

Abstract

We report in this article the synthesis, the X-ray crystal structure of compound syn-2, and its binding properties with cesium and thallium in aqueous solution under basic conditions. Compound syn-2 is the diastereomeric compound of anti-1 that shows very high affinity for cesium and thallium in aqueous solution under the same conditions. Despite the close structural similarities that exist between the syn-2 and anti-1 compounds, they show large discrepancy in their ability to bind cesium and thallium cations in the same conditions. Indeed, the syn-2 derivative has a lower affinity for these two cationic species and the binding constants are several orders of magnitude lower than those found for its congener. The large differences in affinity observed with these two compounds can be explained by the relative position of the six hydroxyl groups to each other.
Introduction
The development of human activities all over the world has strongly modified our planet and the ecosystems in many ways. For instance, it significantly contributed to the pollution of groundwater, soil, and air by toxic substances such as metallic species. Of the various metals involved, thallium has not received as much publicity as mercury and lead, although its toxicity to human health is considered higher.1 However, nowadays, the danger caused by thallium pollution is well recognized by governmental agencies in different countries and the elimination of this metallic species from groundwater and soil has become a top priority.2
Several options have been proposed to recover thallium salts, mainly thallium(I), dispersed in the environment. For instance, mineral matrices seem to be interesting materials to extract thallium from groundwater.3 These materials are easier to prepare than organic compounds, and they can be produced in larger quantities more easily. On the other hand, non-natural compounds, mainly macrocyclic compounds, with a high affinity for thallium salts have been specifically developed for this purpose.4 Interestingly, the physical properties of these molecular hosts can be tuned more easily in order to maximize interactions with the cations of interest.
On the other hand, the 133Cs cation is not as toxic as thallium, but the radioactive isotopes of cesium (mainly 137Cs) can represent a significant environmental problem as observed during the Fukushima (2011) and Chernobyl (1986) nuclear accidents. When released in the atmosphere, this species can spread over great distances and contaminate large areas for a long period of time. Many methods to recover 137Cs and decontaminate soils and groundwater have been proposed. For example, different sorbents such as clays, zeolites, and organic compounds including macrocyclic derivatives have been developed for this purpose.5,6
Covalently bound molecular capsules, called cryptophanes, decorated with phenol functions exhibit very high affinity for cesium and thallium cations in aqueous solutions under basic conditions.7 For instance, cryptophane anti-1 bearing six phenol functions shows high binding properties for alkali cations (Rb+ and Cs+) and Tl+ in LiOH/H2O or NaOH/H2O (Chart 1). These complexes have been thoroughly studied by different techniques such as 133Cs and 205Tl NMR spectroscopy, electronic and vibrational circular dichroism spectroscopy, or isothermal titration calorimetry (ITC) experiments.
Chart 1. Chemical Structures of Compound Anti-Cryptophane-A, Syn-Cryptophane-B, and Cryptophanes Anti-1 and Syn-2.

All the cryptophanes studied so far for cesium and thallium complexation possess the relative configuration anti. This means that the linkers connecting the two cyclotribenzylene units adopt a helical arrangement. Consequently, the two cyclotribenzylene (CTB) units are tilted from each other and these structures possess an inherent chirality regardless of the nature of the substituents grafted on the six benzene rings. This particular arrangement of the linkers allows the cryptophane to adopt a higher flexibility and to maximize interactions with the encapsulated species.8 On the other hand, cryptophanes with a small cavity (Vvdw < 100 Å3) and the syn configuration have been little described and only a few examples have been reported in the literature.9 Recently, a new synthetic approach has allowed us to extend the synthesis to cryptophanes with the syn configuration. For example, syn-cryptophane-B, the diastereomer of anti-cryptophane-A, was prepared in this way (see Chart 1).10
Interestingly, the synthetic strategy developed for syn-cryptophane-B can also be used to prepare the new syn-2 derivative (Scheme 1), which is nothing but the diastereomer of anti-1 (see Chart 1). At first glance, considering that cesium and thallium cations do not interact with the alkyl chains and that the cavities of anti-1 and syn-2 possess similar volumes, it seems reasonable to assume that compound syn-2 will behave like compound anti-1 in the presence of these two cationic species. Thus, high-binding constants for the Cs+@syn-2 and Tl+@syn-2 complexes are also expected with this compound as observed with the anti-1 derivative.
Scheme 1. Synthesis of Compound syn-2 from the Tris-phenol Derivative syn-3.

In this work, we report the binding properties of both the Cs+@syn-2 and Tl+@syn-2 complexes by means of NMR spectroscopy and ITC. These conditions are identical as those reported in a previous article for the study of the Cs+@anti-1 and Tl+@anti-1 complexes. Finally, we compare the results obtained with these two compounds to evaluate the impact of the spatial arrangement of the three O–CH2–CH2–O linkers on the molecular recognition properties of this new host.
Results
Synthesis
Hexa-phenol 2 was prepared in a single step from the known derivative syn-3, the latter being prepared in eight steps from the commercially available 3,4-dihydroxybenzaldehyde.10
A hydrogenation step in the presence of a catalytic amount of palladium on charcoal in a CH2Cl2/MeOH mixture (50/50: v/v) provided rapidly the desired syn-2 derivative. Extraction with a NaOH (0.5 M)/H2O solution followed by acidification at 0 °C with few drops of a concentrated HCl solution gave rise to compound syn-2 with 83% yield. This compound can be recrystallized from a hot acetone solution. Compound syn-2 was fully characterized by 1H and 13C NMR spectroscopy and HRMS. The 1H NMR spectrum (3.0–9.0 ppm; DMSO-d6) of 2 is shown Figure 1 and compared to the known hexa-phenol derivative anti-1. The two spectra look very similar, with the exception of the diastereotopic protons of compound syn-2, which appear as two sets of signals instead of one for its congener. 1H and 13C NMR spectra of compound syn-2 are reported in Figures S1 and S2.
Figure 1.
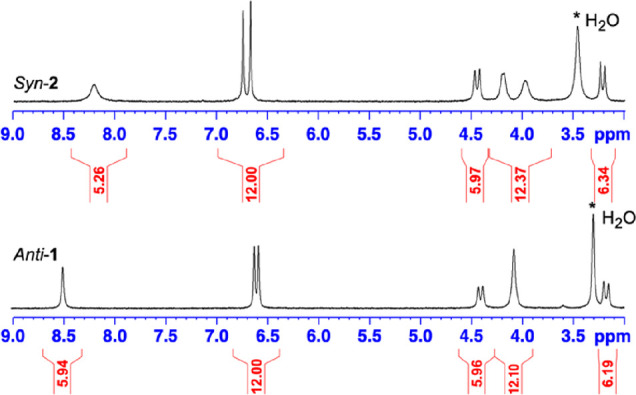
1H NMR spectra of compounds anti-1 and syn-2 recorded in DMSO-d6 at 298 K. The full spectrum of syn-2 (0–10 ppm) is reported in Figure S1.
Experimental Procedures
Compound syn-3 (0.38 g, 0.35 mmol) prepared according to a known procedure was introduced in a 50 mL round-bottom flask and dissolved in a mixture of CH2Cl2 (12 mL) and MeOH (12 mL) at room temperature.10 Then, a catalytic amount of Pd/C (10%) was introduced into the flask. The balloon was closed with a septum. Then, hydrogen gas was bubbled into the flask for several minutes and the reaction was kept for 16 h at room temperature under a hydrogen atmosphere. The mixture was filtered over celite, and the solid was washed four times with CH2Cl2 and then with a few milliliters of DMSO. The solution was poured into a separating funnel, and the product was extracted twice with a NaOH (0.5 M)/H2O solution. The aqueous solution was finally washed four times with CH2Cl2 and then cooled down at 0 °C. Acidification with few drops of a concentrated HCl solution gave rise to a white precipitate, which was collected on a frit. The solid was washed with water, dried, and finally washed with diethyl ether to give compound syn-2 as a white powder (0.24 g, 85%). Compound syn-2 crystallized in acetone. Mp: 84 °C. UV–vis (LiOH-0.1 M): λmax (log ε) 340 (4.5 shoulder), 300 (4.27). 1H NMR (300 MHz, DMSO-d6, 25 °C): δ 8.20 (s, 6H, broad), 6.74 (s, 6H), 6.66 (s, 6H), 4.44 (d, J = 13.2 Hz), 3.35 (m, 12 H, broad), 3.21 (d, J = 13.2 Hz). 13C{1H} NMR (MHz, CDCl3, 25 °C): δ 146.1 (6C), 143.7 (6C), 134.1 (6C), 130.2 (6C), 119.1 (6C), 117.2 (6C), 67.8 (6C), 35.1 (6C). HRMS (ESI) m/z: [M + Na]+ calcd for C48H42NaO12: 833,2577; found: 833,2568.
Despite their structural similitude, compounds anti-1 and syn-2 show significant differences in their physical properties. For instance, compared to compound anti-1, the new derivative syn-2 appears to be only soluble in LiOH/H2O or NaOH/H2O solution at a concentration around 0.1 M. At a higher concentration, compound syn-2 precipitates and at a lower LiOH concentration, it gives rise to nano-aggregates as evidenced by diffusion light scattering (DLS). For instance, compound syn-2 dissolved at a concentration of c = 3 × 10–5 M in LiOH/H2O (10–3 M) gives rise to small aggregates whose size is estimated to be 90 nm (Figure S3). This contrasts with compound anti-1 that shows better solubility under these conditions. Thus, all the binding experiments reported with syn-2 in this article will be performed at a LiOH/H2O and NaOH/H2O concentration close to 0.1 M.
133Cs NMR Spectroscopy
The ability of compound syn-2 to encapsulate cesium in water under basic conditions was assessed by 133Cs NMR spectroscopy. Syn-2 was dissolved in a 0.1 M NaOD aqueous solution in the presence of an excess of CsNO3 (Figure 2), and 133Cs NMR spectra were recorded as a function of the temperature (275–340 K). At 290 K, the 133Cs NMR spectrum reveals a broad signal at δ = −10 ppm, which has been identified as Cs+ dissolved in the bulk. We also distinguish another NMR signal located at δ = −130 ppm. Decreasing the temperature up to 275 K allows us to distinguish more easily the second 133Cs NMR signal and to assign this resonance signal to the spectral signature of the Cs+@syn-2 complex. At 275 K, the Cs+@syn-2 complex resonates at a higher frequency than the NMR signal observed for the anti-1 congener (δ = −270 ppm). Increasing the temperature decreases the chemical shift splitting between free and bound cesium signals and at 310 K, they coalesce to give a single resonance signal located at δ = −55 ppm. Due to the Eyring relationship (Figure S3), this allows us to calculate an exchange constant kTc ≈ 1.8 × 104 s–1 and an activation barrier of ΔG# = 12.1 kcal.mol–1 at this temperature.
Figure 2.

133Cs NMR spectra of syn-2 dissolved in NaOD (0.1 M)/D2O in the presence of an excess of CsNO3. Spectra were recorded as a function of temperature (275–340 K).
UV–visible Spectra
Spectroscopic tools such as UV–visible and electronic circular dichroism (ECD) spectroscopy have been very useful to study compound syn-1 and its congeners upon binding with cationic species. However, in contrast to the anti-1 derivative, the achiral structure (C3h symmetry) of this syn-2 prevents us to use ECD spectroscopy to characterize this compound and its complexes. Nevertheless, cryptophanes are made of six benzene chromophores that absorb light below 350 nm, which makes recording their UV–visible spectrum under different conditions possible. Thus, it is possible to detect the symmetry-forbidden 1Lb transitions of the six benzene rings of cryptophanes that appears near 300 nm. The symmetry-forbidden 1La transition of the benzene chromophore, which is located at lower wavelengths, is also accessible but more difficult to detect because it is buried below the strong symmetry-allowed 1Bb transition. The UV–visible spectrum of syn-2 in LiOH (0.1 M)/H2O is reported in Figure S4. This spectrum is very similar to that observed for its congener under the same conditions. For instance, the UV–visible spectrum shows a transition with a maximum at 305 nm that corresponds to the 1Lb transitions. A shoulder is also observed at lower wavelengths and corresponds to the 1La transitions of the benzene rings. Upon addition of Cs+ to the solution, the UV–visible spectrum of syn-2 is modified and a slight bathochromic shift of the spectrum is observed. This effect is more pronounced when Cs+ is replaced by the Tl+ cation as this was previously observed with its anti-1 congener. Thus, the UV–visible spectra in the presence of these two cations allow us to ascertain that encapsulation of Cs+ and Tl+ takes place under these conditions. However, a quantitative description of this phenomenon seems difficult to achieve with this technique and ITC experiments are more appropriate to measure the association constants in this case.
X-ray Structures of Compounds 1–3
X-ray-quality single crystals have been obtained for compounds 1–3 in different solvents. Small colorless plate crystals of compound anti-1 suitable for X-ray analysis have been obtained from slow evaporation of a CH2Cl2/CH3CN mixture. Compound anti-1 crystallizes in a monoclinic crystal system and in the P21/c space group (Table S1). The unit cell contains four molecules, and each cavity of the host molecules is filled with an acetonitrile molecule. From the ORTEP structure it can be seen that the three O–CH2–CH2–O linkers adopt dihedral angles 71.7, −67.8, and 62.2° that are close to those observed for a gauche (g) conformation (Figure 3, Figure S5, and Table S2). This conformation results in a flattened structure of the anti-1 compound where the two CTB caps are staggered with respect to each other. The volume of the internal cavity has been calculated to be 73 Å3 for this conformation, and the intermolecular cavity has been estimated to be 92 Å3 (Table S3). It is noteworthy that compound anti-1 also showed residual intermolecular electronic density that could not be modeled by a solvent molecule. The contribution of this disordered solvent to the structure factors was taken into account with the SQUEEZE algorithm.11
Figure 3.
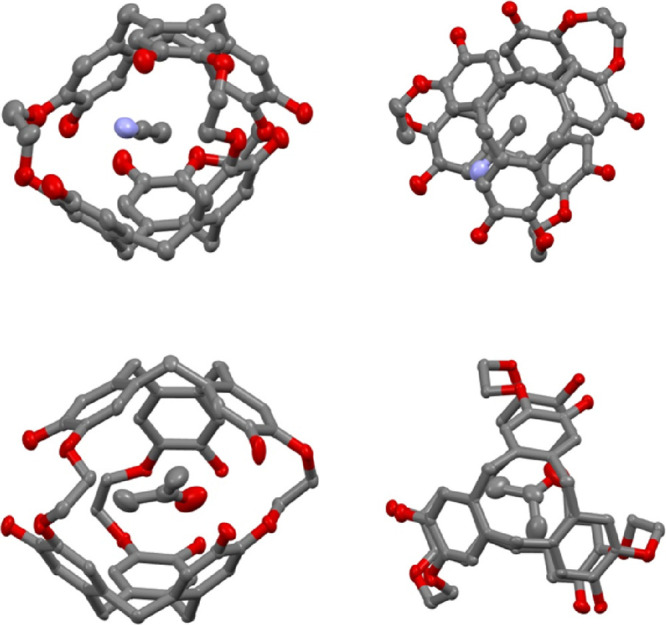
ORTEP representation of compounds CH3CN@anti-1 (top view) and acetone@syn-2 (bottom view). Two spatial representations are given for each structure. Hydrogen atoms have been removed for clarity. The displacement ellipsoids were plotted at a 30% probability level.
We successfully obtained X-ray-quality crystals of compound syn-2 in two different solvents (acetone and acetonitrile). For instance, slow evaporation of an acetone solution provides colorless plate-shaped single crystals of syn-2 suitable for X-ray analysis. In this solvent, compound syn-2 crystallizes in an orthorhombic crystal system and in the Pbca space group (Table S1). The unit cell contains eight molecules of syn-2, and the cavity of the host is filled with an acetone molecule. The interstitial space between molecules has been estimated to be 298 Å3 and is filled with acetone and water molecules (Table S3). As observed for compound anti-1, the conformation of the three O–CH2–CH2–O linkers is close to that observed for a gauche conformation and dihedral angles of −56.3, 48.3, and 62.6° have been measured from the ORTEP structure (Figure 3, Figure S6, and Table S2). This results in a measured internal cavity V = 93 Å3 for syn-2, a value 25% higher than that calculated for the anti-1 compound (Table S3). In pure acetonitrile, compound syn-2 also gives rise to single colorless plate crystals suitable for X-ray analysis. In this solvent, this compound crystallizes in a monoclinic crystal system and in the P21/c space group with four molecules per unit cell (Table S1). Both the cavities of the host molecules and the interstitial spaces are filled with acetonitrile molecules. From the ORTEP representation, the internal cavity of the host and the intermolecular voids have been estimated to be V = 91 Å3 and V = 180 Å3, respectively (Table S3). This representation (Figure S7) allows us to measure three dihedral angles of 51.9, 62.6, and −54.2° for the three O–CH2–CH2–O linkers resulting to angle values very close to those determined previously for the same compound (Table S2). It can be noticed that if we exclude the nature of the guest molecule present in the cavity of syn-2, the two structures of compound syn-2 in acetone and acetonitrile look very similar.
Finally, single crystals of the intermediate syn-3 suitable for X-ray analysis have been obtained by slow evaporation of a CH2Cl2/EtOH solution. Interestingly, in this example, we observed that spontaneous resolution takes place. Compound syn-3 crystallizes in an orthorhombic crystal system and in a Pca21 space group (Table S1). Near-zero values of the Flack (0.05) and Hooft (0.063) parameters indicate that a single enantiomer is present into the crystal lattice. In our example, the ORTEP representation (Figure 4 and Figure S8) represents the syn-3 derivative with the MP absolute configuration, the first letter corresponding to the stereodescriptor of the CTB unit decorated with the OH group. Four molecules are present per unit cell, and the internal cavity of the host molecule (V = 95 Å3) is filled with a CH2Cl2 solvent molecule (Table S3). Large intermolecular voids (V = 305 Å3) calculated from the ORTEP representation are filled with CH2Cl2 molecules. As observed with the previous X-ray structures of compounds anti-1 and syn-2, the three linkers of syn-3 adopt conformation angles −53.4, 51.0, and 56.8° close to that observed for a gauche (g) conformation (Table S2).
Figure 4.

ORTEP representation of compound CH2Cl2@MP-syn-3. The first stereodescriptor refers to CTB decorated with the three phenol groups. The two spatial representations are given for each structure. Hydrogen atoms have been removed for clarity. The displacement ellipsoids were plotted at a 30% probability level. In all these structures, the ORTEP representation reveals the presence of intra- and inter-molecular hydrogen bonding, which have been listed in Tables S4 and S5.
ITC Experiments
ITC is a very efficient method to characterize host–guest interactions under different conditions. This method gives access, in a single experiment, to the stoichiometry of the complex, the binding constant, and the enthalpy of complexation ΔH0. Thus, the entropy of complexation ΔS0 and the free energy of complexation ΔG0 can be easily deduced from these parameters, thereby giving a complete map of the binding process. For instance, titration experiments have been used with success in the case of cryptophane anti-1 and its congeners in the presence of alkali cations and thallium(I) under different conditions.7b
In order to compare the binding properties of compound anti-1 with compound syn-2, a series of ITC experiments were first undertaken with syn-2 in LiOH (0.1 M)/H2O in the presence of rubidium chloride (RbCl), cesium hydroxide (CsOH), and thallium acetate (TlOAc). The experimental conditions used to study compound syn-2 are similar to those used previously for the anti-1 derivative. Contrary to compound anti-1 that shows good recognition properties for Rb+, the titration experiments of syn-2 in the presence of Rb+ in LiOH (0.1 M)/H2O reveal that this compound does not show any affinity for this cation (Figure S9). In the same solvent, compound syn-2 shows a moderate affinity for the Cs+ cation and an association constant K = 1.3 × 104 M–1 has been measured from the titration experiment (Figure S10). This value appears to be lower by five orders of magnitude than that observed for the Cs+@anti-1 complex under the same conditions.7b In the presence of Tl+, the situation appears to be different. Indeed, the syn-2 compound shows an enthalpogram that presents the classical S-shape (Figure S11), thus suggesting a strong interaction between syn-2 and this cation. Nevertheless, we failed to fit these experimental points regardless of the fitting model chosen. This difficulty in fitting the experimental data point arises from the fact that the enthalpy of dilution of the Tl+ cation in LiOH (0.1 M)/H2O is lower from the enthalpy of dilution of the Tl+ cation in the presence of syn-2. Thus, the subtraction of these two enthalpograms results in a small negative enthalpic term that still remains once the cage is fully saturated. Two independent ITC experiments performed under the same conditions give similar results (Figure S11). It is noteworthy that ignoring this small contribution (data points set to zero) leads to a curve that fits perfectly the experimental data to give a binding constant K = 6.9 × 105 M–1 (ΔG0 = −8000 cal.mol–1) for the Tl+@syn-2 (Figure 5). As observed in the case of the Cs+@syn-2 complex, the binding constant observed in this case is five orders of magnitude lower than the one observed under the same conditions for the Tl+@anti-1 complex.
Figure 5.

(Left) Isothermal calorimetric titration data (forward titration) of compound syn-2 in LiOH (0.1 M)/H2O at 298 K. The host solution (c = 0.993 mmol) was placed into the calorimeter cell (1.4 mL), and 28 successive aliquots (10 μL) of TlOAc (c = 0.099 mmol) were added at 3 min intervals (n = 0.8; K = 6.9 × 105 M–1; ΔH0 = −8500 cal·mol–1; ΔS0 = −1.9 cal·mol–1·K–1; ΔG0 = −7950 cal·mol–1). (right) Isothermal calorimetric titration data (reverse titration) of compound syn-2 recorded under the same conditions. TlOAc (c = 0.10 mmol) was placed into the calorimeter cell, and 28 successive aliquots (10 μL) of syn-2 (c = 1.02 mmol) were added at 3 min intervals (n = 1.2; K = 3.25 × 105 M–1; ΔH0 = −8100 cal·mol–1; ΔS0 = −2.0 cal·mol–1·K–1; ΔG0 = −7500 cal·mol–1).
Although this procedure has a small impact on the measured binding constant, it remains questionable. Indeed, this small contribution may reflect additional interactions between the Tl+ cation and syn-2. In such a case, it is recommended to perform the reverse titration, which requires introduction of the ligand (Tl+) in the cell and the host molecule (syn-2) in the syringe.12 In the case of a classic 1:1 complex such as the one encountered here, we expect the forward and reverse isothermal titrations experiment to be identical. In contrast, a difference between them may reveal a more complex host–guest binding process or additional interactions such as aggregation. It is interesting to note that this procedure was not used with the anti-1 host because in all previously reported examples, the experimental ITC data were fully consistent with a 1:1 complex, regardless of the nature of the cation. However, for comparison, we decided to report in this article the reverse titration of anti-1 with the Rb+, Cs+, and Tl+ performed under the same conditions. A comparison of the reverse titration between these two host molecules seems interesting and can provide additional information about their behavior in the presence of these cationic species.
The reverse titration performed with syn-2 in the presence of Cs+ shows an experimental ITC curve that fits well with a one-site model. From this experiment, a moderate binding constant K = 7.4 × 103 M–1 (ΔG0 = −5300 cal·mol–1) was measured, a value close to that measured for the forward titration (Figure S10). In contrast, replacing Cs+ with Tl+ leads to a titration curve more difficult to interpret since the theoretical curve does not fit perfectly well the experimental data (Figure 5). Nevertheless, it should be pointed out that the binding constant K = 3.3 × 105 M–1 (ΔG0 = −7500 cal·mol–1) extracted from this experiment is in good agreement with the binding constant measured for the forward titration.
On the other hand, the forward and reverse titrations of compound anti-1 were found identical regardless of the nature of the cation. Thus, the forward and reverse titrations of the Rb+@anti-1, the Cs+@anti-1, and the Tl+@anti-1 complexes fit perfectly with a 1:1 complex (Figures S12–S14). For instance, the reverse titration gives a binding constant K = 7.3 × 105 M–1 (ΔG0 = −8000 cal·mol–1) for the Rb+@anti-1 complex, whereas an association constant K = 9.2 × 105 M–1 (ΔG0 = −8200 cal·mol–1) was previously determined for the forward titration.5b Similarly, an association constant K = 8.9 × 108 M–1 (ΔG0 = −12200 cal·mol–1) was measured for the Cs+@anti-1 complex instead of K = 2.7 × 109 M–1 (ΔG0 = −12900 cal·mol–1) for the reverse reaction. Finally, a binding constant K = 6.7 × 1010 M–1 (ΔG0 = −14800 cal·mol–1) was measured for the reverse titration of the Tl+@anti-1 complex, a value close to that measured for the forward titration.7b Note that in the latter case, the presence of a competitor (Rb+) was mandatory to accurately measure the association with Tl+. All together, these results show that compound syn-2 seems to behave differently from compound anti-1 toward cationic species and, upon complexation with thallium, give enthalpograms that are more complex to interpret.
It is important to point out that the titration experiments described above are not performed at constant pH. Indeed, small injection of ligand during the forward and reverse titration experiments slightly modifies the pH of the solution. Thus, a possible change in the pH of the solution may affect the isothermal titration of the syn-2 compound compared to its congener anti-1. To test this hypothesis, a new series of experiments were performed in a NaOH/KCl buffer to maintain the pH of the solution around pH = 13. For comparison, the titration experiments (forward and reverse) of anti-1 were also performed under the same conditions.
The forward titration of syn-2 shows exothermic heat release upon injection of a small amount of Cs+ cation in the NaOH/KCl buffer. Using these conditions, a one-site model fits perfectly the data points and allows an estimation of the binding constant K = 4.1 × 103 M–1 (ΔG0 = −4900 cal·mol–1) (Figure S15). The reverse titration also results in an enthalpogram that fits well with a one-site model and a binding constant K = 2.2 × 103 M–1 (ΔG0 = – 4600 cal·mol–1) was extracted from this experiment. A change in the nature of the cation leads to a more complex situation since the addition of Tl+ to a buffer solution of syn-2 leads to a titration whose data points can only be correctly fitted with two sequential binding sites. Thus, this leads to the determination of two binding constants with different magnitudes K1 = 1.1 × 105 M–1 (ΔG0 = −6900 cal·mol–1) and K2 = 1.8 × 103 M–1 (ΔG0 = −4400 cal·mol–1) (Figure 6). In contrast, the data point fits well with a one-site model for the reverse titration and gives a binding constant of K = 6.8 × 104 M–1. We can point out that the first binding constant is consistent with the data measured in LiOH (0.1 M)/H2O for the forward and reverse experiments.
Figure 6.

(Left) Isothermal calorimetric titration data (forward titration) of compound syn-2 in NaOH/H2O-KCl buffer (pH = 12.9) at 298 K. The host solution (c = 0.099 mmol) was placed into the calorimeter cell (1.4 mL), and 28 successive aliquots (10 μL) of TlOAc (c = 3.02 mmol) were added at 3 min intervals (sequential binding site: K1 = 1.1 × 105 M–1; ΔH10 = −6100 cal·mol–1; ΔS10 = 2.6 cal·mol–1·K–1; ΔG10 = −6900 cal·mol–1; K2 = 1.8 × 103 M–1; ΔH20 = −15200 cal·mol–1; ΔS20 = −36.1 cal·mol–1·K–1; ΔG20 = −1100 cal·mol–1). (Right) Isothermal calorimetric titration data (reverse titration) of compound syn-2 recorded under the same conditions. TlOAc (c = 0.1 mmol) was placed into the calorimeter cell, and 28 successive aliquots (10 μL) of syn-2 (c = 0.986 mmol) were added at 3 min intervals (n = 0.8; K = 6.8 × 104 M–1; ΔH0 = −8900 cal·mol–1; ΔS0 = −7.8 cal·mol–1·K–1; ΔG0 = −6600 cal·mol–1).
Under the same conditions, the forward and reverse titrations have been performed with the host anti-1 and the ligands Rb+, Cs+, and Tl+ (Figures S16–S18). In all cases, we noticed that the experimental points fit very well with a 1:1 complex. In addition, both the forward and reverse titrations give similar results. It should be noticed that under these conditions, the binding constants deduced from these experiments are smaller than those previously reported in pure NaOH (0.1 M)/H2O.5b This is because the buffer contains potassium cations present in large excess. Indeed, K+ shows a moderate affinity (K = 500–1000 M–1) for the host anti-1 and acts as a competitor, thus resulting in lower affinity for the Rb+@anti-1, Cs+@anti-1 and Tl+@anti-1 complexes (Figure S19) in this solvent.
Discussion
The intermediate syn-3 appears to be a useful derivative to prepare a wide range of C3-symmetric derivatives with the syn configuration due to the presence of three unprotected phenol groups and three other protected phenol functions that allow us to easily modify its structure. For example, recent studies have suggested that cryptophanes with the syn configuration possess superior characteristics for the design of 129Xe NMR-based biosensors compared to their anti-congeners.13 Therefore, the interest for the syn-compounds should rapidly grow in the future. Interestingly, compound syn-3 allows us to give access to the syn-2 derivative in good yield and in a single step. In our case, this approach was preferred to the demethylation step of syn-cryptophane-B with PPh2Li in THF. Indeed, although this procedure has been successfully used to prepare the anti-1 derivative, the purification step for the syn-2 compound would be more difficult with this approach. For instance, the presence of the large excess of PPh2Li present in the reaction mixture and the lower solubility of the syn-2 compound would make its purification process more difficult. In addition, syn-cryptophane-B also requires the synthesis of the syn-3 derivative to be prepared. Thus, a direct synthesis of syn-2 from compound syn-3 is fully justified in our case. It is worth mentioning that, if necessary, compound anti-1 can also be prepared by this way. Indeed, both anti-3 and syn-3 diastereomers are produced at the same time during the second ring closure reaction and they can be separated from the crude product.10 However, the synthesis of anti-1 is easier starting from cryptophane-A and larger quantities of this compound can be obtained by this way.
The fact that spontaneous resolution occurs with the syn-3 compound during the crystallization process is also an interesting feature worth discussing. Indeed, the formation of conglomerate has rarely been observed with cryptophanes since most of them crystallize as racemic compounds.14 Thus, the production of larger quantities of syn-3 would allow us to rapidly isolate the two enantiomers of this cryptophane without the use of costly HPLC methods.15 It is noteworthy that a large range of new enantiopure derivatives could be obtained by this way.
NMR spectroscopy appeared to be the spectroscopic tool of choice to investigate Cs+. Due to its large electronic cloud, this heavy atom is known to be very sensitive to a change of their surrounding environment. For instance, in the presence of these two cations, the water-soluble anti-cryptophanes show a specific signature, which is usually observable at room temperature. The Cs+@cryptophane complexes give a specific spectral signature shifted by several hundreds of ppm toward low frequencies on the 133Cs spectrum as a consequence of the strong shielding effect induced by the aromatic ring current. Here, the chemical shift observed for the Cs+@syn-2 complex is shifted by δ = −120 ppm toward a high frequency, a value significantly lower than the chemical shift observed with the anti-1 congener and other cryptophanes of anti-configuration.7b,7c The higher rigidity of the cryptophane backbone and the larger cavity of the syn-2 allows us to explain this result. Indeed, the syn-2 compound is not able to modify its structure to maximize the interaction with the cesium cation as easily as the anti-1 compound. As a consequence, the shielding effect induced by the aromatic ring current is not as effective as that observed with its congener.
A fast in–out exchange dynamics is also observed with the Cs+@syn-2 complex above room temperature, leading to a unique 133Cs NMR signal. Decreasing the temperature allows us to slow down the exchange of 133Cs and two distinct NMR signals start to appear at 285 K and below. Thus, in the presence of cesium, compound syn-2 differs significantly from the anti-1 congener. Previously, two distinct behaviors were observed with the water-soluble cryptophanes in the presence of cesium salt.7c For cryptophanes showing very high affinity for the Cs+ cation, a sharpening of the 133Cs NMR signal was observed when increasing the temperature. This is the case of the Cs+@anti-1 complex that gives rise to a very sharp 133Cs NMR signal at 345 K compared to that observed at room temperature. The sharpening of the 133Cs NMR signal depends on the molecular symmetry of the host. For instance, we have observed that the higher the symmetry of the cryptophane, the sharper the 133Cs NMR signal.7b,7d,16 An opposite effect was observed for cryptophanes showing a moderate or low affinity for the Cs+ cation, and a sharpening of the 133Cs NMR signal is observed upon decreasing the temperature. For, instance, the Cs+@cryptophane-233 complex shows this behavior as a function of the temperature.7b Our results reveal that the Cs+@syn-2 complex behaves similarly. Thus, 133Cs NMR spectroscopy allows us to predict whether water-soluble cryptophanes show high or moderate affinity for this cation. Similar experiments were attempted with syn-2 and thallium salts. Unfortunately, in each case, we observed the formation of a precipitate upon addition of a thallium(I) salt, and the liquid-state NMR experiments were impossible.
Despite the structural similarities that exist between the anti-1 and syn-2, these two compounds differ greatly in their physical properties from a general perspective. Thus, a little change in the arrangement of the O–CH2–CH2–O linkers has a strong impact on the overall physical properties of the syn-2 derivative compared to anti-1. For instance, the solubility properties of syn-2 are significantly modified compared to those of anti-1 under the same conditions and this compound is only soluble in a limited range of pH. On the other hand, the arrangement of the three linkers also modifies significantly the binding properties of compound syn-2 with respect to the anti-1 derivative. Indeed, we observe that the affinity of this host for Cs+ and Tl+ cations is decreased by five orders of magnitude with respect to anti-1. In addition, compound syn-2 does not show any affinity for the Rb+ cation, whereas a relatively high binding constant K = 9.2 × 105 M–1 has been measured with the diastereomer anti-1 in LiOH (0.1 M)/H2O. Thus, despite the presence of six phenol functions grafted on its skeleton, the syn-2 compound behaves like the recently studied C3-cryptophane derivative whose skeleton is composed only of three phenol functions and three carboxylic acid functions.7a
At first sight, a change in the volume of the cavity could be invoked to explain this difference in behavior between these two cryptophanes but we have previously shown that this parameter is not crucial to explain the recognition properties of cationic species by the water-soluble cryptophanes. Indeed, we have shown that anti-cryptophanes with cavity volume larger than that of syn-2 show high affinity for the Rb+, Cs+, and Tl+ cations. For instance, a cryptophane-233 derivative possessing one O–CH2–CH2–O linker and two O–CH2–CH2–CH2–O linkers still shows good affinity for Rb+, Cs+, and Tl+ with measured binding constants K = 3.2 × 104, 4.1 × 107, and 3.7 × 108 M–1, respectively.7b Thus, another explanation has to be found to explain this difference of behavior. A previous study has revealed that the number of phenol groups grafted onto the cryptophane skeleton is pivotal to observe good complexation constants with the Rb+, Cs+, and Tl+ cations.7c Thus, a decrease in the number of phenolic functions has a strong impact on the ability of these hosts to bind cationic species. For instance, the water-soluble cryptophane decorated with three phenol groups and three carboxylic acid functions binds moderately the Cs+ and Tl+ cations in LiOH (0.1 M)/H2O.7a On the other hand, DFT calculations have suggested that coulombic interactions between the cationic species and the phenolate group play an important role in the stabilization of these complexes.16 Herein, both anti-1 and syn-2 possess the same number of OH groups and only the spatial arrangement of the phenol functions is modified. Therefore, we must assume that a change in the arrangement of the three O–CH2–CH2–O bonds affects the chemical properties of the phenol groups to some extent, making the ionization state of the OH groups more difficult in the case of the syn-2 derivative. The difficulties to deprotonate in part the six OH groups in compound syn-2 could explain both the lower solubility of this derivative compared to the anti-1 derivative and the large difference in the association constants observed between these two derivatives. This assumption seems to be supported by a comparative study of the structure of these two compounds. In the case of syn-2 derivative, it can be observed that the OH groups grafted on its skeleton are facing each other. In contrast, the six phenol functions are eclipsed in the structure of the anti-1 derivative, making them less likely to interact with each other. Consequently, a greater difference in the pKa of the OH groups should result in the case of the syn-2 derivative compared to the anti-1 derivative. This interpretation seems reasonable and allows us to explain the isothermal titration experiments measured for both compounds anti-1 and syn-2. Our assumption is also supported by the fact that the forward and reverse titration experiments of compound syn-2 are more sensitive to a small pH change of the solution compared to the anti-1 diastereomer. In addition, different enthalpograms are observed in LiOH (0.1 M)/H2O and in the NaOH/H2O-KCl buffer in the case of the thallium complexation for compound syn-2. In contrast, compound anti-1 does not show such variation and the forward and reverse titration are found to be similar in shape. Unfortunately, this assumption remains difficult to verify experimentally owing to the low solubility of syn-2 in aqueous solutions that prevents an accurate determination of the pKa of the phenol functions. Furthermore, in the case of anti-1 and syn-2, the quantum calculations did not allow for accurate determination of the pKa values of the phenolic functions. The difficulties in fitting the experimental ITC data in the case of the Tl+@syn-2 complex also raise some questions. Indeed, this problem does not seem to arise for the Cs+@syn-2 whatever the experimental conditions. It is therefore possible that additional interactions take place with the Tl+@syn-2 due to its lower solubility, as was the case in the 205Tl NMR studies. Nevertheless, precipitation of this complex does not seem to occur in the cell since the enthalpogram shows a very characteristic pattern in this case.17 Instead, small aggregates are formed in solution, resulting in additional interactions.
Overall, our results show that a change in the conformation of the O–CH2–CH2–O linkers of cryptophanes not only affects the cavity volume and the size of the portals but also modifies the chemical properties of the chemical functions grafted onto the cryptophane skeleton. In the example reported here, a change in the chemical properties of the phenol groups strongly affects the ability of compound syn-2 to accommodate cationic species in basic solutions as easily as its diastereomer anti-1. Thus, the difficulty for the syn-2 compound to have all six phenol groups fully deprotonated can be suggested to explain the poor binding properties of this compound. As a result, in its ability to bind cationic species, the syn-2 compound has more similarities with a previous cryptophane decorated with only three phenolic functions that moderately binds Cs+ and Tl+ than with its anti-1 diastereomer that shows a very high affinity for these two species.
conclusions
We report the synthesis and the characterization of cryptophane syn-2 as well as its crystal structure. This compound is the diastereomer molecule of cryptophane anti-1, which is known to bind very well the cesium and thallium(I) cations in an aqueous solvent. However, despite the structural similarities that exist between these two compounds, our study reveals that from a general point of view, the physical properties of these two derivatives are significantly different. We noticed, for instance, that these two hosts show large differences in their binding properties toward the Cs+ and the Tl+ cations. Indeed, isothermal calorimetric titration experiments performed in LiOH (0.1 M)/H2O or in NaOH/H2O-KCl buffer reveal that the syn-2 compound has a decreased affinity of several orders of magnitude for these two cations compared to the anti-1 diastereomer studied under the same conditions. In addition, forward and reverse titration experiments of new derivative syn-2 were found more sensitive to small pH variations compared to the anti-1 derivative. To explain this behavior, a larger difference in the pKa of the phenol functions grafted onto the syn-2 derivative is assumed compared to the anti-1 derivative in which the phenol functions can be more easily ionized at the same pH. This result shows that a change in the linker arrangement not only alters the cavity size and flexibility of the host but also changes the chemical properties of the substituents grafted onto the cryptophane backbone. In turn, this leads to a significant change in the molecular recognition properties of the host.
Overall, the results presented in this paper, in combination with previous studies, show that cryptophanes with an anti-configuration exhibit binding of Cs+ and Tl+ cations stronger than that of syn-congeners. Indeed, while the greater rigidity of syn-congeners has most often been cited to explain the weaker binding properties of syn-cryptophanes, a change in the physical properties of the substituents grafted onto the benzene rings may also have a strong impact on the binding properties of these derivatives.
Experimental Details
Mass Spectroscopy
Mass spectra (HRMS) were obtained by the Centre de Spectrométrie de Masse, University of Lyon. Analyses were performed with a hybrid quadrupole-time-of-flight mass spectrometer, microToF QII equipped with an electrospray ion source. Data Analysis 4.0 was used for instrument control, data collection, and data treatment. HRMS analyses were performed in full-scan MS with a mass range from 50 to 2000 Da at an acquisition rate of 1 Hz. Transfer parameters were as follows: RF Funnel 1, 200 V; RF Funnel 2, 200 V; hexapole, 50 V; transfer time, 70 μs; and PrePulse storage time, 1 μs. Before each acquisition batch, external calibration of the instrument was performed with a sodium formate clusters solution. 1H and 13C NMR spectra were recorded at 300 and 75.5 MHz, respectively. Chemical shifts were referenced to Me4Si (1H and 13C). Column chromatographic separations were carried out over Merck silica gel 60 (0.040–0.063 mm). Analytical thin-layer chromatography (TLC) was performed on Merck silica gel TLC plates, F-254. The solvents were distilled prior to use: DMF and CH2Cl2 from CaH2, THF from Na/benzophenone, and pyridine from KOH.
Single-Crystal X-ray Diffraction
Single-crystals were selected and mounted on an Xcalibur Gemini kappa-geometry diffractometer equipped with an Atlas CCD. Data collections were achieved at 150 K using copper X-ray radiation (l = 1.54184 Å) for anti-1, syn-2, and syn-3 and molybdenum radiation (l = 0.71073 Å) for the second X-ray structure of syn-2. Intensities were collected by means of the CrysalisPro software.18 Reflection indexing, unit-cell parameter refinement, Lorentz-polarization correction, peak integration, and background determination were carried out with the CrysalisPro software.18 An analytical absorption correction was applied using the modeled faces of the crystal.19 The resulting set of hkl was used for structure solution and refinement. The structures were solved with the ShelXT structure solution program using the intrinsic phasing solution method and by using Olex2 as the graphical interface.20,21 The model was refined with version 2018/3 of ShelXL using least-squares minimization.22
CCDC 2152416-2152417-2152418-2152419 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Date Centre via www.ccdc.cam.ac.uk/data_request/cif.
133Cs NMR Spectroscopy
These experiments were performed on a 11.7 T narrow-bore Bruker spectrometer equipped with a 5 mm BBI probehead and z-axis gradient.
Acknowledgments
The French National Research Agency (ANR) is acknowledged for financial support (project ANR19-CE19-0024 PHOENIX).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c06570.
1H and 13C NMR spectra of compound syn-2, DLS of syn-2 in basic solution, and ITC experiments of anti-1 and syn-2 in LiOH (0.1 M)/H2O and NaOH/H2O-KCl (buffer) (forward and reverse experiments) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Campanella B.; Colombaioni L.; Benedetti E.; Di Ciaula A.; Ghezzi L.; Onor M.; D’Orazio M.; Giannecchini R.; Petrini R.; Bramanti E. Toxicity of Thallium at Low Doses: A Review. Int. J. Environ. Res. Public Health 2019, 16, 4732–4737. 10.3390/ijerph16234732. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Xiao T.; Yang F.; Li S.; Zheng B.; Ning Z. Thallium Pollution in China: A Geo-Environmental Perspective. Sci. Total Environ. 2012, 421–422, 51–58. 10.1016/j.scitotenv.2011.04.008. [DOI] [PubMed] [Google Scholar]; c Peter A. L. J.; Viraraghavan T. Thallium: A Review of Public Health and Environmental Concerns. Environ. Int. 2005, 31, 493–501. 10.1016/j.envint.2004.09.003. [DOI] [PubMed] [Google Scholar]
- a Cvjetko P.; Cvjetko I.; Pavlica M. Thallium Toxicity in Humans. Arch. Environ. Con. Toxicol. 2010, 61, 111–118. 10.2478/10004-1254-61-2010-1976. [DOI] [PubMed] [Google Scholar]; b Saha A. Thallium Toxicity: A Growing Concern. Indian J. Occup. Health 2005, 9, 53–56. 10.4103/0019-5278.16741. [DOI] [Google Scholar]; c Kazantzis G. Thallium in the Environment and Health Effects. Environ. Geochem. Health 2000, 22, 275–280. 10.1023/A:1006791514080. [DOI] [Google Scholar]
- a López-García I.; Muñoz-Sandoval M. J.; Hernández-Córdoba M. Dispersive Micro-Solid Phase Extraction with a Magnetic Nanocomposite Followed by Electrothermal Atomic Absorption Measurement for the Speciation of Thallium. Talanta 2021, 228, 122206 10.1016/j.talanta.2021.122206. [DOI] [PubMed] [Google Scholar]; b Liu J.; Luo X.; Sun Y.; Tsang D. C. W.; Qi J.; Zhang W.; Li N.; Yin M.; Wang J.; Lippold H.; Chen Y.; Sheng G. Thallium Pollution in China and Removal Technologies for Waters: A Review. Environ. Int. 2019, 126, 771–790. 10.1016/j.envint.2019.01.076. [DOI] [PubMed] [Google Scholar]; c Li H.; Li X.; Chen Y.; Long J.; Zhang G.; Xiao T.; Zhang P.; Li C.; Zhuang L.; Huang W. Removal and Recovery of Thallium from Aqueous Solutions via a Magnetite-Mediated Reversible Adsorption-Desorption Process. J. Cleaner Prod. 2018, 199, 705–715. 10.1016/j.jclepro.2018.07.178. [DOI] [Google Scholar]; d Li H.; Li X.; Xiao T.; Chen Y.; Long J.; Zhang G.; Zhang P.; Li C.; Zhuang L.; Li K. Efficient Removal of Thallium(I) from Wastewater Using Flower-Like Manganese Dioxide Coated Magnetic Pyrite Cinder. Chem. Eng. J. 2018, 353, 867–877. 10.1016/j.cej.2018.07.169. [DOI] [Google Scholar]; e Huangfu X.; Jiang J.; Lu X.; Wang Y.; Liu Y.; Pang S.-Y.; Cheng H.; Zhang X.; Ma J. Adsorption and Oxidation of Thallium(I) by a Nanosized Manganese Dioxide. Water, Air, Soil Pollut. 2014, 226, 2272. 10.1007/s11270-014-2272-7. [DOI] [Google Scholar]
- a Frei A.; Rigby A.; Yue T. T.; Firth G.; Ma M. T.; Long N. J. To Chelate Thallium(I) - Synthesis and Evaluation of Kryptofix-Based Chelators for 201Tl. Dalton Trans. 2022, 51, 9039–9048. 10.1039/D2DT01074G. [DOI] [PubMed] [Google Scholar]; b Zhai H.; Xiong S.; Peng S.; Sheng W.; Xu G.; Sessler J. L.; He Q. Thallium(I) Salts: New Partners for Calix[4]Pyrroles. Org. Lett. 2021, 23, 2638–2642. 10.1021/acs.orglett.1c00555. [DOI] [PubMed] [Google Scholar]; c Zhao Z.; Xiong Y.; Cheng X.; Hou X.; Yang Y.; Tian Y.; You J.; Xu L. Adsorptive Removal of Trace Thallium(I) from Wastewater: A Review and New Perspectives. J. Hazard. Mater. 2020, 393, 122378 10.1016/j.jhazmat.2020.122378. [DOI] [PubMed] [Google Scholar]; d Zhao Z.; Tian H.; Zhang M.; Yang Y.; Zhang H. Molecular Design of Macrocyclic Compounds for Complete Removal of Thallium(I) from Wastewater. Environ. Sci. Pollut. Res. 2018, 25, 34550–34558. 10.1007/s11356-018-3393-0. [DOI] [PubMed] [Google Scholar]; e Makrlík E.; Dybal J.; Vaňura P. Interaction of the Thallium Cation with 1,3-Alternate-25,27-Bis(1-Octyloxy)Calix[4]Arene-Crown-6: Experimental and Theoretical Study. J. Mol. Struct. 2013, 1042, 73–77. 10.1016/j.molstruc.2013.03.025. [DOI] [Google Scholar]; f Chester R. T.; Couton D.; Lobler R.; Mocerino M.; Ogden M. I.; Pettersen J. K.; Skelton B. W.; White A. H. The extraction of thallium(I) and silver(I) ions with 1,3-alternate calix[4]arene derivatives. J. Inclusion Phenom. Macrocyclic Chem. 2011, 71, 471–477. 10.1007/s10847-011-0010-y. [DOI] [Google Scholar]; g Haddadi H.; Alizadeh N.; Shamsipur M.; Asfari Z.; Lippolis V.; Bazzicalupi C. Cation-π interaction in Complex Formation Between Tl+ Ion and calix[4]crown-6 and Some calix[4]biscrown-6 Derivatives: Thallium-203 NMR, Proton NMR, and X-ray Evidence. Inorg. Chem. 2010, 49, 6874–6882. 10.1021/ic100097v. [DOI] [PubMed] [Google Scholar]; h Cuc D.; Bouguet-Bonnet S.; Morel-Desrosiers N.; Morel J.-P.; Mutzenhardt P.; Canet D. Behavior of Cesium and Thallium Cations inside a Calixarene Cavity as Probed by Nuclear Spin Relaxation. Evidence of Cation−π Interactions in Water. J. Phys. Chem. B 2009, 113, 10800–10807. 10.1021/jp902787n. [DOI] [PubMed] [Google Scholar]; i Roper E. D.; Talanov V. S.; Butcher R. J.; Talanova G. G. Selective Recognition of Thallium(I) by 1,3-Alternate Calix[4]arene-bis(crown-6 Ether) : A New Talent of the Known Ionophore. Supramol. Chem. 2008, 20, 217–229. 10.1080/10610270701803074. [DOI] [Google Scholar]; j Talanova G. G.; Roper E. D.; Buie N. M.; Gorbunova M. G.; Bartsch R. A.; Talanov V. S. Novel Fluorogenic Calix[4]arene-bis(crown-6-Ether) for Selective Recognition of Thallium(I). Chem. Commun. 2005, 5673–5675. 10.1039/b510348g. [DOI] [PubMed] [Google Scholar]; k Matthews S. E.; Rees N. H.; Felix V.; Drew M. G. B.; Beer P. D. Thallium π-Cation Complexation by Calix[4]tubes: 205Tl NMR and X-ray Evidence. Inorg. Chem. 2003, 42, 729–734. 10.1021/ic025884x. [DOI] [PubMed] [Google Scholar]; l Parham H.; Shamsipur M. Spectrofluorometric Study of Thallium (I) Complexes with Several Macrocyclic Ligands in Methanol Solution. Talanta 1993, 40, 1353–1356. 10.1016/0039-9140(93)80209-A. [DOI] [PubMed] [Google Scholar]; m Yang-Chih L.; Allison J.; Popov A. I. Thallium-205 NMR Study of Tl(I) Macrocyclic Complexes in Nonaqueous Solvents. Polyhedron 1985, 4, 441–445. 10.1016/S0277-5387(00)87006-0. [DOI] [Google Scholar]; n Shamsipur M.; Popov A. I. Multinuclear Nmr Study of Some Alkali and Thallium Complexes with 1,10-Diaza-18-Crown-6 in Nonaqueous Solutions. Inorg. Chim. Acta 1980, 43, 243–247. 10.1016/S0020-1693(00)90537-1. [DOI] [Google Scholar]; o Shamsipur M.; Rounaghi G.; Popov A. I. Sodium-23, Cesium-133 and Thallium-205 NMR Study of Sodium, Cesium and Thallium Complexes with Large Crown Ethers in Nonaqueous Solutions. J. Solution Chem. 1980, 9, 701–714. 10.1007/BF00645898. [DOI] [Google Scholar]
- a Izosimova Y.; Gurova I.; Tolpeshta I.; Karpukhin M.; Zakusin S.; Zakusina O.; Samburskiy A.; Krupskaya V. Adsorption of Cs(I) and Sr(II) on Bentonites with Different Compositions at Different PH. Minerals 2022, 12, 862. 10.3390/min12070862. [DOI] [Google Scholar]; b Jiménez-Reyes M.; Almazán-Sánchez P. T.; Solache-Ríos M. Radioactive Waste Treatments by Using Zeolites. A Short Review. J. Environ. Radioact. 2021, 233, 106610 10.1016/j.jenvrad.2021.106610. [DOI] [PubMed] [Google Scholar]; c Estelrich J.; Busquets M. A. Prussian Blue: A Safe Pigment with Zeolitic-Like Activity. Int. J. Mol. Sci. 2021, 22, 780. 10.3390/ijms22020780. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wang J.; Zhuang S. Removal of Cesium Ions from Aqueous Solutions Using Various Separation Technologies. Rev Environ Sci Biotechnol 2019, 18, 231–269. 10.1007/s11157-019-09499-9. [DOI] [Google Scholar]; e Figueiredo B. R.; Cardoso S. P.; Portugal I.; Rocha J.; Silva C. M. Inorganic Ion Exchangers for Cesium Removal from Radioactive Wastewater. Sep. Purif. Rev. 2018, 47, 306–336. 10.1080/15422119.2017.1392974. [DOI] [Google Scholar]; f Johan E.; Yamada T.; Munthali M. W.; Kabwadza-Corner P.; Aono H.; Matsue N. Natural Zeolites as Potential Materials for Decontamination of Radioactive Cesium. Procedia Environ. Sci. 2015, 28, 52–56. 10.1016/j.proenv.2015.07.008. [DOI] [Google Scholar]; g Zhaoyi T.; Zhaoya H.; Dong Z.; Xiaolin W. Structural Characterization of Ammonium Molybdophosphate with Different Amount of Cesium Adsorption. J. Radioanal. Nucl. Chem. 2014, 299, 1165–1169. 10.1007/s10967-013-2843-x. [DOI] [Google Scholar]
- see for instance:; a Wang J.; Zhuang S. Cesium Separation from Radioactive Waste by Extraction and Adsorption Based on Crown Ethers and Calixarenes. Nucl. Eng. Technol. 2020, 52, 328–336. 10.1016/j.net.2019.08.001. [DOI] [Google Scholar]; b Pichierri F. Macrocycles for the Complexation of Radiocesium: A Concise Review of Crystallographic and Computational Studies. J. Radioanal. Nucl. Chem. 2017, 311, 1251–1263. 10.1007/s10967-016-4968-1. [DOI] [Google Scholar]; c Albelda M. T.; Frías J. C.; García-España E.; Schneider H.-J. Supramolecular Complexation for Environmental Control. Chem. Soc. Rev. 2012, 41, 3859–3877. 10.1039/c2cs35008d. [DOI] [PubMed] [Google Scholar]; d Mokhtari B.; Pourabdollah K.; Dallali N. A Review of Calixarene Applications in Nuclear Industries. J. Radioanal. Nucl. Chem. 2010, 287, 921–934. [Google Scholar]; e Wintergerst M. P.; Levitskaia T. G.; Moyer B. A.; Sessler J. L.; Delmau L. H. Calix[4]Pyrrole: A New Ion-Pair Receptor As Demonstrated by Liquid–Liquid Extraction. J. Am. Chem. Soc. 2008, 130, 4129–4139. 10.1021/ja7102179. [DOI] [PubMed] [Google Scholar]; f Casnati A.; Pochini A.; Ungaro R.; Ugozzoli F.; Arnaud F.; Fanni S.; Schwing M.-J.; Egberink R. J. M.; de Jong F.; Reinhoudt D. N. Synthesis, Complexation, and Membrane Transport Studies of 1,3-Alternate Calix[4]Arene-Crown-6 Conformers: A New Class of Cesium Selective Ionophores. J. Am. Chem. Soc. 1995, 117, 2767–2777. 10.1021/ja00115a012. [DOI] [Google Scholar]; g Mei E.; Popov A. I.; Dye J. L. Complexation of the Cesium Cation by Macrocyclic Polyethers in Various Solvents. A Cesium-133 Nuclear Magnetic Resonance Study of the Thermodynamics and Kinetics of Exchange. J. Phys. Chem. 1977, 81, 1677–1681. 10.1021/j100532a015. [DOI] [Google Scholar]
- a Brotin T.; Berthault P.; Pitrat D.; Mulatier J.-C. Selective capture of Thallium and Cesium by a Cryptophane Soluble at neutral pH. J. Org. Chem. 2020, 85, 9622–9630. 10.1021/acs.joc.0c00950. [DOI] [PubMed] [Google Scholar]; b Brotin T.; Goncalves S.; Berthault P.; Cavagnat D.; Buffeteau T. Influence of the Cavity Size of Water-Soluble Cryptophanes on Their Binding Properties for Cesium and Thallium Cations. J. Phys. Chem B 2013, 117, 12593–12601. 10.1021/jp4074009. [DOI] [PubMed] [Google Scholar]; c Brotin T.; Cavagnat D.; Berthault P.; Montserret R.; Buffeteau T. Water-Soluble Molecular Capsule for the Complexation of Cesium and Thallium Cations. J. Phys. Chem. B 2012, 116, 10905–10914. 10.1021/jp305110k. [DOI] [PubMed] [Google Scholar]; d Brotin T.; Montserret R.; Bouchet A.; Cavagnat D.; Linares M.; Buffeteau T. High Affinity of Water-Soluble Cryptophanes for Cesium Cations. J. Org. Chem. 2012, 77, 1198–1201. 10.1021/jo202259r. [DOI] [PubMed] [Google Scholar]
- Brotin T.; Dutasta J.-P. Cryptophanes and Their Complexes—Present and Future. Chem. Rev. 2009, 109, 88–130. 10.1021/cr0680437. [DOI] [PubMed] [Google Scholar]
- a Doll M.; Berthault P.; Léonce E.; Boutin C.; Jeanneau E.; Brotin T.; De Rycke N. Study of syn and anti Xenon-Cryptophanes Complexes Decorated with Aromatic Amine Groups: Chemical Platforms for Accessing New Cryptophanes. J. Org. Chem. 2022, 87, 2912–2920. 10.1021/acs.joc.1c02774. [DOI] [PubMed] [Google Scholar]; b Doll M.; Berthault P.; Léonce E.; Boutin C.; Buffeteau T.; Daugey N.; Vanthuyne N.; Jean M.; Brotin T.; De Rycke N. Are the Physical Properties of Xe@Cryptophane Complexes Easily Predictable? The Case of syn- and anti-Tris-aza-Cryptophanes. J. Org. Chem. 2021, 86, 7648–7658. 10.1021/acs.joc.1c00701. [DOI] [PubMed] [Google Scholar]; c Canceill J.; Césario M.; Collet A.; Guilhem J.; Riche C.; Pascard C. Selective recognition of neutral molecules: 1H n.m.r. Study of the complexation of CH2Cl2 and CH2Br2 by cryptophane-D in solution and crystal structure of its CH2Cl2 cavitate. J. Chem. Soc., Chem. Commun. 1986, 339–341. 10.1039/c39860000339. [DOI] [Google Scholar]
- Brotin T.; Jeanneau E.; Berthault P.; Léonce E.; Pitrat D.; Mulatier J.-C. Synthesis of Cryptophane-B: Crystal Structure and Study of Its Complex with Xenon. J. Org. Chem. 2018, 83, 14465–14471. 10.1021/acs.joc.8b02246. [DOI] [PubMed] [Google Scholar]
- Spek L. A. PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr., Sect. A: Found. Crystallogr.allogr., Sect. C: Struct. Chem. 2015, C71, 9–18. 10.1107/S2053229614024929. [DOI] [PubMed] [Google Scholar]
- a Herrera I.; Winnik M. A. Differential Binding Models for Direct and Reverse Isothermal Titration Calorimetry. J. Phys. Chem. B 2016, 120, 2077–2086. 10.1021/acs.jpcb.5b09202. [DOI] [PubMed] [Google Scholar]; b Grolier J.-P. E.; Del Rio J. M. Isothermal Titration Calorimetry: Application of the Gibbs–Duhem Equation to the Study of the Relationship Between Forward and Reverse Titrations. J. Solution Chem. 2015, 44, 987–1003. 10.1007/s10953-014-0243-7. [DOI] [Google Scholar]
- Léonce E.; Brotin T.; Berthault P. syn-Cryptophanes: macrocyclic compounds with optimized characteristics for the design of 129Xe NMR-based biosensors. Phys.Chem.Chem.Phys. 2022, 24, 24793–24799. 10.1039/D2CP03714A. [DOI] [PubMed] [Google Scholar]
- Canceill J.; Cesario M.; Collet A.; Guilhem J.; Riche C.; Pascard C. Selective Recognition of Neutral Molecules: 1H NMR Study of the Complexation of CH2Cl2 and CH2Br2 by Cryptophane-D in Solution and Crystal Structure of Its CH2Cl2 Cavitate. J. Chem. Soc., Chem. Commun. 1986, 339–341. 10.1039/c39860000339. [DOI] [Google Scholar]
- Jacques J.; Collet A.; Wilen S. H.. Enantiomers, Racemates, and Resolutions; Wiley: New York, 1981. [Google Scholar]
- Chapellet L. L.; Dognon J.-P.; Jean M.; Vanthuyne N.; Berthault P.; Buffeteau T.; Brotin T. Experimental and Theoretical Study of the Complexation of Cesium and Thallium Cations by a Water-Soluble Cryptophane. ChemistrySelect 2017, 2, 5292–5300. 10.1002/slct.201700979. [DOI] [Google Scholar]
- Eggers D. K.; Fu S.; Ngo D. V.; Vuong E. H.; Brotin T. Thermodynamic Contribution of Water in Cryptophane Host–Guest Binding Reaction. J. Phys. Chem. B 2020, 124, 6585–6591. 10.1021/acs.jpcb.0c05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaku Oxford Diffraction, CrysAlis(Pro) Software system, version 1.171.40.67a, Rigaku Corporation: Oxford, UK. 2019 [Google Scholar]
- Clark R. C.; Reid J. S. The analytical calculation of absorption in multifaceted crystals. Acta Crystallogr., Sect. A: Found. Crystallogr. 1995, A51, 887–897. 10.1107/S0108767395007367. [DOI] [Google Scholar]
- Sheldrick G. M. SHELXT – Integrated space-group and crystal-structure determination. Acta Crystallogr., Sect. A: Found. Crystallogr. 2015, A71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. A: Found. Crystallogr.allogr., Sect. C: Struct. Chem. 2015, C27, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.