

Abstract
Antidepressants have been shown to improve longevity in C. elegans. It is plausible that orthologs of genes involved in mood regulation and stress response are involved in such an effect. We sought to understand the underlying biology. First, we analyzed the transcriptome from worms treated with the antidepressant mianserin, previously identified in a large-scale unbiased drug screen as promoting increased lifespan in worms. We identified the most robust treatment-related changes in gene expression, and identified the corresponding human orthologs. Our analysis uncovered a series of genes and biological pathways that may be at the interface between antidepressant effects and longevity, notably pathways involved in drug metabolism/degradation (nicotine and melatonin). Second, we examined which of these genes overlap with genes which may be involved in depressive symptoms in an aging non-psychiatric human population (n = 3577), discovered using a genome-wide association study (GWAS) approach in a design with extremes of distribution of phenotype. Third, we used a convergent functional genomics (CFG) approach to prioritize these genes for relevance to mood disorders and stress. The top gene identified was ANK3. To validate our findings, we conducted genetic and gene-expression studies, in C. elegans and in humans. We studied C. elegans inactivating mutants for ANK3/unc-44, and show that they survive longer than wild-type, particularly in older worms, independently of mianserin treatment. We also show that some ANK3/unc-44 expression is necessary for the effects of mianserin on prolonging lifespan and survival in the face of oxidative stress, particularly in younger worms. Wild-type ANK3/unc-44 increases in expression with age in C. elegans, and is maintained at lower youthful levels by mianserin treatment. These lower levels may be optimal in terms of longevity, offering a favorable balance between sufficient oxidative stress resistance in younger worms and survival effects in older worms. Thus, ANK3/unc-44 may represent an example of antagonistic pleiotropy, in which low-expression level in young animals are beneficial, but the age-associated increase becomes detrimental. Inactivating mutations in ANK3/unc-44 reverse this effect and cause detrimental effects in young animals (sensitivity to oxidative stress) and beneficial effect in old animals (increased survival). In humans, we studied if the most significant single nucleotide polymorphism (SNP) for depressive symptoms in ANK3 from our GWAS has a relationship to lifespan, and show a trend towards longer lifespan in individuals with the risk allele for depressive symptoms in men (odds ratio (OR) 1.41, P = 0.031) but not in women (OR 1.08, P = 0.33). We also examined whether ANK3, by itself or in a panel with other top CFG-prioritized genes, acts as a blood gene-expression biomarker for biological age, in two independent cohorts, one of live psychiatric patients (n = 737), and one of suicide completers from the coroner’s office (n = 45). We show significantly lower levels of ANK3 expression in chronologically younger individuals than in middle age individuals, with a diminution of that effect in suicide completers, who presumably have been exposed to more severe and acute negative mood and stress. Of note, ANK3 was previously reported to be overexpressed in fibroblasts from patients with Hutchinson–Gilford progeria syndrome, a form of accelerated aging. Taken together, these studies uncover ANK3 and other genes in our dataset as biological links between mood, stress and longevity/aging, that may be biomarkers as well as targets for preventive or therapeutic interventions. Drug repurposing bioinformatics analyses identified the relatively innocuous omega-3 fatty acid DHA (docosahexaenoic acid), piracetam, quercetin, vitamin D and resveratrol as potential longevity promoting compounds, along with a series of existing drugs, such as estrogen-like compounds, antidiabetics and sirolimus/rapamycin. Intriguingly, some of our top candidate genes for mood and stress-modulated longevity were changed in expression in opposite direction in previous studies in the Alzheimer disease. Additionally, a whole series of others were changed in expression in opposite direction in our previous studies on suicide, suggesting the possibility of a “life switch” actively controlled by mood and stress.
INTRODUCTION
Age is an issue of mind over matter. If you don’t mind, it doesn’t matter.
—Mark Twain
The merits of longevity and the perils of aging are the subject of active debate at a societal level, and of concerted scientific research. Longevity and aging may be influenced by, and in turn influence, both positive and negative mood and response to stress, due to teleological evolutionary reasons or mundane lifestyle consequences. On the negative side, individuals with mood disorders1 and stress disorders2 have a significantly shorter life expectancy. Aging can lead to depression, attributable at least in part to physical health problems and related disability.3 This bidirectional relationship may have a genetic basis, and be susceptible to therapeutic interventions. We sought to understand the mediating genes by conducting a series of translational studies and analyses (Figure 1).
Figure 1.

Flowchart of studies. Integration of C. elegans and human data for discovery, prioritization and testing. CFG, convergent functional genomics.
The atypical antidepressant mianserin, which treats depression and stress disorders,4,5 was first identified as a longevity promoting compound in a large-scale screen for compounds that extend C. elegans lifespan.6 More recently, we have shown that it increases longevity in C. elegans by preventing transcriptional drift.7 We first analyzed the human orthologs of genes that were significantly and consistently changed in expression by mianserin treatment in C. elegans, and identified biological pathways involved in longevity. Second, we focused on the subset of the above genes that also had cross-validating genetic evidence in a genome-wide association study (GWAS) of depressive symptoms in aging (Figure 2). Third, we conducted a convergent functional genomics (CFG) analysis of this subset of genes for involvement in mood disorders and stress disorders, prioritizing our findings based on the whole body of work in the field to date (Figure 3). Fourth, in order to test whether ANK3, the top gene thus prioritized by CFG, is indeed involved in longevity/aging, we conducted studies with C. elegans ANK3/unc-44 inactivating mutants (Figure 4), as well as gene-expression analyses (Supplementary Figure 2). Fifth, we conducted human genetic analyses with ANK3 (Figure 5). Sixth, we conducted blood gene-expression analyses with ANK3 (Figure 6) and a panel of the top genes (Supplementary Figure 1), showing their potential as biomarkers for age. Lastly, we sought to derive translational medicine insights by understanding the underlying biology, implications for disease and identification/repurposing of drugs. A mechanistic model is proposed (Figure 7).
Figure 2.
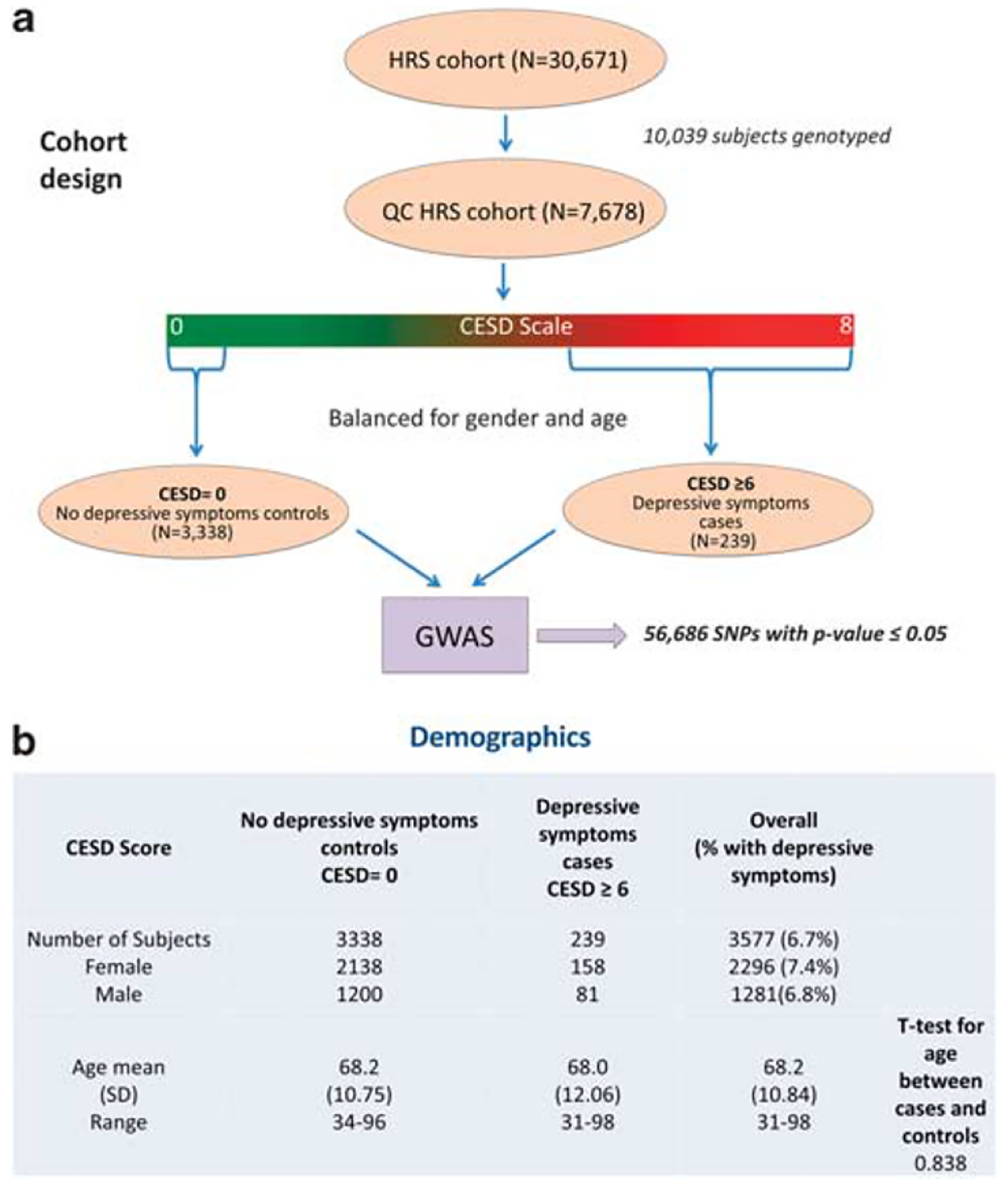
Human GWAS. Identifying SNPs associated with depressive symptoms in an aging cohort using an extreme trait design. (a) Design of the study. (b) Demographics. CESD scale, Center for Epidemiological Studies Depression Scale; GWAS, genome-wide association study; HRS, Health and Retirement Study; SNP, single nucleotide polymorphism.
Figure 3.
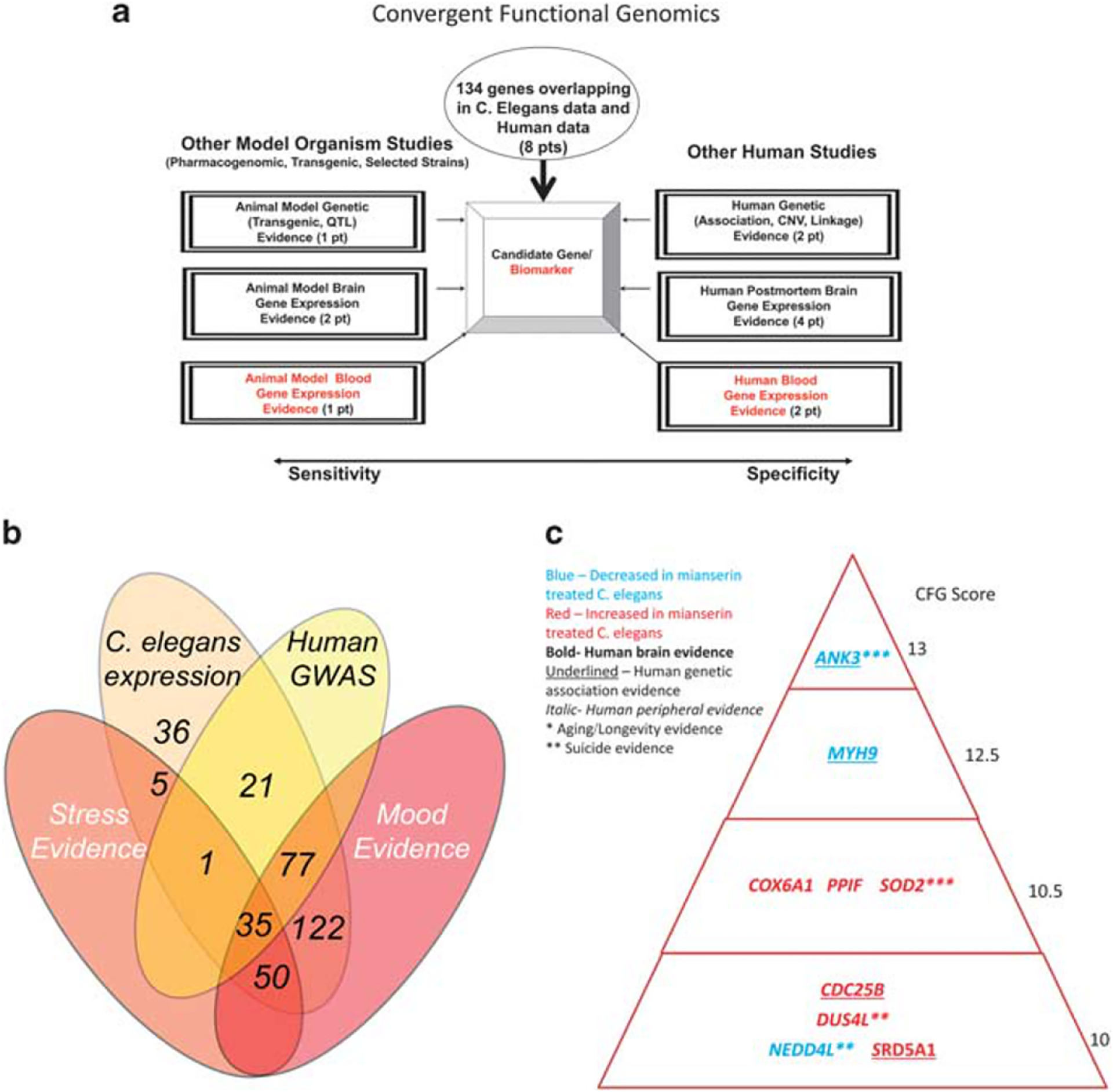
Convergent functional genomics. (a) Prioritizing the 134 genes that overlap between C. elegans discovery and human GWAS discovery, using convergent evidence for involvement in mood disorders and stress disorders. (b) Number of genes in the different evidentiary overlaps. (c) Top candidate genes for mood and stress-modulated longevity (CFG score ≥ 10). GWAS, genome-wide association study.
Figure 4.

The antidepressant mianserin requires ANK3/unc-44 to protect C. elegans from oxidative stress and to extend lifespan. (a) Mianserin-induced protection from oxidative stress requires ANK3/unc-44, the C. elegans homolog of mammalian ANK3. Wild-type (wt) N2 strain (dotted lines) or ANK3/unc-44 mutants (bold lines), at day 1 adult stage, were treated with water (black) or 50 μM mianserin (red), followed by increasing concentrations of paraquat 5 days later. Survival of animals was determined 24 h after paraquat addition and plotted in (%) (Y axis) as a function of paraquat concentration (mM) (X axis). Parallel wt (N2) control experiments (dotted lines) are shown for each graph. Mianserin failed to increase resistance to oxidative stress in three independent alleles (e362, e1197, e1260) of ANK3/unc-44. All error bars show s.e.m. for 3 to 4 independent experiments. (b) Lifespan curves of wt and unc-44(e362) animals treated water or 50 μM mianserin. Graph shows animals alive (%) (Y axis) as a function of time (days) (X axis). Dotted lines represent wt (N2) animals and bold lines represent unc-44(e362) mutants. Black: solvent control; red: mianserin 50 μM. In wt animals, mianserin increases lifespan by +40%, whereas it does not (−9%) in unc-44(e362) mutant animals. Asterisks indicate P values (**P < 0.01, ***P < 0.001). (c) Graph shows mean increase in lifespan (%) (Y axis) as a function of mianserin concentration (μM) (X axis). Solid red line represents unc-44(e362) animals. Dotted red line represents the parallel control experiment of mianserin-treated wt (N2) animals. Error bars show s.d. for experimental replicates. No lifespan extension is observed in ANK3/unc-44(e362) mutants at any mianserin concentration. (d) ANK3/unc-44 expression with age.
Figure 5.

ANK3 and survival. (a) Mapping of the nominally significant SNPs in ANK3 for association with depressive symptoms in our GWAS. Circled is a cluster of SNPs around the most significant SNP (rs10994379; OR: 1.457, P-value: 0.0053). (b) ANK3 association with survival, in the individuals who were alive at follow-up vs the individuals who died, using rs10994379 frequency (shown as a fraction of 1, whereas 1 is everybody in that group). GWAS, genome-wide association study; OR, odds ratio; SNP, single nucleotide polymorphism.
Figure 6.
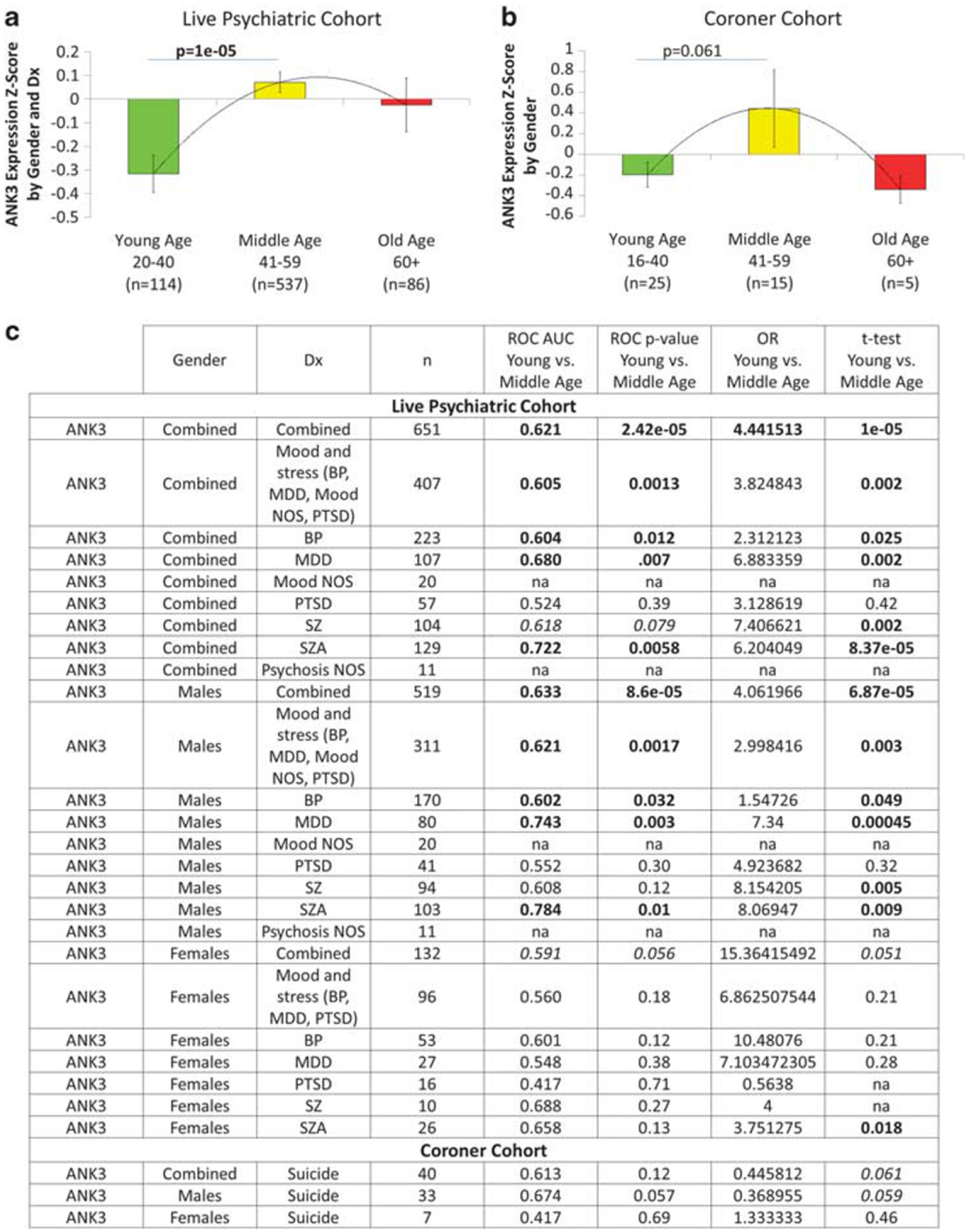
ANK3 as a gene expression biomarker for biological age. ANK3 gene expression in whole blood. (a) In live psychiatric patients. (b) In individuals who committed suicide. (c) Results by gender and diagnosis. ANK3 expression levels are Z-scored by gender and diagnosis for the live psychiatric participants cohort, and Z-scored by gender for the coroner suicide cohort. Bold values are nominally significant. Italic values trend to significance. AUC, areas under the curve, for younger age vs middle age; BP, bipolar; MDD, major depressive disorder; mood NOS, mood disorder not otherwise specified; psychosis NOS, psychotic disorder not otherwise specified; OR, odds ratio; PTSD, posttraumatic stress disorder; ROC, receiver operating characteristic.
Figure 7.
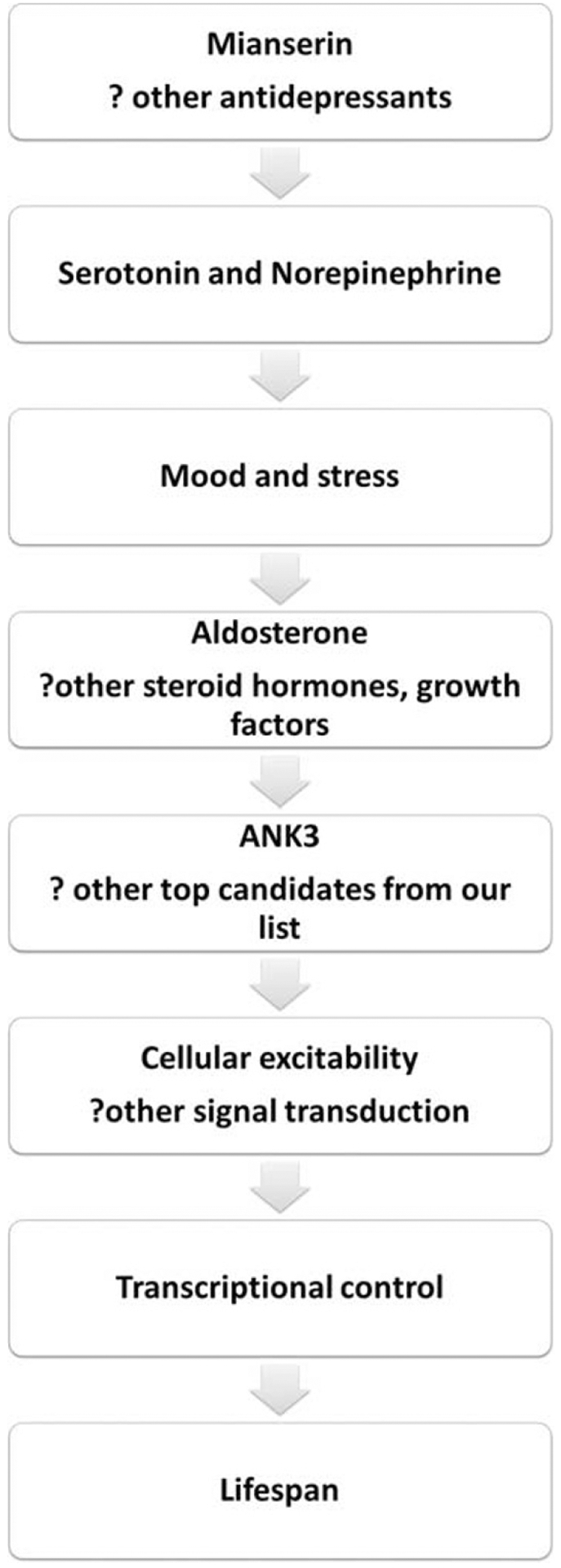
Proposed mechanistic cascade. ANK3 may be a nodal point bringing together key biological processes related to cellular excitability, activity and connectivity.
MATERIALS AND METHODS
C. elegans studies
Mianserin-induced gene-expression changes.
Mianserin-treated C. elegans whole-genome transcriptomic data were obtained as described by us.7 Overall 6701 genes were changed in expression with at least nominal significance (P < 0.05, false discovery rate < 10%) in mianserin vs water-treated worms. To ensure stringency in our analyses, we further applied to their P values a Bonferroni correction for number of genes in the genome (~21 035 genes), and carried forward in our analyses genes with a P value of < 2.4 × 10−6. A total of 1068 worm genes survived correction. Out of these, 971 were consistently upregulated or downregulated at the three time-points tested (days 3, 5 and 10).
Assignment of human orthologs.
To identify their corresponding orthologs we used OrthoList (www.greenwaldlab.org/ortholist/), a compendium of four orthology prediction programs, and manually searched for the human orthologs of the 971 worm genes. We only retained genes which had at least two out of the four prediction programs agreeing on assignments to human orthologs. We did that to ensure reliability of annotation, so we do not rely just on one software package/method. Out of 971 worm genes, 231 satisfied the above criteria. There were 347 human orthologs corresponding to these 231 worm genes. We used GeneCards (www.genecards.org) to confirm their gene symbol, name and chromosomal location.
Internal score for C. elegans data.
We assigned the internal score to the 971 Bonferroni-corrected C. elegans genes according to the distribution of P values. The top 0.1% genes received an internal score of 4, top 5% received a score of 2 and remaining C. elegans genes received a score of 1. The corresponding human orthologs received same scores assigned to their C. elegans counterparts.
Human studies
GWAS for depressive symptoms in an ageing population.
A new GWAS was conducted on data from 2006 Health and Retirement Study (HRS) wave 8, containing 30 671 subjects, which had previously been analyzed by us in a different fashion.8 Subjects were assessed for depressive symptoms using the Center for Epidemiologic Studies Depression Scale comprising eight items. The score range is from 0 to 8, a score of 0 suggesting no depressive symptoms and a score of 6 and above suggesting severe depressive symptoms. The study sample only included non-Hispanic Caucasians without a history of psychiatric or memory disorders. After balancing for age and gender, the study sample contained a total of 3577 subjects. We used an extreme trait design, wherein the control group consists of 3338 subjects without any depressive symptoms (1200 males, 2138 females) and cases consist of 239 subjects with severe depressive symptoms (81 males and 158 females). Genotype data was filtered using stringent quality control criteria. Association testing was performed using the Plink software package. Filtering resulted in a set of significant 56 499 single nucleotide polymorphisms (SNPs) (P value < 0.05).
Assignment of SNPs to genes.
Initially the genes corresponding to the SNPs with P < 0.05 were identified using the annotation file from the UCSC Genome Browser website. A total of 8823 unique genes were identified. Thereafter, genes were cross checked using GeneCards to ensure that each gene symbol was current.
Internal score for the GWAS data.
For the GWAS data the internal score was assigned based on the percentage of nominally significant SNPs (ratio of the number of significant SNPs over total number of SNPs tested per gene, multiplied by 100). We obtained a distribution of values. Genes in the top 0.1% of the distribution were assigned a score of 4, those in the top 5% were assigned a score of 2, whereas the remaining genes received a score of 1. We have successfully used a similar methodology in previous work.9
Convergent functional genomics
Databases:
We have established in our laboratory (Laboratory of Neurophenomics, Indiana University School of Medicine, www.neurophenomics.info) manually curated databases of all the human gene expression (postmortem brain, blood and cell cultures), human genetics (association, copy number variations and linkage), and animal model gene expression and genetic studies published until now on psychiatric disorders. Only the findings deemed significant in the primary publication, by the study authors, using their particular experimental design and thresholds, are included in our databases. Our databases include only primary literature data and do not include review papers or other secondary data integration analyses to avoid redundancy and circularity. These large and constantly updated databases have been used in our CFG prioritization (Figure 3). For this study, data from 1556 papers on mood and on stress were present in the databases at the time of the CFG analyses (February 2015) (human: genetic studies—761, brain studies—226, peripheral fluids—311; non-human: genetic studies—41, brain studies—195, peripheral fluids—22).
Human postmortem brain gene-expression evidence:
Converging evidence was scored for a gene if there were published reports of human postmortem data showing changes in expression of that gene or changes in protein levels in brains from subjects with mood or stress disorders.
Human blood and other peripheral tissue gene-expression data:
Converging evidence was scored for a gene if there were published reports of human blood, lymphoblastoid cell lines, cerebrospinal fluid or other peripheral tissue data showing changes in expression of that gene or changes in protein levels in participants with mood or stress disorders.
Human genetic evidence (association and linkage):
To designate convergence for a particular gene, the gene had to have independent published evidence of association or linkage for mood disorders or stress disorders. For linkage, the location of each gene was obtained through GeneCards (http://www.genecards.org), and the sex averaged centimorgan location of the start of the gene was then obtained through http://compgen.rutgers. edu/mapinterpolator. For linkage convergence, the start of the gene had to map within 5 centimorgan of the location of a marker linked to the disorder.
Animal model brain and blood gene-expression evidence:
For animal model brain and blood gene-expression evidence, we have used our own datasets,10–12 as well as published reports from the literature curated in our databases.
Animal model genetic evidence:
To search for mouse genetic evidence (transgenic and quantitative trait locus (QTL)) for our candidate genes, we utilized PubMed and the Jackson Laboratory database.
CFG scoring:
For CFG analysis (Figure 3), the external cross-validating lines of evidence were weighted such that the findings in human postmortem brain tissue, the target organ, were prioritized over peripheral tissue findings and genetic findings, by giving them twice as many points. Human brain expression evidence was given 4 points, whereas human peripheral evidence was given 2 points, and human genetic evidence was given a maximum of 2 points for association, and 1 point for linkage. The scoring for the corresponding non-human lines of evidence were half of those in human (genetic—1 point, brain—2 points, peripheral—1 point). Each line of evidence was capped in such a way that any positive findings within that line of evidence result in maximum points, regardless if it came from mood or stress (as the two may be interrelated in some studies), and regardless of how many different studies support that single line of evidence (to avoid potential popularity biases). In addition to our external CFG score, we also prioritized genes based upon the internal score from the discovery analyses used to identify them, in mianserin-treated C. elegans and the depressive symptoms GWAS study. Genes identified in the discovery could receive a maximum of 8 points (4 from C. elegans, 4 from GWAS). Thus, the maximum possible total CFG score for each gene was 20 points (8 points for the internal score, 12 points for external CFG score) (Table 1 and Supplementary Table 2). The scoring system was decided on before the analyses were carried out. We sought to give more weight to external score as to internal in order to increase generalizability and avoid fit-to-cohort of the prioritized genes.13 It has not escaped our attention that other ways of scoring the lines of evidence may give slightly different results in terms of prioritization, if not in terms of the list of genes per se. Nevertheless, we feel this simple scoring system provides a good separation of genes based on internal discovery evidence and on external independent cross-validating evidence in the field (Figure 3). In the future, with multiple large datasets, machine learning approaches could be used and validated to assign weights to CFG.
Table 1.
Top candidate genes for mood and stress-modulated longevity (n = 9, CFG score ≥ 10)
| Human gene symbol/gene name/(worm gene) | Direction of change in mianserin C. elegans | Discovery score | Human genetic evidence | Human brain expression evidence | Human peripheral expression evidence | Animal model genetic evidence | Animal model brain expression evidence | Animal model peripheral expression evidence | Prioritization total CFG score |
|---|---|---|---|---|---|---|---|---|---|
| ANK3 ankyrin 3, node of Ranvier (ankyrin G) (unc-44) | D | C. elegans 1 Human 1 | Bp40–44 ptsd45 | (I) BP Brain29 | (I) BP Blood30 Lymphoblasts31 |
ANK3 KO32 | (I) DBP Mice AMY11 (D) DBP Mice Omega-3 fatty acids AMY46 | 13 | |
| MYH9 myosin, heavy chain 9, non-muscle (nmy-1) | D | C. elegans 1 Human 1 | BP47 | (D) MDD Choroid plexus48 | (D) BP Blood49 | (I) DBP Mice AMY11 Stress AMY, HIP50 | (I) Stimulants Blood10 (D) DBP Mice Omega-3 fatty acids Blood46 | 12.5 | |
| PPIF peptidylprolyl isomerase F (cyn-7) | I | C. elegans 1 Human 1 | (D) BP Brain29,51 (D) PTSD PFC52 | (I) Chronic Stress Blood53 | (D) BP Brain51 | (D) DBP Mice Omega-3 fatty acids Blood46 | 10.5 | ||
| SOD2 superoxide dismutase 2, mitochondrial (sod-2) | I | C. elegans 1 Human 1 | Stimulants54 Linkage55 |
(D) MDD AMY and cingulate cortex56 PFC57 | (D) Antidepressants Blood58 | (D)Depression Cingulate cortex59 | (I) DBP Mice Omega-3 fatty acids Blood46 | 10.5 | |
| COX6A1 cytochrome c oxidase subunit Via polypeptide 1 (tag-174) | I | C. elegans 1 Human 1 | Linkage60 | (D) BP, MDD PFC, cingulate cortex61 | (I) Relaxation Response Blood62 | (I) Mood Stabilizers Cerebral Cortex63 | (I) DBP Mice Omega-3 fatty acids Blood46 | 10.5 | |
| SRD5A1 steroid-5-alpha-reductase, alpha polypeptide 1 (3-oxo-5 alpha-steroid delta 4-dehydrogenase alpha 1) (F19H6.4) | I | C. elegans 1 Human 1 | BP, Psychological stress64 | (D) MDD Brain29 | (D) PTSD AMY,HIP,PFC65 | 10 | |||
| NEDD4L neural precursor cell expressed, developmentally downregulated 4-like, E3 ubiquitin protein ligase (Y92H12A.2) | D | C. elegans 1 Human 1 | Linkage66 | (D) BP Brain29 PFC67 | (D) BP Blood68 | (D) DBP Mice Omega-3 fatty acids HIP (females)46 (I) DBP Mice Omega-3 fatty acids NAC (females)46 | 10 | ||
| CDC25B cell division cycle 25B (cdc-25.3) | I | C. elegans 1 Human 1 | BP47 | (D) BP Brain29 | (D) Psychological Distress Blood69 (I) Social Isolation Blood70 | 10 | |||
| DUS4L dihydrouridine synthase 4-like (S. cerevisiae) (C45G9.2) | I | C. elegans 1 Human 1 | (I) MDD PFC71 | (D) BP Blood68 | (D) Social Isolation HIP72 | 10 |
Abbreviations: AMY, amygdala; BP, bipolar; CP, caudate-putamen; (D), decreased in expression; HIP, hippocampus; (I), increased in expression; MDD, major depressive disorder; NAC, nucleus accumbens; PBMC, peripheral blood mononuclear cells; PFC, prefrontal cortex; PTSD, posttraumatic stress disorder.
Clock gene database
We compiled a database of genes associated with circadian function, by using a combination of review papers (Zhang et al.,14 McCarthy and Welsh15) and searches of existing databases CircaDB (http://circadb.hogeneschlab.org), GeneCards (http://www.genecards.org) and GenAtlas (http://genatlas.medecine.univ-paris5.fr). Using the data we compiled from these sources, we identified a total of 1468 genes that show circadian functioning. We further classified genes into ‘core’ clock genes, that is, those genes that are the main engine driving circadian function (n = 18), ‘immediate’ clock genes, that is, the genes that directly input or output to the core clock (n = 331), and ‘distant’ clock genes, that is, genes that directly input or output to the immediate clock genes (n = 1119).
Genetic testing of ANK3
We tested the most significant SNP associated with depressive scores in ANK3 (rs10994379; OR 1.457, P value 0.0053) for association with survival in our human GWAS cohorts (Figure 5). A one-tailed t-test with unequal variance was performed between subjects that were alive vs deceased at follow-up, which were carefully matched by gender and age at time of initial testing.
Human gene-expression studies
Cohorts.
We derived our data from two cohorts: a live psychiatric participants cohorts, and a postmortem coroner’s office cohort of individuals who died by suicide (Supplementary Table 1). The live psychiatric participants are part of a large longitudinal cohort that we are continuously collecting.16 Participants are recruited from the patient population at the Indianapolis VA Medical Center and Indiana University School of Medicine through referrals from care providers, the use of brochures left in plain sight in public places and mental health clinics, and through word of mouth. All participants understood and signed informed consent forms detailing the research goals, procedure, caveats and safeguards. Participants completed diagnostic assessments by an extensive structured clinical interview—Diagnostic Interview for Genetic Studies—at a baseline visit, followed by up to six testing visits, 3–6 months apart or whenever a new psychiatric hospitalization occurred. At each testing visit, they received a series of psychiatric rating scales, and the blood was drawn. Our postmortem cohort consisted of a demographically matched cohort of 45 violent suicide completers obtained through the Marion County coroner’s office (Supplementary Table 1B). We required a last observed alive postmortem interval of 24 h or less, and the cases selected had completed suicide by means other than overdose, which could affect gene expression. Next of kin signed informed consent at the coroner’s office for donation of blood for research. The samples were collected as part of our INBRAIN initiative (Indiana Center for Biomarker Research in Neuropsychiatry).
In both cohorts, whole blood (10 ml) was collected in two RNA-stabilizing PAXgene tubes, labeled with an anonymized ID number, and stored at −80 °C in a locked freezer until the time of future processing. Whole-blood (predominantly lymphocyte) RNA was extracted for microarray gene expression studies from the PAXgene tubes, as previously described.16
Gene expression analyses
We imported all Affymetrix microarray data as .cel files into Partek Genomic Suites 6.6 software package (Partek, St Louis, MI, USA). Using only the perfect match values, we ran a robust multi-array analysis, background corrected with quantile normalization and a median polish probeset summarization, to obtain the normalized expression levels of all probesets for each chip. Robust multi-array analysis was performed independently for each of the diagnoses used in the study, to avoid potential artefacts due to different ranges of gene expression in different diagnoses. Then the participants’ normalized data were extracted from these robust multi-array analyses and assembled for the different cohort analyses. Gene expression data was then z-scored by gender and diagnosis, to avoid potential artefacts due to different ranges of gene expression in different gender and diagnoses when combining cohorts, and to be able to combine different markers into a panel.
Statistical analyses
Receiver-operating characteristic analyses were calculated using the pROC function of the R studio, and double-checked using IBM SPSS Statistics 22. Diagnosis was converted to a binary call of 0 (middle and old age, above 40 years old) or 1 (young age, 20–40 years old) and entered as the state variable, with gene-expression levels entered as the test variable. In addition, Student’s t-test was performed between young age (20–40 years old) and middle age (40–60 years old). For the top nine candidate genes of the effects of mood and stress on longevity (CFG score of 10 and above), a Pearson R correlation (one-tail) was calculated between age and gene-expression levels. On the Affymetrix HG-U133 Plus 2.0 GeneChip (Affymetrix, Santa Clara, CA, USA), there were 25 probesets total in the nine genes. Four out of five probesets in ANK3, and 16 probesets in five genes (ANK3, PPIF, SOD2, NEDD4L, DUS4L) (Supplementary Table 6), were inversely correlated with age compared with their direction of change in the mianserin-treated worms data, that is, if a gene was increased in expression in mianserin-treated worms, it should correlate inversely with age; if a gene was decreased in expression in mianserin-treated worms, it should correlate directly with age. The single best correlated probeset in each gene were selected for future analyses, for ANK3 (Figure 6) and for the panel of five genes (BioM-5) (Supplementary Figure 1). The probesets were combined in the panel by computing the average of the z-scores of the increased markers minus the average of the z-scores of the decreased markers.
Pathway analyses
IPA (Ingenuity Systems, www.ingenuity.com, Redwood City, CA, USA), GeneGO MetaCore (Encinitas, CA, USA) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (through the Partek Genomics Suite 6.6 software package) were used to analyze the biological roles, including top canonical pathways, and diseases, of the candidate genes resulting from our work, as well as to identify genes in our dataset that are the target of existing drugs (Table 2; Supplementary Table 4). We ran the pathway analyses together for all the 347 human orthologs, then for those of them that had some evidence for the human GWAS data (n = 134), then for those that had a total CFG score of 5 and above (n = 67), and finally for those that had a CFG score of 10 and above (n = 9) (Table 2; Supplementary Table 5).
Table 2.
Biological pathways
| A. | Ingenuity pathways | KEGG pathways | GeneGO pathways | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| # | Top canonical pathways | P value | Ratio | Pathway name | Ratio | Enrichment P value | Process networks | Ratio | P value | |
| N = 347, C. elegans genes-corresponding unique human orthologs | 1 | Nicotine degradation III | 1.42E–17 | 28.8% 17/59 |
Retinol metabolism | 29.7956 | 1.15E–13 | Transcription_Chromatin modification | 5.686E–04 | 9/127 |
| 2 | Melatonin degradation I | 2.03E–16 | 25.0% 17/68 |
Drug metabolism—cytochrome P450 | 27.5974 | 1.03E–12 | Cardiac development_FGF_ErbB signaling | 2.108E–03 | 8/124 | |
| 3 | Nicotine degradation II | 7.46E–16 | 23.3% 17/73 |
Metabolism of xenobiotics by cytochrome P450 | 25.6469 | 7.27E–12 | Cell adhesion_Cell-matrix interactions | 5.836E–03 | 10/211 | |
| 4 | Superpathway of melatonin degradation | 6.04E–15 | 20.7% 17/82 |
Drug metabolism— other enzymes | 24.7258 | 1.83E–11 | Development_Skeletal muscle development | 1.767E–02 | 7/144 | |
| 5 | Estrogen biosynthesis | 9.71E–11 | 23.4% 11/47 |
Steroid hormone biosynthesis | 22.9864 | 1.04E–10 | Cardiac development_Wnt_beta-catenin, Notch, VEGF, IP3 and integrin signaling | 2.160E–02 | 7/150 | |
| N = 134, genes that have GWAS evidence | 1 | Nicotine degradation III | 7.44E–11 | 15.3% 9/59 |
Arachidonic acid metabolism | 12.2523 | 4.77E–06 | Development_Skeletal muscle development | 1.237E–03 | 6/144 |
| 2 | Melatonin degradation I | 2.79E–10 | 13.2% 9/68 |
Metabolism of xenobiotics by cytochrome P450 | 12.2158 | 4.95E–06 | DNA damage_MMR repair | 1.451E–03 | 4/59 | |
| 3 | Nicotine degradation II | 5.36E–10 | 12.3% 9/73 |
Drug metabolism—cytochrome P450 | 11.5538 | 9.60E–06 | Cardiac development_Wnt_beta-catenin, Notch, VEGF, IP3 and integrin signaling | 1.526E–03 | 6/150 | |
| 4 | Superpathway of melatonin degradation | 1.55E–09 | 11.0% 9/82 |
Retinol metabolism | 11.4604 | 1.05E–05 | Cardiac development_FGF_ErbB signaling | 3.705E–03 | 5/124 | |
| 5 | Bupropion degradation | 5.23E–07 | 18.5% 5/27 |
Starch and sucrose metabolism | 11.3695 | 1.15E–05 | Transcription_Chromatin modification | 4.104E–03 | 5/127 | |
| N = 67, genes CFG ≥ 5 | 1 | Mitochondrial dysfunction | 1.80E–05 | 3.2% 6/188 |
Linoleic acid metabolism | 10.7199 | 2.21E–05 | Signal transduction_Androgen receptor nuclear signaling | 4.711E–03 | 4/126 |
| 2 | Bupropion degradation | 6.57E–05 | 11.1% 3/27 |
Arachidonic acid metabolism | 7.58419 | 0.000508 | Transcription_Chromatin modification | 4.845E–03 | 4/127 | |
| 3 | Acetone degradation i (to methylglyoxal) | 1.21E–04 | 9.1% 3/33 |
Glycine, serine and threonine metabolism | 7.06642 | 0.000853 | Development_Skeletal muscle development | 7.537E–03 | 4/144 | |
| 4 | Cysteine biosynthesis/homocysteine degradation | 2.31E–04 | 25.0% 2/8 |
Viral myocarditis | 6.46753 | 0.001553 | Transport_Synaptic vesicle exocytosis | 1.468E–02 | 4/175 | |
| 5 | Estrogen biosynthesis | 3.49E–04 | 6.4% 3/47 |
Linoleic acid metabolism | 10.7199 | 2.21E–05 | Cytoskeleton_Actin filaments | 1.497E–02 | 4/176 | |
| N = 9, genes CFG ≥ 10 | 1 | Mitochondrial dysfunction | 1.97E–03 | 1.1% 2/188 |
Huntington’s disease | 7.03869 | 0.000877 | Cytoskeleton_Actin filaments | 1.107E–03 | 3/176 |
| 2 | Superoxide radicals degradation | 3.64E–03 | 10.0% 1/10 |
Parkinson’s disease | 4.76458 | 0.008526 | Signal transduction_Androgen receptor nuclear signaling | 1.025E–02 | 2/126 | |
| 3 | Androgen biosynthesis | 9.43E–03 | 3.8% 1/26 |
Aldosterone—regulated sodium reabsorption | 2.98928 | 0.050324 | Cardiac development_Role of NADPH oxidase and ROS | 1.154E–02 | 2/134 | |
| 4 | Cell cycle: G2/M DNA damage checkpoint regulation | 9.43E–03 | 2.0% 1/49 |
Androgen and estrogen metabolism | 2.98358 | 0.050611 | Cell cycle_Mitosis | 2.002E–02 | 2/179 | |
| 5 | Mitotic roles of polo-like kinase | 2.38E–02 | 1.5% 1/66 |
Steroid hormone biosynthesis | 2.53105 | 0.079575 | Cytoskeleton_Regulation of cytoskeleton rearrangement | 2.088E–02 | 2/183 | |
Abbreviations: CFG, convergent functional genomics; GWAS, genome-wide association study.
Connectivity map analyses
To elucidate which drugs may induce a gene-expression signature similar to the nine top candidate genes for longevity from our current work (Supplementary Table 7), we utilized the Connectivity Map v2 (also known as cmap), which comprises a collection of genome-wide transcriptional expression data from cultured human cells treated with bioactive small molecules and simple pattern-matching algorithms that together enable the discovery of functional connections between drugs, genes and diseases through the transitory feature of common gene-expression changes.17 The cmap (http://www.broad.mit.edu/cmap) contains more than 7 000 expression profiles representing 1 309 compounds. We used the Affymetrix website to obtain the probesets ID corresponding to these nine top candidate genes in the HGU133A array chip that cmap used. We then used the quick query selection and uploaded the increased in expression genes probeset id in the up tag file, and the decreased in expression genes probeset id in the down tag file.
RESULTS
We first analyzed the human orthologs of genes that were significantly and consistently changed in expression by mianserin treatment in C. elegans, and identified biological pathways involved in longevity (Table 2). Notably, nicotine degradation, melatonin degradation, retinol metabolism, drug metabolism by cytochrome P450, chromatin modification and FGF-Erb signaling, were the top biological pathways identified.
Second, we focused on the subset of genes that also had genetic evidence in a GWAS of depressive symptoms in aging (Figure 2). We conducted a CFG analysis of those genes for involvement in mood disorders and stress disorders, prioritizing our findings based on the whole body of work in the field to date (Figure 3). Out of the 347 human orthologs of genes from the worm analysis, 134 had some nominal significant evidence for association with depressive symptoms in an aging population. Nine genes had a score of CFG 10 and above (⩾50% of the maximum possible CFG score of 20) (Table 1), and 67 genes had a CFG score of 5 and above (⩾25% of the maximum possible CFG score of 20) (Table 2). The top scoring gene from the CFG analysis was ANK3 (Table 1).
Third, in order to validate whether ANK3 is involved in longevity and aging, we conducted studies in ANK3/unc-44 inactivating mutants in C. elegans, and demonstrate that they are longer lived than wt worms (Figure 4b). We also demonstrated using three different mutants that some ANK3/unc-44 expression was necessary for the effects of mianserin on prolonging lifespan and survival in the face of oxidative stress, particularly in younger worms (Figures 4a and c). Wt ANK3/unc-44 increases in expression with age in C. elegans, and is maintained at lower youthful levels by mianserin treatment (Figure 4d). These lower levels may be optimal in terms of longevity, offering a favorable balance between sufficient oxidative stress resistance in younger worms and survival effects in older worms.
We examined if genetic variants for depressive symptoms in ANK3 are associated with longevity. Specifically, we focused on the strongest SNP from our GWAS analysis, rs10994379 (OR 1.457, P value 0.0053). Men with the risk allele for depressive symptoms had higher likelihood of survival (OR 1.41, P value 0.031), but not women (OR 1.08, P value 0.33). It is possible that SNPs that are associated with depressive symptoms may actually have a protective role in the non-depressed elderly in terms of lifespan, perhaps by reducing the level of activities that may be taxing in older people, or having some other non-mood related protective role. Consistent with that, individuals with bipolar disorder are known to have a shorter lifespans than individuals with depression.1
Fourth, we examined whether ANK3, by itself or in a panel with four other top CFG prioritized genes, acts as a blood gene-expression biomarker for age, in two independent cohorts, one of live psychiatric patients (n = 737), and one of suicide completers from the coroner’s office (n = 45). We show significantly lower levels of ANK3 expression in younger individuals, with another decrease in levels occurring in older age. ANK3 also increases in expression with age in worms (Figure 4d). Importantly, ANK3 was previously reported to be overexpressed in fibroblasts from patients with Hutchinson–Gilford progeria syndrome, a form of accelerated aging. We also show similar results for a panel of five top genes (Supplementary Figure 1; Supplementary Table 6).
Lastly, we sought to derive translational medicine insights. Previous work by our group had shown that ANK3 was increased in expression in the amygdala of a mouse model of mood disorders and stress,11 and that ANK3 was decreased in expression in that model by treatment with the omega-3 fatty acid DHA (docosahexaenoic acid), similar to the effects of mianserin in worms. A number of other top scoring genes (MYH9, SOD2, COX6A1, NEDD4L, SYT1, TROVE2, H3F3A, PEBP1, PLA2G6, SCD5) had evidence of modulation by DHA in the same direction with mianserin (Supplementary Table 4), suggesting that omega-3 fatty acids may have longevity promoting effects as well. In fact, one of the top biological pathways is linoleic acid metabolism, related to omega-3 fatty acids (Table 2).
A number of top biomarkers identified by us have biological roles that are related to the circadian clock. To be able to ascertain all the genes in our dataset that were circadian and do estimates for enrichment, we compiled from the literature a database of all the known genes that fall into these three categories: core clock, immediate input or output and distant input or output, numbering a total of 1468 genes. Using an estimate of about 21 000 genes in the human genome, that gives about 7% of genes having some circadian pattern. Out of our 67 top longevity biomarker genes, 11 had circadian evidence (16.4%) (Supplementary Table 2), suggesting an over twofold enrichment for circadian genes. Circadian clock abnormalities are related to mood disorders11,15 and neurodegenerative disorders;18 sleep abnormalities have been implicated in aging.19
We have identified a series of biomarkers that seem to be changed in expression opposite direction in longevity vs in Alzheimer disease (Supplementary Table 3). These biomarkers could potentially suggest targets for early intervention and preventive approaches. COX6A1 (cytochrome c oxidase subunit VIa polypeptide 1, the terminal enzyme of the mitochondrial respiratory chain), and CYB5R3 (cytochrome b5 reductase 3, which functions in desaturation and elongation of fatty acids, in cholesterol biosynthesis, and in drug metabolism), are increased in longevity, and decreased in blood of Alzheimer disease individuals.20 KAT2B (K(lysine) acetyltransferase 2B, which has histone acetyl transferase activity with core histones and nucleosome core particles, indicating that this protein plays a direct role in transcriptional regulation) is increased in longevity and decreased in the hippocampus of Alzheimer diseases individuals.21
One of the other biomarkers increased in longevity is SRD5A1 (steroid-5-alpha-reductase alpha polypeptide 1). Inhibitors of this enzyme, such as those used in prostate disorders, lead to androgenic blockade, which has been associated with a higher rate of Alzheimer disease.22 Interestingly, one of the top biological pathways identified by us is androgen receptor signaling (Table 2).
We have also identified a series of biomarkers that seem to be changed in opposite direction in longevity vs suicide: ANK3, SOD2, DUS4L, NEDD4L, MYH11, NAV3, YIPF5 and 15 other genes (Supplementary Table 3). In particular, EPHX1, increased in longevity, is decreased in both brain23 and blood24 in suicidality. The other genes that have blood evidence in suicide in opposite direction to longevity are: ANK3, DUS4L, NEDD4L, MYH11, NAV3, YIPF5, TROVE2, CLASP2, MSH2, POLH, FAM184A, CBS, CYB5R3, CYP2C9, and NLGN2. Such genes may be blood biomarkers for a biological switch implicated in survival, that is, a ‘life switch’.
Pharmacogenomics and therapeutics
We have identified a series of biomarkers that seem to be changed in the same direction in longevity vs in treatments with mood stabilizing agents, such as lithium, valproate and omega-3 fatty acids (Supplementary Table 4). These biomarkers could potentially be used to stratify patients to different treatment approaches, and monitor their response. COX6A1, SYT1, TROVE2 and NLGN2 are changed in expression by two of these three treatments, suggesting they may be core to the mood and longevity mechanisms of these drugs. MYH9, SOD2, COX6A1, TROVE2, H3F3A, PLA2G6 and PEBP1 may be useful blood pharmacogenomic markers of response to omega-3 fatty acids. Two existing drugs used for other indications have been identified as targeting top longevity biomarkers identified by us (Supplementary Table 4), and them or their derivatives could potentially be repurposed for testing for prolonging life: quinacrine (inhibiting PLA2G6), and sulfinpyrazone (inhibiting ABCC1).
In addition, Connectivity Map analyses17 identified compounds that induce gene-expression signatures that are similar to those of the top nine mood and stress-modulated longevity genes (Supplementary Table 7), and might generate leads and/or be tested for use in prolonging lifespan: antidiabetic medications (troglitazone, gliquidone, pioglitazone, rosiglitazone), immuno-suppressant/anti-transplant rejection medications with known longevity effects across species (sirolimus/rapamycin, mycophenolic acid), nootropic (piracetam) and non-drug flavonoid antioxidant/vitamin compounds (quercetin, resveratrol, ergocalci-ferol/vitamin D). Known mood modulating drugs identified by the Connectivity Map analyses are: antidepressants (minaprine, amox-apine), antihistamines (homochlorcyclizine), calcium-channel blockers (nifedipine) and female sex hormone-like compounds (diethylstilbestrol, estradiol). Of note, females tend to live longer than males in humans, and estradiol has direct prior experimental evidence of extending lifespan in worms.25
We have also looked using Connectivity Map at compounds that have similar gene-expression signatures to the 22 genes that are changed in opposite direction in longevity and suicide, and identified additional flavonoid antioxidants (apigenin, luteolin, acacetin) and vitamins (vitamin K, folic acid), along with resveratrol, estradiol, antidiabetics and antineoplastics. Moreover, some of the genes in this ‘life switch’ are modulated by omega-3 fatty acids, lithium and valproate (Supplementary Table 8).
DISCUSSION
ANK3, the top gene at the overlap of longevity, mood and stress identified by our convergent approach, is a scaffolding protein, involved in assembly and trafficking of receptors and channels, notably at the nodes of Ranvier and in the synaptic compartment,26,27 with a strong genetic association with several psychiatric disorders. It has been implicated in anhedonia, stress, novelty seeking and cognition in humans. ANK3 is a top genome-wide significant hit from GWAS studies of bipolar disorder.28 Other work in the field has identified ANK3 as increased in expression in individuals with bipolar disorder, in brain29 and blood.30,31 Mice with deletions of ANK3 display a hyperactive phenotype, that is responsive to treatment with the mood stabilizer lithium, and switches to a more inactive phenotype in response to chronic stress, as described in elegant studies by Petryshen and colleagues.32 At a cellular level, over-expression of ANK3 increases lysosomal-mediated down-regulation of cell-surface receptors.33 ANK3 has been shown to regulate the Wnt pathway signaling and cell proliferation.34 ANK3 has also been shown to be regulated by aldosterone, and in turn to regulate the activity of sodium channels that promote sodium retention.35 Sodium homeostasis may be a very primitive cellular mechanism that has evolutionarily been co-opted for increasingly complex organismal functions and responses, including neuronal excitability.
Our data shows that ANK3 seems to be expressed at lower levels in younger individuals (worms and humans) (Figures 4d, 6a and b), and that mianserin may have longevity effects primarily by maintaining ANK3 at lower/youthful levels (Figure 4), which in turn leads to a tighter transcriptional control (Figure 7) and prevents transcriptional drift.7 The observation that inactivating mutations in ANK3/unc-44 cause hypersensitivity to oxidative stress in young animals but extend lifespan later in life suggest ANK3/unc-44 may be an example of antagonistic pleiotropy.36 ANK3/unc-44 provides a fitness advantage in the young organism, but its increased expression becomes detrimental in older animals. Interestingly, the curve for ANK3 expression with age might not be linear but rather bell-shaped, with lower levels of ANK3 again occurring in older age (Figure 6). Whether that represents a survival effect or an age-related effect is unclear at this point. The effects of ANK3 in older age in general, and in Alzheimer disease in particular, are an area of future exploration by our group.
The ability of ANK3 (Figure 6), and of our panel of top biomarkers (BioM-5) (Supplementary Figure 1) to distinguish between younger age and middle age, while significant overall, seems to be even better in certain gender and diagnostic subgroups (males and females with schizoaffective disorder and schizophrenia, males with depression), suggesting avenues for personalized medicine, potential medication effects, as well as potential biological severity and burden of disease. Our human blood gene-expression data are in a psychiatric cohort and a coroner suicide cohort, populations where ANK3 and other biomarkers might be more strongly modulated by mood and stress effects. Additional normative studies in non-psychiatric populations are needed for the magnitude of the effects with age of ANK3, and BioM-5. They may uncover utility for our biomarkers to contribute to a biological age score, as opposed to a chronological age. For example, they may be shifted in a negative direction in individuals exposed to mood and stress disorders. Consistent with that, our coroner cohort, composed of individuals who committed suicide and presumably were exposed to more severe negative mood and stress, appears shifted compared to our live psychiatric cohort (lower OR between young and middle age). It would also be interesting as part of future work to correlate the levels of our candidate biomarkers with telomere length, a known stress-responsive marker of aging.37
The probeset used for ANK3 maps to protein coding transcripts of ANK3 (Supplementary Figure 2), which is a very complex gene. As a caveat, our gene-expression data was determined using probesets for the genes that showed best concordance with the worm directionality in our psychiatric patients test cohort. However, these probesets were enriched in the complete list of probsets from the Affymetrix chip (Supplementary Table 6). Moreover, the results were very similar in the independent coroner cohort.
Independent of this study, we recently observed somewhat similar effects to ANK3/unc-44 for mutants of SYT1/snt-1,7 one of our top genes from this study (Supplementary Table 2). It would be interesting in the future to test other mutants of top genes identified by us, and see if they converge on synaptic pathology.
Other biological insights (Alzheimer disease implications, circadian clocks, longevity/suicide ‘life switch’), as well as pharmacogenomics and therapeutics leads, notably antidiabetic/insulin modulating drugs and flavonoid antioxidant/vitamin compounds, are listed in the Results section. Of note, as reassuring positive controls, the gene-expression signature of our top genes for mood and stress modulated longevity has similarity with the gene-expression signature of resveratrol, a known longevity promoting compound, and of sirolimus/rapamycin, an anti-transplant rejection medication with known longevity effects across species (Supplementary Table 7).38 Conversely, the mechanistic target of rapamycin pathway it targets was a top pathway identified in previous work by us on suicide biomarkers, pointing to its role as a potential life/death switch.16
Mitochondrial dysfunction was the top biological pathway where our top candidate genes for mood and stress-modulated longevity mapped (Table 2). Over the last decade, accumulating evidence has suggested a causative link between mitochondrial dysfunction and major phenotypes associated with aging. Besides free radicals, other possible mechanisms, including insulin signaling and mechanistic target of rapamycin (mTOR) pathways connect mitochondria to aging.39
The fact that ANK3 and other genes are changed in expression in opposite directions in longevity vs suicide datasets is of speculative evolutionary interest in terms of a ‘life switch’. One essential program that every organism might have is an inherent will to survive, a program that must be biologically encoded. That program may be inactivated/switched off, that is, a breakdown of the drive to survive may occur in suicide. Suicide is, de facto, a form of reduced longevity. Such genes may be tested in the future for use in a polygenic gene-expression ‘viability score’, to quantify the likelihood of longevity vs suicide in psychiatric patients (and non-psychiatric individuals), as well as to monitor response to treatments in individuals at risk.
Overall, we propose a model, whereas positive or negative mood and stress responses, reflecting the organism’s state of health and perception of the environment as favorable or hostile, and their influence on behaviors, may be involved in the active modulation of lifespan, with ANK3 and other genes as mediators. In particular, ANK3 may be a nodal point in the regulation of overall cellular excitability, signal transduction and transcriptional control (Figure 7).
Supplementary Material
ACKNOWLEDGMENTS
This work is, in essence, a field-wide collaboration. We acknowledge our debt of gratitude for the efforts and results of the many other groups, cited in our paper, who have conducted and published studies (clinical, genetic and biological) in mood disorders, stress, longevity and aging. With their arduous and careful work, a convergent approach such as ours is possible. We thank Farnoosh Khan, Naga Vanipenta and Eddie Stage for help with building literature databases. We also would particularly like to thank the subjects who participated in these studies, their families and their caregivers. Without their contribution, such work to advance the understanding of aging would not be possible. This work was supported by two NIH Directors’ New Innovator Awards (1DP2OD007363 to ABN and 1DP2OD008398 to MP), as well as NIH U19 A1063603 to DRS, NIH R00 LM011384 to KN and IADC P30 AG010133 to AJS.
Footnotes
Supplementary information is available at the journal website and from the Niculescu Laboratory website (www.neurophenomics.info).
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp).
CONFLICT OF INTEREST
The authors declare no conflict of interest. ABN, MP, DRS and AJS are listed as inventors on a patent application being filed by Indiana University and Scripps.
REFERENCES
- 1.Chesney E, Goodwin GM, Fazel S. Risks of all-cause and suicide mortality in mental disorders: a meta-review. World Psychiatry 2014; 13: 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lohr JB, Palmer BW, Eidt CA, Aailaboyina S, Mausbach BT, Wolkowitz OM et al. Is post-traumatic stress disorder associated with premature senescence? A review of the literature. Am J Geriatr Psychiatry 2015; 23: 709–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts RE, Kaplan GA, Shema SJ, Strawbridge WJ. Does growing old increase the risk for depression? Am J Psychiatry 1997; 154: 1384–1390. [DOI] [PubMed] [Google Scholar]
- 4.Coppen A, Kopera H. Workshop on the clinical pharmacology and efficacy of mianserin. Br J Clin Pharmacol 1978; 5: 91 S–99 S. [PMC free article] [PubMed] [Google Scholar]
- 5.Moreau JL, Bourson A, Jenck F, Martin JR, Mortas P. Curative effects of the atypical antidepressant mianserin in the chronic mild stress-induced anhedonia model of depression. J Psychiatry Neurosci 1994; 19: 51–56. [PMC free article] [PubMed] [Google Scholar]
- 6.Petrascheck M, Ye X, Buck LB. An antidepressant that extends lifespan in adult Caenorhabditis elegans. Nature 2007; 450: 553–556. [DOI] [PubMed] [Google Scholar]
- 7.Rangaraju S, Solis GM, Thompson RC, Gomez-Amaro RL, Kurian L, Encalada SE et al. Suppression of transcriptional drift extends C. elegans lifespan by post-poning the onset of mortality. eLife 2015; 4: e08833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nho K, Ramanan VK, Horgusluoglu E, Kim S, Inlow MH, Risacher SL et al. Comprehensive gene- and pathway-based analysis of depressive symptoms in older adults. J Alzheimers Dis 2015; 45: 1197–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levey DF, Le-Niculescu H, Frank J, Ayalew M, Jain N, Kirlin B et al. Genetic risk prediction and neurobiological understanding of alcoholism. Transl Psychiatry 2014; 4: e391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogden CA, Rich ME, Schork NJ, Paulus MP, Geyer MA, Lohr JB et al. Candidate genes, pathways and mechanisms for bipolar (manic-depressive) and related disorders: an expanded convergent functional genomics approach. Mol Psychiatry 2004; 9: 1007–1029. [DOI] [PubMed] [Google Scholar]
- 11.Le-Niculescu H, McFarland MJ, Ogden CA, Balaraman Y, Patel S, Tan J et al. Phenomic, convergent functional genomic, and biomarker studies in a stress-reactive genetic animal model of bipolar disorder and co-morbid alcoholism. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 134–166. [DOI] [PubMed] [Google Scholar]
- 12.Le-Niculescu H, Case NJ, Hulvershorn L, Patel SD, Bowker D, Gupta J et al. Convergent functional genomic studies of omega-3 fatty acids in stress reactivity, bipolar disorder and alcoholism. Transl Pychiatry 2011; 1: e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niculescu AB, Le-Niculescu H. Convergent functional genomics: what we have learned and can learn about genes, pathways, and mechanisms. Neuropsychopharmacology 2010; 35: 355–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang EE, Liu AC, Hirota T, Miraglia LJ, Welch G, Pongsawakul PY et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell 2009; 139: 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarthy MJ, Welsh DK. Cellular circadian clocks in mood disorders. J Biol Rhythms 2012; 27: 339–352. [DOI] [PubMed] [Google Scholar]
- 16.Niculescu AB, Levey DF, Phalen PL, Le-Niculescu H, Dainton HD, Jain N et al. Understanding and predicting suicidality using a combined genomic and clinical risk assessment approach. Mol Psychiatry 2015; 20: 1266–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006; 313: 1929–1935. [DOI] [PubMed] [Google Scholar]
- 18.Musiek ES. Circadian clock disruption in neurodegenerative diseases: cause and effect? Front Pharmacol 2015; 6: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolkove N, Elkholy O, Baltzan M, Palayew M. Sleep and aging: 1. Sleep disorders commonly found in older people. CMAJ 2007; 176: 1299–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maes OC, Xu S, Yu B, Chertkow HM, Wang E, Schipper HM. Transcriptional profiling of Alzheimer blood mononuclear cells by microarray. Neurobiol Aging 2007; 28: 1795–1809. [DOI] [PubMed] [Google Scholar]
- 21.Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res 2002; 70: 462–473. [DOI] [PubMed] [Google Scholar]
- 22.Nead KT, Gaskin G, Chester C, Swisher-McClure S, Dudley JT, Leeper NJ et al. Androgen deprivation therapy and future alzheimer’s disease risk. J Clin Oncol 2016; 34: 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim S, Choi KH, Baykiz AF, Gershenfeld HK. Suicide candidate genes associated with bipolar disorder and schizophrenia: an exploratory gene expression profiling analysis of post-mortem prefrontal cortex. BMC Genomics 2007; 8: 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le-Niculescu H, Levey DF, Ayalew M, Palmer L, Gavrin LM, Jain N et al. Discovery and validation of blood biomarkers for suicidality. Mol Psychiatry 2013; 18: 1249–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carretero M, Gomez-Amaro RL, Petrascheck M. Pharmacological classes that extend lifespan of Caenorhabditis elegans. Front Genet 2015; 6: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith KR, Kopeikina KJ, Fawcett-Patel JM, Leaderbrand K, Gao R, Schurmann B et al. Psychiatric risk factor ANK3/ankyrin-G nanodomains regulate the structure and function of glutamatergic synapses. Neuron 2014; 84: 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tseng WC, Jenkins PM, Tanaka M, Mooney R, Bennett V. Giant ankyrin-G stabilizes somatodendritic GABAergic synapses through opposing endocytosis of GABAA receptors. Proc Natl Acad Sci USA 2015; 112: 1214–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rueckert EH, Barker D, Ruderfer D, Bergen SE, O’Dushlaine C, Luce CJ et al. Cis-acting regulation of brain-specific ANK3 gene expression by a genetic variant associated with bipolar disorder. Mol Psychiatry 2013; 18: 922–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen H, Wang N, Zhao X, Ross CA, O’Shea KS, McInnis MG. Gene expression alterations in bipolar disorder postmortem brains. Bipolar Disord 2013; 15: 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wirgenes KV, Tesli M, Inderhaug E, Athanasiu L, Agartz I, Melle I et al. ANK3 gene expression in bipolar disorder and schizophrenia. Br J Psychiatry 2014; 205: 244–245. [DOI] [PubMed] [Google Scholar]
- 31.Kato T, Hayashi-Takagi A, Toyota T, Yoshikawa T, Iwamoto K. Gene expression analysis in lymphoblastoid cells as a potential biomarker of bipolar disorder. J Hum Genet 2011; 56: 779–783. [DOI] [PubMed] [Google Scholar]
- 32.Leussis MP, Berry-Scott EM, Saito M, Jhuang H, de Haan G, Alkan O et al. The ANK3 bipolar disorder gene regulates psychiatric-related behaviors that are modulated by lithium and stress. Biol Psychiatry 2013; 73: 683–690. [DOI] [PubMed] [Google Scholar]
- 33.Ignatiuk A, Quickfall JP, Hawrysh AD, Chamberlain MD, Anderson DH. The smaller isoforms of ankyrin 3 bind to the p85 subunit of phosphatidylinositol 3’-kinase and enhance platelet-derived growth factor receptor down-regulation. J Biol Chem 2006; 281: 5956–5964. [DOI] [PubMed] [Google Scholar]
- 34.Durak O, de Anda FC, Singh KK, Leussis MP, Petryshen TL, Sklar P et al. Ankyrin-G regulates neurogenesis and Wnt signaling by altering the subcellular localization of beta-catenin. Mol Psychiatry 2015; 20: 388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edinger RS, Coronnello C, Bodnar AJ, LaFramboise WA, Benos PV, Ho J et al. Aldosterone regulates microRNAs in the cortical collecting duct to alter sodium transport. J Am Soc Nephrol 2014; 25: 2445–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kapahi P Protein synthesis and the antagonistic pleiotropy hypothesis of aging. Adv Exp Med Biol 2010; 694: 30–37. [DOI] [PubMed] [Google Scholar]
- 37.Blackburn EH, Epel ES, Lin J. Human telomere biology: a contributory and inter-active factor in aging, disease risks, and protection. Science 2015; 350: 1193–1198. [DOI] [PubMed] [Google Scholar]
- 38.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature 2013; 493: 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Investig 2013; 123: 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen DT, Jiang X, Akula N, Shugart YY, Wendland JR, Steele CJ et al. Genome-wide association study meta-analysis of European and Asian-ancestry samples identifies three novel loci associated with bipolar disorder. Mol Psychiatry 2013; 18: 195–205. [DOI] [PubMed] [Google Scholar]
- 41.Takata A, Kim SH, Ozaki N, Iwata N, Kunugi H, Inada T et al. Association of ANK3 with bipolar disorder confirmed in East Asia. Am J Med Genet B 2011; 156B: 312–315. [DOI] [PubMed] [Google Scholar]
- 42.Psychiatric GCBDWG. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 2011; 43: 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim CH, Zain SM, Reynolds GP, Zain MA, Roffeei SN, Zainal NZ et al. Genetic association of LMAN2L gene in schizophrenia and bipolar disorder and its interaction with ANK3 gene polymorphism. Prog Neuropsychopharmacol Biol Psychiatry 2014; 54: 157–162. [DOI] [PubMed] [Google Scholar]
- 44.Lee MT, Chen CH, Lee CS, Chen CC, Chong MY, Ouyang WC et al. Genome-wide association study of bipolar I disorder in the Han Chinese population. Mol Psychiatry 2011; 16: 548–556. [DOI] [PubMed] [Google Scholar]
- 45.Logue MW, Solovieff N, Leussis MP, Wolf EJ, Melista E, Baldwin C et al. The ankyrin-3 gene is associated with posttraumatic stress disorder and externalizing comorbidity. Psychoneuroendocrinology 2013; 38: 2249–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le-Niculescu H, Case NJ, Hulvershorn L, Patel SD, Bowker D, Gupta J et al. Convergent functional genomic studies of omega-3 fatty acids in stress reactivity, bipolar disorder and alcoholism. Transl Psychiatry 2011; 1: e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wellcome Trust Case Control C. Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 2007; 447: 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turner CA, Thompson RC, Bunney WE, Schatzberg AF, Barchas JD, Myers RM et al. Altered choroid plexus gene expression in major depressive disorder. Front Hum Neurosci 2014; 8: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beech RD, Lowthert L, Leffert JJ, Mason PN, Taylor MM, Umlauf S et al. Increased peripheral blood expression of electron transport chain genes in bipolar depression. Bipolar Disord 2010; 12: 813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gray JD, Rubin TG, Hunter RG, McEwen BS. Hippocampal gene expression changes underlying stress sensitization and recovery. Mol Psychiatry 2014; 19: 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kubota M, Kasahara T, Iwamoto K, Komori A, Ishiwata M, Miyauchi T et al. Therapeutic implications of down-regulation of cyclophilin D in bipolar disorder. Int J Neuropsychopharmacol 2010; 13: 1355–1368. [DOI] [PubMed] [Google Scholar]
- 52.Su YA, Wu J, Zhang L, Zhang Q, Su DM, He P et al. Dysregulated mitochondrial genes and networks with drug targets in postmortem brain of patients with posttraumatic stress disorder (PTSD) revealed by human mitochondria-focused cDNA microarrays. Int J Biol Sci 2008; 4: 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R et al. A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry 2008; 64: 266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakamura K, Chen CK, Sekine Y, Iwata Y, Anitha A, Loh el W et al. Association analysis of SOD2 variants with methamphetamine psychosis in Japanese and Taiwanese populations. Hum Genet 2006; 120: 243–252. [DOI] [PubMed] [Google Scholar]
- 55.McAuley EZ, Blair IP, Liu Z, Fullerton JM, Scimone A, Van Herten M et al. A genome screen of 35 bipolar affective disorder pedigrees provides significant evidence for a susceptibility locus on chromosome 15q25–26. Mol Psychiatry 2009; 14: 492–500. [DOI] [PubMed] [Google Scholar]
- 56.Gaiteri C, Guilloux JP, Lewis DA, Sibille E. Altered gene synchrony suggests a combined hormone-mediated dysregulated state in major depression. PLoS One 2010; 5: e9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhurov V, Stead JD, Merali Z, Palkovits M, Faludi G, Schild-Poulter C et al. Molecular pathway reconstruction and analysis of disturbed gene expression in depressed individuals who died by suicide. PLoS One 2012; 7: e47581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martins-de-Souza D, Maccarrone G, Ising M, Kloiber S, Lucae S, Holsboer F et al. Blood mononuclear cell proteome suggests integrin and Ras signaling as critical pathways for antidepressant treatment response. Biol Psychiatry 2014; 76: e15–e17. [DOI] [PubMed] [Google Scholar]
- 59.Surget A, Wang Y, Leman S, Ibarguen-Vargas Y, Edgar N, Griebel G et al. Corticolimbic transcriptome changes are state-dependent and region-specific in a rodent model of depression and of antidepressant reversal. Neuropsychopharmacology 2009; 34: 1363–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ewald H, Flint T, Kruse TA, Mors O. A genome-wide scan shows significant linkage between bipolar disorder and chromosome 12q24.3 and suggestive linkage to chromosomes 1p22–21, 4p16, 6q14–22, 10q26 and 16p13.3. Mol Psychiatry 2002; 7: 734–744. [DOI] [PubMed] [Google Scholar]
- 61.Vawter MP, Tomita H, Meng F, Bolstad B, Li J, Evans S et al. Mitochondrial-related gene expression changes are sensitive to agonal-pH state: implications for brain disorders. Mol Psychiatry 2006; 11: 663–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhasin MK, Dusek JA, Chang BH, Joseph MG, Denninger JW, Fricchione GL et al. Relaxation response induces temporal transcriptome changes in energy metabolism, insulin secretion and inflammatory pathways. PLoS One 2013; 8: e62817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McQuillin A, Rizig M, Gurling HM. A microarray gene expression study of the molecular pharmacology of lithium carbonate on mouse brain mRNA to understand the neurobiology of mood stabilization and treatment of bipolar affective disorder. Pharmacogenet Genomics 2007; 17: 605–617. [DOI] [PubMed] [Google Scholar]
- 64.Velders FP, Kuningas M, Kumari M, Dekker MJ, Uitterlinden AG, Kirschbaum C et al. Genetics of cortisol secretion and depressive symptoms: a candidate gene and genome wide association approach. Psychoneuroendocrinology 2011; 36: 1053–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pibiri F, Nelson M, Guidotti A, Costa E, Pinna G. Decreased corticolimbic allopregnanolone expression during social isolation enhances contextual fear: A model relevant for posttraumatic stress disorder. Proc Natl Acad Sci USA 2008; 105: 5567–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zubenko GS, Maher B, Hughes HB 3rd, Zubenko WN, Stiffler JS, Kaplan BB et al. Genome-wide linkage survey for genetic loci that influence the development of depressive disorders in families with recurrent, early-onset, major depression. Am J Med Genet B Neuropsychiatr Genet 2003; 123: 1–18. [DOI] [PubMed] [Google Scholar]
- 67.Doyle AE, Biederman J, Ferreira MA, Wong P, Smoller JW, Faraone SV. Suggestive linkage of the child behavior checklist juvenile bipolar disorder phenotype to 1p21, 6p21, and 8q21. J Am Acad Child Adolesc Psychiatry 2010; 49: 378–387. [PMC free article] [PubMed] [Google Scholar]
- 68.Le-Niculescu H, Kurian SM, Yehyawi N, Dike C, Patel SD, Edenberg HJ et al. Identifying blood biomarkers for mood disorders using convergent functional genomics. Mol Psychiatry 2009; 14: 156–174. [DOI] [PubMed] [Google Scholar]
- 69.Morita K, Saito T, Ohta M, Ohmori T, Kawai K, Teshima-Kondo S et al. Expression analysis of psychological stress-associated genes in peripheral blood leukocytes. Neurosci Lett 2005; 381: 57–62. [DOI] [PubMed] [Google Scholar]
- 70.Cole SW, Hawkley LC, Arevalo JM, Sung CY, Rose RM, Cacioppo JT. Social regulation of gene expression in human leukocytes. Genome Biol 2007; 8: R189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kang HJ, Adams DH, Simen A, Simen BB, Rajkowska G, Stockmeier CA et al. Gene expression profiling in postmortem prefrontal cortex of major depressive disorder. J Neurosci 2007; 27: 13329–13340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ibi D, Takuma K, Koike H, Mizoguchi H, Tsuritani K, Kuwahara Y et al. Social isolation rearing-induced impairment of the hippocampal neurogenesis is associated with deficits in spatial memory and emotion-related behaviors in juvenile mice. J Neurochem 2008; 105: 921–932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.