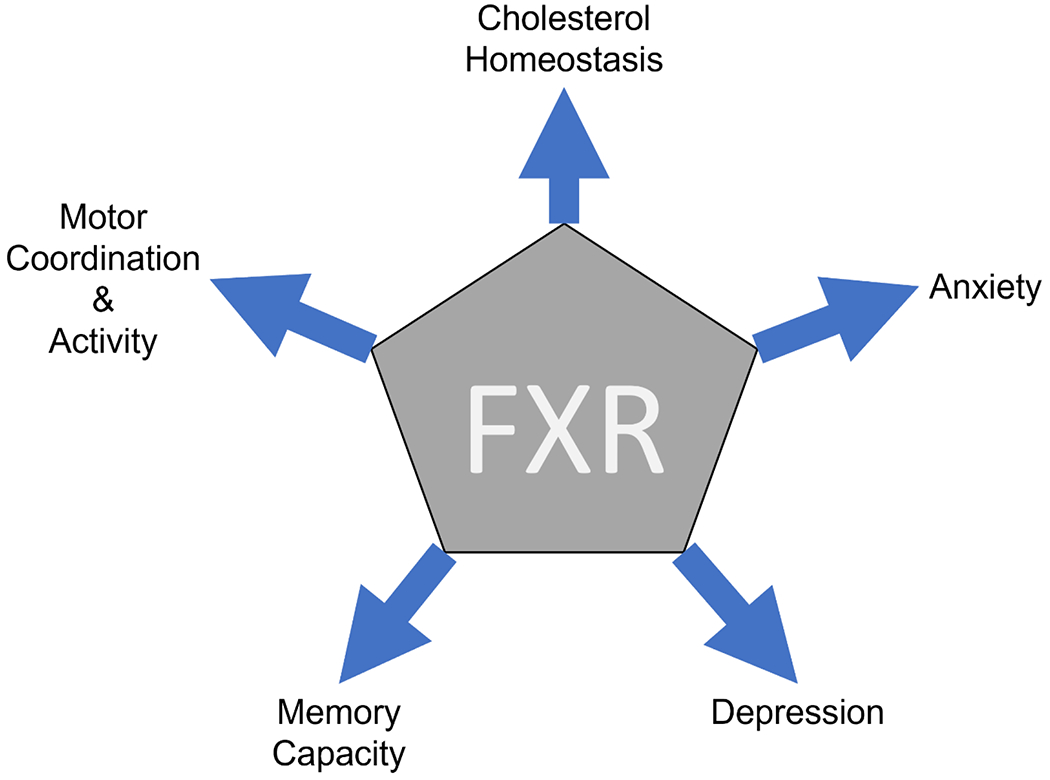
FXR signaling response. Modulation of FXR activation can have influences on mood and memory, as well as, playing a role in movement. FXR also plays a large role in cholesterol homeostasis and the production of bile acids. Aberrant bile acid signaling in HE has been observed to impact these pathways.
In support of the notion that FXR signaling is involved in the pathogenesis of HE, deletion of FXR has been observed to have many different effects, and recently was shown to have a disruptive effect on neurotransmitter systems and well as altered neurobehavior53. FXR knockout mice displayed increased motor activity with anxiety-related and depressive-like behavior, and biochemically there was observed alterations in norepinephrinergic, serotoninergic, GABAergic and glutamatergic neurotransmissions in the cerebellum and hippocampus respectively53. Many of these neurotransmitter system alterations have also been identified in HE and it is conceivable to believe that they could result from aberrant FXR activation54.
While it has yet to be studied in depth in the brain, SHP inactivation due to aberrant FXR signaling may also play a role in the pathogenesis of HE. In a double knockout mouse, for FXR and SHP, mice develop a range of cholestatic dysfunctions55. These double knockout mice lack the FXR/SHP mediated bile acid negative feedback loop, and result in increased serum bile acids55. This double knockout mouse is used as a model for progressive familial intrahepatic cholestasis 1, a severe liver disease that impacts children in infancy. According to The Childhood Liver Disease Research Network (childrennetwork.org), patients that live to early adulthood experienced problems with balance, learning, and memory. These observations are consistent with HE showcasing an increased amount of bile acids in the serum along with problems with balance, learning, and memory. Therefore, it is conceivable to link the observed effects of SHP deletion, or downregulation, in progressive familial intrahepatic cholestasis 1 to those found in HE when viewing aberrant FXR signaling in the brain.
The downstream consequences of FXR signaling in HE is not yet clear, however dysregulated cholesterol homeostasis could also be another factor in the propagation of HE via aberrant FXR activation in the brain. Due to fluctuations in cholesterol found in the diet, the brain tightly regulates its cholesterol levels independent of the rest of the body, with approximately 25% of the body’s cholesterol can be found in the brain56. Cerebral cholesterol is incorporated into the plasma membrane of cells, regulating signal transduction pathways, or is used to influence synapse formation, neurotransmitter release and action potentials57. Additionally, cholesterol can serve as the precursor for many neurosteroids, such as allopregnanolone, that are synthesized in the brain57. Clearance of cholesterol can occur via the conversion to 24-(S)-hydroxycholesterol, a major clearance pathway catalyzed by Cytochrome p450 46A1 (Cyp46A1)58. After being converted, 24-(S)-hydroxycholesterol then exits the brain and is integrated into the de novo synthesis of bile acids in the liver. Previously, the expression of the enzyme Cyp46A1 was shown to be suppressed in the frontal cortex of a Type A HE mouse model, leading to an accumulation of predominantly un-esterified cholesterol found both in the plasma membrane and intracellularly54. The downregulation of Cyp46A1, cholesterol accumulation and neurological symptoms of HE can be attenuated via strategies to eliminate the dysregulation of bile acids in the brain and the associated accumulation of brain cholesterol54. These strategies included intracerebroventricular infusion of FXR antagonists and cholestyramine feeding to attenuate aberrant bile acid signaling and intracerebroventricular infusion of a cholesterol sequestrant54. Understanding the roles of cholesterol in the brain and the interplay with bile acid signaling, it is conceivable that this pathway is responsible for the observed alterations to neurosteroid synthesis and neurotransmitter release in HE59, 60.
6.2. Takeda G-Protein Coupled Receptor 5 (TGR5)
Takeda G-Protein Coupled Receptor 5 is a membrane bound receptor and the first G-protein coupled receptor identified to be bile acid responsive61. TGR5 can be found in neurons and glial cells62 in the brain as well as many other organ systems and tissues.
In the azoxymethane model of Type A HE, TGR5 has an initial increase in expression prior to the onset of neurological symptoms followed by a marked decrease at late stages of neurological decline63. In contrast to FXR, central infusion of a TGR5 agonist delays the neurological decline and attenuated the reflex impairment and presence of ataxia63. Interestingly treatment of astrocytes with ammonia was found to reduce the expression of TGR562. Ammonia levels in the above mentioned model of Type A HE are not significantly increased until the later stages of the disease64, 65. Taken together, these observations suggest that there is an initial increase in TGR5 expression, perhaps as an attempt to protect the brain, but once ammonia levels increase TGR5 expression is attenuated.
Specifically, how TGR5 activation plays a role in the attenuation of symptoms of HE is not clear. However, TGR5 has been observed to play a role in neuroinflammation via interactions with neurons and microglia in a rodent model of HE63. Expression of CCL2, a proinflammatory chemokine, was downregulated in neurons treated with a TGR5 agonist63. The conditioned media from these neurons was then used to treat microglia and a decrease in cytokine production and phagocytic activity was observed63. These data suggest that TGR5 activation may initiate the anti-inflammatory actions via the downregulation of CCL2 in neurons, resulting in dampened activation of microglia rather than TGR5 acting directly on microglia63.
6.3. Sphingosine-1 Phosphate Receptor-2 (S1P2R)
Sphingosine-1 Phosphate Receptor-2 is a plasma membrane bound receptor found in neurons and astrocytes in the brain44, 66, the liver and in the immune system67. S1P2R is a G-protein coupled receptor responsive to conjugated bile acids and has been observed to play roles in neuroinflammation in other metabolic diseases68.
S1P2R has not been greatly studied in HE, but a link between S1P2R and the neuroinflammation observed in HE has been suggested. Firstly, the expression of S1P2R is increased, specifically in neurons in a mouse model of Type A HE44. Mice fed a cholestyramine diet to reduce the buildup of bile acids in the brain, attenuates microglial activation and expression of proinflammatory cytokines43, 44. Similarly, the expression of pro-inflammatory cytokines, microglial activation and the subsequent neurological impairment found in HE can be attenuated with an infusion of a S1P2R antagonist44. Taken together these reports suggest that bile acids potentially regulate neuroinflammation through the activation of S1P2R. To further examine this process, primary microglia were treated with the bile acid taurocholic acid, but activation of a pro-inflammatory phenotype was not observed, suggesting that bile acids are not directly responsible for microglial activation44. However, activation of neuronal S1PR2 lead to an increase in CCL2 expression and subsequent activation of microglia and expression of proinflammatory cytokines and further propagation of the neuroinflammation observed in HE44.
6.4. Other Bile Acid Receptors
Currently, in the case of HE, there is not any information on the other bile acid receptors, as this is still a novel area of research. More research is needed to elucidate if the other receptors play a role in the propagation of HE and if they could be potential therapeutic options for the treatment of HE.
7. CONCLUSIONS AND FUTURE DIRECTIONS
While the full breadth of the effects of bile acid signaling in the brain during HE has not yet been appreciated, there is mounting evidence indicating that aberrant bile acid signaling is likely contributing to the pathogenesis of HE. The majority of the mechanistic studies have focused on HE due to acute liver failure, however there is evidence suggesting that bile acids play a role in the neurological complications of chronic liver diseases as well. Further research is needed to elucidate the role and/or contribution of individual bile acids and their corresponding receptors in the development of HE. Furthermore, given that the etiology of HE is multifactorial, further research is required to identify potentially synergistic interaction of these bile acids with ammonia and other molecules known to be involved in HE pathogenesis and the effects of these molecules on bile acid receptors. Due to the pleiotropic roles of bile acids and bile acid receptors in normal physiological and metabolic processes, directly targeting bile acid signaling in the brain may not be a feasible target for the development of therapeutic targets. However, further research into the downstream consequences and off target effects of aberrant bile acid signaling in the brain may identify viable therapeutic options for the management of HE resultant from acute and chronic liver failure.
HIGHLIGHTS.
There is an association with blood bile acid profiles and the development of HE.
The effects of aberrant bile acid signaling in the brain during HE are discussed.
There is differential bile acid receptor involvement in the pathogenesis of HE.
ACKNOWLEDGEMENTS
This material is the result of work supported with resources and the use of facilities at the Central Texas Veterans Health Care System, Temple, Texas and was funded by a VA merit award (BX002638) from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Service (BLR&D) and NIH R01 awards (DK082435 and DK112803) to Dr. DeMorrow. The content is the responsibility of the author(s) alone and does not necessarily reflect the views or policies of the Department of Veterans Affairs or the United States Government.
ABBREVIATIONS:
- HE
Hepatic Encephalopathy
- ALF
Acute Liver Failure
- CCL2
Chemokine Ligand 2
- FXR
Farnesoid X Receptor
- SHP
Small Heterodimer Partner
- Cyp46A1
Cytochrome p450 46A1
- TGR5
Takeda-G Coupled Protein Receptor 5
- S1P2R
Sphingosine-1 Phosphate Receptor-2
Footnotes
CONFLICTS OF INTEREST
The authors have none to declare.
References
- 1.American Association for the Study of Liver D, European Association for the Study of the L. Hepatic encephalopathy in chronic liver disease: 2014 practice guideline by the European Association for the Study of the Liver and the American Association for the Study of Liver Diseases. J Hepatol 2014;61:642–59. [DOI] [PubMed] [Google Scholar]
- 2.Rakoski MO, McCammon RJ, Piette JD, et al. Burden of cirrhosis on older Americans and their families: analysis of the health and retirement study. Hepatology 2012;55:184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lima LCD, Miranda AS, Ferreira RN, et al. Hepatic encephalopathy: Lessons from preclinical studies. World J Hepatol 2019;11:173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romero-Gomez M, Montagnese S, Jalan R. Hepatic encephalopathy in patients with acute decompensation of cirrhosis and acute-on-chronic liver failure. J Hepatol 2015;62:437–47. [DOI] [PubMed] [Google Scholar]
- 5.Liere V, Sandhu G, DeMorrow S. Recent advances in hepatic encephalopathy. F1000Res 2017;6:1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aldridge DR, Tranah EJ, Shawcross DL. Pathogenesis of hepatic encephalopathy: role of ammonia and systemic inflammation. J Clin Exp Hepatol 2015;5:S7–S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahboucha S, Butterworth RF. The neurosteroid system: an emerging therapeutic target for hepatic encephalopathy. Metab Brain Dis 2007;22:291–308. [DOI] [PubMed] [Google Scholar]
- 8.Butterworth RF. Hepatic Encephalopathy in Cirrhosis: Pathology and Pathophysiology. Drugs 2019;79:17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.European Association for the Study of the Liver. Electronic address eee, Clinical practice guidelines p, Wendon J, et al. EASL Clinical Practical Guidelines on the management of acute (fulminant) liver failure. J Hepatol 2017;66:1047–1081. [DOI] [PubMed] [Google Scholar]
- 10.Montrief T, Koyfman A, Long B. Acute liver failure: A review for emergency physicians. Am J Emerg Med 2019;37:329–337. [DOI] [PubMed] [Google Scholar]
- 11.Weissenborn K Hepatic Encephalopathy: Definition, Clinical Grading and Diagnostic Principles. Drugs 2019;79:5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ridola L, Riggio O, Gioia S, et al. Clinical management of type C hepatic encephalopathy. United European Gastroenterol J 2020;8:536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scott TR, Kronsten VT, Hughes RD, et al. Pathophysiology of cerebral oedema in acute liver failure. World J Gastroenterol 2013;19:9240–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiss N, Rosselli M, Mouri S, et al. Modification in CSF specific gravity in acutely decompensated cirrhosis and acute on chronic liver failure independent of encephalopathy, evidences for an early blood-CSF barrier dysfunction in cirrhosis. Metab Brain Dis 2017;32:369–376. [DOI] [PubMed] [Google Scholar]
- 15.Chastre A, Belanger M, Nguyen BN, et al. Lipopolysaccharide precipitates hepatic encephalopathy and increases blood-brain barrier permeability in mice with acute liver failure. Liver Int 2014;34:353–61. [DOI] [PubMed] [Google Scholar]
- 16.McMillin MA, Frampton GA, Seiwell AP, et al. TGFbeta1 exacerbates blood-brain barrier permeability in a mouse model of hepatic encephalopathy via upregulation of MMP9 and downregulation of claudin-5. Lab Invest 2015;95:903–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen JH, Yamamoto S, Steers J, et al. Matrix metalloproteinase-9 contributes to brain extravasation and edema in fulminant hepatic failure mice. J Hepatol 2006;44:1105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamamoto S, Nguyen JH. TIMP-1/MMP-9 imbalance in brain edema in rats with fulminant hepatic failure. J Surg Res 2006;134:307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McMillin M, Grant S, Frampton G, et al. Fractalkine suppression during hepatic encephalopathy promotes neuroinflammation in mice. J Neuroinflammation 2016;13:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMillin M, Frampton G, Thompson M, et al. Neuronal CCL2 is upregulated during hepatic encephalopathy and contributes to microglia activation and neurological decline. J Neuroinflammation 2014;11:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med 1999;159:2647–58. [DOI] [PubMed] [Google Scholar]
- 22.Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res 2015;56:1085–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeMorrow S Bile Acids in Hepatic Encephalopathy. J Clin Exp Hepatol 2019;9:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budmiger H, Kocher F. [Boletus luridus and alcohol. Case report]. Schweiz Med Wochenschr 1982;112:1179–81. [PubMed] [Google Scholar]
- 25.Horvatits T, Drolz A, Roedl K, et al. Serum bile acids as marker for acute decompensation and acute-on-chronic liver failure in patients with non-cholestatic cirrhosis. Liver Int 2017;37:224–231. [DOI] [PubMed] [Google Scholar]
- 26.Mousa OY, Juran BD, McCauley BM, et al. Bile Acid Profiles in Primary Sclerosing Cholangitis and their Ability to Predict Hepatic Decompensation. Hepatology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wegermann K, Howe C, Henao R, et al. Serum Bile Acid, Vitamin E, and Serotonin Metabolites Are Associated With Future Liver-Related Events in Nonalcoholic Fatty Liver Disease. Hepatol Commun 2021;5:608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie G, Wang X, Jiang R, et al. Dysregulated bile acid signaling contributes to the neurological impairment in murine models of acute and chronic liver failure. EBioMedicine 2018;37:294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kakiyama G, Pandak WM, Gillevet PM, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol 2013;58:949–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acharya C, Bajaj JS. Gut Microbiota and Complications of Liver Disease. Gastroenterol Clin North Am 2017;46:155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Islam KB, Fukiya S, Hagio M, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011;141:1773–81. [DOI] [PubMed] [Google Scholar]
- 32.Ramirez-Perez O, Cruz-Ramon V, Chinchilla-Lopez P, et al. The Role of the Gut Microbiota in Bile Acid Metabolism. Ann Hepatol 2017;16:s15–s20. [DOI] [PubMed] [Google Scholar]
- 33.Dalal R, McGee RG, Riordan SM, et al. Probiotics for people with hepatic encephalopathy. Cochrane Database Syst Rev 2017;2:CD008716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dhiman RK, Rana B, Agrawal S, et al. Probiotic VSL#3 reduces liver disease severity and hospitalization in patients with cirrhosis: a randomized, controlled trial. Gastroenterology 2014;147:1327–37 e3. [DOI] [PubMed] [Google Scholar]
- 35.Pratap Mouli V, Benjamin J, Bhushan Singh M, et al. Effect of probiotic VSL#3 in the treatment of minimal hepatic encephalopathy: A non-inferiority randomized controlled trial. Hepatol Res 2015;45:880–9. [DOI] [PubMed] [Google Scholar]
- 36.Jones ML, Tomaro-Duchesneau C, Prakash S. The gut microbiome, probiotics, bile acids axis, and human health. Trends Microbiol 2014;22:306–8. [DOI] [PubMed] [Google Scholar]
- 37.Rackayova V, Flatt E, Braissant O, et al. Probiotics improve the neurometabolic profile of rats with chronic cholestatic liver disease. Sci Rep 2021;11:2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McMillin M, DeMorrow S. Effects of bile acids on neurological function and disease. FASEB J 2016;30:3658–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naqvi SH, Herndon BL, Kelley MT, et al. Detection of monohydroxy “bile” acids in the brains of guinea pigs afflicted with experimental allergic encephalomyelitis. J Lipid Res 1969;10:115–20. [PubMed] [Google Scholar]
- 40.Naqvi SH, Nicholas HJ. Conversion of 3-keto-5-beta-cholanoic acid to lithocholic acid by guinea pig brain tissue, in vitro. Steroids 1970;16:297–316. [DOI] [PubMed] [Google Scholar]
- 41.Bron B, Waldram R, Silk DB, et al. Serum, cerebrospinal fluid, and brain levels of bile acids in patients with fulminant hepatic failure. Gut 1977;18:692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiss N, Barbier Saint Hilaire P, Colsch B, et al. Cerebrospinal fluid metabolomics highlights dysregulation of energy metabolism in overt hepatic encephalopathy. J Hepatol 2016;65:1120–1130. [DOI] [PubMed] [Google Scholar]
- 43.McMillin M, Frampton G, Quinn M, et al. Bile Acid Signaling Is Involved in the Neurological Decline in a Murine Model of Acute Liver Failure. Am J Pathol 2016;186:312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McMillin M, Frampton G, Grant S, et al. Bile Acid-Mediated Sphingosine-1-Phosphate Receptor 2 Signaling Promotes Neuroinflammation during Hepatic Encephalopathy in Mice. Front Cell Neurosci 2017;11:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schnelle ANW, Richardson LT, Pettit ME, et al. Trihydroxycholanoyl-taurine in brains of rodents with hepatic encephalopathy. J Mass Spectrom 2021;56:e4729. [DOI] [PubMed] [Google Scholar]
- 46.Czarnecka AM, Milewski K, Albrecht J, et al. The Status of Bile Acids and Farnesoid X Receptor in Brain and Liver of Rats with Thioacetamide-Induced Acute Liver Failure. Int J Mol Sci 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grant S, McMillin M, Frampton G, et al. Direct Comparison of the Thioacetamide and Azoxymethane Models of Type A Hepatic Encephalopathy in Mice. Gene Expr 2018;18:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMillin M, Frampton G, Quinn M, et al. Suppression of the HPA Axis During Cholestasis Can Be Attributed to Hypothalamic Bile Acid Signaling. Mol Endocrinol 2015;29:1720–30. 4664228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tripodi V, Contin M, Fernandez MA, et al. Bile acids content in brain of common duct ligated rats. Ann Hepatol 2012;11:930–4. [PubMed] [Google Scholar]
- 50.Quinn M, McMillin M, Galindo C, et al. Bile acids permeabilize the blood brain barrier after bile duct ligation in rats via Rac1-dependent mechanisms. Dig Liver Dis 2014;46:527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang C, Wang J, Hu W, et al. Identification of functional farnesoid X receptors in brain neurons. FEBS Lett 2016;590:3233–42. [DOI] [PubMed] [Google Scholar]
- 52.Yan N, Yan T, Xia Y, et al. The pathophysiological function of non-gastrointestinal farnesoid X receptor. Pharmacol Ther 2021;226:107867. [DOI] [PubMed] [Google Scholar]
- 53.Huang F, Wang T, Lan Y, et al. Deletion of mouse FXR gene disturbs multiple neurotransmitter systems and alters neurobehavior. Front Behav Neurosci 2015;9:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McMillin M, Grant S, Frampton G, et al. FXR-Mediated Cortical Cholesterol Accumulation Contributes to the Pathogenesis of Type A Hepatic Encephalopathy. Cell Mol Gastroenterol Hepatol 2018;6:47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anakk S, Watanabe M, Ochsner SA, et al. Combined deletion of Fxr and Shp in mice induces Cyp17a1 and results in juvenile onset cholestasis. J Clin Invest 2011;121:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orth M, Bellosta S. Cholesterol: its regulation and role in central nervous system disorders. Cholesterol 2012;2012:292598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cartocci V, Servadio M, Trezza V, et al. Can Cholesterol Metabolism Modulation Affect Brain Function and Behavior? J Cell Physiol 2017;232:281–286. [DOI] [PubMed] [Google Scholar]
- 58.Lund EG, Xie C, Kotti T, et al. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem 2003;278:22980–8. [DOI] [PubMed] [Google Scholar]
- 59.Butterworth RF. Neurosteroids in hepatic encephalopathy: Novel insights and new therapeutic opportunities. J Steroid Biochem Mol Biol 2016;160:94–7. [DOI] [PubMed] [Google Scholar]
- 60.Hazell AS, Butterworth RF. Hepatic encephalopathy: An update of pathophysiologic mechanisms. Proc Soc Exp Biol Med 1999;222:99–112. [DOI] [PubMed] [Google Scholar]
- 61.Duboc H, Tache Y, Hofmann AF. The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig Liver Dis 2014;46:302–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keitel V, Gorg B, Bidmon HJ, et al. The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia 2010;58:1794–805. [DOI] [PubMed] [Google Scholar]
- 63.McMillin M, Frampton G, Tobin R, et al. TGR5 signaling reduces neuroinflammation during hepatic encephalopathy. J Neurochem 2015;135:565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Belanger M, Cote J, Butterworth RF. Neurobiological characterization of an azoxymethane mouse model of acute liver failure. Neurochem Int 2006;48:434–40. [DOI] [PubMed] [Google Scholar]
- 65.Matkowskyj KA, Marrero JA, Carroll RE, et al. Azoxymethane-induced fulminant hepatic failure in C57BL/6J mice: characterization of a new animal model. Am J Physiol 1999;277:G455–62. [DOI] [PubMed] [Google Scholar]
- 66.Chen Z, Doyle TM, Luongo L, et al. Sphingosine-1-phosphate receptor 1 activation in astrocytes contributes to neuropathic pain. Proc Natl Acad Sci U S A 2019;116:10557–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Aoki H, Yang J, et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017;65:2005–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang YH, Fehrenbacher JC, Vasko MR, et al. Sphingosine-1-phosphate via activation of a G-protein-coupled receptor(s) enhances the excitability of rat sensory neurons. J Neurophysiol 2006;96:1042–52. [DOI] [PubMed] [Google Scholar]