

SUMMARY
Recent work showed that the dominant post-menopausal estrogen, estrone, cooperates with nuclear factor κB (NF-κB) to stimulate inflammation, while pre-menopausal 17β-estradiol opposes NF-κB. Here, we show that post-menopausal estrone, but not 17β-estradiol, activates epithelial-to-mesenchymal transition (EMT) genes to stimulate breast cancer metastasis. HSD17B14, which converts 17β-estradiol to estrone, is higher in cancer than normal breast tissue and in metastatic than primary cancers and associates with earlier metastasis. Treatment with estrone, but not 17β-estradiol, and HSD17B14 overexpression both stimulate an EMT, matrigel invasion, and lung, bone, and liver metastasis in estrogen-receptor-positive (ER+) breast cancer models, while HSD17B14 knockdown reverses the EMT. Estrone:ERα recruits CBP/p300 to the SNAI2 promoter to induce SNAI2 and stimulate an EMT, while 17β-estradiol:ERα recruits co-repressors HDAC1 and NCOR1 to this site. Present work reveals novel differences in gene regulation by these estrogens and the importance of estrone to ER+ breast cancer progression. Upon loss of 17β-estradiol at menopause, estrone-liganded ERα would promote ER+ breast cancer invasion and metastasis.
In brief
Qureshi et al. show that the dominant pre- and post-menopausal estrogens E1 and E2 recruit different co-regulators to ERα to induce or repress SNAI2, respectively. E1 exposure and increased intracellular E2-to-E1 conversion upregulate EMT transcription profiles, promoting tumor invasion and multi-organ ER+ breast cancer metastasis in vivo.
Graphical Abstract
INTRODUCTION
Despite major advances in targeted therapy and early detection, breast cancer is the most common cancer and the second leading cause of cancer death in women. Breast-cancer-related mortality is caused by metastasis to distant organs.1 Hormone receptor-positive, HER2-negative breast cancer is the most common subtype, accounting for approximately 65% of all cases, and causes the most breast cancer-related deaths.2 Despite a favorable prognosis relative to other breast cancer subtypes, these cancers can recur many years later, and outcomes of metastatic hormone receptor-positive breast cancer remain poor, with a median overall survival of 36 months.3
Estrogen receptor-positive (ER+) breast cancer incidence increases with age.4,5 Among women with metastatic breast cancer (MBC), those under age 50 have a better prognosis than women over 50 years old (χ2 = 69.8, p < 0.001), which is not the case for patients with hormone receptor-negative MBC.6 While ovarian 17β-estradiol (E2) is the primary estrogen in pre-menopausal women, most breast cancers are diagnosed after menopause, when ovarian E2 production is minimal7 and estrone (E1) is the major post-menopausal hormone. Here, we investigated how the different estrogens before and after menopause might contribute to the greater ER+ breast cancer incidence after menopause and the adverse survival of ER + MBC in post- versus pre-menopausal women.
The development of metastasis is associated with morphological changes characterized by an epithelial-to-mesenchymal transition (EMT).8 EMT changes cell polarity, adhesion, and migratory properties and is characterized by upregulation of mesenchymal markers and loss of epithelial markers such as E-cadherin.9–14 EMT endows cells with a more motile, invasive phenotype, supporting local invasion and metastasis.14–17
Breast cancer stromal cells include fibroblasts, endothelial and immune cells, and adipocytes.18,19 Adipocytes are the most abundant breast cancer microenvironment component and secrete hormones, growth factors, and cytokines that promote cancer invasion and metastasis.20,21 Adipose tissue is the major component of the post-menopausal breast and the major source of E1 production. E1 is produced via aromatization of adrenal androstenedione largely in adipose tissue but also in bone, breast, and brain tissues.22 After menopause, E1 dominates, and circulating and tissue levels rise as adipose biomass increases in obesity.23 Breast cancer cell:adipocyte interaction upregulates cytokines and activates Src to expand cancer stem cells (CSCs).20 This would facilitate breast cancer progression upon invasion into local fat.20 We recently made the novel observation that E1- and E2-stimulated transcriptomes are not identical.24 In contrast to the anti-inflammatory action of E2,25,26 E1 is pro-inflammatory.24 E1-liganded ERα is co-recruited with nuclear factor κB (NF-κB) to upregulate pro-inflammatory cytokine drivers of CSCs, while E2 opposes this. Both E1 and overexpression of 17β-hydroxysteroid dehydrogenase B14 (HSD17B14), which converts E2 to E1, increased intratumor E1 and tumor-initiating stem cells to mediate greater ER+ cancer growth in vivo. Thus, E1 has a pro-oncogenic, pro-inflammatory role, cooperating with NF-κB to promote cytokine drivers of CSCs. After menopause, this is unopposed by ovarian E2.
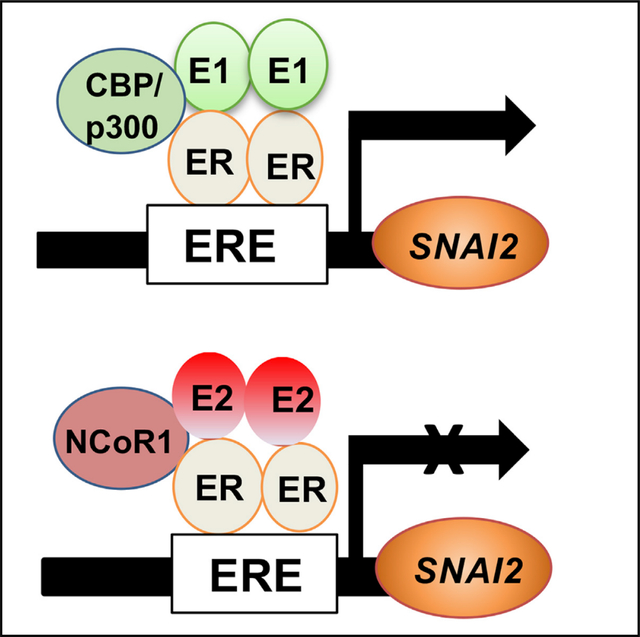
Here, we investigated whether and how the dominant post-menopausal estrogen E1 contributes to EMT and metastasis. We found that co-culture with adipocytes, a major source of E1, stimulates ER+ breast cancer cell EMT. E1, but not E2, activates gene-expression profiles of EMT and signatures characteristic of lung, brain, and bone metastasis. E1 and overexpression of HSD17B14 both promote ER+ breast cancer EMT, invasion, and multi-organ metastasis in vivo. E1-bound ERα recruits co-activator CBP/p300 to a promoter estrogen response element (ERE) in the snail family transcriptional repressor 2 (SNAI2) gene to induce its expression, while E2 opposes this, recruiting Nuclear receptor corepressor 1 (N-CoR1) with ERα to repress SNAI2.
RESULTS
Estrone drives invasion and metastasis in ER+ breast cancer cells
In a prior study, we reported that E1 stimulated orthotopic MCF7 tumor formation with shorter latency and greater final tumor volume than did E2.24 Here, we investigated the effects of these two estrogens on ER+ cancer invasion and metastasis. Estrogen effects on matrigel invasion were assayed by hanging drop spheroid cultures as in Berens et al.27 Spheroids formed in three-dimensional (3D) culture without estrogen (in 5% charcoal-stripped fetal bovine serum [cFBS]) were smaller and less invasive than those formed in the presence of estrogen. Of the two estrogens, E1 stimulated greater sphere growth and significantly increased the area invaded compared with no-estrogen controls or E2 treated cells in 4 independent ER+ cancer lines: MCF7, T47D (Figures 1A–1D), MDA-MB-361, and ZR75-1 (Figures S1A–S1D). Furthermore, E1 supported greater migration in wound-closure assays than E2 following wounding of a confluent lawn of estrogen-starved MCF7 (p < 0.05; Figure S1E).
Figure 1. Estrone promotes ER+ breast cancer invasion and metastasis.

(A–D) MCF7 and T47D spheroid invasion in 5% cFBS (no estrogen), 10 nM E1, or 10 nM E2 for indicated times. Representative photomicrographs (A and C). Quantitative analysis (B and D) of sphere area is graphed as mean (±SEM) from at least 3 biological repeat assays, with p values from Student’s t test and ANOVA; see also data for MDA-MB-361 and ZR75–1 in Figures S1A–S1D.
(E) Bioluminescence (BLI) 8 weeks after intravenous (i.v.) injection of luciferase-tagged MCF7-luc cells into NSG mice supplemented with E1, E2, or placebo control pellets; representative images of n = 6 mice/group; see also Figure S1F.
(F) Mean normalized photon flux of BLI (±SEM) of lung metastases at 8 weeks after i.v. injection of mice in (E).
(G) Quantitation of lung and liver metastatic nodules in mice in (E).
(H) Table shows organ metastasis in mice supplemented with E1, E2, or placebo control.
(I) Representative hematoxylin and eosin (H&E)-stained lung and liver. The arrows highlight metastases; see also Figure S1G.
(J) Representative H&E-stained bone is shown with arrow highlighting metastases.
(K) Mean volume/time of orthotopic E0771 tumors from C57BL/6 mice supplemented with E1, E2, or placebo control (C) pellets (n = 5/group), left. Quantification of the lung metastasis in EO771, right graph; see also Figure S1H.
For all graphed data, *p < 0.05, **p < 0.001, and ***p < 0.0001. Scale bars indicate microns.
To quantitate metastasis stimulated by estrogens in MCF7, parental MCF7 was luciferase tagged and then injected via tail vein (intravenously [i.v.]) into either E1- or E2-supplemented NOD/SCID gamma (NSG) mice and assayed by bioluminescent imaging (BLI) every 2 weeks. As previously reported for MCF7, few distant metastases of luciferase-positive MCF7 were detected following i.v. injection into E2-supplemented NSG mice, and none formed in no-estrogen controls with sham pellets. In contrast, E1-supplemented mice showed much greater tumor bioluminescence within 8 weeks than E2-supplemented hosts (Figures 1E, 1F, and S1F). E1 supplements also led to widespread dissemination of i.v.-injected MCF7 to common sites of clinical breast cancer metastasis (lung, liver, and bone) (Figures 1G–1J). Histopathologic confirmation of lung, liver, and bone metastasis is shown in Figures 1I, 1J, and S1G. Thus, the major post-menopausal hormone E1 promotes widespread ER+ breast cancer metastasis in vivo, which is not detected in E2-treated mice.
To validate the unprecedented finding that the dominant post-menopausal estrogen E1 is more pro-metastatic than E2, these hormones were assayed further in a second independent estrogen-sensitive breast cancer model, E0771. Orthotopic injection of ER+ E0771 into syngeneic, oophorectomized mice yielded little tumor growth in the absence of estrogen. Primary E0771 cancers grew faster with E1 than E2 supplementation in vivo (Figure 1K, left). Primary tumors were removed at 1,000 m3, and mice were followed for metastasis. The number of metastatic lung nodules and histopathologic evidence of lung metastasis from primary E0771 tumors was significantly greater in E1-treated than E2-treated mice or in mice with sham no-estrogen control pellets (Figures 1K, right, and S1H).
E1 drives a program of EMT and pro-metastatic gene expression
To compare the effects of these estrogens on gene programs of EMT and tumor invasion, global expression profiling was compared in E1- or E2-stimulated MCF7 orthotopic xenografts recovered at 1,000 mm3 (3 tumors/group) as in Qureshi et al.24 Taube et al. established an “EMT core signature” common to human mammary epithelial cells overexpressing several different master EMT regulators and showed that it is predictive of early metastasis in women with aggressive breast cancers.28 Genes differentially expressed in E1- versus E2-stimulated MCF7 xenografts (fold change [FC] >2× up or <0.5× down, Q < 0.05) were compared with this EMT core signature. EMT driver genes from this signature were highly expressed in our E1-driven ER+ MCF7 cancers, while genes downregulated in this EMT signature were also reduced by E1 but not by E2 (Figure 2A). Hypergeometric tests revealed that the overlap of 65/136 genes in the EMT up signature (p = 0.00033), and 35/79 genes in the EMT down signature (p = 0.0277) with genes differentially expressed between E1- and E2-treated tumors was significant. Genes associated with ECM, GAP junctions, focal adhesion, actin cytoskeleton, and vascular endothelial growth factor (VEGF) signaling were significantly upregulated in E1- compared with E2-stimulated cancers (Figure 2B). Thus, E1 appears to increase ER+ cancer cell motility and invasion in part through induction of EMT programs.
Figure 2. E1 treatment and adipocyte co-culture with MCF7 induce EMT and metastatic gene signatures.
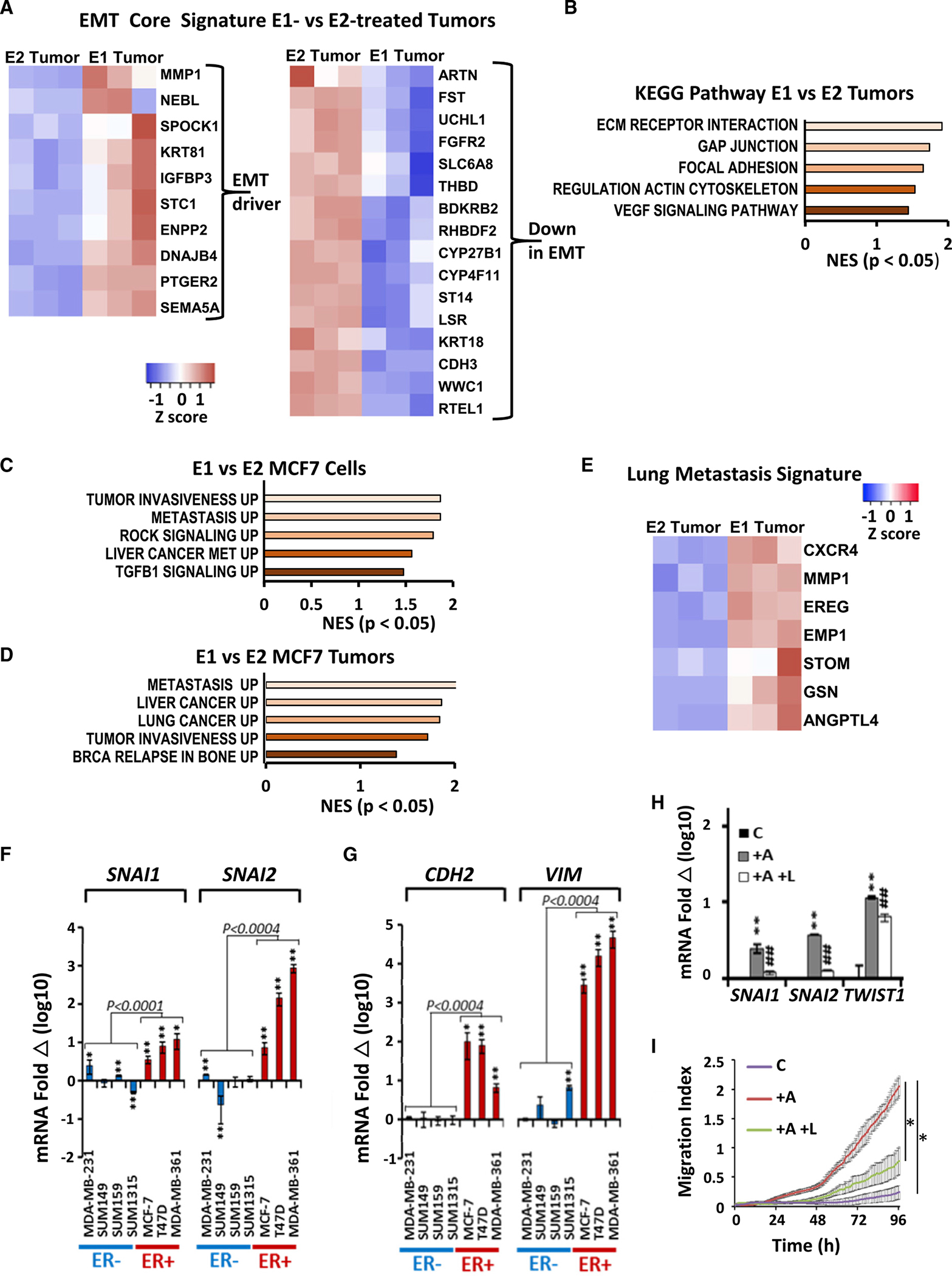
(A) EMT-related gene-expression heatmap from RNA sequencing (RNA-seq) of 3 independent E1- or E2-treated MCF7 xenografts. Fold change (FC) > 2 fold up or < 0.5 fold down, Q < 0.05.
(B) KEGG pathway analysis shows enrichment of EMT-related pathways in E1- versus E2-supplemented tumors, p < 0.05.
(C) Estrogen-starved MCF7 (cFBS) cells were treated with10 nM E1 or E2 for 8 h followed by RNA-seq on 3 biologic repeat samples. Gene Ontology (GO) analysis shows gene profiles upregulated in E1- versus E2-treated cells, FC > 2, Q < 0.05.
(D) GO analysis of E1- or E2-supplemented MCF7 xenograft tumors (n > 3/group) shows gene profiles upregulated in E1- versus E2-treated tumors, FC > 2, Q < 0.05.
(E) Heatmap shows lung metastasis signature genes enriched in E1- versus E2-stimulated tumors (n = 3 tumors).
(F and G) SNAI1 and SNAI2 (F) and CDH2 and VIM (G) expression in ER− and ER+ breast cancer lines ± 7-day co-culture with adipocytes, normalized to 1 for breast cancer monoculture; n > 3 repeat assays with different adipocyte donors; *p < 0.05 and **p < 0.001.
(H) qPCR of indicated genes in MCF7 alone controls, C, or after 7-day co-culture with mammary adipocytes (+A), or with adipocytes plus 10 nM letrozole (+A +L), normalized to 1 for monocultures; n > 3 different adipocyte donors; **p < 0.001 versus C; ##p < 0.001 versus +A.
(I) Transwell migration of MCF7 alone, C, and after 7-day co-culture with mammary adipocytes (+A) or with adipocytes plus 10 nM letrozole (+A +L) analyzed by Real-Time Cell Analysis xCELLigence.
(F–I) Graphs show mean (±SEM) from at least 3 biological repeat and triplicate replicate assays and show p from Student’s t test and ANOVA, *p < 0.05.
Estrogen-starved MCF7 cells were treated with 10 nM E1 or E2 for 8 h followed by gene-expression profiling. Gene set enrichment analysis (GSEA) showed upregulation of programs of tumor invasiveness, metastasis, ROCK, and transforming growth factor B1 (TGFB1) signaling in E1-treated compared with E2-treated MCF7 (Figure 2C). Similarly, GSEA comparing E1- or E2-driven MCF7 xenograft tumor profiles showed greater expression of pathways associated with tumor invasiveness, metastasis, and breast cancer relapse in bone in E1-driven tumors when compared with the E2 tumors (Figure 2D). In a series of key papers, Massague’s group selected, by serial in vivo xenografting, human breast-cancer-derived variant lines with high metastatic tropism for lung29 or bone30 and validated the ability of these signatures to predict metastases to these respective sites in cohorts of patients with breast cancer. Notably, heatmap comparisons of E1- and E2-stimulated tumor expression data showed that E1 tumors strongly overexpressed the lung metastatic gene-expression profile,29 as shown in Figure 2E. Hypergeometric test showed that the overlap of 22/44 genes in the lung metastatic signature with genes differentially expressed in E1- versus E2-treated tumors was significant (p = 0.0119). E1-stimulated cancers had a high expression of chemokine (C-X-C motif) receptor 4 (CXCR4), matrix metalloproteinase (MMP1), and angiopoietin-like 4 (ANGPTL4) (Figure 2E), all of which are known to promote lung metastasis in breast cancer.31–33
Adipocyte:cancer cell contact induces an EMT
Breast cancer cells invading beyond the duct basement membrane are conditioned by contact with peritumoral adipocytes, which are abundant in mammary stroma. Mammary adipocytes are a major source of peritumoral E1. Co-culture with mammary adipocytes stimulates pro-inflammatory cytokines and stem cell markers in ER+ breast cancer lines through cross talk between E1-liganded ERα with NF-κB.20,24 Here, we tested the effects of co-culture with adipocytes on EMT transcription factors (TFs) and cell motility. Co-culture over 7 days of breast cancer cells with mature mammary adipocytes from obese, postmenopausal women induced expression of EMT- TF genes SNAI1 and SNAI2 and upregulated genes encoding N-cadherin (CDH2) and vimentin (VIM) significantly more in ER+ than in ER-negative (ER−) breast cancer lines, indicating a role for estrogens in EMT induction (Figures 2F and 2G). Adipocyte co-culture also upregulated Snail, Slug, and Twist1 protein levels (Figure S1I).
We previously showed that 7 days of adipocyte:breast cancer cell co-culture generates high media E1 concentrations, with E1 concentrations up to 30-fold higher than those of E2.24 Here, we showed that inhibition of E1 production by the aromatase inhibitor letrozole impaired co-culture-induced upregulation of SNAIL1, SNAI2, and TWIST1 in MCF7 (Figure 2H). Transwell migration of MCF7 was faster following co-culture with adipocytes and was impaired by pre-treatment with letrozole (Figure 2I). These findings suggest that adipocyte E1 production can stimulate EMT in local cancer cells.
HSD17B isoforms that upregulate E1 are greater in cancer than normal tissue, increase with metastasis, and correlate with early ER+ breast cancer relapse
In addition to local E1 production by peritumoral adipocytes, high intratumor E1 can arise in breast cancer cells through changes in the balance of 17β-hydroxysteroid dehydrogenase (HSD17B) family members34 that either convert intracellular E1 to E2 or E2 to E1. We previously showed that HSD17B14 overexpression in ER+ breast cancer models leads to high intracellular E1 and is pro-inflammatory and pro-oncogenic.24 Here, we investigated the associations between HSD17B enzymes and metastasis. Analysis of breast cancers from The Cancer Genome Atlas (TCGA) showed that HSD17B14 expression is increased significantly in cancer compared with normal breast tissue and that it rises with increasing disease stage, particularly between stage 4 (metastatic cancer) and all others (Figure S2A). HSD17B14 expression was also higher in metastatic compared with primary human ER+ breast cancers (Figure 3A). The prognostic importance of HSD17B family members was evaluated using the Kaplan-Meier (KM) plotter primary human breast cancer database, which permits univariate analysis of the prognostic importance of gene-expression data pooled from many different large patient cohorts. HSD17B14 expression was prognostic of greater risk of distant metastasis in women with ER+ breast cancer (hazard ratio [HR] for relapse 1.59, p = 0.0074; Figure 3B). Notably, for two additional HSD17B family members that convert E2 to E1, HSD17B2 and HSD17B10, high expression also associated with a greater risk of ER+ breast cancer metastasis (HR 1.27, p = 0.0024, and HR 1.31, p = 0.055, respectively) (Figures S2B and S2C). In contrast, high intratumor levels of three other HSD17B family members that convert E1 to E2, HSD17B1, HSD17B7, and HSD17B5, associated inversely with disease recurrence (HR for recurrence 0.83, p = 0.029; HR 0.81, p = 0.0082; and HR 0.65 p = 0.027, respectively; Figures S2D–S2F).
Figure 3. HSD17B14 induces EMT and increases motility and invasion.
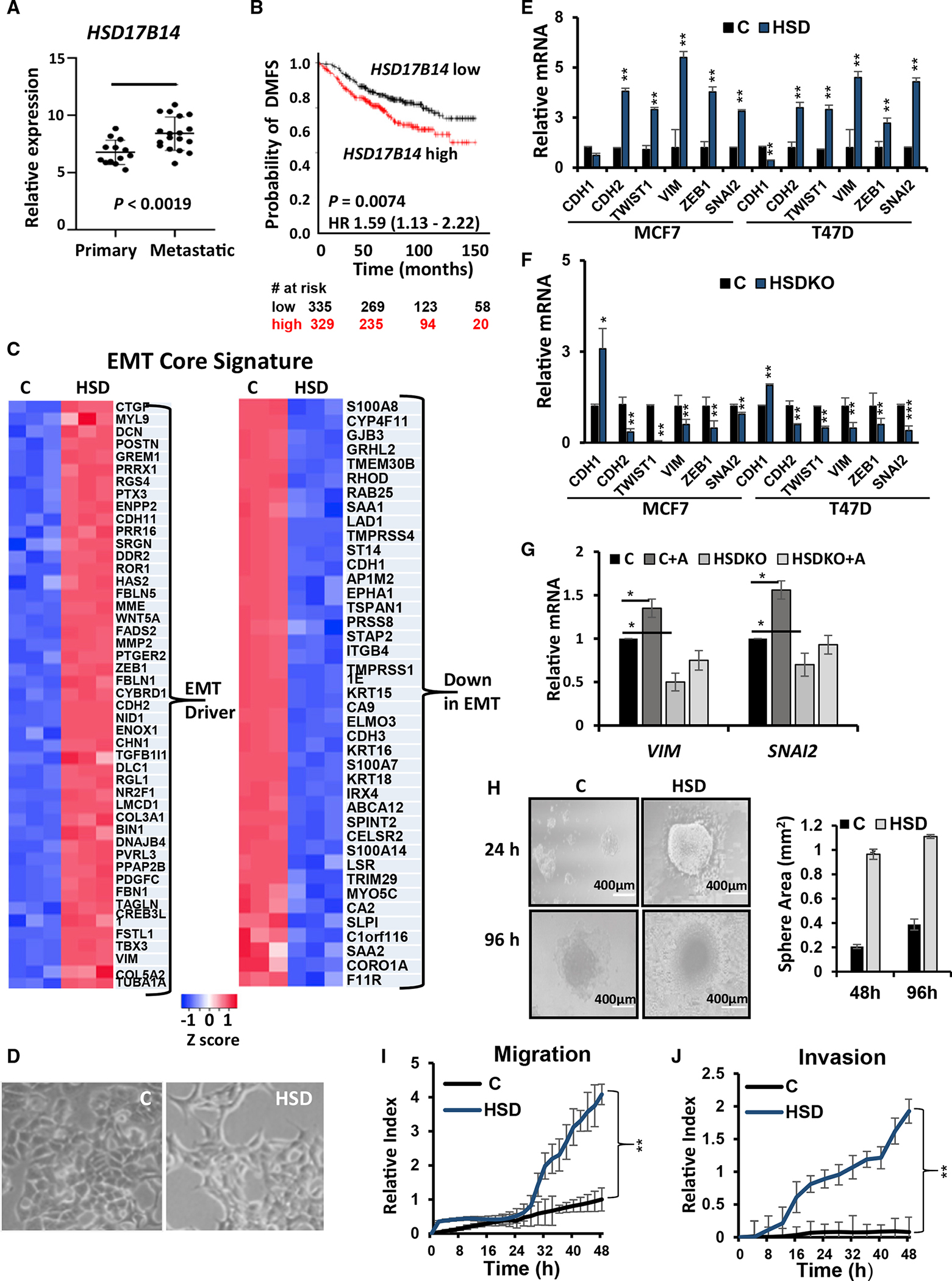
(A) Boxplot comparing HSD17B14 expression in primary (n = 13) and metastatic breast cancer tissues (n = 18) from GEO: GSE32531 analyzed using Geo2R; Student’s t test, p < 0.0019.
(B) Kaplan-Meier plots for distant metastasis free survival (DMFS) in ER+ breast cancers with high or low HSD17B14 expression (above or below the median); log rank p value and HR indicated; see also Figure S2.
(C) RNA-seq heatmap shows EMT core signature gene expression in control versus MCF7HSD. FC >2, Q < 0.05. n = 3.
(D) HSD17B14 overexpression in MCF7 promotes morphologic EMT.
(E) EMT markers and TFs were compared in MCF7 and T47D vector controls and lines overexpressing HSD17B14. Data are graphed as mean (±SEM) ratio of expression versus GAPDH normalized to 1 for vehicle control (n = 3).
(F) EMT marker and TF expression were compared in HSD17B14 CRISPR knockout (HSDKO) and MCF7 and T47D vector control lines and data graphed as mean (±SEM) ratio of expression versus GAPDH normalized to 1 for vehicle control (n = 3).
(G) MCF7-HSDKO was co-cultured with human mammary adipocytes for 7 days, then VIM and SNAI2 expression was assayed by qPCR (n = 3).
(H) Representative photomicrographs and quantitative analysis of spheroid areas at indicated times after plating hanging drop spheroid invasion assay in control MCF7 and MCF7HSD (n = 3).
(I and J) Analysis of migration (I) and invasion (J) of MCF7 controls compared with MCF7HSD for 48 h by Real-Time Cell Analysis xCELLigence system (n = 3). All graphs show mean (±SEM) from at least 3 biological repeat and >triplicate replicate assays; p from Student’s t test and ANOVA, *p < 0.05, **p < 0.001, and ***p < 0.0001; see also Figure S3. Scale bars indicate microns.
HSD17B14 overexpression promotes EMT in ER+ breast cancer cell
To investigate further links between HSD17B14 and tumor metastasis, global gene expression from MCF7 vector controls and the HSD17B14-overexpressing MCF7 line (MCF7HSD) described in Qureshi et al.24 were compared with the same EMT core signature28 used in Figure 2. Genes downregulated in EMT were decreased, while gene drivers of EMT were highly expressed in MCF7HSD (Figure 3C). Hypergeometric tests comparing EMT profiles with genes differentially expressed in MCF7HSD versus MCF7 showed that 109/136 of genes whose downregulation associates with EMT were down in MCF77HSD versus MCF7 (p = 0) and that 70/79 upregulated in the EMT profile were up in MCF7HSD versus MCF7 (p = 3.97e–08). Notably, MCF7HSD showed morphological transformation from an epithelial to a more mesenchymal phenotype (Figure 3D). Both MCF7 and T47D lines overexpressing HSD17B14 show upregulation of mesenchymal genes and EMT-TFs including CDH2, SNAI2, TWIST, VIM, and ZEB1 when compared with controls and reduced expression of CDH1 encoding the epithelial marker E-cadherin (Figure 3E). Findings were confirmed at the protein level, with immunofluorescence microscopy showing a loss of E-cadherin and increased vimentin in MCF7HSD (Figure S3A), and immunoblots also showed that E-cadherin was reduced and mesenchymal markers N-cadherin, vimentin, Twist1 and Slug were increased in MCF7HSD (Figure S3B). Similar results were observed in HSD17B14-transduced MDA-MB-361 compared with controls (Figure S3C). MCF7 and T47D lines with CRISPR knockout of HSD17B14, first described in Qureshi et al.,24 showed decreased expression of mesenchymal markers and increased CDH1 (Figure 3F). Finally, loss of HSD17B14 in MCF7-HSD17B14 CRISPR knockout (HSDKO) prevented the significant induction of genes encoding vimentin and SNAI2 observed in control MCF7 cells after 7 days of co-culture with mammary adipocytes (Figure 3G).
MCF7HSD showed a significantly greater area of invasion after 96 h in 3D spheroid assays compared with vector controls (morphology shown in Figure 3H), with similar results observed in MDA-MB-361-HSD (Figure S3D). Notably, both MCF7HSD and MDA-MB-361-HSD also showed significantly greater migration and matrigel invasion on Transwell assays than their vector controls (Figures 3I, 3J, S3E, and S3F). Wound-healing assays confirmed that HSD17B14 overexpression promoted faster MCF7 migration over 48 h and that HSD17B14 KO inhibited MCF7 migration (Figure S3G).
HSD17B14 overexpression increases intracellular estrone and cancer metastasis
Since HSD17B variants that upregulate E1 are associated with early metastasis in women with ER+ breast cancers, we next tested effects of HSD17B14 on ER+ breast cancer metastasis in vivo. Orthotopic primary MCF7HSD xenografts were grown in NOD/SCID mice (n = 8/group). These showed a significant increase in gene profiles associated with early cancer recurrence, TGF-β1 targets, breast cancer relapse in the brain, and breast tumor EMT compared with MCF7 control tumors (Figure 4A). Differentially expressed genes in control and HSD17B14-overexpressing tumors (up by FC = 2 or down < 0.5, Q < 0.05) were compared with the breast cancer lung metastasis gene signature (Figure 4B) as defined in Minn et al.29 HSD17B14 strongly upregulated this lung metastasis gene signature. The overlap between these gene lists was significant on hypergeometric test, with 34/43 lung metastasis signature genes differentially expressed in MCF7HSD versus MCF7 control (p = 0.0087). Increased expression of genes encoding CXCR4, MMP2, and prostaglandin-endoperoxide synthase 2 (PTGS2) was confirmed by qPCR (Figure 4C). GSEA revealed not only upregulation of the Taube EMT signature but a trend toward upregulation of genes involved in angiogenesis and in metastasis in MCF7HSD versus MCF7 control lines and/or tumors derived from them (Figures S4A–S4D).
Figure 4. HSD17B14 increases intracellular E1:E2 ratio, leading to multi-organ metastasis.
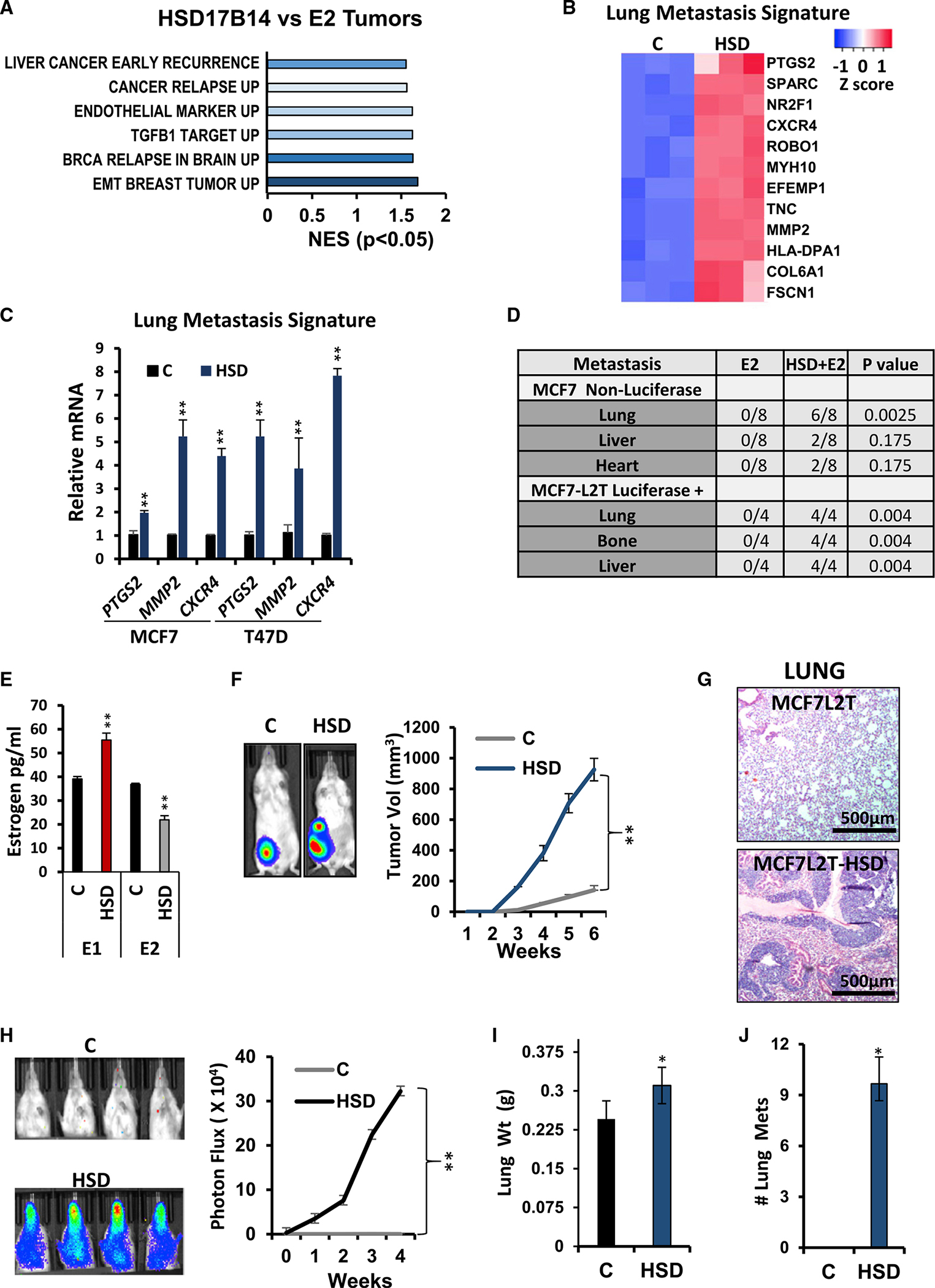
(A) GO analysis of differentially expressed genes in E2-treated mice injected with MCF7HSD (HSD17B14) or MCF7 control (E2 tumors) (n = 3 tumors).
(B) MCF7HSD shows high expression of breast cancer lung metastasis signature genes (n = 3 tumors).
(C) qPCR validates overexpression of lung metastasis mediators in HSD17B14 (HSD) overexpressing MCF7 and T47D compared with vector controls (n = 3 tumors).
(D) Table showing numbers of E2-supplemented mice with metastasis to different organs after injection with non-luciferase-tagged MCF7 controls versus MCF7HSD (non-luciferase, top) and with luciferase-expressing MCF7L2T controls versus MCF7L2T-HSD (luciferase+, bottom).
(E) E1 and E2 concentrations (pg/mL) in MCF7L2T control, C, or MCF7L2T-HSD (HSD), (n = 3).
(F) MCF7L2T, C, or MCF7L2T-HSD (HSD) were orthotopically injected into E2-supplemented NOD-SCID mice. See representative BLI at 6 weeks (left) and mean tumor volumes/time (right) (n = 3).
(G) Representative H&E-stained lung sections.
(H) Primary tumors were removed at 1,000 mm3, and mice followed for metastasis. Representative BLI from mice orthotopically injected with MCF7L2T Cs or MF7L2T-HSD (HSD) showing lung metastasis. Mean ± SEM normalized photon flux/second from tumor metastases is graphed. p from ANOVA, **p < 0.001.
(I and J) Mean ± SEM lung weights (I) and lung metastatic nodules (J) in mice injected orthotopically with control or HSD17B14-overexpressing MCF7 cells. p from ANOVA, *p < 0.05, **p < 0.001, and ***p < 0.0001, see also Figure S4. All graphs show mean (±SEM) from at least 3 biological repeat and >triplicate replicate assays; p from Student’s t test and ANOVA, *p < 0.05, **p < 0.001, and ***p < 0.0001. Scale bars indicate microns.
When orthotopic primary tumors from MCF7 control and MCF7HSD-injected mice reached 1,000 mm3, tumors were excised, and mice were followed for metastasis. Notably, on histopathologic analysis, metastasis to lungs, liver, and bone was detected only in mice bearing MCF7HSD tumors and not from MCF7 control tumors (summarized for non-luciferase-tagged MCF7 derived lines, top of table in Figure 4D).
To more readily quantitate metastasis generated by HSD17B14-overexpressing MCF7, the parental MCF7 line was luciferase and tdTomato tagged to generate the luciferase-positive control line MCF7L2T and then infected with an HSD17B14 lentiviral vector to generate MCF7L2T-HSD. HSD17B14 overexpression was confirmed by qPCR and western blot (Figures S4E and S4F). Concentrations of intracellular E1 increased and E2 decreased in MCF7L2T-HSD compared with MCF7L2T (Figure 4E), as previously reported in MCF7HSD compared with MCF7.24 NOD/SCID mice supplemented with E2 were injected orthotopically with either vector control MCF7L2T or MCF7L2T-HSD (n = 4/group). Both lines remained estrogen dependent since control mice implanted with sham no-estrogen pellets failed to generate tumors by week 12. MCF7L2T-HSD generated larger tumors than MCF7L2T (Figure 4F). As above, primary tumors were removed at 700–1,000 mm3, and mice were monitored for metastasis from the primary site by weekly BLI. Extensive lung metastasis from primary MCF7L2T-HSD tumors was detected by H and E staining and by BLI (Figures 4G, 4H, and S4G). Lung weights and numbers of lung metastases were increased significantly after HSD-overexpressing MCF7 injections compared with controls (Figures 4I and 4J). Notably, one MCF7L2T-HSD tumor showed extensive invasion into cardiac muscle (Figure S4H). Taken together, these data strongly indicate that microenvironmental changes that increase local E1 or changes in HSD17B enzymes that upregulate intracellular E1 stimulate a pro-metastatic profile of gene expression to drive ER+ breast cancer metastasis in vivo.
HSD17B14 overexpression leads to multi-organ metastasis in vivo
While all breast cancer subtypes can metastasize to bone, breast cancers expressing both ER and progesterone receptor (PR) proteins have the highest propensity to do so.35 In contrast to the bone metastatic signature described by Kang et al., which was identified in pre-clinical models and then shown to predict bone metastasis in humans, Savci-Heijink et al. identified a 15-gene signature predictive of bone metastasis from among over 500 primary breast cancers and validated its independent predictive value for bone metastasis in multi-variate analysis.30,36 To better understand the role of HSD17B14 in bone metastasis, we evaluated expression of both signatures above in our xenograft models. When compared with controls, HSD17B14-overexpressing orthotopic MCF7 tumors were enriched for the breast cancer bone metastasis signatures identified by these two groups.30,36 HSD17B14 upregulates bone metastasis mediators (Figure 5A). Upregulation of known bone metastasis mediator37,38 genes selected from the bone metastasis signature,30,36 including connective tissue growth factor (CTGF), dual specificity phosphatase1 (DUSP1), and fibroblast growth factor 5 (FGF5), was confirmed by qPCR in our HSD17B14-overexpressing ER+ MCF7-derived lines (Figure 5B). Primary orthotopic MCFL2T-HSD cancers generated bone metastasis readily detected by ex vivo IVIS, while MCF7L2T-injected controls did not (Figure 5C). These data corroborate the MCF7 bone metastasis shown by both IVIS and confirmed by histopathology in E1-treated mice in Figure 1.
Figure 5. HSD17B14 overexpression mediates bone and liver metastasis.
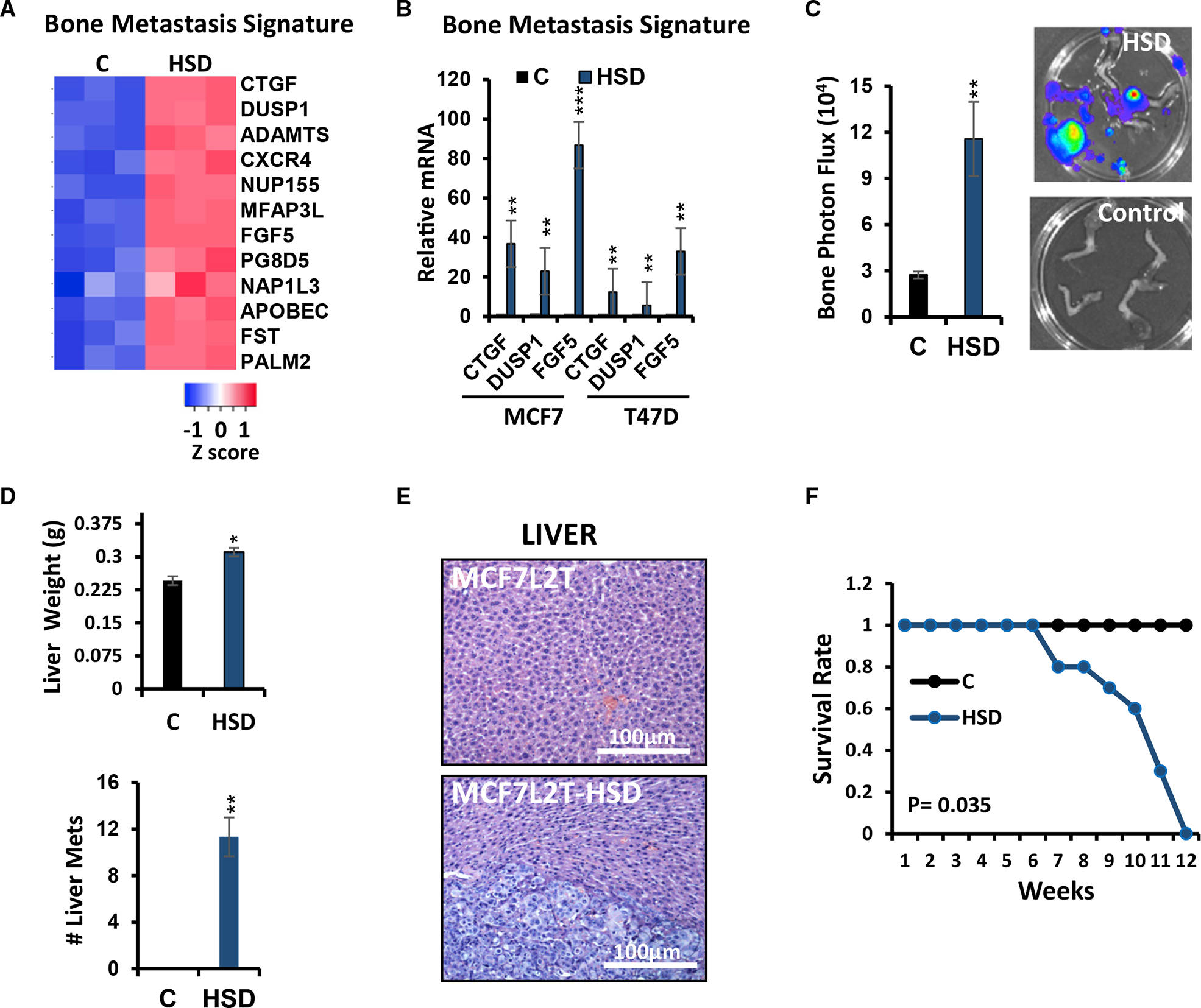
(A) Heatmap of bone-metastasis-related genes in primary orthotopic MCF7 control, C, versus MCF7HSD (HSD) tumors (n = 3 tumors of each used for RNA-seq, FC > 2, Q < 0.05).
(B) qPCR validation of selected bone metastasis mediators overexpressed in MCF7HSD tumors compared with MCF7 controls (n = 3).
(C) Bone metastasis arose from primary orthotopic MCF7L2T-HSD (HSD), but not from MCF7L2T controls, by 4 weeks after primary tumor removal at 1,000 mm3 (shown by BLI in four representative excised leg bones/group). Mean normalized photon flux/second from bone metastases is graphed (±SEM); Student’s t test, **p < 0.001.
(D) Quantification of liver weights (top) and metastatic liver nodules (bottom) in mice injected with HSD17B14 overexpressing MCF7 (HSD) or MCF7 controls; Student’s t test, *p < 0.05 and **p < 0.001.
(E) Representative H&E sections show normal liver from a mouse orthotopically injected with control MCF7L2T (top) and an MCF7L2T-HSD-generated liver metastasis (bottom).
(F) Mean survival (±SEM) is plotted over time from orthotopic injection of control or HSD17B14-overexpressing MCF7 cells; *p < 0.05. All graphs show mean (±SEM) from at least 3 biological repeat and >triplicate replicate assays; p from Student’s t test and ANOVA, *p < 0.05, **p < 0.001, and ***p < 0.0001; see also Figure S3. Scale bars indicate microns.
HSD17B14-expressing tumors also generated liver metastasis, with a significant increase in mean liver weight and mean number of liver metastasis over MCF7L2T-derived controls (Figure 5D). This was also confirmed histologically (Figure 5E). Mice bearing HSD-overexpressing tumors showed a significant increase in multi-organ metastasis and reduced survival (p = 0.035; Figure 5F). Thus, treatment with E1 or overexpression of HSD17B14, which increases intracellular E1, both mediate metastases to lung, bone, and liver, sites commonly affected by metastasis in women with ER+ breast cancer.
Estrone induces EMT through direct transcriptional activation of SNAI2
Since E1 stimulates pro-metastatic gene signatures, we next sought to identify key target genes differently regulated by E1- and E2-bound ERα that could contribute critically to E1-driven EMT. Notably, E1 significantly induced expression of genes encoding EMT markers N-cadherin and vimentin and upregulated the EMT mediator SNAI2 in MCF7 within 24 h compared with estrogen-starved controls, while E2 did not (Figure 6A). SNAI2 upregulation by E1 was also greater than by E2 in a second independent ER+ cancer line, T47D (Figure 6B). E1 increased Slug protein more than E2 within 48 h in both MCF7 and T47D. E1-treated MCF7 had lower Ecadherin and higher N-cadherin than E2-treated cells, (Figure 6C). Primary MCF7 xenograft tumors supplemented with E1 also showed higher CDH2 and SNAI2 and reduced CDH1 expression compared with E2-stimulated tumors (Figure 6D). Western analysis confirmed the elevated E-cadherin and lower expression of Slug and N-cadherin in E2- compared with E1-treated tumors (Figure 6E). Small interfering RNA (siRNA)-mediated SNAI2 knockdown (confirmed in Figures S5A and S5B) significantly decreased the excess migration and invasion in MCF7HSD cells (Figures S5C and S5D). SNAI2 overexpression in both vector controls and MCF7HSD increased invasion and migration (Figures S5E–S5G).
Figure 6. E1:ER promotes EMT through SNAI2 induction, while E2:ER represses SNAI2.

(A) CDH2, SNAI2, and VIM expression by qPCR in MCF7 control and after addition of 10 or 100 nM E1 or E2 for 24 h (n = 3).
(B) SNAI2 expression assayed by qPCR in T74D control and after addition of 10 nM E1 or E2 for 24 h (n = 3).
(C) Western blot of indicated proteins in MFC7 (left) and T47D (right) control and after 10 nM E1 or E2 treatment for 45 min.
(D and E) qPCR analysis of CDH1, CDH2, and SNAI2 expression (D) and western blot of E-cadherin and Slug (E) in E1- or E2-stimulated orthotopic MCF7 tumors (n = 3).
(F–H) Quantitative chromatin immunoprecipitation (ChIP)-qPCR assays of ERα (F), CBP/p300 (G), and NCoR1 (H) at an ERE half-site −464 bp of the SNAI2 transcription start site in estrogen-starved MCF7 controls or after 10 nM E1 or E2 for 45 min (n = 3).
(A–H) All graphs show mean (±SEM) from at least 3 biological repeat and triplicate replicate assays and p from Student’s t test and ANOVA, *p < 0.05, **p < 0.001, and ***p < 0.0001.
(I and J) Kaplan Meier plots from METABRIC data of overall survival (OS) (I) and disease-free survival (DFS) (J) in patients whose ER+ breast cancers show SNAI2 expression above or below the median; log rank p and hazard ratio, HR, (± confidence interval [CI]) indicated.
(K) Model shows different E1- and E2-mediated ERs and co-regulator recruitment at pro-oncogenic target gene sites such as SNAI2.
We next tested if E1- and E2-liganded ERα might direct different receptor/co-regulator recruitment to an ERE half-site at −467 in the SNAI2 promoter. Chromatin immunoprecipitation (ChIP) assays showed both E1 and E2 stimulate ERα recruitment to this ERE half-site in MCF7 (Figure 6F). A SNAI2 promoter region that flanked the ERE half-site at −467 served as a negative control. Notably, E1-liganded ERα recruits the co-activator CBP/p300 to the −467 ERE half-site (Figure 6G), while E2-bound ERα did not. Not only did it fail to recruit CBP to this target gene, E2-bound ERα showed robust recruitment of co-repressor NCoR1 to the SNAI2 promoter, which was not observed in E1-stimulated cells (Figure 6H). Thus, E1 and E2 can direct recruitment of different ERα co-regulators to ERE-bearing target genes. E1 and E2 also had different actions on NF-κB:ERα-co-regulated cytokine genes at κB response elements.24
Analysis of the Metabric database v.4.6, comprising n = 1,498 ER+ breast cancers, revealed that high SNAI2 expression is prognostic of poor overall ER+ breast cancer survival (HR 1.22, p = 0.0039; Figure 6I) and shorter time to metastasis (HR for disease-free survival 1.14, p = 0.05; Figure 6J), supporting a key role for this gene in metastatic ER+ disease progression. Thus, SNAI2 is positively regulated by E1-bound ERα through recruitment of CBP/p300, while E2:ERα and NCoR1 repress SNAI2, disrupting co-activator CBP/p300 recruitment (see model in Figure 6K).
DISCUSSION
Estrogens are master transcriptional regulators of normal development and tissue homeostasis, and their oncogenic roles, particularly in breast cancer, have been extensively studied.22 Differences in oncogenic effects of the dominant pre- and post-menopausal estrogens E2 and E1, respectively, have only recently come to light.24 E1 was shown to drive more rapid ER+ breast cancer development than E2 through cooperation of E1-liganded ERα with NF-κB to induce proinflammatory cytokines and expand the stem-like cell population in ER+ breast cancer models. While E1-liganded ERα stimulates NF-κB-driven gene activation, this is opposed by E2.24 Loss of the restraining effect of E2:ERα on oncogenic NF-κB-driven gene programs might contribute not only to the greater incidence of ER+ breast cancer after menopause but also to its adverse outcome. Present work suggests that the increase in ER+ breast cancer incidence and mortality after menopause is also due in part to E1-driven activation of gene programs of EMT that promote invasion and metastasis.
While ER+ breast cancer accounts for the greatest number of breast cancer deaths worldwide, it is heterogeneous, and metastases can arise within months of diagnosis or decades later. Since breast cancer was first shown to be estrogen responsive in the 1800s,39 there have been tremendous efforts to prevent and treat metastasis through endocrine intervention.1 While tumor dedifferentiation (high histologic grade), nodal spread, and tumor size predict early ER+ breast cancer metastasis,40,41 there is no association between serum E2 levels and cancer metastasis.42 Notably, age >50 years is a significant risk factor for worse outcome of metastatic ER+ breast cancer.6 While age-related factors contributing to excess metastasis include DNA damage, reduced immune surveillance,43 and chronic inflammation,44 the worse prognosis of metastatic ER+ breast cancer with age might not simply result from greater co-morbidities but rather reflect the loss of E2 and the dominance of E1 after menopause.
Transgressing the basement membrane into the extracellular matrix (ECM) is an essential step in progression to invasion and metastasis. While others have shown that E2 does not stimulate ER+ breast cancer cell motility and invasion,45–47 we make the unprecedented observation that the dominant pre- and post-menopausal estrogens have different effects on ER+ cancer cell invasion. In four independent ER+ models, E1 increased EMT and matrigel invasion in 3D sphere assays, but E2 did not. Production of the E1 precursor, androstenedione, is similar before and after menopause,23 but weight gain after menopause is extremely prevalent20,48,49 and increases androstenedione conversion to E1 in breast and adipose tissue. Tumor-associated adipocytes secrete cytokines and FABP4 to promote cancer metastasis.50 Present data indicate that high mammary adipocyte E1 synthesis and chronic inflammation in obesity would not only drive tumorigenesis but also the acquisition of mesenchymal invasive features, promoting local invasion and subsequent metastasis. Thus, cancer-associated adipocytes, particularly in obesity, are critical mediators of E1-stimulated ER+ breast cancer progression. Adipocyte-rich metastatic niches such as the bone marrow, with high local E1, would also facilitate establishment of aggressive intravasating metastatic subclones.
Increased breast cancer exposure to E1 can arise not only through local synthesis in peritumoral adipocytes but also through intratumoral E1 production and/or reduced conversion of E1 to E2. 17β-hydroxysteroid enzymes, such as HSD17B14, which converts E2 to E1, are present in the mammary epithelium and fat.34 We recently identified an oncogenic role for HSD17B14. HSD17B14 overexpression in MCF7 increased intratumor conversion of E2 to E1, expanded stem cells, and caused greater tumor growth in both E1- and E2-supplemented mice.24 Here, we find that high intratumor HSD17B14, HSD17B10, and HSD17B2, all of which convert E2 to E1, associate with earlier ER+ breast cancer metastasis. Conversely, high HSD17B5, HSD17B1, and HSD17B7, which convert E1 to E2, each associate with longer metastasis-free ER+ breast cancer survival. E1 treatment or a gain of HSD17B14 promotes mesenchymal characteristics to increase ER+ breast cancer invasion and metastasis. Moreover, loss of HSD17B14 decreased adipocyte co-culture-induced SNAI2 upregulation in cancer cells and reversed EMT.
While E1 stimulated greater primary tumor growth, tumor cells showed only a modest increase in Ki67- compared with E2-stimulated cancers.24 Rather, dissociated E1-stimulated tumors showed greater abundance of ADH1+, sphere forming, and tumor-initiating stem cells on reimplantation into secondary hosts.24 This, together with present findings, suggests that E1 promotes mesenchymal/stem cell features to increase dissemination, likely through expansion of pro-metastatic/stem-like cells. The E1:ERα-induced EMT program appears to stimulate collective invasion in spheroid invasion assays and at metastatic sites (see cardiac muscle). Whether E1:ERα promotes dissemination of single cells, cell clusters in contiguity, or both warrants further investigation in vivo.
Bone, liver, lung, and brain are niches for breast cancer metastasis.51,52 E1 stimulated expression of EMT and lung metastasis signatures, promoting greater metastasis of MCF7 and E0771 ER+ cancers compared with E2. HSD17B14 overexpression also induced expression of lung and bone metastasis gene signatures and markedly increased metastasis to these tissues. Molecules that link migratory signals to the actin cytoskeleton are upregulated in metastatic cancer cells.53 Actin cytoskeletal remodeling profiles were preferentially activated by E1, but not E2. E1 and HSD17B14 lead to activation of genes including CXCR4,31 MMP1/2,32 and/or ANGPTL4,33 which mediate breast cancer metastasis. Thus, E1 appears to drive ER+ breast cancer metastasis in vivo. The balance of estrogens, with the increased E1:E2 ratio in women who are post-menopausal, particularly as occurs with obesity, might be critical for metastasis.
SNAI2 is conserved across species and expressed in the mammary basal/stem-cell-enriched population in both mice and humans.54 Slug, encoded by SNAI2, is one of the most powerful EMT mediators,55,56 and the E-cadherin-snail-slug EMT pathway is critical for human breast cancer invasion and metastasis.57–60 Elevated SNAI2 expression correlates with increased metastasis and shorter survival in a variety of cancers.61–63 Slug promotes metastasis by enhancing cell invasion, supporting metastatic cell survival64 and increasing breast cancer stem cells.65 Here, we show SNAI2 is both necessary and largely sufficient for the gain of invasion observed with E1 stimulation.
A key contribution of the present work lies in demonstrating that in cancer and potentially other cells, E1- and E2-liganded ERα can recruit different complexes to differentially regulate common target genes. The biologic consequences of E1 on ERα-driven gene expression are not fully known. Our prior work showed that E1 and E2 have opposing effects on IL6 and CCL2 regulation, with E1 co-recruiting CBP to ERα/p65 at NF-κB response elements to induce expression and an E2-liganded receptor excluding CBP to co-repress expression of these genes.24 Present work identifies SNAI2 as an ERE-bearing ERα target gene that is differently regulated by E1 and E2. SNAI2 regulation by these steroids was unaffected by NF-κB activation. E1-liganded ERα stimulates recruitment of co-activator CBP/p300 to the SNAI2 promoter to induce its expression, while the E2-bound receptor recruits a repressive NCoR1 complex.
Together, our analyses indicate that E1-bound ERα, in part through SNAI2, serves as a master enforcer of mesenchymal cell fate. In physiological contexts, E1, which is produced lifelong at a fairly constant level in individuals of a normal weight,23 might govern mesenchymal cell identity, whereas in emergent mammary cancers, E1 and its upregulation by HSD17B14 would induce EMT and cancer metastasis. An increase in E1 levels and loss of opposing effects of E2 after menopause would drive critical steps in local invasion and metastatic progression. Obesity in women who are post-menopausal, which upregulates E1 in the context of reduced E2 synthesis, would drive pre-invasive neoplastic breast cells to undergo EMT, invade locally, and metastasize.
Limitations of the study
One limitation of this work is that effects of E1- versus E2-liganded ERα have been evaluated in ER+ breast cancer lines (MCF7, T47D, MDA-MB-361, and ZR-75-1). Further evaluation of E1 and E2 in closer to human cancer models, such as ER+ human breast cancer organoids or patient-derived xenografts (PDXs), and in the normal breast would be desirable. We have not addressed how E1 interacts with obesity and immunomodulatory mechanisms in syngeneic models to promote metastasis. Inhibition or overexpression of HSD17B14 in the mature adipocytes is not possible because they are post-mitotic. Evaluating such effects in fat in vivo in genetically modified mouse models could prove informative. While E1- and E2-bound ERα recruit different co-regulators to induce or repress SNAI2, respectively, the role of E1-regulated SNAI2 was not functionally tested in vivo. Finally, broader analysis of E1- and E2-regulated cistromes and transcriptomes in vivo in the context of obesity and inflammation warrant further study.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Slingerland J (js4915@georgetown.edu).
Materials availability
This study did not generated new unique reagents.
Data and code availability
RNA seq data were submitted to the Gene Expression Omnibus (GEO: GSE132913).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
This study conformed to the principles outlined in the Declaration of Helsinki. All human subjects provided written informed consent prior to donation of adipose tissue samples following Institutional Review Board review. Samples obtained from human subjects were de-identified waste material from reduction mammoplasty, lumpectomy, or mastectomy surgeries performed at the University of Miami Hospital. Donor BMI, age, and menopausal status was recorded.
Cell culture
MDA-MB-231, MCF7, T47D, ZR-75-1, MCF12A, 293T, and MDA-MB-361 were purchased from ATCC and grown per ATCC protocols. SUM149, SUM159, and SUM1315 were provided by Steven Ethiers (Medical University of South Carolina) and grown as described in sumlineknowledgebase.com. HSD17B14 transduced MCF7HSD, T47DHSD and MDA-MB-361HSD were previously described, as was the CRISPR knockout of HSD17B14 in MCF7 and T47D.24 Isolated hASC and mature adipocytes were cultured alone or co-cultured for 7 days with the specified breast cancer cell lines using the corresponding cell line medium. After co-culture, luciferase/GFP tagged cancer lines were flow sorted from adipocytes. Fresh medium was added to the cultures at days 2 and 4 without discarding the old media. For experiments involving in vitro estrogen treatment, cancer lines were estrogen deprived by culture in phenol red-free medium supplemented with 5% charcoal stripped FBS for 48–72 h. Estrogen stimulation used media containing 5% cFBS together with either DMSO vehicle only, or E2 or E1 added at 10 nM, unless otherwise indicated for titration experiments.
Mouse models
All animal experiments and procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee at University of Miami (Protocol #-16-084LF rev).
NOD-SICD Ovariectomized female mice (4–5 week of age) were used for xenograft assays testing the effects of steroids and HSD17B14 expression on tumor growth. NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice were used for tail vein injection of MCF7. 000664 - C57BL/6J mice ovariectomized female 4–5 week old mice were used for syngeneic tumor implantation with the E0771 cell line (details below). Mice were housed in micro-isolator cages, with standard 12hr light/darkness cycle, ambient temperature of 23°C, and were provided a standard rodent diet, unless otherwise indicated, and water ad libitum. Unless otherwise indicated, estrogen pellets containing either E1 or E2 at 0.1mg/90day were used for hormone supplementation. All controls not receiving estrogen supplements had control pellets containing no estrogen inserted. All mice were ovariectomized at 6 weeks of age.
METHOD DETAILS
Adipocyte, SVF, and hASC isolation from fat tissue
Mammary fat was washed 4X with PBS, digested with collagenase 1A 1 g/L in Hank’s solution supplemented with 1% BSA for 30 min at 37°C, and centrifuged at 1500 rpm for 5 min.66 Floating mature adipocytes and pelleted SVF were separated, washed 3X with PBS and filtered using a 100 μm or 70 μm diameter membrane, respectively. hASC were obtained by seeding the SVF in 75 cm2 culture flasks in DMEM medium supplemented with 10% FBS and 1% P/S. After 3 passages, hASC were characterized by flow cytometry as in.20 Mature adipocytes were used immediately after isolation. hASC were used between passages 3–10. Figure 2 used hASC and in Figure 3G, we used mature adipocytes.
Quantitative RT-PCR (qPCR)
Total RNA was isolated using Trizol (Invitrogen) or RNeasy Lipid Tissue Kit (Qiagen) for adipocytes. cDNA was synthesized from the isolated RNA using iScript cDNA synthesis kit (Bio-Rad). qPCR was performed with a LightCycler® 480 Instrument II (Roche) using iQ SYBR Green Supermix (Bio-Rad). All qPCR analyses were performed as both biologic and technical triplicate repeats. Primer sequences are shown in Table S1.
Western blotting
Westerns were performed in at least 3 different biologic repeats and representative shown as in.67 Cells were lysed in RIPA buffer supplemented with 1X protease and phosphatase inhibitor cocktails (G-Biosciences). Usually 20 μg protein/lane were resolved by SDS-PAGE and transferred to PVDF blotting membrane (Bio-Rad). The membranes were incubated with the indicated primary antibodies and HRP-conjugated secondary antibodies (Promega). The immune-reactive bands were visualized using a chemiluminescent substrate (Thermo Scientific) and X-ray film (Phenix Research Products).
Transwell migration and invasion assays
For transwell invasion assays, 105 cells were seeded in the upper chamber of a matrigel (5 mg/mL media)-coated transwell membrane (Corning) and invasion quantitated as described.68 Cells adherent to membrane under-surface were visualized, photographed, and counted, and relative invasion was plotted.68 Automated transwell invasion assays used the Real-Time Cell Analysis (RTCA) system from xCELLigence as described,69 and invasion was plotted as cell index +/− SEM for at least three wells per group.
Scratch assay
Cells were seeded into six-well plates and grown to confluence, and the wound-healing migration assay was performed. The linear wound of cellular monolayer was created by scratching the confluent cell monolayer using a 200 ul plastic pipette tip. The scratched cell monolayer was washed by PBS to remove debris. After incubation at 37°C for 24–48 h, the migration of the cells toward the wound was photographed under a light microscopy. ImageJ was used to determine the migration distance.
siRNA analysis
SiRNA pools of three to five target-specific 19–25 nucleotide siRNAs designed to knockdown and control siRNAs were purchased from Santa Cruz Biotechnology (Dallas, TX, USA) and used per manufacturer.
Chromatin immunoprecipitation assay
For chromatin immunoprecipitation (ChIP) assays for the SNAI2 promoter, soluble chromatin was prepared from 2×107 cells as in.70 The chromatin solution was diluted 10-fold with ChIP dilution buffer (1.1% Triton X-100, 1.2 mm EDTA, 167 mm NaCl, 16.7 mm TrisHCl, pH 8.1, 0.01% SDS, plus protease and phosphatase inhibitors), pre-cleared, and blocked with 2 μg of sheared salmon sperm DNA and pre-immune serum. Pre-cleared chromatin was used in immunoprecipitation assays with anti-anti-ERα (mAb F1 Cell Signaling), anti-NCoR1 (Diagenode), anti-CBP/p300 (Cell Signaling), or an anti-IgG (Santa Cruz) antibody. In addition to IgG controls, all TF binding assays used unrelated promoter specific controls to show binding was specific. The washed antibody-protein-DNA complexes were eluted from the beads in 1% SDS, 0.1 M NaHCO3 at room temperature for 20 min. Twenty μg/μL of proteinase K was used for removal of protein at 2 h at 55°C, and reverse cross-linking was performed with incubating at 65°C overnight. Purified DNA was subjected to qPCR with primers specific for the SNAI2 promoter binding sites. All ChIP analyses were performed as triplicate technical repeats for each of three biologic repeat assays. Primer sequences, including those for non-sequence specific controls, are shown in Key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-Aromatase (D5Q2Y) Rabbit mAb | Cell Signaling Technology | Cat#14528; RRID: AB_2630344 |
| Anti-Nuclear Receptor Corepressor NCoR antibody | abcam | ab3482 |
| Anti-GAPDH (14C10) Rabbit mAb | Cell Signaling Technology | Cat#2118; RRID: AB_561053 |
| E-cadherin (Mouse IgG2a) | BD Transduction | Cat# 610182 |
| N-cadherin (Mouse IgG1) | BD Transduction | Cat# 610920 |
| Vimentin (5G3F10, Mouse mAB) | Cell Signaling Technology | Cat#3390S |
| Anti-SLUG antibody | Abcam | Cat# ab27568 |
| Acetyl-CBP (Lys1535)/p300 (Lys1499, Rabbit IgG) | Cell Signaling | 4771S |
| Anti-ER alpha (F-10) Mouse mAb | Santa Cruz | Cat#SC8002; RRID:AB_627558 |
| Anti-β-Actin (AC-15) Mouse mAb | Sigma | Cat#A1978; RRID:AB_476692 |
| NFkB p65 (C22B4) Rabbit mAb | Cell Signaling Technology | Cat#4764; RRID: AB_823578 |
| Anti-Mouse IgG (H + L), HRP Conjugate | Promega | Cat#W4021; RRID:AB_430834 |
| Anti-Rabbit IgG (H + L), HRP Conjugate | Promega | Cat#W4011; RRID:AB_430833 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| One Shot® MAX Efficiency® DH5α™-T1R Competent Cells | Invitrogen | Cat#12297-016 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Collagenase from Clostridium histolyticum Type IA | Sigma-Aldrich | Cat#C9891 |
| TRIzol Reagent | Invitrogen | Cat#10296–028 |
| iQ™ SYBR® Green Supermix | Bio-Rad | Cat#170–8886 |
| Hydrocortisone | Stemcell Technologies | Cat#7925 |
| Heparin Solution | Stemcell Technologies | Cat#7980 |
| Insulin, human recombinant, zinc solution | Thermo Fisher Scientific | Cat#12585014 |
| B27 Supplement (50X), serum free | Thermo Fisher Scientific | Cat#17504044 |
| Recombinant Human EGF Protein, CF | R&D Systems | Cat#236-EG |
| FGF-Basic (AA 10–155) Recombinant Human | Thermo Fisher Scientific | Cat# PHG0026 |
| RIPA Buffer | Cell Signaling | Cat#9806 |
| PhosphataseArrest™ Phosphatase Inhibitor Cocktail | G-Biosciences | Cat#786-450 |
| ProteaseArrest™ Protease Inhibitor Cocktail | G-Biosciences | Cat#786-331 |
| Pierce™ ECL Western Blotting Substrate | Thermo Scientific | Cat#32106 |
| Letrozole | Sigma-Aldrich | Cat#L6545 |
| B-Estradiol | Sigma-Aldrich | Cat#E8875 |
| Estrone | Sigma-Aldrich | Cat# E9750 |
| BAY 11–7082 | Sigma-Aldrich | Cat# B5556 |
| TNF-α Human | Sigma-Aldrich | Cat# SRP3177 |
| Luciferin | PERKINELMER IN | Cat# 770504 |
|
| ||
| Critical commercial assays | ||
|
| ||
| iScript cDNA Synthesis Kit | Bio-Rad | Cat#1708891 |
| ALDEFLUOR kit | Stem Cell Technologies | Cat#01700 |
| Estradiol ELISA Kit (Competitive EIA) | LifeSpan BioSciences | Cat#LS-F5297 |
| Estrone ELISA Kit (Competitive EIA) | LifeSpan BioSciences | Cat#LS-F10566 |
| Cytokine/Chemokine/Growth Factor 45-Plex Human ProcartaPlex™ Panel 1 | Invitrogen | Cat#EPX450-12171-901 |
| RNeasy Lipid Tissue Kit | Qiagen | Cat#74804 |
| NE-PER Nuclear and cytoplasmic extraction reagents | Thermo Fisher Scientific | Cat#78835 |
| Dual-Luciferase Reporter Assay System | Promega | Cat#E1960 |
|
| ||
| Deposited data | ||
|
| ||
| The accession number for the sequencing data reported in this paper is GSE132913 | Gene Expression Omnibus | GEO: GSE132913 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human: MDA-MB-231 | ATCC | HTB-26 |
| Human: SUM149 | Steven Ethiers | CVCL_3422 |
| Human: SUM159 | Steven Ethiers | CVCL_5423 |
| Human: SUM1315 | Steven Ethiers | CVCL_5589 |
| Human: MCF7 | ATCC | HTB-22 |
| Human: T47D | ATCC | HTB-133 |
| Human: MDA-MB-361 | ATCC | HTB-27 |
|
| ||
| Experimental models: Organisms/Strains | ||
|
| ||
| 000664 - C57BL/6J Ovariectomized | The Jackson Laboratory | JAX -000664 |
| NOD.CB17-Prkdc<scid>/J HOM Homozygous for Prkdc<scid> ovariectomized | The Jackson Laboratory | JAX- 001303 |
| NOD.Cg-Prkdc<scid> Il2rg < tm1Wjl>/SzJ M01 Homozygous for Prkdc<scid>, Homozygous for Il2rg < tm1Wjl > ovariectomized | The Jackson Laboratory | JAX-005557 |
|
| ||
| Oligonucleotides | ||
|
| ||
| QPCR PRIMER | N/A | N/A |
| Primer PCR: GAPDH Forward: 5’-ATCAAGTGGGGCGATGCTG-3’ | This paper | N/A |
| Primer PCR: GAPDH Reverse5’-ACCCATGACGAACATGGGG-3 | This paper | N/A |
| QPCR Primer CDH1-Forward: AATTCCTGCCATTCTGGGGA | This paper | N/A |
| QPCR Primer CDH1-Reverse: TCTTCTCCGCCTCCTTCTTC | This paper | N/A |
| QPCR Primer SNAI1-Forward: ACCCCACATCCTTCTCACTG | This paper | N/A |
| QPCR Primer SNAI1-Reverse: TACAAAAACCCACGCAGACA | This paper | N/A |
| QPCR Primer SNAI2-Forward: TGCGATGCCCAGTCTAGAAA | This Paper | N/A |
| QPCR Primer SNAI2-Reverse: TTCTCCCCCGTGTGAGTTC | This Paper | N/A |
| QPCR Primer TWIST1-Forward: GTCCGCAGTCTTACGAGGAG | This Paper | N/A |
| QPCR Primer TWIST1-Reverse: GCTTGAGGGTCTGAATCTTGCT | This Paper | N/A |
| QPCR Primer VIM Forward: 5’-CGAGTAGGACATGCTGTAGGT-3’ | This Paper | N/A |
| Oligonucleotide continued as Table S1 | N/A | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| Genecopoeia | EX-U0801-Lv224 | ORF expression clone for human HSD17B14 (NM_016246.2)) |
| Santa Cruz | sc-412138 | 17β-HSD14 CRISPR/Cas9 KO Plasmid (h) |
|
| ||
| Software and algorithms | ||
|
| ||
| Adobe Illustrator | Adobe Systems, San Jose, CA | https://www.adobe.com/ca/products/illustrator.html; RRID: SCR_010279 |
| FlowJo software V10 | FlowJo, LLC | https://www.flowjo.com/ |
|
| ||
| Other | ||
|
| ||
| Estradiol 0.1mg/90 day pellet | Innovative Research of America | Cat#NE-121 |
| Estrone 0.1mg/pellet 90 day | Innovative Research of America | Cat#NE-111 |
| Placebo 0.1mg/pellet 90 day | Innovative Research of America | Cat# NC-111 |
Lentivirus production and establishment of HSD17B14 expressing cells
Lentivirus vectors encoding ORF HSD17B14 and ORF control were purchased from GeneCopoeia. Lentivirus vectors encoding different ORFs were co-transfected with DeltaVPR and CMVVSVG plasmids (Addgene) into asynchronous 293T with Lipofectamine 3000 Reagent. Viral supernatants were collected after 48 and 72 h. MCF7L2T cells stably transduced with expression clone were incubated for 8–16 h with the medium containing the virus, supplemented with 4 μg/mL of polybrene (Sigma-Aldrich). Cells were infected twice with polybrene, selected with 2 μg/mL of puromycin, and analyzed 3–5 days post infection by RFP visualization. Overexpression was confirmed by western blotting. Cells were maintained in IMEM, RPMI, or DMEM plus 10% FBS, and 0.2 μg/mL of puromycin was used to maintain the cell line.
RNA sequencing (RNAseq)
Total RNA quality was measured using Bioanalyzer RNA Nano 6000 (Agilent Technologies, Santa Clara, CA, USA). Library preparation was performed by TruSeq Standed Total RNA Library Prep (Illumina, San Diego, USA), and quality confirmed using KAPA qPCR Library Quantification (Kapa Biosystems, Wilmington, MA, USA). Paired end sequencing was performed on Illumina NextSeq platform using 150 cycles 400M kit. All RNAseq experiments were performed in triplicate on independent biologic repeat assays.
Orthotopic xenograft assay
For orthotopic xenograft assays, 5 × 105 cells were suspended in 100 μL Matrigel and injected into the fourth mammary fat-pad of NOD SCID gamma, ovariectomized, female, 4–5 week old mice (4–8/group). Tumor growth was monitored by weekly IVIS, measured twice-weekly, and its volumes calculated as (long-side × short-side2)/2. Primary tumors in the inguinal mammary fat pads were removed at 700–1000mm3, and mice were monitored weekly by IVIS for metastasis. The IVIS quantification of metastasis excludes the inguinal region to disregard the recurrent primary tumor. The mean normalized photon flux is plotted/time +/− SEM. Animal work was compliant with University of Miami Institutional Animal Care and Use Committee.
Experimental lung metastasis assay
MCF7-luc and controls cells were used for injection of 1 × 106 cells via tail vein into 4–5 week old, ovariectomized, female NOD SCID (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) (NSG) mice. Each experimental group contained 8 animals. The mice were imaged by in vivo imaging system (Xenogen, Caliper, Hopkinton, MA, USA), and bioluminescence (photon flux) was quantified with time as described. All animal work was carried out in compliance with the Institutional Animal Care and Use Committee in the University of Miami.
IVIS imaging and data quantification
Animals were imaged weekly through the IVIS imaging system. Prior to imaging, animals were injected with Xenolight D-luciferin K+ salt (PerkinElmer) and anesthetized with 2.5% isoflurane. After 8 min of incubation with luciferin, bioluminescence was quantified for each animal. Tumor volumes were monitored as bioluminescence (photon flux/second) by IVIS using Living Image software. At sacrifice, the final tumor was measured using Vernier calipers and the volume calculated using the formula (long-side × short-side2)/2. For in vivo metastasis, the primary tumor sites were covered, and bioluminescence was quantified as photon flux by IVIS.
Hanging drop invasion assay
Tumor cell invasion was assessed using a three-dimensional (3D) spheroid invasion assay.27 ZR-75-1, MDA-MB-361 or MCF7 cells grown in suspension in 40 hanging drops of culture medium on the lid of cell culture dishes (approximate 500–1000 cells per 20 μL drop of complete media). After 72 h, cells were collected and resuspended in 200 μL of a mix of rat tail type I collagen (final concentration is 2.3 μg/mL) mixed 1:1 with matrigel, and embedded in 24 well plates (40μL/well) for 3D culture. Suspended cells were then supplemented with 1 mL media containing either 5%cFBS alone or with 10 nM E1 or E2 estrogen. Invasion was measured at 96 hours-7 days cells. Invasion was quantitated by measuring the maximal invaded area using ImageJ software.
Immunohistochemistry
Primary xenograft tumors were fixed in 10% neutral buffered formalin for 24 h and then paraffin embedded. Tumor sections were cut at 4 μM and stained with hematoxylin and eosin by immunohistochemistry as in.71
Immunofluorescence
Control and HSD17B14 overexpressing cells were seeded on coverslips for 24 h. Cells were then washed with cold PBS and fixed for 10 min at room temperature with 4% paraformaldehyde, permeabilized for 5 min with 0.2% Triton X-100 PBS, and blocked for 30 min with 5% BSA. This was followed by incubation with the primary antibodies (1:500) overnight at 4°C, PBS wash, and then by the secondary antibodies at 1:1000 dilution for another hour at room temperature. Primary antibodies used in this study are included in the Key resources table. To stain the nucleus, cells were incubated with 40 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) for 20 min at room temperature. Coverslips were mounted on glass slides and cells imaged using a Zeiss LSM 710 confocal microscope.
Gene expression microarray datasets from a primary breast tumor and metastatic tissue
Gene expression microarray datasets from NCBI public data GSE32531 entry were analyzed with Geo2R. Data from thirteen primary breast cancer and eighteen unmatched metastatic breast cancer and tissue samples were analyzed. Probes from the Agilent GSE32531-based cohort were filtered using the intensity values obtained in each sample for the HSD17B14 gene. The data were subjected to an outlier examination and comparison of variances before performing the T test, to determine the difference in HSD17B14 gene expression between the groups analyzed.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
All graphed data are presented as mean ± SEM from at least three biological replicate experiments done in triplicate technical repeats. Student’s t-test was used for experiments with two groups. Comparisons of >2 groups used one-way analysis of variance (ANOVA) followed by Dunnett′s or Tukey′s post hoc analysis. Some experiments used two-way ANOVA followed by Tukey′s post hoc tests. Statistical significance values were set as *p < 0.05. **p < 0.01.***p < 0.001. A p value less than 0.05 would be considered statistically significant, ns = not significant. p value and n can be found in main and supplementary figure legends. Statistical differences between tumor growth curves used ‘Compare Growth Curves’ function statmod software package (http://bioinf.wehi.edu.au/software/compareCurves/). TGCA data analysis used UALCAN http://ualcan.path.uab.edu/index.html to study expression of HSD17B14 in primary breast cancers and normal breast tissues from the TCGA database. For analysis of differences in the HSD17B14 expression in primary breast cancers and normal breast tissues, t test was performed using a PERL script with Comprehensive Perl Archive Network (CPAN)module “Statistics:Test” (http://search.cpan.org/~yunfang/Statistics-TTest-1.1.0/TTest.pm).
RNAseq bioinformatic analysis
Quality and adapter trimming was performed using cutadapt 1.15. Transcriptome alignment and quantification was performed using RSEM 1.3.0 and STAR 2.0.6c against human transcriptome (GRCh38_no_alt_analysis_set_GCA_000001405.15 and GENCODE v28). Differentially expressed genes were identified using DESeq2 1.18.1 with median-ratio normalization, and heat maps, clustering, and PCA plots were generated using sample blind variance stabilized log2 gene counts. To further evaluate genes identified as uniquely up or downregulated by E1 or E2 in the initial analysis, differential expression was evaluated by DESeq2 analysis after combining all E1 and E2 data together and comparing this against the cFBS group. Most genes identified as uniquely regulated by either E1 or E2, with FDR<0.05 following the comparison of each group versus cFBS were also confirmed in the analysis of the combined E1 and E2 data versus cFBS. Gene Set Enrichment Analysis (GSEA 3.0.1) was performed using Wald statistic ranked genes lists.72 Gene sets that enriched with a BH FDR<0.05, that had the top 20 positive or negative NES scores, and that were relevant for breast cancer pathways were presented in figures. KEGG 2016 pathway enrichment was performed using Enrichr. Pathways were presented if BH FDR<0.05, they were in the top ten significant pathways, and they were relevant for breast cancer.
Supplementary Material
Highlights.
Gene profiles governed by the dominant pre- and post-menopausal estrogens differ
Estrone, but not estradiol, stimulates EMT genes, invasion, and metastasis in vivo
Changes in HSD17Bs increase estrone to drive multi-organ metastatic cancer progression
Estrone:ERα:CBP promotes invasion via SNAI2, while 17β-estradiol:ERα:NCoR1 represses this
ACKNOWLEDGMENTS
We acknowledge helpful discussions with Marc E. Lippman and Gray Pearson, the services of Sylvester Comprehensive Cancer Center Oncogenomics and Biospecimen Shared Resources, the Lombardi Comprehensive Cancer Center Histopathology & Tissue Shared Resource, and grants from NIH (1R01CA210440-01A1; J.S.); Florida Breast Cancer Foundation (R.Q.); Breast Cancer Research Foundation (J.S.); Susan G. Komen Foundation (PDF16380958; M.P.-R. and J.S.); the Spanish Ministry of Science and Innovation (PID2020-119502RJ-I00; M.P.-R.); and the UGR-FEDER program (E-CTS-654-UGR20; M.P.-R.).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111672.
REFERENCES
- 1.Ma CX, Reinert T, Chmielewska I, and Ellis MJ (2015). Mechanisms of aromatase inhibitor resistance. Nat. Rev. Cancer 15, 261–275. 10.1038/nrc3920. [DOI] [PubMed] [Google Scholar]
- 2.Obradović MMS, Hamelin B, Manevski N, Couto JP, Sethi A, Coissieux MM, Münst S, Okamoto R, Kohler H, Schmidt A, and Bentires-Alj M (2019). Glucocorticoids promote breast cancer metastasis. Nature 567, 540–544. 10.1038/s41586-019-1019-4. [DOI] [PubMed] [Google Scholar]
- 3.Amanatullah DF, Tamaresis JS, Chu P, Bachmann MH, Hoang NM, Collyar D, Mayer AT, West RB, Maloney WJ, Contag CH, and King BL (2017). Local estrogen axis in the human bone microenvironment regulates estrogen receptor-positive breast cancer cells. Breast Cancer Res. 19, 121. 10.1186/s13058-017-0910-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kolečková M, Kolář Z, Ehrmann J, Kořínková G, and Trojanec R (2017). Age-associated prognostic and predictive biomarkers in patients with breast cancer. Oncol. Lett. 13, 4201–4207. 10.3892/ol.2017.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenkins EO, Deal AM, Anders CK, Prat A, Perou CM, Carey LA, and Muss HB (2014). Age-specific changes in intrinsic breast cancer subtypes: a focus on older women. Oncologist 19, 1076–1083. 10.1634/theoncologist.2014-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen MT, Sun HF, Zhao Y, Fu WY, Yang LP, Gao SP, Li LD, Jiang HL, and Jin W (2017). Comparison of patterns and prognosis among distant metastatic breast cancer patients by age groups: a SEER population-based analysis. Sci. Rep. 7, 9254. 10.1038/s41598-017-10166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brodowska A, Brodowski J, Laszczyńska M, Słuczanowska-Głąbowska S, Rumianowski B, Rotter I, Starczewski A, and Ratajczak MZ (2014). Immunoexpression of aromatase cytochrome P450 and 17beta-hydroxysteroid dehydrogenase in women’s ovaries after menopause. J. Ovarian Res. 7, 52. 10.1186/1757-2215-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alves CL, Elias D, Lyng MB, Bak M, and Ditzel HJ (2018). SNAI2 upregulation is associated with an aggressive phenotype in fulvestrant-resistant breast cancer cells and is an indicator of poor response to endocrine therapy in estrogen receptor-positive metastatic breast cancer. Breast Cancer Res. 20, 60. 10.1186/s13058-018-0988-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, and Weinberg RA (2008). Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell 14, 818–829. 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Lee JM, Dedhar S, Kalluri R, and Thompson EW (2006). The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J. Cell Biol. 172, 973–981. 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomaskovic-Crook E, Thompson EW, and Thiery JP (2009). Epithelial to mesenchymal transition and breast cancer. Breast Cancer Res. 11, 213. 10.1186/bcr2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Acloque H, Ocaña OH, Matheu A, Rizzoti K, Wise C, Lovell-Badge R, and Nieto MA (2011). Reciprocal repression between Sox3 and snail transcription factors defines embryonic territories at gastrulation. Dev. Cell 21, 546–558. 10.1016/j.devcel.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nieto MA (2011). The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 27, 347–376. 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 14.Thiery JP, Acloque H, Huang RYJ, and Nieto MA (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890. 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Thompson EW, and Williams ED (2008). EMT and MET in carcinoma–clinical observations, regulatory pathways and new models. Clin. Exp. Metastasis 25, 591–592. 10.1007/s10585-008-9189-8. [DOI] [PubMed] [Google Scholar]
- 16.Thiery JP (2002). Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442–454. 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 17.Polyak K, and Weinberg RA (2009). Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer 9, 265–273. 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 18.Mao Y, Keller ET, Garfield DH, Shen K, and Wang J (2013). Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 32, 303–315. 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Place AE, Jin Huh S, and Polyak K (2011). The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res. 13, 227. 10.1186/bcr2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Picon-Ruiz M, Pan C, Drews-Elger K, Jang K, Besser AH, Zhao D, Morata-Tarifa C, Kim M, Ince TA, Azzam DJ, et al. (2016). Interactions between adipocytes and breast cancer cells stimulate cytokine production and drive Src/Sox2/miR-302b-mediated malignant progression. Cancer Res. 76, 491–504. 10.1158/0008-5472.CAN-15-0927. [DOI] [PubMed] [Google Scholar]
- 21.Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, Wang YY, Meulle A, Salles B, Le Gonidec S, et al. (2011). Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 71, 2455–2465. 10.1158/0008-5472.CAN-10-3323. [DOI] [PubMed] [Google Scholar]
- 22.Yue W, Wang JP, Hamilton CJ, Demers LM, and Santen RJ (1998). In situ aromatization enhances breast tumor estradiol levels and cellular proliferation. Cancer Res. 58, 927–932. [PubMed] [Google Scholar]
- 23.Grodin JM, Siiteri PK, and MacDonald PC (1973). Source of estrogen production in postmenopausal women. J. Clin. Endocrinol. Metab. 36, 207–214. 10.1210/jcem-36-2-207. [DOI] [PubMed] [Google Scholar]
- 24.Qureshi R, Picon-Ruiz M, Aurrekoetxea-Rodriguez I, Nunes de Paiva V, D’Amico M, Yoon H, Radhakrishnan R, Morata-Tarifa C, Ince T, Lippman ME, et al. (2020). The major pre- and postmenopausal estrogens play opposing roles in obesity-driven mammary inflammation and breast cancer development. Cell Metab. 31, 1154–1172.e9. 10.1016/j.cmet.2020.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Giraud SN, Caron CM, Pham-Dinh D, Kitabgi P, and Nicot AB (2010). Estradiol inhibits ongoing autoimmune neuroinflammation and NFkappaB-dependent CCL2 expression in reactive astrocytes. Proc. Natl. Acad. Sci. USA 107, 8416–8421. 10.1073/pnas.0910627107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalaitzidis D, and Gilmore TD (2005). Transcription factor cross-talk: the estrogen receptor and NF-kappaB. Trends Endocrinol. Metab. 16, 46–52. 10.1016/j.tem.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Berens EB, Holy JM, Riegel AT, and Wellstein A (2015). A cancer cell spheroid assay to assess invasion in a 3D setting. J. Vis. Exp. 10.3791/53409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, et al. (2010). Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 107, 15449–15454. 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, and Massagué J (2005). Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524. 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, Guise TA, and Massagué J (2003). A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549. 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 31.Xu C, Zhao H, Chen H, and Yao Q (2015). CXCR4 in breast cancer: oncogenic role and therapeutic targeting. Drug Des. Devel. Ther. 9, 4953–4964. 10.2147/DDDT.S84932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merdad A, Karim S, Schulten HJ, Dallol A, Buhmeida A, Al-Thubaity F, Gari MA, Chaudhary AG, Abuzenadah AM, and Al-Qahtani MH (2014). Expression of matrix metalloproteinases (MMPs) in primary human breast cancer: MMP-9 as a potential biomarker for cancer invasion and metastasis. Anticancer Res. 34, 1355–1366. [PubMed] [Google Scholar]
- 33.Zhao J, Liu J, Wu N, Zhang H, Zhang S, Li L, and Wang M (2020). ANGPTL4 overexpression is associated with progression and poor prognosis in breast cancer. Oncol. Lett. 20, 2499–2505. 10.3892/ol.2020.11768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hilborn E, Stål O, and Jansson A (2017). Estrogen and androgen-converting enzymes 17beta-hydroxysteroid dehydrogenase and their involvement in cancer: with a special focus on 17beta-hydroxysteroid dehydrogenase type 1, 2, and breast cancer. Oncotarget 8, 30552–30562. 10.18632/oncotarget.15547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang XHF, Giuliano M, Trivedi MV, Schiff R, and Osborne CK (2013). Metastasis dormancy in estrogen receptor-positive breast cancer. Clin. Cancer Res. 19, 6389–6397. 10.1158/1078-0432.CCR-13-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savci-Heijink CD, Halfwerk H, Koster J, and van de Vijver MJ (2016). A novel gene expression signature for bone metastasis in breast carcinomas. Breast Cancer Res. Treat. 156, 249–259. 10.1007/s10549-016-3741-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hellinger JW, Schömel F, Buse JV, Lenz C, Bauerschmitz G, Emons G, and Gründker C (2020). Identification of drivers of breast cancer invasion by secretome analysis: insight into CTGF signaling. Sci. Rep. 10, 17889. 10.1038/s41598-020-74838-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smid M, Wang Y, Klijn JGM, Sieuwerts AM, Zhang Y, Atkins D, Martens JWM, and Foekens JA (2006). Genes associated with breast cancer metastatic to bone. J. Clin. Oncol. 24, 2261–2267. 10.1200/JCO.2005.03.8802. [DOI] [PubMed] [Google Scholar]
- 39.Beatson GT (1896). On the treatment of inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment, with illustrative cases. Trans. Med. Chir. Soc. Edinb. 15, 153–179. [PMC free article] [PubMed] [Google Scholar]
- 40.Soerjomataram I, Louwman MWJ, Ribot JG, Roukema JA, and Coebergh JWW (2008). An overview of prognostic factors for long-term survivors of breast cancer. Breast Cancer Res. Treat. 107, 309–330. 10.1007/s10549-007-9556-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chia S, Norris B, Speers C, Cheang M, Gilks B, Gown AM, Huntsman D, Olivotto IA, Nielsen TO, and Gelmon K (2008). Human epidermal growth factor receptor 2 overexpression as a prognostic factor in a large tissue microarray series of node-negative breast cancers. J. Clin. Oncol. 26, 5697–5704. 10.1200/JCO.2007.15.8659. [DOI] [PubMed] [Google Scholar]
- 42.Kim JY, Han W, Moon HG, Ahn SK, Kim J, Lee JW, Kim MK, Kim T, and Noh DY (2013). Prognostic effect of preoperative serum estradiol level in postmenopausal breast cancer. BMC Cancer 13, 503. 10.1186/1471-2407-13-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang J, Li X, Liu X, and Liu Y (2015). The role of tumor-associated macrophages in breast carcinoma invasion and metastasis. Int. J. Clin. Exp. Pathol. 8, 6656–6664. [PMC free article] [PubMed] [Google Scholar]
- 44.Hugo HJ, Saunders C, Ramsay RG, and Thompson EW (2015). New insights on COX-2 in chronic inflammation driving breast cancer growth and metastasis. J. Mammary Gland Biol. Neoplasia 20, 109–119. 10.1007/s10911-015-9333-4. [DOI] [PubMed] [Google Scholar]
- 45.Laidlaw IJ, Clarke RB, Howell A, Owen AW, Potten CS, and Anderson E (1995). The proliferation of normal human breast tissue implanted into athymic nude mice is stimulated by estrogen but not progesterone. Endocrinology 136, 164–171. 10.1210/endo.136.1.7828527. [DOI] [PubMed] [Google Scholar]
- 46.Rochefort H, Platet N, Hayashido Y, Derocq D, Lucas A, Cunat S, and Garcia M (1998). Estrogen receptor mediated inhibition of cancer cell invasion and motility: an overview. J. Steroid Biochem. Mol. Biol. 65, 163–168. 10.1016/s0960-0760(98)00010-7. [DOI] [PubMed] [Google Scholar]
- 47.Al Saleh S, Al Mulla F, and Luqmani YA (2011). Estrogen receptor silencing induces epithelial to mesenchymal transition in human breast cancer cells. PLoS One 6, e20610. 10.1371/journal.pone.0020610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Picon-Ruiz M, Morata-Tarifa C, Valle-Goffin JJ, Friedman ER, and Slingerland JM (2017). Obesity and adverse breast cancer risk and outcome: mechanistic insights and strategies for intervention. CA. Cancer J. Clin. 67, 378–397. 10.3322/caac.21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Renehan AG, Tyson M, Egger M, Heller RF, and Zwahlen M (2008). Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 371, 569–578. 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 50.Wu Q, Li B, Li Z, Li J, Sun S, and Sun S (2019). Cancer-associated adipocytes: key players in breast cancer progression. J. Hematol. Oncol. 12, 95. 10.1186/s13045-019-0778-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foulkes WD, Smith IE, and Reis-Filho JS (2010). Triple-negative breast cancer. N. Engl. J. Med. 363, 1938–1948. 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 52.Gerratana L, Fanotto V, Bonotto M, Bolzonello S, Minisini AM, Fasola G, and Puglisi F (2015). Pattern of metastasis and outcome in patients with breast cancer. Clin. Exp. Metastasis 32, 125–133. 10.1007/s10585-015-9697-2. [DOI] [PubMed] [Google Scholar]
- 53.Yamaguchi H, and Condeelis J (2007). Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta 1773, 642–652. 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim E, Wu D, Pal B, Bouras T, Asselin-Labat ML, Vaillant F, Yagita H, Lindeman GJ, Smyth GK, and Visvader JE (2010). Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res. 12, R21. 10.1186/bcr2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hajra KM, Chen DYS, and Fearon ER (2002). The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 62, 1613–1618. [PubMed] [Google Scholar]
- 56.Barrallo-Gimeno A, and Nieto MA (2005). The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 132, 3151–3161. 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 57.Nieto MA (2002). The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 3, 155–166. 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 58.Martin TA, Goyal A, Watkins G, and Jiang WG (2005). Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann. Surg Oncol. 12, 488–496. 10.1245/ASO.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 59.Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, Gilmore H, Tung N, Naber SP, Schnitt S, et al. (2011). Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 8, 149–163. 10.1016/j.stem.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu YN, Abou-Kheir W, Yin JJ, Fang L, Hynes P, Casey O, Hu D, Wan Y, Seng V, Sheppard-Tillman H, et al. (2012). Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor beta-initiated prostate cancer epithelial-mesenchymal transition. Mol. Cell Biol. 32, 941–953. 10.1128/MCB.06306-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shih JY, Tsai MF, Chang TH, Chang YL, Yuan A, Yu CJ, Lin SB, Liou GY, Lee ML, Chen JJW, et al. (2005). Transcription repressor slug promotes carcinoma invasion and predicts outcome of patients with lung adenocarcinoma. Clin. Cancer Res. 11, 8070–8078. 10.1158/1078-0432.CCR-05-0687. [DOI] [PubMed] [Google Scholar]
- 62.Shioiri M, Shida T, Koda K, Oda K, Seike K, Nishimura M, Takano S, and Miyazaki M (2006). Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Br. J. Cancer 94, 1816–1822. 10.1038/sj.bjc.6603193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alves CC, Carneiro F, Hoefler H, and Becker KF (2009). Role of the epithelial-mesenchymal transition regulator Slug in primary human cancers. Front. Biosci. 14, 3035–3050. 10.2741/3433. [DOI] [PubMed] [Google Scholar]
- 64.Kim S, Yao J, Suyama K, Qian X, Qian BZ, Bandyopadhyay S, Loudig O, De Leon-Rodriguez C, Zhou ZN, Segall J, et al. (2014). Slug promotes survival during metastasis through suppression of Puma-mediated apoptosis. Cancer Res. 74, 3695–3706. 10.1158/0008-5472.CAN-13-2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zürrer-Härdi U, Bell G, et al. (2012). Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148, 1015–1028. 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Picon-Ruiz M, Marchal JA, and Slingerland JM (2020). Obtaining human breast adipose cells for breast cancer cell Co-culture studies. STAR Protoc. 1, 100197. 10.1016/j.xpro.2020.100197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sandhu C, Garbe J, Bhattacharya N, Daksis J, Pan CH, Yaswen P, Koh J, Slingerland JM, and Stampfer MR (1997). Transforming growth factor á stabilizes p15 INK4B protein, increases p15 INK4B -cdk4 complexes and inhibits cyclin D1/cdk4 association in human mammary epithelial cells. Mol. Cell Biol. 17, 2458–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, Smith JA, and Slingerland JM (2009). RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc. Natl. Acad. Sci. USA 106, 9268–9273. 10.1073/pnas.0805057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao D, Besser AH, Wander SA, Sun J, Zhou W, Wang B, Ince T, Durante MA, Guo W, Mills G, et al. (2015). Cytoplasmic p27 promotes epithelial-mesenchymal transition and tumor metastasis via STAT3-mediated Twist1 upregulation. Oncogene 34, 5447–5459. 10.1038/onc.2014.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nettles KW, Gil G, Nowak J, Métivier R, Sharma VB, and Greene GL (2008). CBP Is a dosage-dependent regulator of nuclear factor-kappaB suppression by the estrogen receptor. Mol. Endocrinol. 22, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen Y, Guggisberg N, Jorda M, Gonzalez-Angulo A, Hennessy B, Mills GB, Tan CK, and Slingerland JM (2009). Combined Src and aromatase inhibition impairs human breast cancer growth in vivo and bypass pathways are activated in AZD0530-resistant tumors. Clin. Cancer Res. 15, 3396–3405. [DOI] [PubMed] [Google Scholar]
- 72.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA seq data were submitted to the Gene Expression Omnibus (GEO: GSE132913).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.