

Abstract
The respiratory system is continuously exposed to variety of biological and chemical irritants that contain reactive oxygen species, and these are well known to cause oxidative stress responses in lung epithelial cells. There is a clinical need to identify biomarkers of oxidative stress which could potentially support early indicators of disease and health management. To identify volatile biomarkers of oxidative stress, we analyzed the headspace above human bronchial epithelial cell cultures (HBE1) before and after hydrogen peroxide (H2O2) and cigarette smoke extract (CSE) exposure. Using stir bar and headspace sorptive extraction-gas chromatography-mass spectrometry, we searched for volatile organic compounds (VOC) of these oxidative measures. In the H2O2 cell peroxidation experiments, four different H2O2 concentrations (0.1, 0.5, 10, 50 mM) were applied to the HBE1 cells, and VOCs were collected every 12 h over the time course of 48 h. In the cigarette smoke extract cell peroxidation experiments, four different smoke extract concentrations (0, 10, 30, 60 %) were applied to the cells, and VOCs were collected every 12 h over the time course of 48 h. We used partial-least squares (PLS) analysis to identify putative compounds from the mass spectrometry results that highly correlated with the known applied oxidative stress. We observed chemical emissions from the cells that related to both the intensity of the oxidative stress and followed distinct time courses. Additionally, some of these chemicals are aldehydes, which are thought to be non-invasive indicators of oxidative stress in exhaled human breath. Together, these results illustrate a powerful in situ cell culture model of oxidative stress that can be used to explore the putative biological genesis of exhaled breath biomarkers that are often observed in human clinical studies.
Keywords: breath analysis, aldehydes, oxidative stress, peroxidation, mass spectrometry
Introduction
The lungs are continuously exposed to a variety of environmental chemicals and potential insults, ranging from particulate matter to potentially oxidizing compounds [1]. This includes subsets of reactive oxygen species (ROS) such as superoxide anions (O2・−), alkoxyl (RO・), alkylperoxyl (ROO・) hydroxyl (・OH) free radicals and hydrogen peroxide (H2O2) [2]. Hydrogen peroxide is one the most stable of the reactive oxygen species and tightly regulated under physiological conditions along with other ROSs. These endogenously produced ROSs play important roles in various biological processes under low concentration, such as intracellular signaling [3], respiration [4], immune cell activation and vascular remodeling in mammals [5, 6]. However, exogenously produced H2O2 exposes cells to high concentrations of ROS, and is implicated as an important mediator of cellular toxicity [7]. Previous studies have shown that oxidants like H2O2 cause oxidative stress in airway epithelial cells [2, 7, 8]. Oxidative stress, which refers to a state of elevated levels of ROS, occurs from a variety of conditions that produce unbalanced ROS production or a decline in antioxidant defenses [9]. These oxidants are reported to damage cell components including proteins, lipids, and nucleic acids [10–12]. Moreover, proteins can be modified by aldehydes and ketones produced during reactions of ROS with lipids [13–15].
One critically important source of exogenous H2O2 into the lung is cigarette smoke [2, 8, 16], a well-studied contributor that initiates development of many smoking-related diseases. It disrupts many biological processes partially by exposing the lung to ROS, including H2O2 in a complex smoke matrix [2, 8, 16–18]. Prior research has looked at many different cellular changes and damage that occurs due to cigarette smoke exposure [19–23], but very little is known about the cellular level volatile organic compound (VOC) metabolite emissions that result from this exposure.
Oxidative stress is implicated with host immune-inflammatory processes and infectious diseases as well [24, 25]. In earlier work, we cultured human B-cells in situ and monitored VOCs that were released before and after infection with various influenza virus strains [26, 27]. We observed several biomarkers (alcohols, ketones, aldehydes, and other aromatic compounds) that were produced after virus infection, and some of these were specific to the infecting strain of virus. Some of the VOCs were emitted over specific time courses, suggesting that viral infection stimulated certain cell pathways as a mechanism of action. A key finding was oxidative stress markers that was present in all virus infections, and the abundance was correlated to the multiplicity of infection (MOI) used to infect the cells. Similar generic oxidative stress biomarkers were also observed by Phillips, et al. after influenza vaccines were administered to normal healthy subjects [28]. In this latter case, a live attenuated vaccine was used to stimulate the immune response, which may have contributed to minor variations in the specific oxidative stress biomarker signals measured. Ahmed et al. observed significant upregulation of heptanol, propanal and benzeneacetaldehyde active in Crohn’s disease patients [29], and those VOCs may have been produced during the inflammatory processes as a result of lipid peroxidization and oxidative stress [30]. However, the detailed pathway and response mechanism that produced these exact VOC biomarkers at the cellular level is still unclear. Other researchers have replicated some of these findings from in situ cell culture studies [31, 32] and in vivo animal models [33]. We seek to further define a cell-based assay to elucidate some of these mechanisms that translate between cell damage and VOC release.
In the field of breath analysis, we exploit both volatile and non-volatile exhaled biomarkers when they associate with disease or health states. These breath biomarkers are thought to be endogenously produced and partitioned into the breath through two potential processes: diffusion equilibrium between blood and exhaled respiratory gasses in the capillary bed compartment of the lung (with the origin of the biomarker systemically elsewhere in the body) and generation originating directly from the airway epithelial cell surfaces/ in the lung itself. We believe both pathways are probably active, and our present work aims to reveal the potential oxidative stress markers that can be generated directly from lung cells themselves. Given that the airway epithelium is the first line of defense in the lung against agents such as cigarette smoke, this paper investigates the effects of H2O2 and cigarette smoke directly on airway epithelial cells.
Material and Methods
We previously published an initial protocol paper with details of our experimental procedure for growing an immortalized line of human bronchial epithelial cells and the process of monitoring emitted volatile organic compounds [34]. These are briefly described again below, along with new details on the protocols we developed for the H2O2 and smoke extract oxidative stress assays. Research Cigarettes (3R4F) were purchased from the Tobacco Research Institute (University of Kentucky, Kentucky, USA).
Cell Culture Conditions
Immortalized human bronchial epithelial HBE1 cells [35] were plated on Transwell (Coring Costar, Corning, NY) 12 mm chambers at 1–2 × 104 cells/cm2 in LHC Basal Medium (Life Technologies, Carlsbad, CA) supplemented with: insulin (5 µg/mL), transferrin (10 µg/mL), epidermal growth factor (24 ng/mL), hydrocortisone (0.1 µM), triiodothyronine (0.01 µM), bovine hypothalamus extract (10 µg/mL), bovine serum albumin (0.5 mg/mL), epinephrine (0.6 µg/mL), phosphorylethanolamine (0.5 µM), ethanolamine (0.5 µM), zinc sulfate (3 µM), ferrous sulfate (1.5 µM), magnesium chloride (0.6 mM), calcium chloride (0.11 mM) and trace elements (selenium, manganese, silicone, molybdenum, vanadium, nickel sulfate and tin) [36]. All additives were purchased from Sigma Aldrich (St. Louis, MO). Confluent HBE1 cultures were transferred to ALI culture conditions in Pneumacult ALI medium (StemCell Technologies, Vancouver, BC) for 5–7 days before experiments began.
Cell metabolic activity was quantified using an alamarBlue® cell viability reagent per manufacturer recommendations (ThermoFisher) to ensure cells were alive and viable for measurements.
VOC Extraction Protocol
We utilized the same VOC extraction protocol previously published [34], and is briefly described here. Confluent cells were placed into sterile borosilicate glass jars and capped, with the apical and basolateral surfaces bathed in their appropriate media, Opti-MEM (Life Technologies, Carlsbad, CA). The internal standard dacane-22 was dissolved at 1 µL of standard solution per 5 mL of cell media. Twisters® (Part 011222-001-00, Gerstel, Inc., Linthicum, MD) were conditioned and sterilized in 70% aqueous ethanol prior to introduction to cultures, and sampling was achieved using the magnetized surface of the jar lids at 37 °C. Experiments were started at t=0 h. Twelve hours extractions were removed at t=12 h, etc. Cells were confluent at t=0 but were also undergoing normal senescence over the duration of each experiment. During the sampling period, the lids were left ajar for 3 min every 12 h to allow for normal cellular respiration.
Hydrogen Peroxide Oxidative Stress Assay
Concentrated H2O2 stock solution (30%, EMD Millipore) was diluted into Opti-MEM medium to concentrations of 0.1, 0.5, 10, and 50 mM H2O2. Monolayer cultures in transwells were exposed to the various H2O2 concentrations to measure VOC dose responses over timepoints of 12, 24, 36 and 48 hours at 37°C. (Figure 1A). Cell VOCs were collected with Twisters® every 12 h until 48 h elapsed (Figure 1B). Two medium-only controls were used: Opti-MEM medium and Opti-medium with H2O2. Two cell culture treatments were also used: cell cultures with Opti-medium only and cell culture with Opti-medium with H2O2. Jar blank samples were also collected at each time point to provide background controls. Each treatment included two jar biological replicates and two Twister® technical replicates. The cell viability assay was conducted after 24 h in each concentration of H2O2 exposure, and was measured three times among three transwells.
Figure 1.
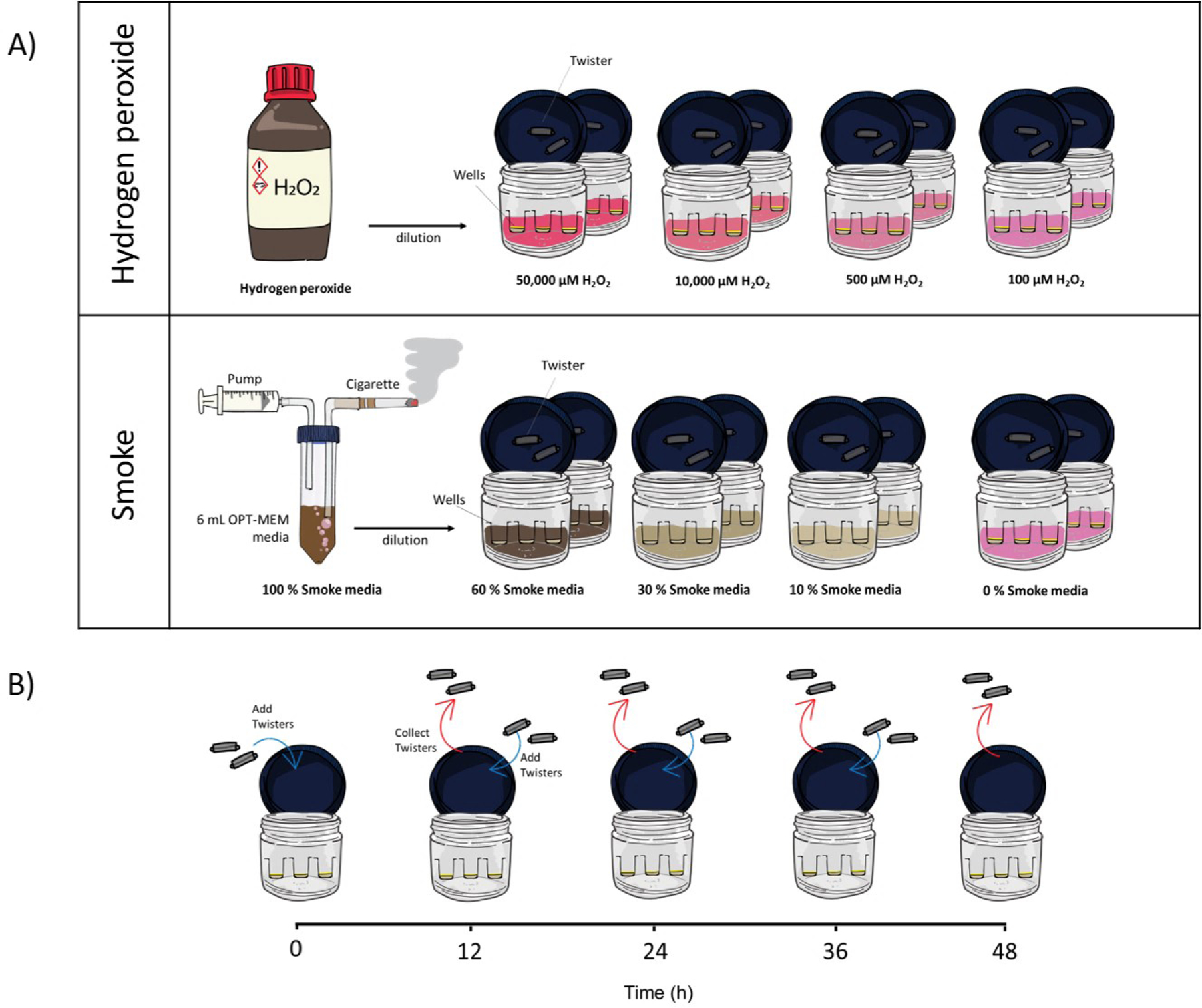
Scheme of the work
Cigarette Smoke Extract (CSE) Oxidative Stress Assay
Researchers have previously reported methods to generate cigarette smoke extract (CSE) medium that can be used to expose cell cultures [37, 38], and we adopted our method from these prior studies. Cigarette smoke extract (CSE) was prepared daily in a custom designed smoking apparatus. Briefly, a cigarette was attached to a 3-way stopcock attached to a 60 mL syringe containing 6 mL of Opti-medium, once the cigarette was lit, mainstream smoke was withdrawn from the cigarette under negative pressure from the syringe and passed through the 6 mL of Opti-medium within the syringe. The cigarette was smoked to within 3 mm of the cigarette filter, with an average burn time of 7 min. After the cigarette was removed, the medium solution containing the extra was incubated at 37 °C for 30 min, and the solution was filtered (0.22 μm) and used immediately. The CSE absorbance was measured (OD 1.1, 320 nM) using a spectrophotometer, and this resulting extract was designated as 100 % CSE. Dilutions were performed in Opti-medium to 10, 30, 60 % CSE (Figure 1A). Cell VOCs were collected with Twister® for every 12 h until 48 h (Figure 1B). The medium control was defined as 0 % CSE (100 % Opti-medium). Two cell culture treatments were also applied to the experiment; cell culture exposed to Opti-medium alone and cell culture exposed to CSE at various concentrations. Each treatment had two jar biological replicates and two Twister® technical replicates. The cell viability assay was conducted after 48 h in each concentration of CSE exposure, and was measured three times among three transwells.
GC-MS Analysis
After VOC extractions, Twisters® were loaded into thermal desorption tubes. A 1.0 µL aliquot of a 10 ppm naphthalene-d8 solution in 100% ethanol was added to each tube as a second internal standard.
Cellular VOCs are desorbed from Twisters® using a thermal desorption unit (TDU) and cooled injection system (Gerstel, Inc.) and are analyzed using a GC-MS system as previously noted [34]. Briefly, the TDU heats a Twister® to 300 °C and holds for 5 min. As VOCs desorb from the Twister®, they are lead via a flow of helium to the CIS, which is held at −80 °C. After desorption, the CIS quickly heats to 300 °C, splitlessly injecting the VOCs onto the GC column.
Analytes are separated on an Agilent 7890B GC (Agilent Technologies Inc., Santa Clara, CA) with a DB-5ms column (20 m x 180 μm x 0.18 μm, Part 221-5522LTM, Agilent Technologies, Inc.). The oven was programmed as follows: initial temperature 35 °C for 3 min, then a ramp of 5 °C/min to 75 °C, then 2 °C/min to 220 °C and a final ramp of 30 °C/min to 300 °C, holding for 3 min. The transfer line was set at 280 °C, leading into an Agilent 5977A mass spectrometer. There was a solvent delay of 5 min. The MS scanned the range from 33 to 520 m/z. The MS source was set to 230 °C and the MS quad set to 150 °C. A standard mix of C8-C20 alkanes was analyzed with every batch of samples to calculate the Kovats Retention Indices and also to monitor the performance of the TDU-CIS-GC-MS system, in addition to the two internal standards used in every sample.
Data Analysis Workflow
The GC-MS data files were deconvoluted, integrated and aligned using MassHunter Profinder B.08.00 (Agilent Technologies, Inc.). Compounds with siloxane base peaks (73, 147, 207, 221 and 281 m/z) were removed as they are artefacts from the PDMS sorbent. Compounds found in less than 60% of replicates from one treatment were also excluded from analysis. Peak areas were normalized to the naphthalene-d8 internal standard. Compounds were tentatively identified by calculating their Kovats Retention Index in comparison to reported literature values and by comparison of extracted mass spectra to the NIST 2014 mass spectral library.
Data analyses were performed on MATLAB R2017a software (Mathworks, Natick, MA), PLS_Toolbox (Version 8.6, Eigenvector Research Inc., Manson, WA) and Agilent’s GeneSpring (Version B.14.9), with p-value = 0.05 throughout. Specifically, PLS regressions were built using PLS_Toolbox. Presented are the results of cross validation only, in which the PLS model predicts the concentration of the oxidative stressor in the media (e.g. 10% CSE or 50 mM H2O2) based on the volatile profile of that sample. Predicted concentrations are plotted against the known, or actual, concentration.
Results and Discussion
H2O2 Effects on VOC Emissions
Viability Assay for H2O2 Exposure
The alamarBlue® Assay incorporates a fluorometric/colorimetric growth indicator based on detection of oxidation-reduction in cell metabolic activity. An ANOVA was applied to 6 treatments (5 H2O2 concentrations and one control; Figure 2). The assay found that the 0, 0.1 and 0.5 mM peroxidation exhibited similar levels of metabolic activity, indicating that there was no mortality in these three peroxidation treatments. Cells that received the 10 mM level of exposure exhibited a significant drop in metabolic activity relative to healthy controls, indicated that this concentration of H2O2 was stressful enough to cause mortality in some cells.
Figure 2.
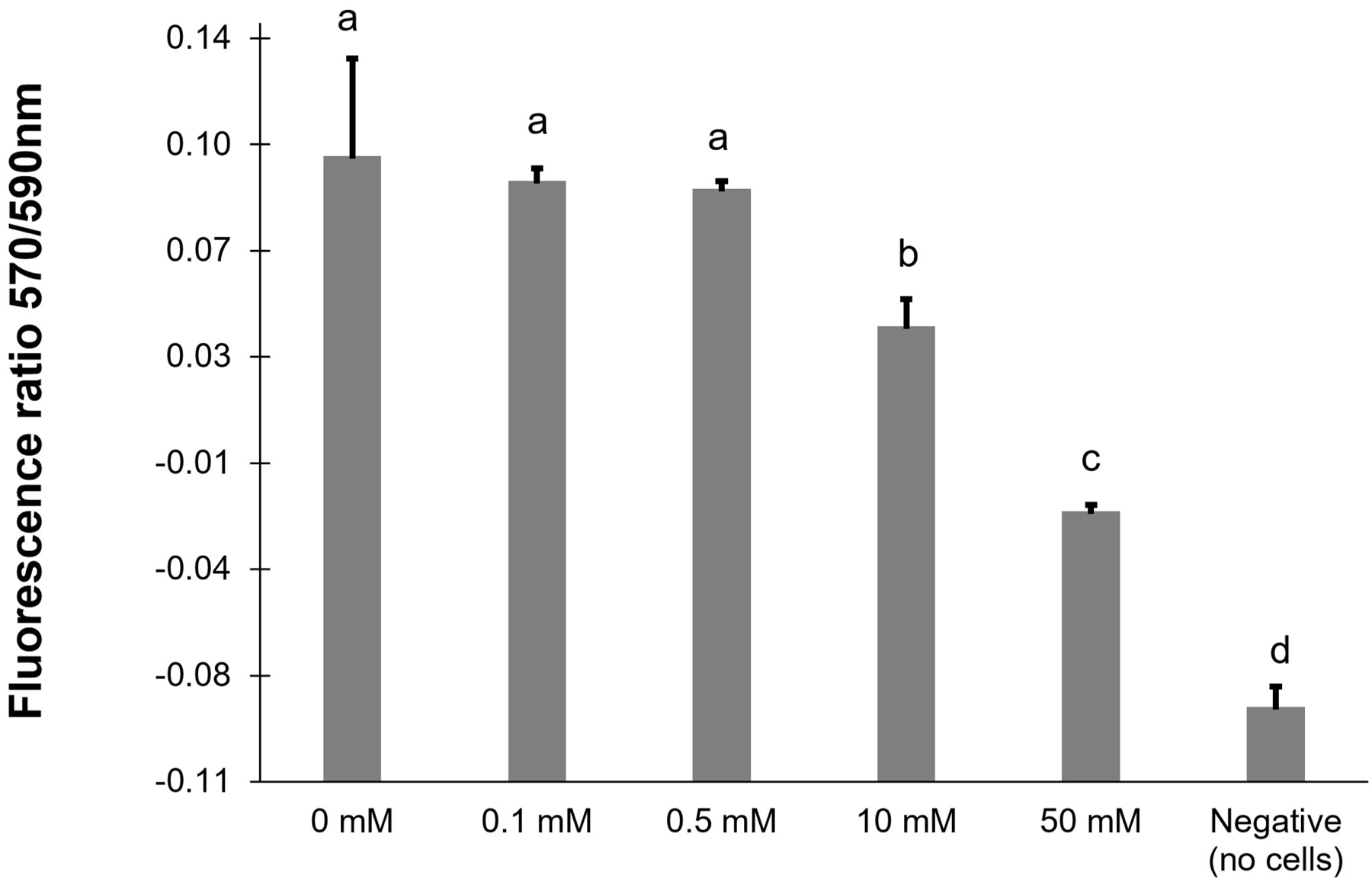
Cell viability from an alamarBlue assay taken after 24 h of hydrogen peroxide exposure analyzed using ANOVA for differences (letters indicate significance grouping).
The viability assay results indicated significant cell death after 24 h in cells exposed to 50 mM H2O2 (absorbance value <0). Similar results in cell viability in response to oxidative stress were observed in various cell culture systems. For example, Ballinger et al. found that mitochondria DNA lesions occurred from 0.05 mM H2O2 in human retinal pigment epithelium cells [39]. Imlay and Linn reported that 10–30 mM H2O2 was significant to kill Escherichia coli due to the DNA damage incurred [40]. It is possible that a higher concentration of H2O2 caused major mitochondria DNA damage in our H2O2 peroxidation experiment as well. Also, Yamada et al. found exposure of human tracheal epithelial cells to 1 mM H2O2 resulted in an increase of permeability, likely related to cell stress and viability loss [7].
PLS Regression Model for H2O2 Exposure
We first wanted to determine the time point at which the HBE1 cells exhibited the strongest volatile response to H2O2 peroxidation. To do so, PLS regression models were built from the four measured time points (Figure 3) for the four concentrations of H2O2 (0.1, 0.5, 10, 50 mM). The t=12 h time point exhibited the best linear fit, indicating the strongest VOC profile exposure response to H2O2. The 36 and 48 h time points do not include the 50 mM exposure, as these cells were found to have suffered mortality after 24 h peroxidation (Figure 2).
Figure 3.
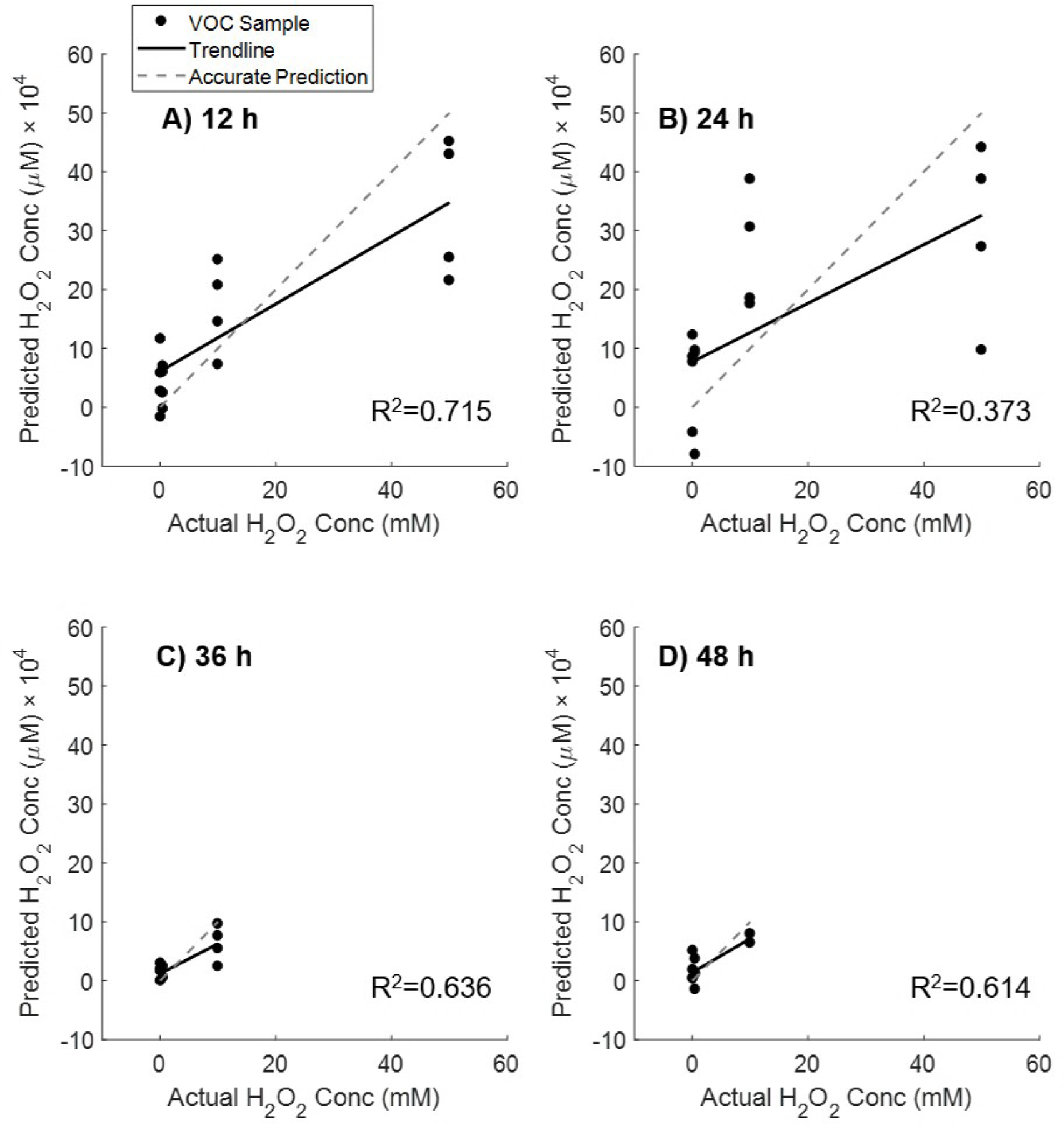
PLS regression models built from VOC profiles at four time points from cells exposed to four concentrations of H2O2 (100, 500, 10,000, 50,000 µM). Cells exposed to 50 mM experienced mortality after 24 h. The t=12 h time point exhibited the best linear fit, indicating the strongest VOC profile response to exposure to hydrogen peroxide.
The t=24 h mark resulted in the poorest linear correlation between predicted and measured H2O2 peroxidation. The 10 mM concentration was indistinguishable from the 50 mM concentration, suggesting these two peroxidation treatments had experienced similar volatile changes after 24 h of peroxidation. It was not possible to investigate this trend after 24 h since the 50,000 µM peroxidation treatment was removed from the rest of the analysis due to cell death. In our cell culture models, the H2O2 treatment would likely first affect the cellular membranes. Shestivska et al. stressed synthetic membranes with a H2O2 solution and observed volatile changes through the first 15 h of exposure [41].
In PLS regression, each variable (metabolite compound) is given a Variable Importance in Project (VIP) score, which estimates the importance of each variable to build the PLS model. Typically, compounds with a VIP score >1 are considered relevant to the PLS regression. In our analysis, 43 compounds were found to have a VIP score >1. These VOC metabolites were identified in the following compound chemical classes: alkanes or/an alkenes (20 %) and aldehydes (12 %), alcohols (8 %), aromatic hydrocarbons (8 %), cycloalkanes (4 %), and acids (3 %). Hydrocarbons, aldehydes, alcohols and ketones are chemical classes known to be related to the lipid oxidation pathways [42, 43]. Therefore, the majority of the compounds which were important to build the PLS model may have strong relation to known oxidative stress response pathways.
Table 1 shows the top 10 VIPscore compounds tentatively identified from cell samples exposed to H2O2. The compounds we detected generally agree with the types of compounds found in similar cell culture models [26, 32, 34]. The alkane (2,9-dimethyl decane), certain alcohols [2-(2-Hydroxyphenoxy)-1-phenylethanol, 3-Methylbutyraldehyde oxime, 3,4-Dimethyl-2-hexanol], and a ketone (Oxolan-2-one) were identified as the top 10 VIP scored compounds. These compounds were potentially produced from lipid fragmentation as a result of the peroxidation treatment. Some of the compounds have been previously reported as oxidative stress biomarkers. Benzenecarbaldehyde and phenylacetonitrile have previously been reported in the headspace of cell cultures infected with viruses due to the lipid fragmentation by the oxidative stress [32, 42, 43]. Benzenecarbaldehyde is a aldehyde known to be produced during the inflammatory processes as a result of lipid peroxidation and oxidative stress [30].
Table 1.
Top 10 VIP score compounds tentatively identified from cell samples exposed to H2O2
| Compound number | Base m/z | Retention Index (RI) | Name | Formula | MS Score | VIP H2O2 |
|---|---|---|---|---|---|---|
| 20 | 117 | 1136 | Phenylacetonitrile | C8H7N | 64.41 | 3.176 |
| 270 | 77 | 963 | Benzenecarbaldehyde | C7H6O | 91.55 | 2.858 |
| 83 | 40 | 900 | 1-Nitropentane | C5H11NO2 | 78.64 | 2.709 |
| 3 | 104.1 | 1297 | 1-Nitro-2-phenylethane | C8H9NO2 | 97.28 | 2.683 |
| 293 | 91.1 | 1050 | 2-Phenylacetaldehyde | C8H8O | 91.97 | 2.646 |
| 4 | 104.1 | 1297 | 2-(2-Hydroxyphenoxy)-1-phenylethanol | C14H14O3 | 58.09 | 2.471 |
| 173 | 43.1 | 887 | 3-Methylbutyraldehyde oxime | C5H11NO | 78.83 | 2.397 |
| 84 | 40 | 923 | Oxolan-2-one | C4H6O2 | 61.48 | 2.250 |
| 234 | 45 | 887 | 3,4-Dimethyl-2-hexanol | C8H18O | 73.8 | 2.068 |
| 77 | 43 | 1272 | 2,9-dimethyl decane | C12H26 | 94.52 | 1.701 |
To demonstrate the effects of H2O2 on HBE1 volatile emissions, the ten compounds with the highest VIP scores were plotted and grouped by concentration exposure (Figure 4). In these ten examples, we saw a majority exhibit a trend to increase in volatile abundance with peroxidation (compounds 270, 83, 4, 173, 176). There were also examples of VOCs that suddenly gained abundance or only appeared in the final 50 mM treatment (compounds 293, 84, 234). Compound 254 decreased with exposure concentration. We examined how these 10 compounds changed over 48 h within the 10 mM peroxide treatment (data not shown). The majority of these compounds peaked in abundance at either the 24 or 36 h time point.
Figure 4.
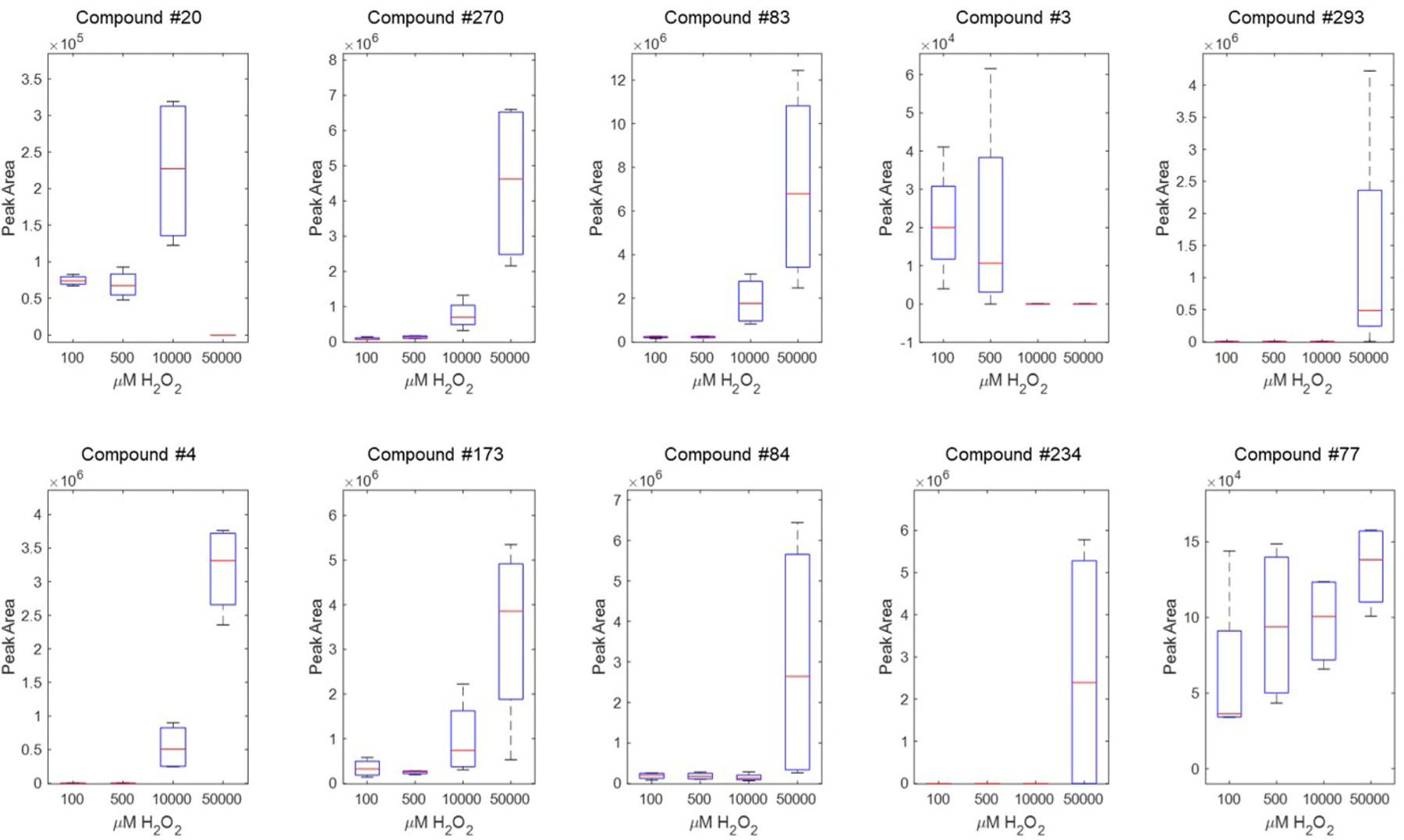
Ten compounds associated with H2O2 exposure per a PLS regression at 12 h H2O2 exposure. Compounds are identified in Table 01
We expect peroxidation of the cell culture media to produce artefact VOCs that are not endogenous to HBE1 cells. Both media controls and media with hydrogen peroxide controls were included in this work. Our cell culture analyses only included compounds that were measured with an abundance of at least three times greater in cell cultures compared to respective media controls. Examples of the types of VOCs found in media with hydrogen peroxide are presented in Supplemental Table 1 for reference by future researchers.
Smoke Extract Effects on VOC Emissions
Viability Assay for CSE Exposure
Per the viability assay, the four CSE peroxidation conditions (0, 10, 30, 60 %) demonstrated similar levels of metabolic activity (Figure 5). An ANOVA was performed on the assay data and found that cells did not show any differences in metabolic activity or cell death or metabolic differences after 48 h in the CSE concentrations (p<0.05 significance level). Chang et al. also reported that they did not see any tissue damage in 5 % and 10 % CSE exposure among non-COPD lung cells using a lactate dehydrogenase detection assay [44]. However, when they measured mRNA expression, they observed upregulation of the pro-inflammatory cytokine IL-17A after 3 h of 5 % CSE peroxidation [44]. Thus, although our cell viability assay did not show any mortality, it is possible that cell metabolites are experiencing robust changes which could lead to alterations in VOC emissions.
Figure 5.
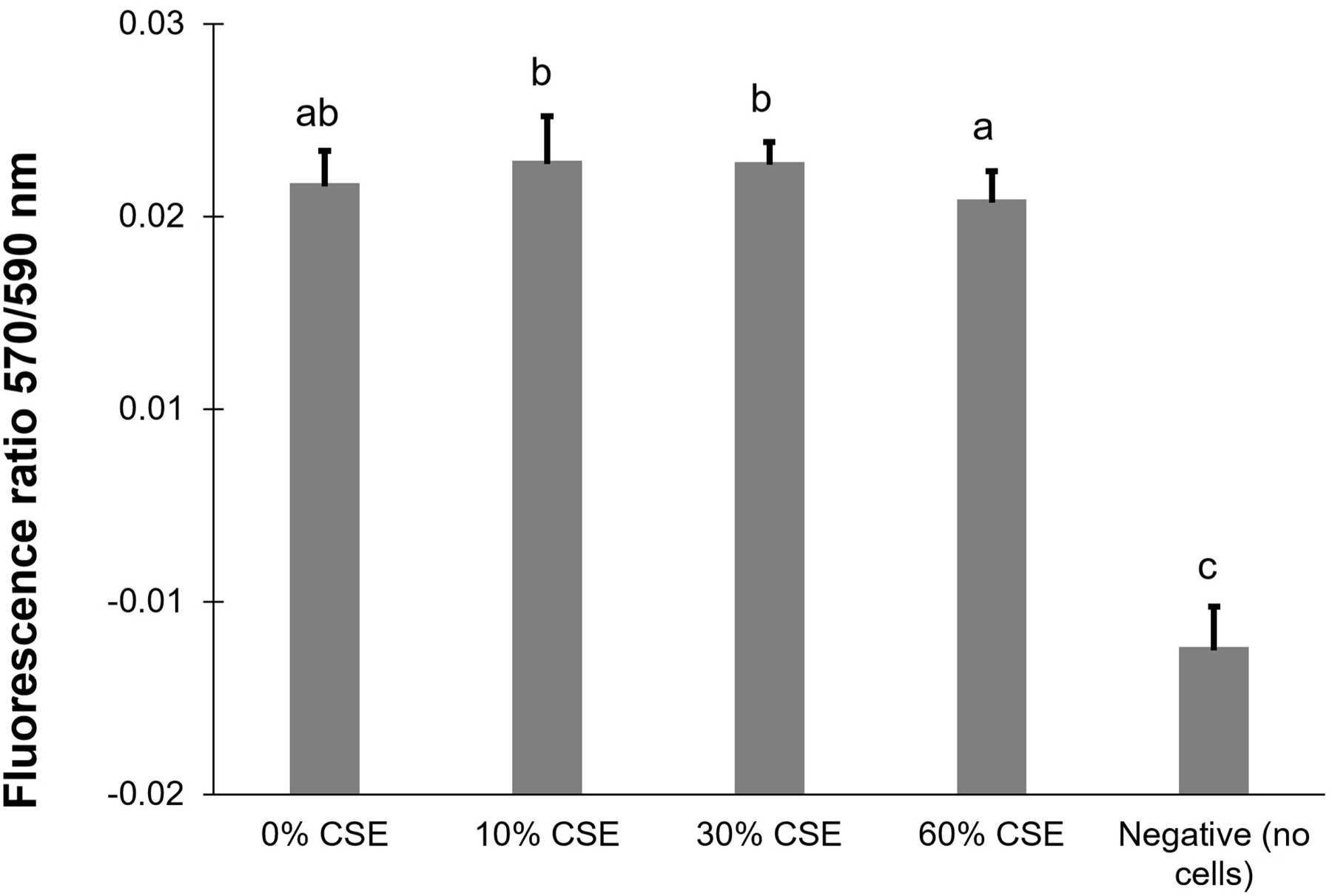
Cell viability from an alamarBlue assay taken after 48 h of cigarette smoke exposure (CSE). Letters indicate significance grouping per an ANOVA (letters indicate significance grouping)
Our viability assay did not demonstrate any differences in metabolic activity, as measured by AlamarBlue assay, for all four CSE peroxidation conditions (0, 10, 30, 60 %) after 48h (Figure 5). Similarly, Chang et al. reported that they did not see any tissue damage in 5 % and 10 % CSE exposure among non-COPD lung cells using a lactate dehydrogenase detection assay [44]. However, when they measured mRNA expression, they observed upregulation of the pro-inflammatory cytokine IL-17A after 3 h of 5 % CSE peroxidation [44]. Therefore, we anticipated robust metabolic changes, with significant alterations in VOC emissions, at the doses of CSE we used despite the uniform results from the viability assay.
PLS Regression Model for CSE Exposure
As in the H2O2 peroxidation experiment, we built PLS regression models to determine the time point at which the HBE1 cells exhibited the strongest change in their volatile emissions that relates to CSE. PLS regression models built at four time points post exposure to four concentrations of CSE (0, 10, 30, 60%) are shown (Figure 6). The t=12 h time point exhibited the best linear fit, indicating the strongest VOC profile response. The 36 h time point exhibited the worst linear correlation, and it is currently unclear why we observed this result. We suspect that some cellular repair mechanisms may be activated that are helping to remediate the oxidative stress effects on the metabolite profiles. However, additional future work is likely needed to understand these phenomena.
Figure 6.
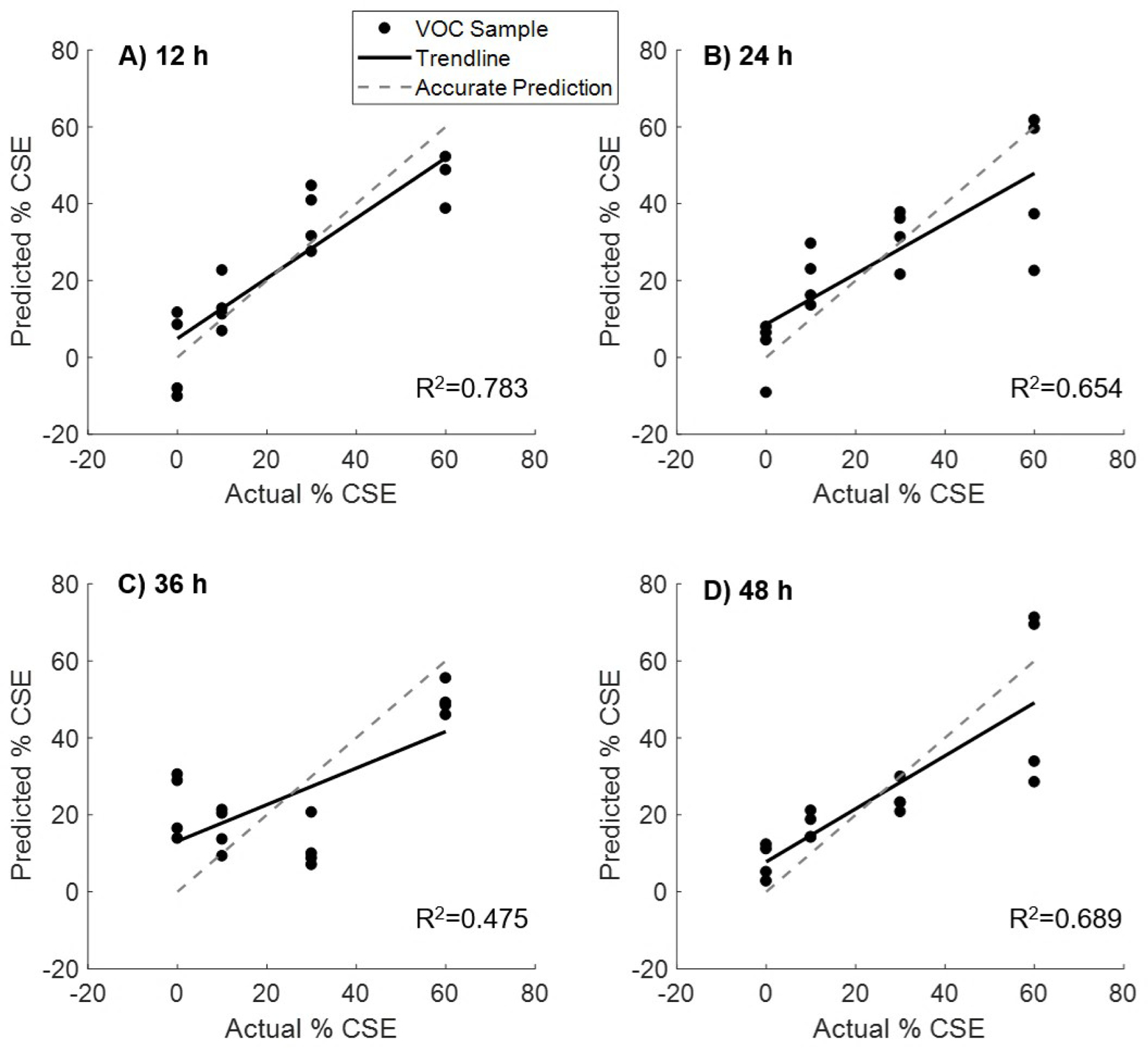
PLS regression models built from VOC profiles at four time points from cells exposed to four concentrations of cigarette smoke extract (0, 10, 30, 60%). The t=12 h time point exhibited the best linear fit, indicating the strongest VOC profile response to exposure to cigarette smoke.
Cigarette smoke itself is a complex aerosol containing more than 4,400 chemicals [45]. Thus, the smoke extract itself confounds the volatile profile of the cell cultures. Using the CSE media controls, we were able to distinguish compounds that were emitted from the cell cultures and not exclusively originating from the smoke extract. We included only compounds that had an abundance of three times greater in cell culture samples than in respective media control samples. Supplemental Table 1 includes examples of the types of compounds found in CSE media controls. There were 35 compounds that had: a) a VIP score >1 per a PLS regression model at 12 h post exposure; and b) were found to be three times greater abundance in HBE1 cell culture samples than in respective CSE media controls. Most of these compounds are alkane or/an alkene (20 %) and aldehydes (20 %). Also identified were: aromatic hydrocarbons (14 %), cycloalkanes (9 %), heterocyclic compounds (9 %), alcohol (11 %), acids (6 %), ketones (3 %), and terpenes (6 %).
Similar to the H2O2 peroxidation experiments, known oxidative stress-related chemical classes were found in these CSE peroxidation experiments such as: hydrocarbons, aldehydes, alcohols and ketones. In contrast, terpenes were not found in the H2O2 peroxidation experiments, but were found in the CSE exposures. Terpene compounds have been reportedly related to antioxidant activity. Oxidative stress impairs the prooxidant-antioxidant balance in favor of the former by increasing the production of reactive oxygen species, in part through a NAD(P)H oxidase-dependent mechanism [9]. Additionally, CSE is a known activator of the Nrf-2/Keap signaling pathway and AP-1 transcription factor[46], both of which are highly responsive to oxidative changes in the environment of the cell. Time-sensitive biphasic responses to CSE have been seen with inflammatory cell profiles and Nrf-2/Keap-dependent heme oxygenase 1 regulation in the lung[47, 48], consistent with our biphasic dose-response observed in these experiments. The precise link between Nrf2 or AP1-induced signaling pathways and specific metabolic compounds remains to be determined, but future experiments can be designed to look at specific activation of these pathways and the resultant metabolic profile that results.
A common terpene mammalian metabolomic pathway is isoprene metabolism, namely the reaction of isopentenyl pyrophosphate and dimethylallyl pyrophosphate to form the 10-carbon geranyl pyrophosphate [49]. It is possible that terpenes were produced from cellular metabolism due to the defense response of peroxidation treatment. However, the terpene antioxidant defense response of mammalian cells has not been reported and further experiments will be needed to confirm the identity of these potential cell defense compounds.
The top 10 VIP score compounds from cell samples exposed to CSE were tentatively identified (Table 2). Aldehydes (5-Methyl-2-phenylhex-2-enal), ketones (isoshyobunone), and alcohols (4-(4-methylphenyl)pentanal) were identified as the top 10 VIP-scored compounds. These compounds are potentially produced due to the lipid fragmentation of peroxidation treatment. Also, 4,6,8-trimethylnon-1-ene was reported as a potential breath biomarker of active pulmonary tuberculosis by Phillips et al. [50], and the alkanes and methylated alkane are putatively products of oxidative stress. Ramya et al. reported that plant extract 2-methyltetracosane had strong activity as a free radical scavenger [51]. Phenolic compounds also have been reported as efficient antioxidant compound due to the function of free radical terminators and metal chelators [52]. Yoon et al. reported that 2,4-Di-tert-butylphenol showed efficient antioxidant effects in thiobarbituric acid reactive substance assay [53].
Table 2.
Top 10 VIP score compounds tentatively identified from cell samples exposed to cigarette smoke extract (CSE).
| Compound number | Base m/z | Retention Index (RI) | Name | Formula | MS Score | VIP H2O2 |
|---|---|---|---|---|---|---|
| 98 | 41.1 | 1322 | 3,7,11,15-Tetramethylhexadec-1-en-3-ol | C20H40O | 77.72 | 2.113 |
| 161 | 43.1 | 1903 | 2-Methyltetracosane | C25H52 | 83.08 | 2.057 |
| 145 | 43.1 | 1322 | 4,6,8-trimethylnon-1-ene | C12H24 | 79 | 1.879 |
| 258 | 104 | 1418 | 5-Methyl-2-phenylhex-2-enal | C13H16O | 85.87 | 1.771 |
| 228 | 57.1 | 1475 | 2,6-Di-tert-butylphenol | C14H22O | 88.48 | 1.436 |
| 275 | 81.1 | 1776 | Dodecan-3-ylbenzene | C18H30 | 65.4 | 1.271 |
| 148 | 43.1 | 1389 | (3S,6S)-6-Isopropyl-2-isopropylidene-3-methyl-3-vinylcyclohexanone | C15H24O | 80.62 | 1.27 |
| 242 | 68.1 | 1024 | A terpene compound | 1.255 | ||
| 159 | 43.1 | 1707 | A alkene compound | 1.198 | ||
| 289 | 91.1 | 1821 | 4-(4-methylphenyl)pentanal | C12H16O | 76.66 | 1.173 |
A PLS regression showed ten compounds at t=12 h CSE peroxidation that were deemed important (Figure 7), and these are tentatively identified (Table 2). In these examples, there were not as many trends that correlated VOC abundances with CSE concentration. Compound 289 decreased in abundance with exposure, while 161 clearly increased with exposure. Compounds 98, 145, 228, 258, 148 and 159 generally see a trend of VOCs increasing with concentration, but the abundances of these compounds for the 30 % CSE suddenly drop and increase again for the 60 % peroxidation. The internal standards for these samples did not suggest any technical issues.
Figure 7.
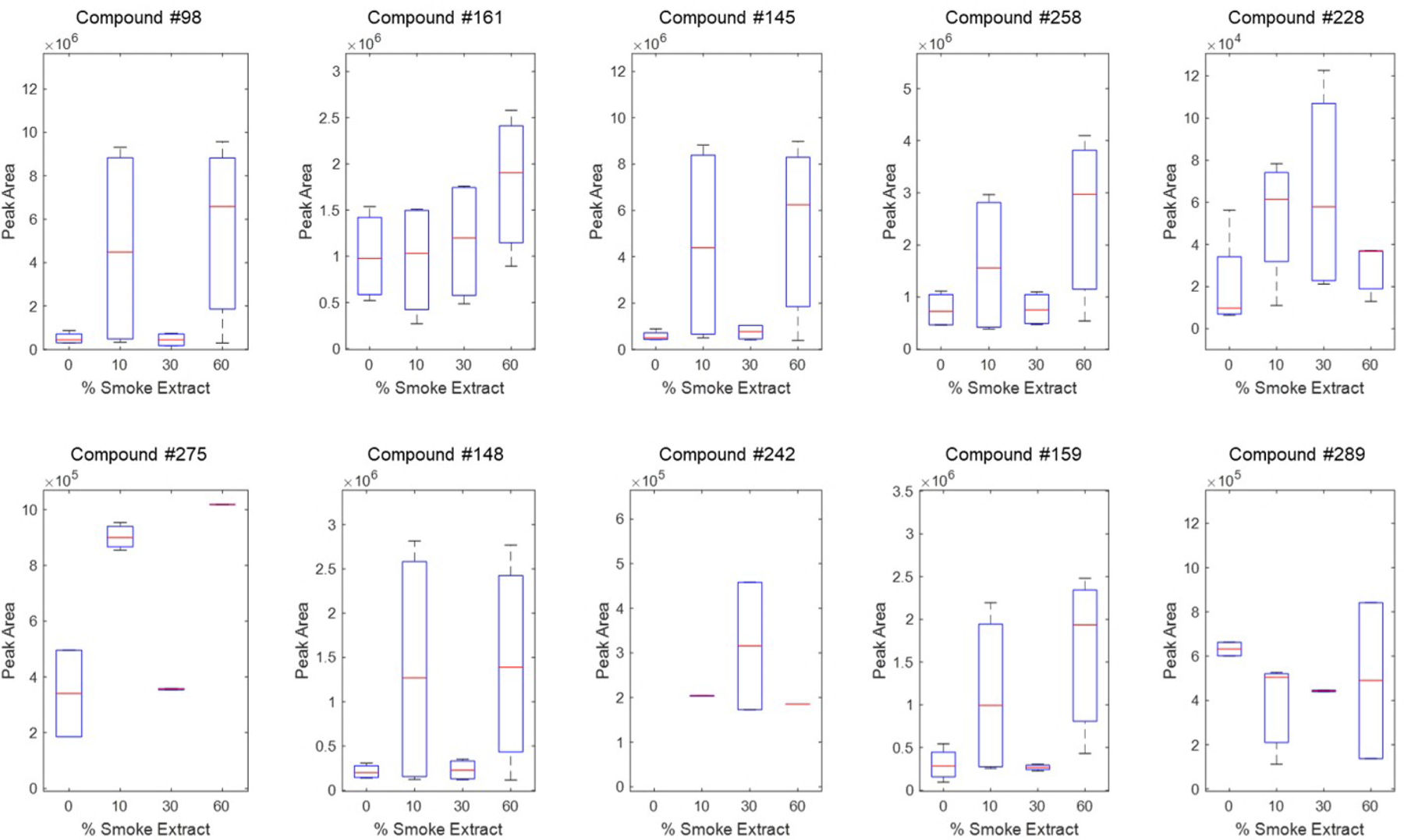
Ten compounds associated with H2O2 exposure per a PLS regression at 12 h CSE exposure. Compounds are identified in Table 02
Comparison of H2O2 and smoke extract on VOC emissions
We saw oxidative stress-related VOCs from both the H2O2 and CSE peroxidation experiments. We found more antioxidant related compounds emitted from the cells in the CSE peroxidation experiment, compared to H2O2 peroxidation. This could partially be due to cell viability differences between the two peroxidation treatments. In H2O2 peroxidation, we observed slightly lower viability compared to the CSE experiments. Hence, cells in H2O2 were experiencing strong oxidative stress and might have contributed to an imbalance of the antioxidative defense response. However, in the CSE peroxidation treatment, cells were still metabolically active and they showed a strong antioxidant defense response, which resulted in the long chain carbon VOCs that we detected. We believe these differences in the cell response contributed to the changes of the VOC profile differences between the two peroxidation experiments. Furthermore, in H2O2 peroxidation, we saw shorter chains and smaller VOCs which are more related to membrane lipids and fatty acid fragmentation, likely due to the high level of peroxidation. In contrast, heavier molecular weight VOCs were emitted in the CSE peroxidation experiment, suggesting these compounds could be related to cytokine or protein damage produced due to the cell antioxidation defense response.
The comparative, parallel findings with H2O2 and CSE was expected and somewhat reassuring. Recent evidence points to the generation of H2O2 by CSE as an important cause of airway epithelial cell injury[54, 55]. Bronchial epithelial cells exposed to five percent CS extract generated maximal amounts of H2O2 at four hours followed by cellular injury and impaired wound healing[55]. Both the antioxidant, N-acetylcysteine, and the transfection of cells with a vector encoding superoxide dismutase to block H2O2 improved wound healing after CS extract exposure. Further studies have revealed that the mechanism of CSE/ H2O2 mediated airway epithelial injury is via upregulation of DUOX1[54]. Given the causal relationship, we would expect similar VOC patterns from such cells.
Conclusions
To our knowledge, our study is the first to show a specific cellular VOC production response to different oxidants concentrations and time points in bronchial epithelial cells, under two different peroxidation paradigms (H2O2 and CSE). From our PLS regression models, we found 12 h was the most effective peroxidation time point for our cell culture system. Previously in the literature, peroxidation studies were typically confined to shorter time courses (< 6 h) and longer duration oxidative stress experiments are not common. However, oxidative stress is likely a chronic cellular response, and our understanding of our data is that longer time course studies, at lower oxidant levels, are needed to more closely mimic a clinical microenvironment for the cells.
From both the H2O2 and CSE peroxidation experiments, known oxidative stress biomarkers were detected in our system. Moreover, in CSE peroxidation, we observed VOCs likely related to the cell antioxidant defense response. The research field of cell VOC oxidative stress markers is still developing, and specific VOCs related to the overall cell antioxidant defense response remains largely unknown. In future studies, we plan to better define the link between specific oxidative stress cellular pathway alterations to specific VOC generation. We believe our initial findings will help steer future VOC oxidative stress research towards understanding the cellular origin of this category of breath biomarkers.
Supplementary Material
Acknowledgments
Partial support was provided by: Revelar, Inc. funded industry contract #A18-1712 [CED, NJK, RHH, SEE, MS]; NIH award U01 EB0220003-01 [CED, NJK]; NIH National Center for Advancing Translational Sciences (NCATS) through grant#UL1 TR000002 [CED, NJK]; NIH award UG3-OD023365 [CED, NJK]; NIH award 1P30ES023513-01A1 [CED, NJK]; NIH-National Heart, Lung and Blood Institute 1K23HL127185 [MS]; and NIH training grant award T32 HL007013 [MSY]. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the funding agencies.
Footnotes
Conflict of interest disclosure
The authors have no conflicts to declare.
References
- [1].Risby TH and Sehnert SS, “Clinical application of breath biomarkers of oxidative stress status,” (in English), Free Radical Biology and Medicine, vol. 27, no. 11–12, pp. 1182–1192, Dec 1999. [DOI] [PubMed] [Google Scholar]
- [2].Liu HL, Sun SH, Zong YL, Li P, and Xie JP, “Analysis of hydrogen peroxide in cigarette smoke from selected Chinese cigarette brands under conventional and intense machine smoking conditions,” (in English), European Food Research and Technology, vol. 235, no. 6, pp. 1107–1115, Dec 2012. [Google Scholar]
- [3].Stone JR and Yang S, “Hydrogen peroxide: a signaling messenger,” Antioxid Redox Signal, vol. 8, no. 3–4, pp. 243–70, Mar-Apr 2006. [DOI] [PubMed] [Google Scholar]
- [4].Gluck MR and Zeevalk GD, “Inhibition of brain mitochondrial respiration by dopamine and its metabolites: implications for Parkinson’s disease and catecholamine-associated diseases,” J Neurochem, vol. 91, no. 4, pp. 788–95, Nov 2004. [DOI] [PubMed] [Google Scholar]
- [5].Cai H, “NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease,” Circ Res, vol. 96, no. 8, pp. 818–22, Apr 29 2005. [DOI] [PubMed] [Google Scholar]
- [6].Geiszt M and Leto TL, “The Nox family of NAD(P)H oxidases: host defense and beyond,” J Biol Chem, vol. 279, no. 50, pp. 51715–8, Dec 10 2004. [DOI] [PubMed] [Google Scholar]
- [7].Yamada N et al. , “Protective effects of heme oxygenase-1 against oxidant-induced injury in the cultured human tracheal epithelium,” Am J Respir Cell Mol Biol, vol. 21, no. 3, pp. 428–35, Sep 1999. [DOI] [PubMed] [Google Scholar]
- [8].Nakayama T, Kodama M, and Nagata C, “Generation of hydrogen peroxide and superoxide anion radical from cigarette smoke,” Gan, vol. 75, no. 2, pp. 95–8, Feb 1984. [PubMed] [Google Scholar]
- [9].Schafer FQ and Buettner GR, “Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple,” (in English), Free Radical Biology and Medicine, vol. 30, no. 11, pp. 1191–1212, Jun 1 2001. [DOI] [PubMed] [Google Scholar]
- [10].Stadtman ER, “Protein oxidation and aging,” (in English), Free Radical Research, vol. 40, no. 12, pp. 1250–1258, Dec 2006. [DOI] [PubMed] [Google Scholar]
- [11].Grimsrud PA, Xie HW, Griffin TJ, and Bernlohr DA, “Oxidative stress and covalent modification of protein with bioactive aldehydes,” (in English), Journal of Biological Chemistry, vol. 283, no. 32, pp. 21837–21841, Aug 8 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Filipiak W et al. , “A Compendium of Volatile Organic Compounds (VOCs) Released By Human Cell Lines,” Curr Med Chem, vol. 23, no. 20, pp. 2112–31, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Esterbauer H, Schaur RJ, and Zollner H, “Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes,” Free Radic Biol Med, vol. 11, no. 1, pp. 81–128, 1991. [DOI] [PubMed] [Google Scholar]
- [14].Esterbauer H, Zollner H, and Lang J, “Metabolism of the Lipid-Peroxidation Product 4-Hydroxynonenal by Isolated Hepatocytes and by Liver Cytosolic Fractions,” (in English), Biochemical Journal, vol. 228, no. 2, pp. 363–373, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Uchida K and Stadtman ER, “Covalent Attachment of 4-Hydroxynonenal to Glyceraldehyde-3-Phosphate Dehydrogenase - a Possible Involvement of Intramolecular and Intermolecular Cross-Linking Reaction,” (in English), Journal of Biological Chemistry, vol. 268, no. 9, pp. 6388–6393, Mar 25 1993. [PubMed] [Google Scholar]
- [16].Pryor WA, “Cigarette smoke and the involvement of free radical reactions in chemical carcinogenesis,” Br J Cancer Suppl, vol. 8, pp. 19–23, Jun 1987. [PMC free article] [PubMed] [Google Scholar]
- [17].Lau WK, Cui LY, Chan SC, Ip MS, and Mak JC, “The presence of serotonin in cigarette smoke - a possible mechanistic link to 5-HT-induced airway inflammation,” Free Radic Res, vol. 50, no. 5, pp. 495–502, 2016. [DOI] [PubMed] [Google Scholar]
- [18].Lau WK, Li X, Yeung DS, Chan KH, Ip MS, and Mak JC, “The involvement of serotonin metabolism in cigarette smoke-induced oxidative stress in rat lung in vivo,” Free Radic Res, vol. 46, no. 11, pp. 1413–9, Nov 2012. [DOI] [PubMed] [Google Scholar]
- [19].Mortaz E et al. , “Effect of cigarette smoke extract on dendritic cells and their impact on T-cell proliferation,” PLoS One, vol. 4, no. 3, p. e4946, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Valdivieso AG, Dugour AV, Sotomayor V, Clauzure M, Figueroa JM, and Santa-Coloma TA, “N-acetyl cysteine reverts the proinflammatory state induced by cigarette smoke extract in lung Calu-3 cells,” Redox Biol, vol. 16, pp. 294–302, Jun 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tejero JD et al. , “Cigarette smoke extract acts directly on CD4 T cells to enhance Th1 polarization and reduce memory potential,” Cell Immunol, vol. 331, pp. 121–129, Sep 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Stabile AM et al. , “Long term effects of cigarette smoke extract or nicotine on nerve growth factor and its receptors in a bronchial epithelial cell line,” Toxicol In Vitro, vol. 53, pp. 29–36, Dec 2018. [DOI] [PubMed] [Google Scholar]
- [23].Somborac-Bacura A et al. , “Differential expression of heat shock proteins and activation of mitogen-activated protein kinases in A549 alveolar epithelial cells exposed to cigarette smoke extract,” Exp Physiol, vol. 103, no. 12, pp. 1666–1678, Dec 2018. [DOI] [PubMed] [Google Scholar]
- [24].Preshaw PM, “Host response modulation in periodontics,” (in English), Periodontology 2000, vol. 48, pp. 92–110, 2008. [DOI] [PubMed] [Google Scholar]
- [25].Lawal O et al. , “Volatile organic compound signature from co-culture of lung epithelial cell line with Pseudomonas aeruginosa,” (in English), Analyst, vol. 143, no. 13, pp. 3148–3155, Jul 7 2018. [DOI] [PubMed] [Google Scholar]
- [26].Aksenov AA et al. , “Cellular scent of influenza virus infection,” Chembiochem, vol. 15, no. 7, pp. 1040–8, May 5 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Aksenov AA et al. , “Characterization of volatile organic compounds in human leukocyte antigen heterologous expression systems: a cell’s “chemical odor fingerprint”,” Chembiochem, vol. 13, no. 7, pp. 1053–9, May 7 2012. [DOI] [PubMed] [Google Scholar]
- [28].Phillips M et al. , “Effect of influenza vaccination on oxidative stress products in breath,” J Breath Res, vol. 4, no. 2, p. 026001, Jun 2010. [DOI] [PubMed] [Google Scholar]
- [29].Ahmed I, Greenwood R, Costello B, Ratcliffe N, and Probert CS, “Investigation of faecal volatile organic metabolites as novel diagnostic biomarkers in inflammatory bowel disease,” (in English), Alimentary Pharmacology & Therapeutics, vol. 43, no. 5, pp. 596–611, Mar 2016. [DOI] [PubMed] [Google Scholar]
- [30].Fritz KS and Petersen DR, “An overview of the chemistry and biology of reactive aldehydes,” (in English), Free Radical Biology and Medicine, vol. 59, pp. 85–91, Jun 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Purcaro G, Rees CA, Melvin JA, Bomberger JM, and Hill JE, “Volatile fingerprinting of Pseudomonas aeruginosa and respiratory syncytial virus infection in an in vitro cystic fibrosis co-infection model,” J Breath Res, vol. 12, no. 4, p. 046001, Jul 3 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Purcaro G et al. , “Volatile fingerprinting of human respiratory viruses from cell culture,” J Breath Res, vol. 12, no. 2, p. 026015, Mar 1 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Traxler S et al. , “VOC breath profile in spontaneously breathing awake swine during Influenza A infection,” Sci Rep, vol. 8, no. 1, p. 14857, Oct 5 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yamaguchi MS, McCartney MM, Linderholm AL, Ebeler SE, Schivo M, and Davis CE, “Headspace sorptive extraction-gas chromatography-mass spectrometry method to measure volatile emissions from human airway cell cultures,” J Chromatogr B Analyt Technol Biomed Life Sci, vol. 1090, pp. 36–42, Jul 15 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yankaskas JR et al. , “Papilloma-Virus Immortalized Tracheal Epithelial-Cells Retain a Well-Differentiated Phenotype,” (in English), American Journal of Physiology, vol. 264, no. 5, pp. C1219–C1230, May 1993. [DOI] [PubMed] [Google Scholar]
- [36].Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, and Randell SH, “Well-Differentiated Human Airway Epithelial Cell Cultures,” in Human Cell Culture Protocols, Picot J, Ed. Totowa, NJ: Humana Press, 2005, pp. 183–206. [DOI] [PubMed] [Google Scholar]
- [37].Parker NS et al. , “Characterisation Of Sub-acute Mouse Model Of Ozone-exacerbated Cigarette Smoke Extract-induced Lung Inflammation,” (in English), American Journal of Respiratory and Critical Care Medicine, vol. 181, 2010. [Google Scholar]
- [38].Cui Y, Liu KW, Liang Y, Ip MS, and Mak JC, “Inhibition of monoamine oxidase-B by selegiline reduces cigarette smoke-induced oxidative stress and inflammation in airway epithelial cells,” Toxicol Lett, vol. 268, pp. 44–50, Feb 15 2017. [DOI] [PubMed] [Google Scholar]
- [39].Ballinger SW, Van Houten B, Jin GF, Conklin CA, and Godley BF, “Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells,” (in English), Experimental Eye Research, vol. 68, no. 6, pp. 765–772, Jun 1999. [DOI] [PubMed] [Google Scholar]
- [40].Imlay JA and Linn S, “Mutagenesis and Stress Responses Induced in Escherichia-Coli by Hydrogen-Peroxide,” (in English), Journal of Bacteriology, vol. 169, no. 7, pp. 2967–2976, Jul 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shestivska V et al. , “Evaluation of lipid peroxidation by the analysis of volatile aldehydes in the headspace of synthetic membranes using selected ion flow tube mass spectrometry,” (in English), Rapid Communications in Mass Spectrometry, vol. 32, no. 18, pp. 1617–1628, Sep 30 2018. [DOI] [PubMed] [Google Scholar]
- [42].Haick H, Broza YY, Mochalski P, Ruzsanyi V, and Amann A, “Assessment, origin, and implementation of breath volatile cancer markers,” Chem Soc Rev, vol. 43, no. 5, pp. 1423–49, Mar 7 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Schulz S and Dickschat JS, “Bacterial volatiles: the smell of small organisms,” Nat Prod Rep, vol. 24, no. 4, pp. 814–42, Aug 2007. [DOI] [PubMed] [Google Scholar]
- [44].Chang Y et al. , “Upregulation of IL-17A/F from human lung tissue explants with cigarette smoke exposure: implications for COPD,” (in English), Respiratory Research, vol. 15, Nov 27 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Polzin GM, Kosa-Maines RE, Ashley DL, and Watson CH, “Analysis of volatile organic compounds in mainstream cigarette smoke,” (in English), Environmental Science & Technology, vol. 41, no. 4, pp. 1297–1302, Feb 15 2007. [DOI] [PubMed] [Google Scholar]
- [46].Sekine T et al. , “Regulation of NRF2, AP-1 and NF-kappaB by cigarette smoke exposure in three-dimensional human bronchial epithelial cells,” J Appl Toxicol, Dec 21 2018. [DOI] [PubMed] [Google Scholar]
- [47].D’Hulst AI, Vermaelen KY, Brusselle GG, Joos GF, and Pauwels RA, “Time course of cigarette smoke-induced pulmonary inflammation in mice,” Eur Respir J, vol. 26, no. 2, pp. 204–13, Aug 2005. [DOI] [PubMed] [Google Scholar]
- [48].Goven D et al. , “Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema,” Thorax, vol. 63, no. 10, pp. 916–24, Oct 2008. [DOI] [PubMed] [Google Scholar]
- [49].Crowell PL, Chang RR, Ren Z, Elson CE, and Gould MN, “Selective-Inhibition of Isoprenylation of 21–26-Kda Proteins by the Anticarcinogen D-Limonene and Its Metabolites,” (in English), Journal of Biological Chemistry, vol. 266, no. 26, pp. 17679–17685, Sep 15 1991. [PubMed] [Google Scholar]
- [50].Phillips M et al. , “Breath biomarkers of active pulmonary tuberculosis,” (in English), Tuberculosis, vol. 90, no. 2, pp. 145–151, Mar 2010. [DOI] [PubMed] [Google Scholar]
- [51].Ramya B, Malarvili T, and Velavan S, “Gc-Ms Analysis of Bioactive Compounds in Bryonopsis Laciniosa Fruit Extract,” (in English), International Journal of Pharmaceutical Sciences and Research, vol. 6, no. 8, pp. 3375–3379, Aug 2015. [Google Scholar]
- [52].Shahidi F, Janitha PK, and Wanasundara PD, “Phenolic Antioxidants,” (in English), Critical Reviews in Food Science and Nutrition, vol. 32, no. 1, pp. 67–103, 1992. [DOI] [PubMed] [Google Scholar]
- [53].Yoon MA et al. , “Antioxidant effects of quinoline alkaloids and 2,4-di-tert-butylphenol isolated from Scolopendra subspinipes,” (in English), Biological & Pharmaceutical Bulletin, vol. 29, no. 4, pp. 735–739, Apr 2006. [DOI] [PubMed] [Google Scholar]
- [54].Tian Z et al. , “Cigarette Smoke Impairs A2A Adenosine Receptor Mediated Wound Repair through Up-regulation of Duox-1 Expression,” Sci Rep, vol. 7, p. 44405, Mar 24 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Allen-Gipson DS et al. , “Smoke extract impairs adenosine wound healing: implications of smoke-generated reactive oxygen species,” Am J Respir Cell Mol Biol, vol. 48, no. 5, pp. 665–73, May 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.