

Abstract
Machine learning (ML) models to predict the toxicity of small molecules have garnered great attention and have become widely used in recent years. Computational toxicity prediction is particularly advantageous in the early stages of drug discovery in order to filter out molecules with high probability of failing in clinical trials. This has been helped by the increase in the number of large toxicology databases available. However, being an area of recent application, a greater understanding of the scope and applicability of ML methods is still necessary. There are various kinds of toxic end points that have been predicted in silico. Acute oral toxicity, hepatotoxicity, cardiotoxicity, mutagenicity, and the 12 Tox21 data end points are among the most commonly investigated. Machine learning methods exhibit different performances on different data sets due to dissimilar complexity, class distributions, or chemical space covered, which makes it hard to compare the performance of algorithms over different toxic end points. The general pipeline to predict toxicity using ML has already been analyzed in various reviews. In this contribution, we focus on the recent progress in the area and the outstanding challenges, making a detailed description of the state-of-the-art models implemented for each toxic end point. The type of molecular representation, the algorithm, and the evaluation metric used in each research work are explained and analyzed. A detailed description of end points that are usually predicted, their clinical relevance, the available databases, and the challenges they bring to the field are also highlighted.
Introduction
Toxicity determination is a challenging process especially due to the complexity of in vivo systems. This makes drug safety one of the leading causes of drug withdrawals at the preclinical or clinical phase.1,2 In recent decades, protection agencies have been experiencing a growing frustration due to the large number of toxicity test failures. It has been noticed that 90% of drug candidates that have entered clinical studies would fail during phases I, II, or III of clinical trials, or drug approval.3−5 In the 2010–2017 period, analyses of clinical trials have shown that unmanageable toxicity is responsible for 30% of failures in drug development. Even after being approved, many drugs are withdrawn from the market for showing health risks. This causes a loss of confidence in the industry for patients, healthcare professionals, investors, and regulators.6 In this scenario, computational toxicity prediction is particularly advantageous in the early stages of drug discovery in order to exclude molecules with a high probability of failing in clinical trials.
Traditionally, quantitative structure–activity relationship (QSAR) models were used to computationally predict drug promiscuity and toxicity. These models correlate biological properties with specific functional groups present in each compound. Although they allow a good mechanistic interpretation of the predictions, models are hard to generate from random and diverse databases.7 In this context, an increased availability of toxicity databases has promoted the use of machine learning (ML) models to predict toxicity of small molecules, which have become widely used in recent years.8−15 These methods use a statistical technique to make predictions based on a model. In 2014, the National Center for Advanced Translational Sciences (NCATS) of the National Institutes of Health (NIH) launched the Tox21 Data Challenge competition. It was intended to analyze ML model performance to identify molecular patterns and predict biological properties using only chemical structural data.16 Promising results were obtained, which generated confidence to apply them in the identification of chemicals with the greatest potential for toxicity. The winning team presented a neural network based model called DeepTox,8 thus gaining a notorious relevance in the field.
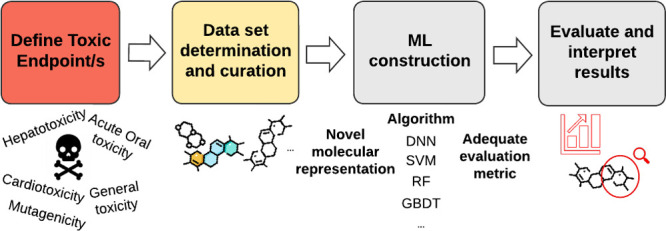
One of the main advantages of ML is that it allows for the modeling and prediction of complex problems, although it generally requires computational power and large amounts of data to learn from. Within ML, two large groups can be distinguished: supervised learning methods and unsupervised learning methods. The former automatically map a set of inputs to a set of outputs from annotated data, and the latter allow learning of underlying relationships directly from a given data set. In the context of drug safety evaluation, supervised learning is commonly used as it can analyze input features associated with compounds to specific outputs such as biological activities or toxic end points. The models that have been mostly used for toxicity evaluation are k-nearest neighbors (kNN), support vector machine (SVM), random forest (RF) and algorithms based on artificial neural networks (NN, neural networks), or deep learning methods (DL, deep learning). DL-based models constitute one of the most common ML methods which are based on multiple layers of neural networks. The number of different architectures and algorithms used in DL is vast and varied. The most common include multilayer perceptron (MLP) networks, recurrent neural networks (RNN, recurrent neural networks) and convolutional networks (CNN, convolutional neural networks). When to use one or the other is problem-dependent and also affected by the chosen molecular representation. Model construction generally involves: data collection and preparation, determination of the molecular representation, building a model by training and validation using a part of the data set, and performing testing on data not previously seen by the model (test set). Figure 1 shows the general pipeline to predict toxicity using ML. Many details can be found in the literature about the construction of ML models and their characteristics,7,17−21 and thus this is not the focus of this work.
Figure 1.

Machine Learning construction outline.
One of the most important steps in computational toxicity prediction is the selection of the type of molecular representation, which has been shown to be highly problem-specific.22 A molecular structure can be represented in terms of a labeled graph with nodes corresponding to atoms and edges corresponding to bonds between these atoms (MGE, molecular graph encoding), by numerical characteristics or molecular descriptors which are calculated from physicochemical properties, by short ASCII strings known as SMILES strings, or by molecular fingerprints which consist on bits representing the presence or absence of particular substructures in a molecule. Molecular Access System keys (MACCS Keys),23 PubChem substructures Fingerprints (PCFP), Klekotha-Roth,24 and Extended Connectivity Fingerprints (ECFP)25 are the most seen in toxicity prediction. Different types of molecular representation and ML algorithms for toxicity prediction can be found nowadays in the literature, and the best performing ones will be described below.
There are various kinds of toxic end points that have been predicted in silico. Acute oral toxicity, hepatotoxicity, cardiotoxicity, endocrine disruption, and the 12 Tox21 Data Challenge end points are among the most common investigated. Obviously, depending on the end point data, one uses a classification model when the toxicity is considered as an active/inactive question, as the case of Tox21 end points, or a regression model when looking for a quantitative prediction, for example, LD50 prediction. Table 1 shows the different sources of data that appear in recent works for ML construction on each toxicity end point. The ML methods show different performances on different data sets due to dissimilar complexity, class distributions or chemical space covered, which makes it hard to compare the performance of algorithms over different toxic end points. The comparison is also made difficult by the different evaluation metrics, whose determination depends mainly on the ML method and the database used. The choice of the evaluation metric is also a crucial step in ML model construction which has been extensively studied.26,27
Table 1. Public Toxicology Databases Available and the Best Performing Ml Models References by Toxicity End Point.
| Toxicity end point | Database Name | ML models references |
|---|---|---|
| Cardiotoxicity (hERG binding) | PubChem bioassay28 and CHEMBL bioactivity29 | (30−34) |
| Acute oral toxicity (LD50) | Li et al. set:35−37 admetSAR, MDL Toxicity, EPA Toxicity Estimation Software Tool. SuperToxic39 | (12, 38) |
| Hepatotoxicity (DILI) | Liver Toxicity Knowledge Base (LTKB)40 LiverTox42 Hepatox43 | (14, 41) |
| Nuclear Receptor and Stress Response panels | Tox2144 | (8, 10, 45, 46) |
| Mutagenicity | Ames data collection47 | (47−49) |
| Carcinogenicity | Carcinogenic Potency Database (CPDB)50 | (51) |
| General Toxicity | TOXNET52 Toxin and Toxin Target Database (T3DB)55 SIDER | (12, 53, 54) |
Moreover, one of the main challenges that computational toxicology faces today is the ability to obtain a mechanistic explanation or understanding of the identified toxic responses. The ML methods, especially DL models, are generally treated as black boxes which can efficiently manage complex problems but often lack an explanation of the prediction. Recent models seek to apply different tools to improve interpretability. Mayr et al., for example, showed that neural networks could learn representations which are comparable to known toxicophores.8 Wenzel et al. introduced response maps that allow researchers to gain an understanding about which are the features most related to toxicity.13 A variety of methods are emerging which could be beneficial for in silico toxicology.56
A list of all the available ML methods developed in the area can be already found in the literature.7,18,20,21 In this contribution, we present a detailed description of the most recent progress in the area and the persistent challenges that need to be addressed in future studies. The state-of-the-art and most representative ML models developed will be analyzed separately for each toxic end point to provide a better understanding and comparison of its performances. The type of molecular representation, the ML algorithms, and the evaluation metrics used in each research work are explained and analyzed. A description of the end points that are usually predicted, their clinical relevance, the available databases, and the challenges they bring are also presented. In addition, we survey the tools used to increase the interpretability of ML models to provide insight for future developments.
Cardiotoxicity: hERG Binding Prediction
The hERG potassium channel plays an extremely important role in heart function through conducting the electrical activity of the heart. Blockade of the channel by small molecules induces the prolongation of the QT interval, which can lead to fatal cardiotoxicity. Many drugs, including cisapride, astemizole, sertindole and terfenadine, have been withdrawn or restricted from the market due to drug-induced arrhythmias and other cardiac side effects.30,57 Therefore, hERG inhibition by drug candidates has become an important concern, and its early evaluation is a desirable step in drug discovery.58
Various ML methods have been proposed in recent years to predict hERG binding compounds.30−34 Most of them are classifier models, and only a few of them are regression models. In 2016, Wang et al. combined pharmacophore modeling with ML to construct classification models for prediction of hERG active compounds.32 They used Naïve Bayes (NB) and SVM algorithms and integrated multiple representative pharmacophore hypotheses identified by a recursive partitioning (RP) approach. RP allows for the identification of the most important pharmacophores creating a decision tree that splits molecules into subsets based on independent properties.59 For model construction and validation, they used a small data set that contained 587 molecules, 527 of which had experimental hERG blocking bioactivities (IC50) and 60 where hERG nonblockers added based on a previous study.60 A threshold of 40 μM was used to define hERG blockers and nonblockers. The data set was divided in training, validation, and testing sets, thus guaranteeing that the selected molecules in the training set have the largest diversity evaluated by the Tanimoto coefficients based on the molecular fingerprints and logP. The best SVM model achieved prediction accuracies of 84.7% for the training set and 82.1% for the test set. One of the big challenges in the development of successful cardiotoxicity models that needs to be addressed is the use of a robust database of compounds with hERG binding affinities measured using uniform experimental conditions. Models are usually developed based on relatively small data sets, and thus their chemical coverage and applicability domains are still limited.
In 2019, Cheng et al. developed a DL based approach called deephERG.33 They used 7,889 compounds with diverse chemical structures and defined hERG inhibition based on experimental data. The database was constructed based on compounds from PubChem and CHEMBL bioactivity databases, and also based on literature information. Compounds without well-defined experimental hERG blocking bioactivities were eliminated, and IC50 values ≤10 μM were considered as hERG blockers. No consensus has yet been reached on how to define a compound as a hERG nonblocker, and different thresholds had been used to distinguish blockers from decoys.32,61 In this work, data were split according to different decoy threshold values (10 μM, 20 μM, 40 μM, 60 μM, 80 μM, and 100 μM) for building multitask models. For each task, data were separated in training, validation, and final evaluation sets. Chemical data were represented by molecular descriptors of the MOE Descriptors tool62 and by vector representation of their chemical structures calculated using the Mol2vec approach.63 As analog to the Word2vec models, where vectors of closely related words are in close proximity in the vector space, Mol2vec learns vector representations of molecular substructures that point in similar directions for chemically related substructures. After systematic comparison, the MT-DNN models outperform other models such as single-task DNN, NB, SVM, RF, and the graph convolutional neural network (GCNN). The AUC-ROC was used as an evaluation metric, reaching a value of 0.967 on the validation set.
In the present years, the availability of the cryo-EM structure of the hERG channel from MacKinnon and co-workers64 encouraged the use of structure-based drug design (SBDD) approaches such as molecular docking, molecular dynamics simulations, or free energy calculations for hERG screening of drug candidates. Mangiatordi et al. presented a hERG binding structure-based classifier which combines docking scores, molecular fingerprints and ML models.34 The data set was constructed from CHEMBL bioactivity database and contains 8,337 entries with high structural diversity measured by the Tanimoto coefficient. Different IC50 inactivity thresholds from 1 μM to 80 μM were used; therefore, positive molecules show IC50 ≤ 1 μM, and negative molecules are those with IC50 values greater than the different inactivity thresholds. Molecular docking simulations were performed using two docking programs on the available cryo-EM structures and two homology models for comparison. A LASSO-regularized SVM model was applied to integrate docking scores and protein–ligand interaction fingerprints as inputs. The best classifiers showed performances comparable to state-of-the-art ligand-based models in terms of AUC-ROC (0.86 ± 0.01) and negative predictive values (0.81 ± 0.01), offering a more interpretable approach. A combination of structure-based strategies with ML models might provide optimal efficacy and interpretability, enhancing its performance as lower resolution structures become available.
Acute Oral Toxicity
Median lethal death, LD50, is a general indicator of chemicals’ acute oral toxicity (AOT). It represents the dose of a chemical that causes a 50% death rate in test animals after administration. Showing AOT information is a standard requirement in several regulatory frameworks, and chemicals are generally classified in toxicity categories based on their LD50 values. Testing on animals, especially when relying on mortality as an end point, is highly controversial. Therefore, alternative in silico methods that provide reliable information about this end point are highly needed and encouraged.65 Many ML models have been proposed to predict AOT in recent years, including regression and classification methods.
Lai et al. presented the deepAOT model which is based on a molecular graph encoding convolutional neural network (MGE-CNN) architecture.38 In MGE-based DL models, the basic chemical information of molecular graphs is used as input, and the DL algorithm automatically learns specific representations using graph convolutions, without the need to manually calculate numerical descriptors or fingerprints.66 They used the AOT database provided by Li et al. in a previous work,67 which consists of the largest data set constructed at the moment for oral LD50 in the rat. It contains a total of ∼12,200 compounds divided in training and validation sets. Data were collected from experimental values from the admetSAR database36 the MDL Toxicity Database (version 2004.1),35 and the Toxicity Estimation Software Tool (TEST version 4.1)37 program from the U.S. EPA. Two external test data sets were used to estimate the predictive power of the models.
The MGE-CNN architecture of deepAOT is shown in Figure 2. The canonical SMILES string of a small molecule is used as an input, and a score capable of describing the LD50 end point is produced. Both regression and multiclassification models were developed. The regression models allow quantitative prediction of the LD50 values for compounds reflecting their toxicity: the smaller the value, the more toxic the compound. For the multiclassification models, data were divided into balanced classes based on the US Environmental Protection Agency (EPA) AOT categories.68 Beginning with the SMILES string, a molecular structural graph is converted, and then the subgraphs from each iteration are encoded into fixed-sized vectors which are then summed to a unique fingerprint representing the molecule. The fingerprint is finally used as the input of a subsequent NN in the output layer.
Figure 2.

DeepAOT architecture for LD50 prediction (Reprinted with permission from ref (38). Copyright 2017. American Chemical Society).
For the regression MGE-CNN models, the root-mean-square error (RMSE), mean absolute error (MAE), and square of Pearson correlation coefficient (PCC2) were used as evaluation metrics. The multiclassification MGE-CNN models were assessed by a confusion matrix, and also the sensitivity, positive predictive value (PPV), and accuracy were reported. The regression model had the best performance, showing higher PCC2 (0.864), lower RMSE (0.268), and lower MAE (0.195) than state-of-art models.69 The deepAOT classifier also demonstrated a good performance with accuracy values of 96%, which are also higher than the best reported results (80% accuracy) from Li et al. AOT classification methods.67 One of the main strengths of the MGE-CNN model is that it uses automatic molecular representations; thus, the AOT prediction can be performed without manual selection of complicated features. The MGE has been shown to be an effective representation of chemical structures without information loss. Moreover, a balanced data set was constructed which facilitates the development of robust predictive ML models. Focus was made in the interpretation and explanation of the predictions made by the model using both a forward exploration and a backward exploration approach. The strategies implemented to enhance interpretability are further analyzed in a later section.
In 2018, Wei et al. introduced topology based descriptors used with multitask deep neural networks (MT-DNN) to predict LC50 and LD50.70 It uses element specific persistent homology (ESPH) as a new molecular representation method for toxicity prediction, which is an algebraic topology approach that retains crucial chemical information during the topological abstraction of geometric complexity. Four benchmark toxicity data sets that involve quantitative measurements are used to validate the proposed approaches (oral rat LD50, 40 h Tetrahymena pyriformis IGC50, 96 h fathead minnow LC50, and 48 h Daphnia magna LC50). The data were downloaded and then curated from the ECOTOX aquatic toxicity database71 and from the ChemIDplus databse.72 The Toxicity Estimation Software Tool (TEST) database37 was used as a final test set to compare results to previous works. Aided with physicochemical descriptors and MT-DNN architecture, ESTDs showed results similar to the state-of-the-art predictions for quantitative toxicity data sets with an R2 value of 0.788. New molecular representation methods such as ESTDs are worth studying to improve the performance of ML in toxicity prediction.
In 2019, Bylinski et al. presented the etoxPred model.12 It employs Extremely Randomized Trees or Extra Trees (ET) algorithm to predict toxicity of small organic compounds. AOT was evaluated using data provided by the SuperToxic database.39 The data set consisted on 12,612 molecules of which 7,392 compounds were labeled as toxic with LD50 values less than 500 mg/kg. The model obtained an AUC value of 0.80 and an accuracy value of 0.854, which are similar results to those obtained by some of the deepAOT multiclassification models.
Hepatotoxicity: Prediction of Drug-Induced Liver Injury
Drug-induced liver injury (DILI) is one toxicological end point of high concern since it has been a leading cause of clinical trials failure and drug withdrawal from the market.40 More than 700 drugs have been reported to be associated with hepatotoxicity in the past years.42 Therefore, in silico DILI prediction models are also encouraged to early estimate DILI potential of drug candidates. There is a particular need to develop models that represent DILI potential in humans, since many drugs that do not show clear hepatotoxicity in animals end up causing severe DILI in humans.73 There are some hepatotoxicity databases that have become available in recent years, for example, the Liver Toxicity Knowledge Base (LTKB),40 the liver toxicological map (LTMap),74 LiverTox,42 or Hepatox,43 as shown in Table 1.
In 2015, Lai et al. presented a DL-based model to predict DILI using data from different sources according to previous works.14 They developed a graph recurrent neural network architecture motivated by previous results on a work by Lusci et al. Molecules were represented by small undirected graphs and then encoded to acyclic graphs for use in RNNs. The best model was trained on 475 compounds and evaluated on an external set of 198 drug-like compounds where it obtained the best performance in terms of sensitivity and specificity until that moment.
In 2018, Li et al. developed binary classification models using five different ML methods using data from 2,144 chemicals collected from FDA approved drugs with known hepatic effects and from human DILI data in the LTKB database.41 They divided the compounds randomly with a ratio of 4:1 into training and validation sets. To further evaluate the ability of the models, they used an external test set of 151 compounds from a study reported by Ivanov et al. in 2017.75 For model building, they calculated 12 types of molecular descriptors based on known physicochemical properties and seven types of commonly used fingerprints including MACCS keys and Klekota-Roth fingerprints. They evaluated the diversity of chemical structures present in the database using the radar chart of five physicochemical descriptors and calculating the Tanimoto similarity index based on the ECFP4 fingerprint. They found that the SVM algorithm combined with MACCS keys obtained the state-of-art results with an accuracy of 80.4%, sensitivity of 88.2%, and specificity of 65.7%, showing similar results in validation and external test sets. It is seen that SVM algorithms usually appear as a suitable approach where only few nonlinear and high dimensional pattern data are available.
There has not been great progress in the development of robust ML models for DILI prediction in recent years, especially since there is still little data available. The available databases contain data on both animal and human DILI class labels for chemical compounds. However, much of the current work looks forward to focus solely on the prediction of human hepatotoxicity. Efforts need to be focused on obtaining more experimental data for this common toxic end point.
Various Toxicities Predictions
Moreover, multiple works are available in the literature that develop models to estimate various toxicities. One of the most well-known and used databases for the prediction of various toxicities is the Tox21 database.44 As from the promising results of the Tox21 competition, diverse ML protocols that use this database have been presented. It contains data on more than 10,000 molecules and their activities against 12 targets associated with different AOPs. These toxic events include nuclear receptor effects (NR) and stress response effects (SR), which are relevant toxic responses in health since the former can affect the endocrine system, and the latter can cause damage to the liver or cancer. Tox21 database is characterized by having class imbalance, where only 5% of annotated data correspond to the toxic class, with some end points having as low as 1% of toxic data. Handling unbalanced data sets is common in ML and entails one of the problems that should be addressed in model construction.
Mayr et al. were the winners of the Tox21 competition implementing a model based on DL which they called DeepTox.8 Based on the assumption that the use of many correlated features is favorable to achieve high performance in DL, Mayr et al. used several thousands of physicochemical descriptors and molecular fingerprints to represent data. Among them, they used the well-known PCFP, ECFPs and MACCS keys. To these they added about 2,500 in-house toxicophore features which represent substructures previously reported as toxicophores. Besides, they included similarity features by using the natural ligands of the receptors (e.g., testosterone and estradiol). A similarity value was calculated using path-kernel-based paths between these ligands and the Tox21 compounds. Approximately 40 similarity features per molecule were included. In order to correct data imbalance, positive samples were enriched from PubChem28 and ChEMBL29 databases. They searched for compounds (structural analogs) and assays in the public data that were similar to compounds and assays of the project data, respectively.
Moreover, the Tox21 database allows for a Multi Task Learning (MTL) approach. In this case, a single molecule has labels for various target activities that can be trained simultaneously by the ML model. Different studies have shown that MT-DNN algorithms can improve the predictive performance.76 In DeepTox, for 10 out of 12 assays MT-DNN models outperformed single-task networks. An average AUC-ROC of 0.858 and 0.826 was obtained in the NR and SR test sets respectively. It should be noted, anyway, that some compounds have not been tested across the entire target set, leading to sparse arrays. To overcome this problem, these compounds were assumed as inactive when the activity value was missing, which can lead to false negatives. The problem of missing data in multitarget matrices is an area of active study within ML.77
In 2020, Peng et al. presented another DL-based ML model called TOP that was trained and evaluated on the Tox21 database.46 The model integrates a RNN based on bidirectional gated recurrent unit (BiGRU) and fully connected neural networks for end-to-end molecular representation learning and chemical toxicity prediction. It uses a mixed molecular representation method combining SMILES strings with few carefully chosen physicochemical descriptors. They obtained better AUC-ROC results than DeepTox model in 11 out of 12 tasks, with an average value of 0.95 (vs 0.85 in DeepTox).
Karim et al. presented another method to predict Tox21 targets which they argue to be simpler, more efficient in computing resource usage, and more powerful to achieve high accuracy levels.45 The approach consisted of a Decision Tree (DT) algorithm that obtains the optimum number of features related to NR and SR toxicities and a shallow NN with one hidden layer to make the final predictions. They calculated 1422 molecular descriptors based on 2D chemical compound structures using the PaDEL tool78 which where then reduced by the feature selection module. This hybrid model was compared with the DeepTox model, achieving similar AUC-ROC values (0.862 vs 0.858 in DeepTox NR panel, and 0.836 vs 0.826 in SR) with less calculated features, thus allowing for better interpretability and lower computational costs.
Recently, Jiang et al. proposed a different approach which they called the geometric graph learning toxicity (GGL-Tox) model.10 It consists of the use of multiscale weighted colored graphs (MWCG) as features and a gradient boosting decision tree (GBDT) algorithm. MWCG has been implemented successfully for protein flexibility analysis and protein–ligand binding prediction.79 The molecular modeling of MWCGs requires only atomic names and coordinates and is characterized by its low-dimensionality, simplicity, and robustness.80 Other ML algorithms were also tested, including RF and SVM, but GBDT performed best in 11 out of the 12 tasks. RF was the second-best, and SVM had the lowest performance. The GGL-Tox model constructed from MWCG features and the GBDT algorithm obtained the best average AUC on both NR and SR data sets (0.875 for NR and 0.871 for SR) until 2021, showing consistently better performance than classic 2D features.
It is essential to consider class imbalance when selecting an evaluation metric that is representative of the model performance. Within the Tox21 competition, the Area Under the Receiver Operating Characteristic Curve (AUC-ROC) was used. It is a widely used metric, and although it might be useful to compare performance between models, it is not the most suitable for unbalanced data and, in particular, for toxicity data. This is due to two main reasons: (1) it is necessary to distinguish between the correct classification of the toxic and nontoxic classes, since it is a more serious problem to classify a toxic compound as if it were not than the opposite case; (2) it does not consider the imbalance problem since the false positive rate (FPR) is reduced due to a large number of true negatives predicted, leading to a high AUC-ROC value and less margin to discriminate between models with different hyperparameters.
Antelo-Collado et al. developed a feature selection method focused on solving the Tox21 data class imbalance problem. The method is based on the use of boosting feature selection ensembles. These ensembles were constructed using two well-known feature selection methods: fast clustering-based feature selection (FAST) and fast correlation-based filter (FCBF).81 They tested the classification performance of two ensemble methods and three ML algorithms (DT, SVM, and RF) using G-mean and MCC as evaluation metrics. These metrics take into account the uneven distribution of class samples. The proposed ensembles of feature selectors achieved better classification performance compared to the use of simple feature selection methods.
Zhang et al. presented a very interesting DNN-based model to predict Tox21 end points in a conformal prediction setting.82 Conformal prediction can be used with any ML model by calibrating the outputs from the predictor using a calibration set and thus have associated quantitative uncertainty measures. Conformal predictors are a convenient way to achieve confident toxicity predictions and especially better predictions of the minority class compared to underlying models. In a binary prediction problem, the output can be either of the two labels, both labels, or no label. They achieved an efficiency greater than 80% for the toxic class at the 90% confidence level using a graph CNN as the underlying model. The efficiency is defined as the fraction of predictions with a single label; a model with a higher fraction of single-label predictions is more efficient. The performance of the models was evaluated using various metrics: balanced accuracy (BA), sensitivity, specificity, AUC-ROC, F1 score, Kappa, precision, and MCC. Even though the obtained AUC-ROC on the Tox21 test set (0.734) is not as high as the obtained in other works, conformal prediction adds both a controllable error rate and better recall of the toxic compounds, compared to the underlying models. In this way, more examples of the minority class are retrieved by the model at the expense of a slightly higher false-positive rate. This is a favorable balance in toxicity prediction as it takes into account the higher importance of the prediction of the toxic class, reducing the number of potentially toxic instances missed by the model. This work shows that if better performing ML models are made into conformal predictors, very robust toxicity prediction models can be achieved.
As seen from these works, ML-based Tox21 toxicity prediction is increasing performance, especially by using novel types of molecular representation and feature selection techniques. Besides, MTL approaches seem to achieve better results than single task models, especially when DNN-based models are used. However, to continue developing effective Tox21 ML models, it is necessary to use appropriate evaluation metrics. Most works still rely on AUC-ROC, probably to be able to compare their results, but due to the great imbalance of classes and the highest importance of the minority (toxic) class it is not an appropriate metric for this data. Metrics such as BA and MCC, which were used in various works, are appropriate for class-imbalance data sets but also do not consider the higher importance of the classification of the toxic class. The Fβ score or the area under the precision recall curve (AUC-PR) is a more consistent metric to address this problem, as well as the use of conformal predictors as shown by Zhang et al.82
Furthermore, the ProTox53 and ProTox-II54 are publicly available web servers that were constructed on a total base of 33 ML models, trained on data sets of various toxicity end points and allow for different toxicity estimates at the same time. The new version, ProTox-II, was an improvement over previous web tools, which incorporates molecular similarity, pharmacophore based, fragment propensities, and most common features information. There are other available ML-based web servers to predict toxicity,36,37 but their performances and applicability domains are still very limited.
General Toxicity Prediction
Some works seek to predict toxic liabilities in a more general manner. In the eToxPred algorithm, for example, ML models were trained and cross-validated against a number of data sets comprising known drugs, potentially hazardous chemicals, natural products, and synthetic bioactive compounds.12 Models were trained and tested on a general toxicity data set constructed using FDA-approved drugs and Kyoto Encyclopedia of Genes and Genomes (KEGG) Drug as nontoxic data and TOXNET52 and the Toxin and Toxin Target Database (T3DB)55 as toxic data comprising a total of ∼10,000 compounds. The ET classifier obtained the best results which show an acceptable accuracy (0.72) and sensitivity (0.63) but a low precision (0.25). Specific toxicities were also addressed with the model.
Another interesting work was presented by Di Filippo et al. in 2021 where a low-dimensional ML model was developed for classifying compounds according to whether they can cross or not the placental barrier, helping to develop safe therapeutic options for pregnancy.83 A data set of 248 molecules was constructed, and a genetic algorithm was used to perform feature selection from an initial group of 5,400 descriptors. A linear discriminant analysis (LDA) model trained with only four features achieved the best results, having only one false positive case across all testing folds.
Other Toxicity End Points
The mutagenicity of a compound is another important studied property that can be responsible for drug candidates failure in drug discovery. Mutagenicity or genetic toxicology is the study of substances that induce DNA damage. Only an isolated positive in vitro genetic toxicology finding can result in the failure of a drug candidate. Damage on the DNA, quantified as the frequency of DNA adducts, strand breaks, mutations, or chromosome aberrations is highly related to carcinogenicity.84 The most known and used assay for testing the mutagenicity of a compound is the Ames test.85 The Ames test consists of a bacterial gene mutation assay with a simulation of mammalian metabolism and is highly sensitive to identify chemicals that can induce genetic damage. However, as in other toxicity end points, experimental mutagenicity identification is inefficient as it requires a lot of chemical resources, too much time to conduct in order to achieve meaningful results, and does not have a 100% rate reproducibility.86 For this reason, mutagenicity computational prediction has attracted attention from several research groups.47−49
In 2021, Berry et al. developed eight classification algorithms, including SVM, RF, and extreme gradient boosting (XGB), which achieved comparable sensitivity and specificity results to the best previous methods.49 They used the Ames data collection from a Xu et al. study47 which consisted of a training set of 7,617 molecules, a validation set of 731 molecules, and a balanced external set of 234 molecules, which they curated to eliminate duplicates. A large number of molecular descriptors and fingerprints was calculated, and it was found that the RF model had the best performance during the 10-fold cross validation training set (AUC-ROC > 0.90) and on the external test set (AUC-ROC > 0.75). On a feature importance analysis, it was seen that physicochemical descriptors including logD, molecular solubility, molecular surface area, number of aromatic rings, number of rings, and number of rotatable bonds presented the highest importance in model training when used in combination with molecular fingerprints.
In 2017, Zhang et al. presented the CarcinoPred-EL tool which consists of three novel ensemble classification models to predict carcinogenicity of chemicals using seven types of molecular fingerprints and three ML methods (SVM, RF, and XGB).51 They used a balanced data set containing 1,003 diverse compounds with rat carcinogenicity data from the Carcinogenic Potency Database (CPDB), which provides information on the measures of carcinogenic potency of compounds on different tissue tumors reported.50 Besides, an external validation data set of 40 compounds from the ISSCAN database87 was used to evaluate the performance of the ensemble models built using the top-seven fingerprint sets. The ensemble models can be formed by combining simple independent classifiers via voting or averaging to produce a more accurate and robust model than any of its constituents. In this work, the three ensemble models achieved higher accuracy and AUC than any basic classifier. Ensemble XGB obtained the best results, with an AUC value of 76.5%, which they indicated were among the best results at the time.
Recently, Mathea et al. proposed an approach to enhance the performance prediction of three in vivo end points (genotoxicity, DILI, and cardiological issues) by using conformal prediction ML models with molecular descriptors (molecular fingerprints and physicochemical properties) and ML-predicted bioactivity assay outcomes as molecular representation. They developed a workflow which they called ChemBioSim based on a conformal prediction framework built on RF models. The bioactivity descriptors for each in vivo end point were preselected with lasso models. The incorporation of bioactivity descriptors increased the mean F1 scores of the genotoxicity model from 0.61 to 0.70 and for the cardiotoxicity model from 0.72 to 0.82, while the mean efficiencies increased by 0.09 and 0.12, respectively, for both end points. For the DILI end point, no significant improvement in model performance was observed. This is a very challenging toxic end point, mainly due to less available data, which combines substances that produce both major and less severe effects as toxic data. This approach shows how the prediction of in vivo end points, which is a highly complex problem due to all the interactions taking place in biological systems, can be improved by the incorporation of bioactivity fingerprints as molecular representation. Besides, the conformal prediction framework increases confidence in model predictions and ensures a defined error rate. As more data becomes available, this approach can be better evaluated, also for other toxicity end points and using other ML models with a higher baseline performance.
Addressing the Problem of ML Interpretability in Computational Toxicology
One of the most widely debated issues in ML is the lack of ability of these models to provide user-interpretable results, especially when using more complex algorithms such as DNNs. Usually, these models are considered black boxes, and although complex algorithms generally perform well in big data sets, the user cannot infer what is actually internally happening. The interpretability of ML models is today an active area of research.88,89
In recent years, the focus of in silico toxicology has shifted from just a mere model building to the use of strategies that help to understand ML model results. Researchers have started to analyze neural unit representations in order to disentangle the neural networks learning process.90 Besides, many works use different techniques to provide information on the data features that the models prioritize the most to estimate toxicity end points.
Mayr et al., for example, trained their DeepTox model and looked for possible associations between neuron activation and known toxicophores.8 To this aim they used a U-test where a neuron was characterized by its activation over the compounds of the training set, and a toxicophore was characterized by its presence or absence in the same compounds. The alternative hypothesis for the test was that compounds containing the toxicophore substructure have different activations than compounds that do not contain the toxicophore substructure. They found that features in higher layers match toxicophores more precisely and that lower layers tend to learn smaller features. This analysis can also be used to identify new substructures related to some type of toxicity (structural alerts identification). In a work by Wenzel et al.13 response maps were introduced. They were intended to evaluate the sensitivity of ML models to a particular substitution in chemical structures and to identify favorable substitutions on the scaffold. They tried different substituents and fragmenting parts of the molecules and checked the predicted property response from a pretrained model. These approaches help to provide more interpretable statistical analyses and useful information to compounds optimization.
In the deepAOT model, Lai et al. used a forward exploration approach to evaluate the extent by which the fingerprints obtained in the deep layer of the MGE-CNN models favored shallow ML models, such as multiple linear regression (MLR) and SVM.38 They also used a backward exploration approach to provide understanding of fingerprint activation by mapping the most relevant features into different substructures and found that most of the highlighted fragments could correspond to reported toxic alerts. The forward exploration was implemented by extracting the values of the Fingerprint layer and constructing MLR and SVM models trained with them. The performance of these models was then compared with previous reported models using application-specific fingerprints or descriptors.
Other works also included backward exploration methods such as Abdul Karim et al. where the 1,422 molecular descriptors used as input were ranked based on their Gini index in the DT classifier. The “path count descriptor” class was the most abundant class in the top features list. The molecular path counts represent the number of unique paths of length k present in the molecular structure. They could see that toxic compunds in the database clustered in a certain range of pipC10 value, leaving a large area as safe zone. A similar approach was carried out by Jiang et al. for the GGL-Tox model. They ranked MWCG and classic 2D features by their Gini index in their GBDT algorithm and then selected subsets of top features to retrain the model and compare results. They found that choosing at least 0.6% or at most 27.8% of the most important features from a total of 1,794 input features optimizes the prediction performance.
Although progress is being made, there is still a lot of space for improvement in the interpretation of these models. Even more, elucidation of mechanistic information which involves the targets and pathways that lead to the different toxicity end points is generally not addressed for pharmaceutical candidates. It has been shown that small molecule drugs bind on average to at least 7–12 distinct targets, with varying affinities.91 These off-target interactions are often unknown and may be linked to a toxicity end point via an adverse outcome pathway (AOP). The availability of more data on AOPs will be crucial to be able to generate robust predictive models. Other approaches such as systems pharmacology could also take advantage of AOP data. System pharmacology considers protein targets in the context of biological networks, which is a more realistic approach as proteins perform their functions within a complicated and integrated system composed of various scales of biological organization. Many reviews and works on this area can be found in the literature.92−95 In 2017, Chua et al. developed MASCOT (ML-based Prediction of Synergistic Combinations of Targets),96 which efficiently predicts synergistic target combinations with desired therapeutic effects and minimum off-target effects in a disease-related signaling network. Zeng et al. presented a network-based model that combines neural networks with heterogeneous networks in which drugs and targets are represented as nodes, and their interactions are represented as edges to predict drug-target interactions.97 Although system pharmacology is beyond the scope of this review and will not be further analyzed, it is interesting to note that the combination of ML methods with biological networks may be an efficient strategy to develop more accurate and understandable predictive models, as more data on AOPs become available.
Conclusions and Future Perspectives
Toxicity estimation of drug candidates is an important issue in drug discovery, being essential to increased costs, failures in late stages, and marked withdrawals. The available evidence shows that ML models, despite its persistent limitations which were addressed in this review, may be a promising approach to function as early filters of toxic compounds within the drug discovery process. This potential ability to be integrated into the natural drug discovery pipeline can be improved as more high-quality data become available and the applicability domain of the methods is expanded. Although the availability of public data has increased in recent years, the collaboration of pharmaceutical companies will be more and more necessary to obtain the amount and quality of data that ML needs to develop reliable predictive toxicology models. This review provides detailed information and analysis on the state-of-art ML methods that have been developed for each toxicity end point, providing insight for future developments.
The main types of toxicity that are in silico predicted are cardiotoxicity, AOT, hepatotoxicity, mutagenicity or genotoxicity, and the nuclear receptor and stress response panels of the Tox21 data, which have been set forth in this review. The various tasks and different databases make it difficult to compare the performance of ML models. In general, it is seen that there is no model that works best for most of the cases. It can be said that complex problems with big data sets can be generally handled correctly using DL-based algorithms such as DNNs or CNNs, while GBDT or SVM algorithms exhibit better results for smaller nonlinear data. It should be noticed that ML models performances are problem-dependent and must therefore be compared only when they have been developed for the same toxicity end point and using similar data for evaluation. The most suitable metric to use in each case is still up to debate, but much care should be taken to have representative results depending mostly on the data set size and class distributions. Conformal prediction frameworks are being used in some recent works, which ensure more confident models with well-defined uncertainties and also as a strategy to handle unbalanced data.
Moreover, the interpretability of ML models for toxicity prediction is an especially desirable aspect to consider in model construction. Most of the work presented in recent years uses techniques to increase the comprehension of model learning, including the understanding of neural units activation and most important features. However, there is much to improve in this area that will be a focal point in future developments. The collection of more data on AOPs will also be essential to allow a mechanistic understanding of drug action by considering targets in the context of biological networks. The synergy between network pharmacology approaches and ML methods could be a potential way of addressing this problem.
Acknowledgments
The authors thank Juan I. Di Filippo for thoroughly reading of the manuscript.
The authors declare no competing financial interest.
References
- Frank C.; Himmelstein D. U.; Woolhandler S.; Bor D. H.; Wolfe S. M.; Heymann O.; Zallman L.; Lasser K. E. Era of Faster FDA Drug Approval has also seen increased Black-Box Warnings and Market Withdrawals. Health Aff 2014, 33, 1453–1459. 10.1377/hlthaff.2014.0122. [DOI] [PubMed] [Google Scholar]
- Giri S.; Bader A. A low-cost, high-quality new drug discovery process. Drug Discovery Today 2015, 20, 37–49. 10.1016/j.drudis.2014.10.011. [DOI] [PubMed] [Google Scholar]
- Biopharmaceutical Research & Development: The Process Behind New Medicines Brochure; Phrma, 2015; http://phrma-docs.phrma.org/sites/default/files/pdf/rd_brochure_022307.pdf (accessed March 2, 2022).
- Dowden H.; Munro J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov 2019, 18, 495–6. 10.1038/d41573-019-00074-z. [DOI] [PubMed] [Google Scholar]
- Takebe T.; Imai R.; Ono S. The current status of drug discovery and development as originated in United States academia: the influence of industrial and academic collaboration on drug discovery and development. Clinical and translational science 2018, 11, 597–606. 10.1111/cts.12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen M. E.; Krewski D. Toxicity Testing in the 21st Century: Bringing the Vision to Life. Toxicol. Sci. 2009, 107, 324–330. 10.1093/toxsci/kfn255. [DOI] [PubMed] [Google Scholar]
- Wang M. W. H.; Goodman J. M.; Allen T. E. H. Machine Learning in Predictive Toxicology: Recent Applications and Future Directions for Classification Models. Chem. Res. Toxicol. 2021, 34, 217–239. 10.1021/acs.chemrestox.0c00316. [DOI] [PubMed] [Google Scholar]
- Mayr A.; Klambauer G.; Unterthiner T.; Hochreiter S. Deep Tox: Toxicity prediction using Deep Learning. Frontiers in Environmental Science 2016, 3, 80. 10.3389/fenvs.2015.00080. [DOI] [Google Scholar]
- Fernandez M.; Ban F.; Woo G.; Hsing M.; Yamazaki T.; LeBlanc E.; Rennie P. S.; Welch W. J.; Cherkasov A. Toxic colors: The use of deep learning for predicting toxicity of compounds merely from their graphic images. J. Chem. Inf. Model. 2018, 58, 1533–1543. 10.1021/acs.jcim.8b00338. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Wang R.; Wei G.-W. GGL-Tox: Geometric Graph Learning for Toxicity Prediction. J. Chem. Inf. Model. 2021, 61, 1691–1700. 10.1021/acs.jcim.0c01294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim A.; Mishra A.; Newton M. A. H.; Sattar A. Efficient Toxicity Prediction via Simple Features Using Shallow Neural Networks and Decision Trees. ACS Omega 2019, 4, 1874–1888. 10.1021/acsomega.8b03173. [DOI] [Google Scholar]
- Pu L.; Naderi M.; Liu T.; Wu H.-C.; Mukhopadhyay S.; Brylinski M. eToxPred: a machine learning-based approach to estimate the toxicity of drug candidates. BMC Pharmacology and Toxicology 2019, 20, 2. 10.1186/s40360-018-0282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel J.; Matter H.; Schmidt F. Predictive multitask deep neural network models for adme-tox properties: Learning from large data sets. Journal of Chemical Information and Modelling 2019, 59, 1253–1268. 10.1021/acs.jcim.8b00785. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Dai Z.; Chen F.; Gao S.; Pei J.; Lai L. Deep Learning for Drug-Induced Liver Injury. Journal of Chemical Information and Modelling 2015, 55, 2085–2093. 10.1021/acs.jcim.5b00238. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Pei J.; Lai L. Deep Learning Based Regression and Multiclass Models for Acute Oral Toxicity Prediction with Automatic Chemical Feature Extraction. Journal of Chemical Information and Modelling 2017, 57, 2672–2685. 10.1021/acs.jcim.7b00244. [DOI] [PubMed] [Google Scholar]
- Huang R.; Xia M.; Nguyen D.-T.; Zhao T.; Sakamuru S.; Zhao J.; Shahane S. A.; Rossoshek A.; Simeonov A.. Tox21challenge to build predictive models of nuclear receptor and stress response pathways as mediated by exposure to environmental chemicals and drugs. Frontiers in Environmental Science 2016, 3. 10.3389/fenvs.2015.00085 [DOI] [Google Scholar]
- Hemmerich J.; Ecker G. F.. In silico toxicology: From structure–activity relationships towards deep learning and adverse outcome pathways. Wires Computational Molecular Science 2020, 10. 10.1002/wcms.1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin I. Machine learning methods in computational toxicology. Methods Mol. Biol. 2018, 1800, 119–139. 10.1007/978-1-4939-7899-1_5. [DOI] [PubMed] [Google Scholar]
- Ciallella H. L.; Zhu H. dvancing Computational Toxicology in the Big Data Era by Artificial Intelligence: Data-Driven and Mechanism-Driven Modeling for Chemical Toxicity. Chem. Res. Toxicol. 2019, 32, 536–547. 10.1021/acs.chemrestox.8b00393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Sun L.; Li W.; Liu G.; Tang Y. In Silico Prediction of Chemical Toxicity for Drug Design Using Machine Learning Methods and Structural Alerts. Frontiers in Chemistry 2018, 6, 30. 10.3389/fchem.2018.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S. Progress in computational toxicology. Journal of pharmacological and toxicological methods 2014, 69, 115–140. 10.1016/j.vascn.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Khan A. U.; et al. Descriptors and their selection methods in QSAR analysis: paradigm for drug design. Drug discovery today 2016, 21, 1291–1302. 10.1016/j.drudis.2016.06.013. [DOI] [PubMed] [Google Scholar]
- Durant J. L.; Leland B. A.; Henry D. R.; Nourse J. G. Reoptimization of MDL keys for use in drug discovery. Journal of chemical information and computer sciences 2002, 42, 1273–1280. 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- Klekota J.; Roth F. P. Chemical substructures that enrich for biological activity. Bioinformatics 2008, 24, 2518–2525. 10.1093/bioinformatics/btn479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Russo D. P.; Zorn K. M.; Clark A. M.; Zhu H.; Ekins S. Comparing multiple machine learning algorithms and metrics for estrogen receptor binding prediction. Mol. Pharmaceutics 2018, 15, 4361–4370. 10.1021/acs.molpharmaceut.8b00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Gandomi A. H.; Chen F.; Holzinger A. Evaluating the quality of machine learning explanations: A survey on methods and metrics. Electronics 2021, 10, 593. 10.3390/electronics10050593. [DOI] [Google Scholar]
- Wang Y.; Xiao J.; Suzek T. O.; Zhang J.; Wang J.; Zhou Z.; Han L.; Karapetyan K.; Dracheva S.; Shoemaker B. A.; et al. PubChem’s BioAssay database. Nucleic acids research 2012, 40, D400–D412. 10.1093/nar/gkr1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez D.; Gaulton A.; Bento A. P.; Chambers J.; De Veij M.; Félix E.; Magariños M. P.; Mosquera J. F.; Mutowo P.; Nowotka M.; et al. ChEMBL: towards direct deposition of bioassay data. Nucleic acids research 2019, 47, D930–D940. 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villoutreix B. O.; Taboureau O. Computational investigations of hERG channel blockers: New insights and current predictive models. Advanced drug delivery reviews 2015, 86, 72–82. 10.1016/j.addr.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Chemi G.; Gemma S.; Campiani G.; Brogi S.; Butini S.; Brindisi M. Computational tool for fast in silico evaluation of hERG K+ channel affinity. Frontiers in chemistry 2017, 5, 7. 10.3389/fchem.2017.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Sun H.; Liu H.; Li D.; Li Y.; Hou T. ADMET evaluation in drug discovery. 16. Predicting hERG blockers by combining multiple pharmacophores and machine learning approaches. Mol. Pharmaceutics 2016, 13, 2855–2866. 10.1021/acs.molpharmaceut.6b00471. [DOI] [PubMed] [Google Scholar]
- Cai C.; Guo P.; Zhou Y.; Zhou J.; Wang Q.; Zhang F.; Fang J.; Cheng F. Deep learning-based prediction of drug-induced cardiotoxicity. J. Chem. Inf. Model. 2019, 59, 1073–1084. 10.1021/acs.jcim.8b00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creanza T. M.; Delre P.; Ancona N.; Lentini G.; Saviano M.; Mangiatordi G. F. Structure-Based Prediction of hERG-Related Cardiotoxicity: A Benchmark Study. J. Chem. Inf. Model. 2021, 61, 4758–4770. 10.1021/acs.jcim.1c00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MDL Toxicity Database (presently Accelrys Toxicity Database); Accelrys Inc., http://www.akosgmbh.de/accelrys/databases/toxicity.htm (accessed Aug. 15, 2022).
- Cheng F.; Li W.; Zhou Y.; Shen J.; Wu Z.; Liu G.; Lee P. W.; Tang Y.. admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties, 2012. [DOI] [PubMed] [Google Scholar]
- Quantitative Structure Activity Relationship; U.S. Environmental Protection Agency, 2012; https://www.epa.gov/chemical-research/toxicity-estimation-software-tool-test (accessed Aug. 15, 2022).
- Xu Y.; Pei J.; Lai L. Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J. Chem. Inf. Model. 2017, 57, 2672–2685. 10.1021/acs.jcim.7b00244. [DOI] [PubMed] [Google Scholar]
- Schmidt U.; Struck S.; Gruening B.; Hossbach J.; Jaeger I. S.; Parol R.; Lindequist U.; Teuscher E.; Preissner R. SuperToxic: a comprehensive database of toxic compounds. Nucleic acids research 2009, 37, D295–D299. 10.1093/nar/gkn850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Vijay V.; Shi Q.; Liu Z.; Fang H.; Tong W. FDA-approved drug labeling for the study of drug-induced liver injury. Drug discovery today 2011, 16, 697–703. 10.1016/j.drudis.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Li X.; Chen Y.; Song X.; Zhang Y.; Li H.; Zhao Y. The development and application of in silico models for drug induced liver injury. RSC Adv. 2018, 8, 8101–8111. 10.1039/C7RA12957B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle J. H.Drug-Induced Liver Disease; Elsevier, 2013; pp 725–732. [Google Scholar]
- Mao Y. Hepatox: a professional web platform for the study of clinical and translational research on drug-induced liver injury in China. Chin. Hepatol. 2014, 19, 575–576. [Google Scholar]
- Krewski D.; Acosta D.; Andersen M.; Anderson H.; Bailar J. C.; Boekelheide K. Toxicity testing in the 21st century: A vision and a strategy. Journal of Toxicology and Environmental Health 2010, 13, 51–138. 10.1080/10937404.2010.483176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim A.; Mishra A.; Newton M. A. H.; Sattar A. Efficient Toxicity Prediction via Simple Features Using Shallow Neural Networks and Decision Trees. ACS Omega 2019, 4, 1874–1888. 10.1021/acsomega.8b03173. [DOI] [Google Scholar]
- Peng Y.; Zhang Z.; Jiang Q.; Guan J.; Zhou S. TOP: a deep mixture representation learning method for boosting molecular toxicity prediction. Methods 2020, 179, 55–64. 10.1016/j.ymeth.2020.05.013. [DOI] [PubMed] [Google Scholar]
- Xu C.; Cheng F.; Chen L.; Du Z.; Li W.; Liu G.; Lee P. W.; Tang Y. In silico prediction of chemical Ames mutagenicity. J. Chem. Inf. Model. 2012, 52, 2840–2847. 10.1021/ci300400a. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Kang Y.-L.; Zhu Y.-Y.; Zhao K.-X.; Liang J.-Y.; Ding L.; Zhang T.-G.; Zhang J. Novel naïve Bayes classification models for predicting the chemical Ames mutagenicity. Toxicology in Vitro 2017, 41, 56–63. 10.1016/j.tiv.2017.02.016. [DOI] [PubMed] [Google Scholar]
- Chu C. S.; Simpson J. D.; O’Neill P. M.; Berry N. G. Machine learning–Predicting Ames mutagenicity of small molecules. Journal of Molecular Graphics and Modelling 2021, 109, 108011. 10.1016/j.jmgm.2021.108011. [DOI] [PubMed] [Google Scholar]
- Gold L. S.; Manley N. B.; Slone T. H.; Rohrbach L.; Garfinkel G. B. Supplement to the Carcinogenic Potency Database (CPDB): results of animal bioassays published in the general literature through 1997 and by the National Toxicology Program in 1997–1998. Toxicol. Sci. 2005, 85, 747–808. 10.1093/toxsci/kfi161. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Ai H.; Chen W.; Yin Z.; Hu H.; Zhu J.; Zhao J.; Zhao Q.; Liu H. CarcinoPred-EL: novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci. Rep. 2017, 7, 1–14. 10.1038/s41598-017-02365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler P. TOXNET: the National Library of Medicine’s toxicology database. American Family Physician 1995, 52, 1677–1678. [PubMed] [Google Scholar]
- Drwal M. N.; Banerjee P.; Dunkel M.; Wettig M. R.; Preissner R. ProTox: a web server for the in silico prediction of rodent oral toxicity. Nucleic acids research 2014, 42, W53–W58. 10.1093/nar/gku401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P.; Eckert A. O.; Schrey A. K.; Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic acids research 2018, 46, W257–W263. 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E.; Pon A.; Djoumbou Y.; Knox C.; Shrivastava S.; Guo A. C.; Neveu V.; Wishart D. S. T3DB: a comprehensively annotated database of common toxins and their targets. Nucleic acids research 2010, 38, D781–D786. 10.1093/nar/gkp934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adadi A.; Berrada M. Peeking inside the black-box: A survey on explainable artificial intelligence. IEEE Access 2018, 6, 52138–52160. 10.1109/ACCESS.2018.2870052. [DOI] [Google Scholar]
- Brown A. Drugs, hERG and sudden death. Cell calcium 2004, 35, 543–547. 10.1016/j.ceca.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Fermini B.; Fossa A. A. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat. Rev. Drug Discovery 2003, 2, 439–447. 10.1038/nrd1108. [DOI] [PubMed] [Google Scholar]
- Rusinko A.; Farmen M. W.; Lambert C. G.; Brown P. L.; Young S. S. Analysis of a Large Structure/Biological Activity Data Set Using Recursive Partitioning. Journal of chemical information and computer sciences 1999, 39, 1017–1026. 10.1021/ci9903049. [DOI] [PubMed] [Google Scholar]
- Wang S.; Li Y.; Wang J.; Chen L.; Zhang L.; Yu H.; Hou T. ADMET evaluation in drug discovery. 12. Development of binary classification models for prediction of hERG potassium channel blockage. Mol. Pharmaceutics 2012, 9, 996–1010. 10.1021/mp300023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didziapetris R.; Lanevskij K. Compilation and physicochemical classification analysis of a diverse hERG inhibition database. Journal of computer-aided molecular design 2016, 30, 1175–1188. 10.1007/s10822-016-9986-0. [DOI] [PubMed] [Google Scholar]
- Vilar S.; Cozza G.; Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Current topics in medicinal chemistry 2008, 8, 1555–1572. 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- Jaeger S.; Fulle S.; Turk S. Mol2vec: unsupervised machine learning approach with chemical intuition. J. Chem. Inf. Model. 2018, 58, 27–35. 10.1021/acs.jcim.7b00616. [DOI] [PubMed] [Google Scholar]
- Wang W.; MacKinnon R. Cryo-EM structure of the open human ether-à-go-go-related K+ channel hERG. Cell 2017, 169, 422–430. 10.1016/j.cell.2017.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlen H.; Worth A. P.; Gabbert S. A tutorial for analysing the cost-effectiveness of alternative methods for assessing chemical toxicity: The case of acute oral toxicity prediction. Alternatives to laboratory animals 2014, 42, 115–127. 10.1177/026119291404200204. [DOI] [PubMed] [Google Scholar]
- Jiang D.; Wu Z.; Hsieh C.-Y.; Chen G.; Liao B.; Wang Z.; Shen C.; Cao D.; Wu J.; Hou T. Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. Journal of cheminformatics 2021, 13, 1–23. 10.1186/s13321-020-00479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Chen L.; Cheng F.; Wu Z.; Bian H.; Xu C.; Li W.; Liu G.; Shen X.; Tang Y. In silico prediction of chemical acute oral toxicity using multi-classification methods. J. Chem. Inf. Model. 2014, 54, 1061–1069. 10.1021/ci5000467. [DOI] [PubMed] [Google Scholar]
- Precautionary Statements. Label Review Manual; U.S. Environmental Protection Agency, 2016; https://www.epa.gov/sites/default/files/2015-03/documents/chap-07-jul-2014.pdf (accessed Aug. 15, 2022).
- Lei T.; Li Y.; Song Y.; Li D.; Sun H.; Hou T. ADMET evaluation in drug discovery: 15. Accurate prediction of rat oral acute toxicity using relevance vector machine and consensus modeling. Journal of Cheminformatics 2016, 8, 1–19. 10.1186/s13321-016-0117-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K.; Wei G.-W. Quantitative toxicity prediction using topology based multitask deep neural networks. J. Chem. Inf. Model. 2018, 58, 520–531. 10.1021/acs.jcim.7b00558. [DOI] [PubMed] [Google Scholar]
- EPA ECOTOX Knowledgebase, 2018; http://cfpub.epa.gov/ecotox/ (accessed Aug. 20, 2022).
- NIH ChemIDplus, 2018; https://chem.nlm.nih.gov/chemidplus/ (accessed Aug. 20, 2022).
- Xu Y.; Dai Z.; Chen F.; Gao S.; Pei J.; Lai L. Deep learning for drug-induced liver injury. J. Chem. Inf. Model. 2015, 55, 2085–2093. 10.1021/acs.jcim.5b00238. [DOI] [PubMed] [Google Scholar]
- Xing L.; Wu L.; Liu Y.; Ai N.; Lu X.; Fan X. LTMap: a web server for assessing the potential liver toxicity by genome-wide transcriptional expression data. Journal of Applied Toxicology 2014, 34, 805–809. 10.1002/jat.2923. [DOI] [PubMed] [Google Scholar]
- Ivanov S.; Semin M.; Lagunin A.; Filimonov D.; Poroikov V. Mol. Inf 2017, 36, 1600142. 10.1002/minf.201600142. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Pérez R.; Bajorath J. Prediction of Compound Profiling Matrices, Part II: Relative Performance of Multitask Deep Learning and Random Forest Classification on the Basis of Varying Amounts of Training Data. ACS Omega 2018, 3, 12033–12040. 10.1021/acsomega.8b01682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Vega de León A.; Chen B.; Gillet V. Effect of missing data on multitask prediction methods. J. Chem. Inf. Model. 2018, 10, 26. 10.1186/s13321-018-0281-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap C. W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. Journal of computational chemistry 2011, 32, 1466–1474. 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- Nguyen D. D.; Wei G. W. AGL-Score: Algebraic Graph Learning Score for Protein-Ligand Binding Scoring, Ranking, Docking, and Screening. J. Chem. Inf. Model. 2019, 59, 3291–3304. 10.1021/acs.jcim.9b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramer D.; Wei G. W. Multiscale weighted colored graphs for protein flexibility and rigidity analysis. J. Chem. Phys. 2018, 148, 054103. 10.1063/1.5016562. [DOI] [PubMed] [Google Scholar]
- Antelo-Collado A.; Carrasco-Velar R.; García-Pedrajas N.; Cerruela-García G. Effective feature selection method for class-imbalance datasets applied to chemical toxicity prediction. J. Chem. Inf. Model. 2021, 61, 76–94. 10.1021/acs.jcim.0c00908. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Norinder U.; Svensson F. Deep learning-based conformal prediction of toxicity. J. Chem. Inf. Model. 2021, 61, 2648–2657. 10.1021/acs.jcim.1c00208. [DOI] [PubMed] [Google Scholar]
- Di Filippo J. I.; Bollini M.; Cavasotto C. N. A Machine Learning Model to Predict Drug Transfer Across the Human Placenta Barrier. Frontiers in Chemistry 2021, 9, 566. 10.3389/fchem.2021.714678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custer L.; Sweder K. The role of genetic toxicology in drug discovery and optimization. Current drug metabolism 2008, 9, 978–985. 10.2174/138920008786485191. [DOI] [PubMed] [Google Scholar]
- McCann J.; Spingarn N. E.; Kobori J.; Ames B. N. Detection of carcinogens as mutagens: bacterial tester strains with R factor plasmids. Proc. Natl. Acad. Sci. U. S. A. 1975, 72, 979–983. 10.1073/pnas.72.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma M.; Kitazawa A.; Cayley A.; Williams R. V.; Barber C.; Hanser T.; Saiakhov R.; Chakravarti S.; Myatt G. J.; Cross K. P.; et al. Improvement of quantitative structure–activity relationship (QSAR) tools for predicting Ames mutagenicity: outcomes of the Ames/QSAR International Challenge Project. Mutagenesis 2019, 34, 3–16. 10.1093/mutage/gey031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benigni R.; Bossa C.; Richard A. M.; Yang C. A novel approach: chemical relational databases, and the role of the ISSCAN database on assessing chemical carcinogenicity. Annali dell’Istituto superiore di sanità 2008, 44, 48–56. [PubMed] [Google Scholar]
- Carvalho D. V.; Pereira E. M.; Cardoso J. S. Machine learning interpretability: A survey on methods and metrics. Electronics 2019, 8, 832. 10.3390/electronics8080832. [DOI] [Google Scholar]
- Stiglic G.; Kocbek P.; Fijacko N.; Zitnik M.; Verbert K.; Cilar L. Interpretability of machine learning-based prediction models in healthcare. Wiley Interdisciplinary Reviews: Data Mining and Knowledge Discovery 2020, 10, e1379 10.1002/widm.1379. [DOI] [Google Scholar]
- Zhang Q.-s.; Zhu S.-C. Visual interpretability for deep learning: a survey. Frontiers of Information Technology & Electronic Engineering 2018, 19, 27–39. 10.1631/FITEE.1700808. [DOI] [Google Scholar]
- Whitebread S.; Dumotier B.; Armstrong D.; Fekete A.; Chen S.; Hartmann A.; Muller P. Y.; Urban L. Secondary pharmacology: screening and interpretation of off-target activities–focus on translation. Drug Discovery Today 2016, 21, 1232–1242. 10.1016/j.drudis.2016.04.021. [DOI] [PubMed] [Google Scholar]
- Barabási A.-L.; Gulbahce N.; Loscalzo J. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran-Frigola M.; Mosca R.; Aloy P. Structural systems pharmacology: the role of 3D structures in next-generation drug development. Chemistry & biology 2013, 20, 674–684. 10.1016/j.chembiol.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Kunimoto R.; Bajorath J. Design of a tripartite network for the prediction of drug targets. Journal of Computer-Aided Molecular Design 2018, 32, 321–330. 10.1007/s10822-018-0098-x. [DOI] [PubMed] [Google Scholar]
- Nagarajan M.; Maadurshni G. B.; Manivannan J. Systems toxicology approach explores target-pathway relationship and adverse health impacts of ubiquitous environmental pollutant bisphenol A. Journal of Toxicology and Environmental Health, Part A 2022, 85, 217–229. 10.1080/15287394.2021.1994492. [DOI] [PubMed] [Google Scholar]
- Chua H. E.; Bhowmick S. S.; Tucker-Kellogg L. Synergistic target combination prediction from curated signaling networks: Machine learning meets systems biology and pharmacology. Methods 2017, 129, 60–80. 10.1016/j.ymeth.2017.05.015. [DOI] [PubMed] [Google Scholar]
- Wan F.; Hong L.; Xiao A.; Jiang T.; Zeng J. NeoDTI: neural integration of neighbor information from a heterogeneous network for discovering new drug–target interactions. Bioinformatics 2019, 35, 104–111. 10.1093/bioinformatics/bty543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- MDL Toxicity Database (presently Accelrys Toxicity Database); Accelrys Inc., http://www.akosgmbh.de/accelrys/databases/toxicity.htm (accessed Aug. 15, 2022).