

Abstract
Rationale
Pediatric pulmonary hypertension is an important cause of childhood morbidity and mortality, but there are limited data on the range of associated diseases, contributions of different pulmonary hypertension subtypes, therapeutic strategies, and clinical outcomes in children.
Objectives
To report the 20-year experience of a large UK National Pediatric Pulmonary Hypertension Service focusing on epidemiology and clinical outcomes.
Methods
Consecutive patients presenting between 2001 and 2021 were included, and survival analysis was performed for incident patients.
Measurements and Main Results
Of 1,353 patients assessed, a pulmonary hypertension diagnosis was made in 1,101 (81.4%) patients (51% female, median age, 2.6 [interquartile range, 0.8–8.2] years). The most common form was pulmonary arterial hypertension in 48%, followed by 32.3% with pulmonary hypertension due to lung disease. Multiple contributory causes of pulmonary hypertension were common, with 16.9% displaying features of more than one diagnostic group. The annual incidence of childhood pulmonary hypertension was 3.5 (95% confidence interval [CI], 3.3–3.8) per 1 million children, and the prevalence was 18.1 (95% CI, 15.8–20.4) per 1 million. The incidence was highest for pulmonary hypertension due to lung disease in infancy (15.0 [95% CI, 12.7–17.2] per 1 million per year). Overall, 82.4% patients received pulmonary arterial hypertension therapy, and escalation to triple therapy during follow-up was required in 13.1%. In 970 (88.1%) incident patients, transplant-free survival was 86.7% (95% CI, 84.5–89%) at 1 and 68.6% (95% CI, 64.7–72.6%) at 10 years. Pulmonary hypertension due to left heart disease had the lowest survival (hazard ratio, 2.0; 95% CI, 1.36–2.94; P < 0.001).
Conclusions
Clinical phenotypes of pediatric pulmonary hypertension are heterogeneous and overlapping, with clinical phenotypes that evolve throughout childhood. Despite widespread use of pulmonary arterial hypertension therapy, the prognosis remains poor.
Keywords: pulmonary arterial hypertension, adolescent medicine, cohort study, epidemiology, survival analysis
At a Glance Commentary
Scientific Knowledge on the Subject
Pediatric pulmonary hypertension represents a heterogeneous group of diseases with overlapping clinical phenotypes that evolve throughout childhood and resemble adult phenotypes in adolescence. Our current understanding of etiology and outcomes in pediatric pulmonary hypertension is based on previous registry studies, which often focus on group 1 pulmonary hypertension and provide survival data to 5 years after diagnosis.
What This Study Adds to the Field
This national cohort study reports on the experience of the single UK referral center for pulmonary hypertension in children, providing the first estimates of incidence and prevalence of all pediatric pulmonary hypertension groups from infancy to late adolescence. This study also uses mortality data from a large cohort of consecutive patients with long-term follow-up and good ascertainment to determine survival for different pediatric pulmonary hypertension groups.
Pulmonary hypertension (PH) is a rare but serious disease in childhood associated with significant morbidity and mortality. Despite sharing common features with adult disease, pediatric PH has many unique characteristics in terms of etiology and presentation, which are highlighted in the sixth World Symposium on PH classification (1). Our current understanding of the etiology of pediatric PH is based on previous registries, which have identified a high proportion of multifactorial and “transient” cases of PH in childhood (2–4). However, given the rarity and heterogeneity of PH in children, only large national datasets with long follow-up can accurately report on the epidemiology and outcomes across the spectrum of pediatric disease.
In the United Kingdom, specialized PH care has been centralized since 2001 to provide a consistent national approach to diagnosis and management. All children with PH are managed by a single center that acts as the hub of a specialist pediatric PH network. This protocolized approach to care and centralization of data offers a unique opportunity to study the epidemiology and outcomes across the breadth of pediatric PH while limiting selection bias. In this study, we report the entire 20-year experience of the UK National Pediatric Pulmonary Hypertension Service, focusing on the incidence, clinical phenotypes, and outcomes of children diagnosed with PH. Some of the results of this study have been previously reported in the form of an abstract (5).
Methods
Study Design
This is a retrospective study of consecutive patients with PH presenting to our service from its inception on January 1, 2001, to January 1, 2021. To reduce selection bias, the initial screening cohort included 1) patients presenting with suspected or confirmed PH to the specialist service, 2) cases referred for a second opinion, and 3) cases requiring specialist inpatient review or advice. Criteria for inclusion into the analysis cohort were age ⩽ 18 years with a firm diagnosis of PH by cardiac catheterization based on established hemodynamic criteria, or on noninvasive clinical assessment with signs of PH on echocardiography and a diagnosis established by the PH mult-disciplinary team (6). This retrospective analysis was covered under ethics approval 17/LO/0008 and based on anonymized data collected for routine clinical care; hence, individual informed consent was waived.
Data Collection
Demographic and clinical characteristics were collected from a dedicated clinical database. Height-for-age and weight-for-age z-scores were calculated using appropriate growth charts (see the online supplement for further details) (7–9). Each patient was assigned to a PH group: group 1 PH or pulmonary arterial hypertension (PAH); group 2, PH associated with left heart disease; group 3, PH associated with lung disease and/or related to hypoxia; group 4, PH due to pulmonary artery obstructions; and group 5, PH with unclear and/or multifactorial mechanisms (10). A PH subgroup was then assigned according to the sixth World Symposium on PH international classification system (1). Secondary and tertiary causes of PH were documented where apparent. If a patient could be allocated to more than one group, this was reviewed by the senior investigator, and a consensus was reached. Resolution of PH was defined as normalization of PA pressure on invasive or noninvasive assessment after cessation of PAH therapy.
Statistical Analysis
Statistical analyses were performed using R version 4.1.0 (R Foundation for Statistical Computing). Continuous variables are presented as mean ± SD or median (interquartile range [IQR]). Categorical variables are presented as number (%). The denominator for percentages was the total number of patients with available data for each variable analyzed. Missing data were not imputed. Comparisons between groups were performed using the Wilcoxon rank sum and chi-square tests. The incidences of groups 1–5 and for overall PH were calculated with reference to annual UK childhood population statistics (11). Additional detail on the method for making these measurements is provided in the online supplement.
Survival analysis was performed in the incident PH group using a composite outcome of all-cause mortality or lung or heart–lung transplantation. Patients were censored at the first occurrence of any of the following: alive on January 1, 2021, reached 18 years of age, transitioned to an adult PH service, emigrated from the United Kingdom, or lost to follow-up. The Kaplan-Meier method was used to generate survival curves (see the online supplement for further details on survival and competing risks analysis). A two-sided P value of less than 0.05 indicated statistical significance.
Results
From January 1, 2001, to January 1, 2021, 1,353 patients were referred to the UK National Pediatric Pulmonary Hypertension Service and were included in this study. From this referral cohort, 252 patients did not have PH (see Table E1 in the online supplement for referral indications), and a diagnosis of PH was made in the remaining 1,101 (81.4%) patients. Most (80.5%) patients in whom PH was excluded at first assessment by the PH service were “at-risk” patients (e.g., reviewed as part of family screening or because of a comorbid condition with a known association to PH).
Overall PH Cohort and International PH Classification Groups
A full list of diagnostic subgroups is show in Table 1, and the baseline characteristics of the PH cohort are shown in Table 2 (for data on each diagnostic PH group, please see Table E2). Overall, 51.0% were female, with a median age at diagnosis of PH of 2.6 (IQR, 0.8–8.2] years (Figure 1). Almost half of patients, 529 (48.0%), belonged to group 1 of the International PH Classification (pulmonary arterial hypertension [PAH]). The majority of these had PAH associated with congenital heart disease (62.9%), followed by idiopathic PAH (22.3%) and then by heritable PAH (5.3%). There were only 8 (1.5%) patients with persistent PH of the newborn.
Table 1.
Diseases Associated with Main Pediatric Pulmonary Hypertension Diagnosis According to the 2018 International Classification System (1)
| Pulmonary Hypertension Group | n | % |
|---|---|---|
| Group 1: PAH | ||
| 1.1 Idiopathic PAH | 63 | 5.7 |
| 1.2 Heritable PAH | 28 | 2.5 |
| 1.3 Drug- and toxin-induced PAH | 10 | 0.9 |
| 1.4.1 APAH connective tissue disease | 13 | 1.2 |
| 1.4.2 APAH HIV infection | 1 | 0.1 |
| 1.4.3 APAH portal hypertension | 10 | 0.9 |
| 1.4.4.1 APAH CHD Eisenmenger Syndrome | 90 | 8.2 |
| 1.4.4.2.1 APAH CHD Left to right Shunt (deemed operable) | 50 | 4.5 |
| 1.4.4.2.2 APAH CHD Left to right Shunt (deemed inoperable) | 36 | 3.3 |
| 1.4.4.3 APAH CHD – PAH with co-incidental CHD | 47 | 4.3 |
| 1.4.4.4 APAH CHD Post-operative PAH | 129 | 11.7 |
| 1.4.4.x APAH CHD Cannot be classified | 28 | 2.5 |
| 1.5 PAH long-term responders to calcium channel blockers | 8 | 0.7 |
| 1.6 PAH with overt features of venous/capillaries (PVOD/PCH) involvement | 8 | 0.7 |
| 1.7 Persistent PH of the newborn syndrome | 8 | 0.7 |
| Group 2: PH due to left heart disease | ||
| 2.1 PH due to heart failure with preserved LVEF | 27 | 2.5 |
| 2.2 PH due to heart failure with reduced LVEF | 10 | 0.9 |
| 2.3 Valvular heart disease | 9 | 0.8 |
| 2.4.1 Pulmonary vein stenosis | 32 | 2.9 |
| 2.4.2 Cor triatriatum | 2 | 0.2 |
| 2.4.3 Obstructed total anomalous pulmonary venous return | 1 | 0.1 |
| 2.4.4 Mitral/aortic valve stenosis (including supra/subvalvular) | 15 | 1.4 |
| 2.4.5 Aorta/great arteries | 1 | 0.1 |
| Group 3: PH due to lung disease and/or hypoxia | ||
| 3.1 Obstructive lung disease | 4 | 0.4 |
| 3.2 Restrictive lung disease | 31 | 2.8 |
| 3.3 Other lung disease with mixed restrictive/obstructive pattern | 48 | 4.4 |
| 3.4 Hypoxia without lung disease | 24 | 2.2 |
| 3.5.1 Bronchopulmonary dysplasia | 172 | 15.6 |
| 3.5.10 Pulmonary lymphangiectasia | 2 | 0.2 |
| 3.5.2 Congenital diaphragmatic hernia | 39 | 3.5 |
| 3.5.3 Down syndrome | 8 | 0.7 |
| 3.5.4 Alveolar capillary dysplasia with “misalignment of veins” (FOXF1) | 6 | 0.5 |
| 3.5.5 Lung hypoplasia, acinar dysplasia | 12 | 1.1 |
| 3.5.6 Surfactant protein abnormalities | 1 | 0.1 |
| 3.5.6 TTF1/NKX2-1 | 1 | 0.1 |
| 3.5.7 TBX4 | 4 | 0.4 |
| 3.5.8 Pulmonary interstitial glycogenesis | 3 | 0.3 |
| 3.5.9 Pulmonary alveolar proteinosis | 1 | 0.1 |
| Group 4: PH due to pulmonary artery obstructions | ||
| 4.1 Chronic thromboembolic PH | 5 | 0.5 |
| 4.2.5 Congenital pulmonary artery stenoses | 15 | 1.4 |
| Group 5 PH with unclear and/or multifactorial mechanisms | ||
| 5.1.1 Hematologic, chronic hemolytic anemia | 4 | 0.4 |
| 5.2.3 Systemic/metabolic, glycogen storage disease | 1 | 0.1 |
| 5.2.4 Systemic/metabolic, neurofibromatosis | 1 | 0.1 |
| 5.2.x Systemic/metabolic - other named | 23 | 2.1 |
| 5.4.1.1 Complex CHD segmental PH isolated PA of ductal origin | 2 | 0.2 |
| 5.4.1.2 Complex CHD segmental PH absent PA | 3 | 0.3 |
| 5.4.1.3 Complex CHD segmental PH PA VSD MAPCAs | 10 | 0.9 |
| 5.4.1.4 Complex CHD segmental PH hemitruncus | 4 | 0.4 |
| 5.4.1.5 Complex CHD segmental PH other | 2 | 0.2 |
| 5.4.2 Single ventricle, including Fontan/Glenn | 37 | 3.4 |
| 5.4.3 Scimitar syndrome | 12 | 1.1 |
Definition of abbreviations: APAH = associated pulmonary arterial hypertension; CHD = congenital heart disease; FOXF1 = Forkhead box F1; LVEF = left ventricular ejection fraction; MAPCA = major aortopulmonary collateral; PA = pulmonary artery; PAH = pulmonary arterial hypertension; PCH = pulmonary capillary hemangiomatosis; PH = pulmonary hypertension; PVOD = pulmonary venoocclusive disease; TBX4 = T-box transcription factor-4; TTF1/NKX2-1 = thyroid transcription factor; VSD = ventricular septal defect.
Table 2.
Baseline Characteristics of Overall Pulmonary Hypertension Cohort and for Pulmonary Hypertension Diagnostic Groups 1 and 3
| Overall (n = 1101) | Group 1 (n = 529) | Group 3 (n = 356) | P Value (Group 1 vs. 3) | |
|---|---|---|---|---|
| Demographic characteristics | ||||
| Incident PH, n (%) | 1061 (96.4) | 497 (94.0) | 355 (99.7) | <0.001 |
| Female, n (%) | 558 (50.7) | 295 (55.8) | 170 (47.8) | 0.02 |
| Age at diagnosis, y | 2.6 [0.8 to 8.2] | 4.8 [1.7 to 11] | 0.85 [0.5 to 2] | <0.001 |
| Perinatal history | ||||
| Gestation at birth, weeks | 39 [32.5 to 40] | 40 [37.7 to 40] | 30.3 [26.1 to 38] | <0.001 |
| Preterm, n (%) | 343 (36.9) | 86 (19.6) | 222 (67.1) | <0.001 |
| Admission to neonatal unit, n (%) | 628 (71.4) | 207 (50.5) | 302 (93.8) | <0.001 |
| Oligohydramnios, n (%) | 14 (1.3) | 3 (0.6) | 11 (3.2) | 0.008 |
| Chorioamnionitis, n (%) | 10 (0.9) | 2 (0.4) | 8 (2.3) | 0.03 |
| History of PPHN, n (%) | 101 (11.1) | 31 (6.8) | 63 (22) | <0.001 |
| History of ECMO, n (%) | 62 (5.7) | 19 (3.6) | 25 (7.1) | 0.03 |
| Comorbidities | ||||
| Family history of PH, n (%) | 24 (3.3) | 17 (4.2) | 3 (1.6) | 0.16 |
| CHD, n (%) | 816 (74.1) | 405 (76.6) | 231 (64.9) | <0.001 |
| Down syndrome, n (%) | 176 (16.0) | 123 (23.3) | 44 (12.4) | <0.001 |
| Associated lung disease, n (%) | 499 (45.4) | 104 (19.7) | 336 (94.4)* | <0.001 |
| Developmental | 337 (30.6) | 45 (8.5) | 256 (71.9) | <0.001 |
| Aspiration | 82 (7.4) | 22 (4.5) | 55 (15.4) | <0.001 |
| Recurrent infection | 35 (3.2) | 8 (1.5) | 24 (6.7) | <0.001 |
| ILD | 33 (3.0) | 7 (1.3) | 25 (7) | <0.001 |
| Other primary lung disease | 18 (1.6) | 6 (1.1) | 11 (3.1) | 0.07 |
| Unclear cause | 31 (2.8) | 11 (2.1) | 14 (3.9) | 0.15 |
| Associated airway disease, n (%) | 132 (12.0) | 42 (7.9) | 71 (19.9) | <0.001 |
| Clinical assessment at presentation | ||||
| Height z-score | −1.1 [−2.1 to 0.1] | −0.8 [−1.8-0.1] | −1.6 [−3.1 to 0.9] | <0.001 |
| Weight z-score | −1.4 [−2.7 to −0.3] | −1.0 [−2.0-0] | −2.3 [−3.7 to 0.9] | <0.001 |
| Resting oxygen saturations, % | 96 [92 to 98] | 96 [90 to 98] | 96 [94 to 98] | 0.11 |
| WHO functional class | <0.001 | |||
| I | 181 (20.3) | 109 (22.9) | 43 (18.1) | |
| II | 247 (27.8) | 146 (30.7) | 46 (19.4) | |
| III | 262 (29.4) | 161 (33.8) | 32 (13.5) | |
| IV | 200 (22.5) | 60 (12.6) | 116 (48.9) | |
| Presenting syncope, n (%) | 68 (6.2) | 57 (10.8) | 5 (1.4) | <0.001 |
| 6MW test completed† | 190 (54.4) | 139 (59.1) | 12 (38.7) | <0.001 |
| 6MW distance, m | 309 ± 118 | 312 ± 119 | 293 ± 128 | 0.59 |
| Catheterization completed | 684 (62.4) | 425 (81.0) | 99 (27.9) | <0.001 |
Definition of abbreviations: 6MW = 6-minute-walk; CHD = congenital heart disease; ECMO = extracorporeal membrane oxygenation; ILD = interstitial lung disease; PH = pulmonary hypertension; PPHN = persistent pulmonary hypertension of the newborn; WHO = world health organization.
Continuous variables are presented as median [interquartile range] or mean ± standard deviation. Categorical variables are presented as n (%). P < 0.05 indicative of statistical significance. P values <0.05 were indicative of statistical significance (shown in bold font).
Patients with group 3 pulmonary hypertension without lung disease had pulmonary hypertension secondary to hypoxia alone.
Reported in patients >6 yr old.
Figure 1.
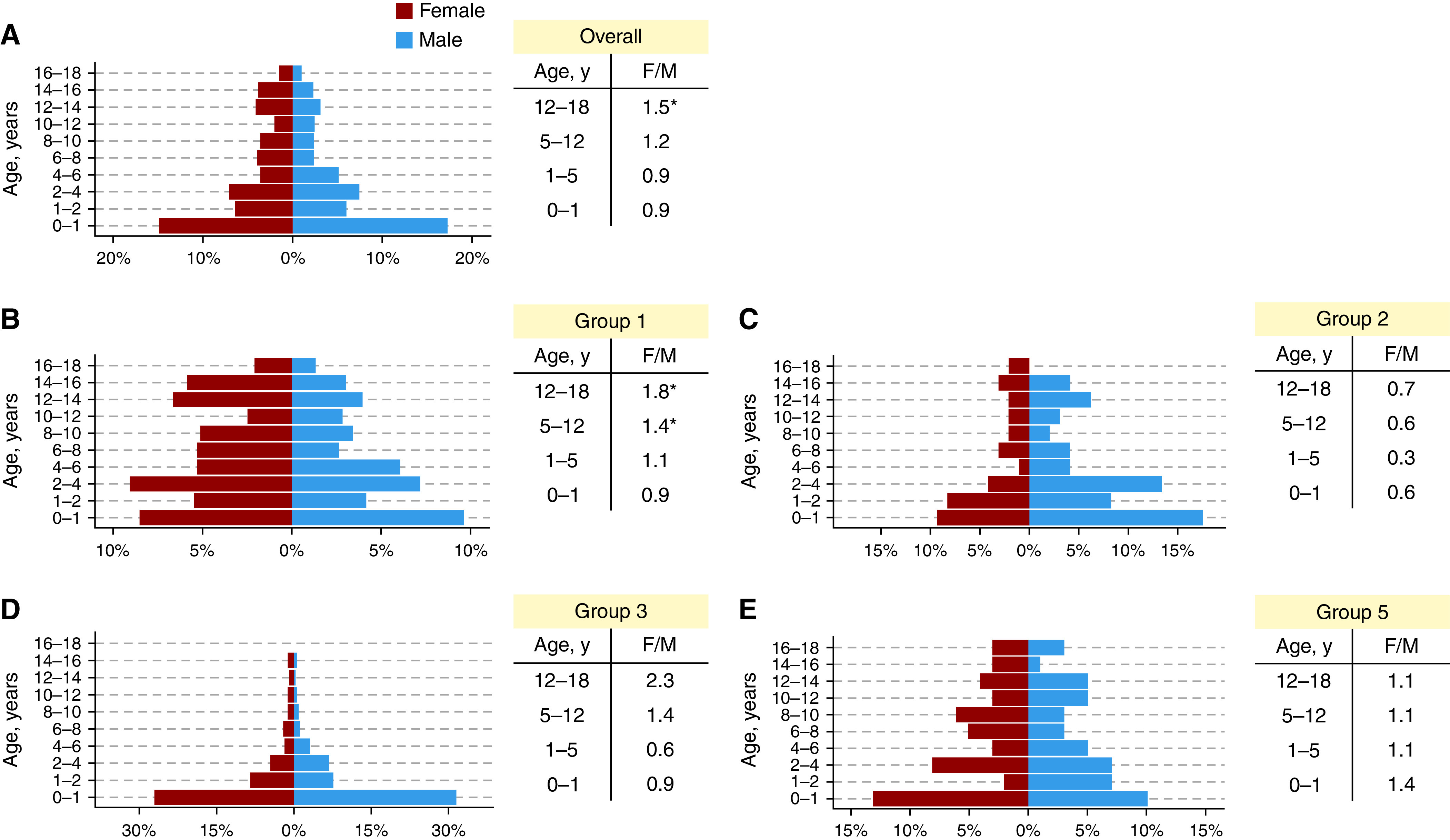
(A–E) Diagnoses by age for the overall pulmonary hypertension cohort (A) and for pulmonary hypertension groups 1, 2, 3, and 5 (B–E) with corresponding sex ratios for infancy, young childhood, middle childhood, and adolescence. *P < 0.05 on chi-square testing compared with expected childhood sex ratio in the United Kingdom.
Group 3 PH (PH due to lung disease) was the second most common diagnosis (32.3% of the total population). Within this category, young children with bronchopulmonary dysplasia and congenital diaphragmatic hernia were the largest subgroups, accounting for 48.3% and 11.0% of group 3 patients, respectively.
Group 2 (left heart disease) and group 5 PH (unclear and/or multifactorial mechanisms) accounted for 8.8% and 9.0%, respectively. Of group 2 patients, 38.1% were related to ventricular disease, 33.0% were because of pulmonary vein stenosis, and 24.7% were secondary to valve disease. Group 5 mainly consisted of patients with congenital heart disease (28.3% Glenn/Fontan, 8.1% unoperated univentricular, 21.0% segmental PH, and 12.0% scimitar syndrome) or patients with systemic/metabolic disorders (29.3%). Group 4 PH was the least common diagnosis (20, 1.8%), with only 5 (0.5%) cases of chronic thromboembolic PH and 15 (1.4%) cases of peripheral pulmonary artery obstructions.
Overall, 186 (16.9%) patients had features fitting more than one PH diagnostic group (“intergroup links”) (Figure E1). Two different PH diagnostic groups were identified in 14.5% and three groups in 2.4%. The most commonly associated diagnostic groups were groups 1 and 3 (12.3%, 113 of 917 patients with groups 1 or 3 PH), bronchopulmonary dysplasia and PAH related to congenital heart disease coexisting in 3.6% of patients.
Incidence, Prevalence, and Sex Ratios by Primary PH Group
The estimated annual incidence of childhood PH in the United Kingdom was 3.5 (95% confidence interval [CI], 3.3–3.8) per 1 million population, varying by age and diagnostic group (Figure 2; see Figure E2 for CIs). The incidence of PH was highest in infancy across all groups, with the highest incidence occurring in group 3 (15.0 [95% CI, 12.7–17.2] cases per 1 million children per year). Beyond infancy, group 1 PH had the highest incidence. The prevalence of childhood PH in the United Kingdom was 18.1 (95% CI, 15.8–20.4) per 1 million population. PAH had the highest prevalence at 9.8 (95% CI, 8.1–11.5) cases per 1 million, whereas the prevalence of group 4 PH was 0.3 (95% CI, 0–0.5) cases per 1 million.
Figure 2.

Estimated annual incidence of childhood pulmonary hypertension (PH) in the United Kingdom (groups 1–5) for infancy (0–1 yr), early childhood (1–5 yr), prepubescence (5–12 yr), and adolescence (12–18 yr). The average point prevalence for the years 2005–2019 inclusive is also given for each PH group and for the overall childhood population. See Figure E2 in the online supplement for confidence intervals.
There was a significant interaction between age group and sex (P = 0.03), deriving mainly from the adolescent age group (12–18 yr). Indeed, in all groups, there was a significant female preponderance in adolescence (12–18 years; 1.5:1; P = 0.006), but not in children <12 years (P = 0.9) (Figure 1). This was exaggerated in PAH, where there was an excess of female patients in the 5- to 12- and 12- to 18-year age groups (P = 0.04 and P = 0.001, respectively). Group 2 PH was significantly more frequent in male children (0–18 years; 1.8:1; P = 0.02), whereas there was a female preponderance in idiopathic or heritable PAH (0–18 years; 1.6:1; P = 0.02).
Heritable PAH and Associated Genetic Syndromes
Genetic abnormalities were detected in 370 (34.1%) patients. Down syndrome was the most commonly associated syndrome, occurring in 176 (16.0%) children. Overall, 17 (4.2% of 724 patients for whom family history was available) patients had a positive family history of PAH. Gene mutations causing heritable PAH along with other genetic abnormalities are indicated in Table E3.
Presenting Clinical Features
In this pediatric cohort, only 677 (61.9%) patients had typical symptoms of PH at presentation. In the remainder, PH was identified during routine evaluation of an underlying condition, such as congenital heart disease (57.0%), or during intercurrent illness (43.0%). Dyspnea on exertion or exercise limitation (n = 329, 30.1%) was the most reported presenting symptom, followed by cardiorespiratory failure (n = 117, 10.7%) (Table E4). In 68 (6.2%) patients, syncope was the main presenting symptom, and this was much more common in patients with PAH (10.8% vs. 1.9% in other PH groups; P < 0.001). Functional class at diagnosis (World Health Organization classification) was available for 890 (80.8%) patients. World Health Organization functional class could not be determined for 92(8.4%) patients with various types of disability. Patients in group 3 had the highest proportion in functional class III or IV (P < 0.001) followed by patients in group 1 (P < 0.001). Growth restriction was observed in the overall PH cohort (median height z-score, −1.1 [IQR, −2.1 to 0.1]; median weight z-score, −1.4 [IQR, −2.7 to 0.3]) and was greater for group 3 than group 1 patients with PH (both P < 0.001). A 6-minute-walk distance at presentation was available for 190 (54.4%) patients over the age of 6 years at diagnosis. The median distance walked was 325 [IQR, 240 to 400] m in the overall PH group.
Diagnostic cardiac catheterization was performed in 684 (62.4%) patients in the overall group. Median pulmonary vascular resistance index was higher in group 1 than group 3 PH (12.0 [IQR, 7.0–19.9] vs. 7.2 [IQR, 4.7–10.1] WU.m2; P < 0.001) (Table E5).
Initial PAH Therapy
The initial PAH therapeutic strategy (within 8 weeks of PH diagnosis) in the overall PH cohort and in PH group 1 and 3 is shown in Table 3 (for details of clinical management and outcomes for all PH diagnostic groups, see Table E6). At 8 weeks, 617 (56.0%) patients were on monotherapy, most commonly with a phosphodiesterase-5 inhibitor (47.7%) or an endothelin receptor antagonist (5.8%). Upfront dual-agent therapy was used in 157 (14.3%) patients, whereas initial triple therapy was used in 46 (4.2%). Patients with group 3 PH were less likely to be started or maintained on combination therapy after initial specialist assessment (12.6% vs. 23.4%; P < 0.001). Conversely, children diagnosed with idiopathic PAH were more likely to be treated aggressively with upfront combination therapy than other patients with PAH (49.3% vs. 13.6%; P < 0.001). Of 146 patients with idiopathic or heritable PAH, 24 (16.4%) were treated with calcium channel blockers as acute vasoresponders.
Table 3.
Clinical Management and Outcome in the Overall Pediatric Pulmonary Hypertension Cohort and for Groups 1 and 3 Pulmonary Hypertension
| Overall (n = 1101) | Group 1 (n = 529) | Group 3 (n = 356) | P Value (Group 1 vs. 3) | |
|---|---|---|---|---|
| Clinical management | ||||
| PH therapy use at any time | 910 (82.7) | 437 (82.6) | 309 (86.8) | 0.11 |
| Initial PH therapeutic strategy | ||||
| Monotherapy | 617 (56) | 266 (50.3) | 238 (66.9) | <0.001 |
| Dual agent combination therapy | 157 (14.3) | 90 (17.0) | 39 (11.0) | 0.02 |
| Triple therapy | 46 (4.2) | 34 (6.4) | 6 (1.7) | 0.002 |
| None | 281 (25.5) | 139 (26.3) | 73 (20.5) | 0.06 |
| Initial monotherapy | ||||
| PDE5 inhibitor | 525 (47.7) | 193 (36.5) | 234 (65.7) | <0.001 |
| ERA | 64 (5.8) | 54 (10.2) | 2 (0.6) | <0.001 |
| Prostanoid | 3 (0.3) | 2 (0.4) | 0 (0) | 0.66 |
| Therapy at end of follow-up | ||||
| Monotherapy | 390 (35.4) | 168 (31.8) | 135 (37.9) | 0.07 |
| Dual agent combination therapy | 197 (17.9) | 135 (25.5) | 29 (8.1) | <0.001 |
| Triple therapy | 102 (9.3) | 83 (15.7) | 14 (3.9) | <0.001 |
| None | 412 (37.4) | 143 (27.0) | 178 (50) | <0.001 |
| Clinical outcomes | ||||
| Resolution within 5 yr of diagnosis, %* | 23.6 | 10.7 | 38.6 | — |
| Transplanted lung or heart within 5 yr of diagnosis, %* | 2.7 | 3.5 | 1.1 | — |
Definition of abbreviations: ERA = endothelin receptor antagonist; PDE5 = phosphodiesterase 5; PH = pulmonary hypertension.
Outcome statistics are based on the competing risks analysis. Continuous variables are presented as median [interquartile range] or mean ± standard deviation. Categorical variables are presented as n (%). P < 0.05 indicative of statistical significance.
Management of PH at the Latest Follow-Up
At the latest follow-up visit, 696 (76.4% of patients started on PAH therapy) patients remained on PAH therapy (monotherapy in 35.0% and combination therapy in 27.2%). During the entire study period, prostanoid therapy was used in 144 (13.1%) patients, in 102 (70.8%) of whom it was required as part of triple therapy. Triple therapy was far more common in patients with idiopathic PAH than other PAH (37.0% vs. 7.6%; P < 0.001). Of patients on prostanoid therapy, 73.6% were on intravenous epoprostenol, 16.7% on inhaled iloprost, 6.9% on subcutaneous Treprostinil, and 3.5% on oral selexipag. Only half (n = 181, 50.8%) of patients in group 3 were maintained on PAH therapy until the end of follow-up and were more likely to receive sildenafil monotherapy than patients with PAH (36.5% vs. 23.3%; P < 0.001). Only 15 (4.2%) patients in group 3 received prostanoid therapy, compared with 113 (21.4%) with PAH overall and 75 (51.4%) with idiopathic PAH (both P < 0.001).
In addition to PAH pharmacotherapy, 57 (5.2%) patients underwent the following interventional procedures for management of PH during the follow-up period: 41 (3.7%) patients received an atrial septostomy or atrial flow regulator, 6 (0.5%) underwent a Potts shunt, and 10 (0.9%) had stenting of an existing patent ductus arteriosus.
Clinical Outcomes
Clinical outcomes were examined for 970 (88.1%) patients with incident PH (median follow-up, 3.0 years [range, 1 day to 16.6 years]; 3,948.6 total patient-years). Resolution of PH occurred in 171 (15.5%) patients. In group 3, one-half of patients experienced PH resolution within 8.6 (95% CI, 8.5–8.7) years of initial PH diagnosis. Resolution was more common in group 3 than all other groups combined (38.6% vs. 16.6% at 5 years after diagnosis; P < 0.001), and patients with PAH (10.7%; P < 0.001). Of patients with group 1 PH who experienced resolution, the most common diagnostic subgroup was PAH related to congenital heart disease (CHD) (21 [5.5% of patients with PAH related to CHD and 61.8% of resolved PAH]), followed by drug- or toxin-related PAH (5 [50.0% of patients with drug-/toxin-related PAH and 14.7% of resolved PAH]) and persistent pulmonary hypertension of the newborn (PPHN) (4 [50.0% of patients with PPHN and 11.8% of resolved PAH]).
During follow-up, there were 268 (24.3%) deaths and 39 (3.5%) bilateral lung or heart–lung transplants after a median time on the active waiting list of 4.8 (IQR, 1.5–10.1) months. A further 20 (1.8%) patients had been listed for transplantation, of whom 55.0% died while on the transplant list after a median 8.2 (IQR, 2.3–15.4) months. In the overall PH group, transplant-free survival was 86.7% (95% CI, 84.5–89.0%) at 1 year, 73.9% (95% CI, 70.7–77.1%) at 5 years, and 68.6% (95% CI, 64.7–72.6%) at 10 years (Figure 3A). PAH related to CHD had the highest transplant-free survival (hazard ratio, 0.58; 95% CI, 0.43–0.79; P < 0.001). Group 2 had the lowest transplant-free survival (hazard ratio, 2.0; 95% CI, 1.36–2.94; P < 0.001), whereas overall survival in groups 3 and 5 did not differ significantly from group 1 PH (Figure 3B). Although early survival appeared to diverge between groups 1 and 3 PH, this difference was not statistically significant (P = 0.07; see Figure E3 for competing outcomes).
Figure 3.
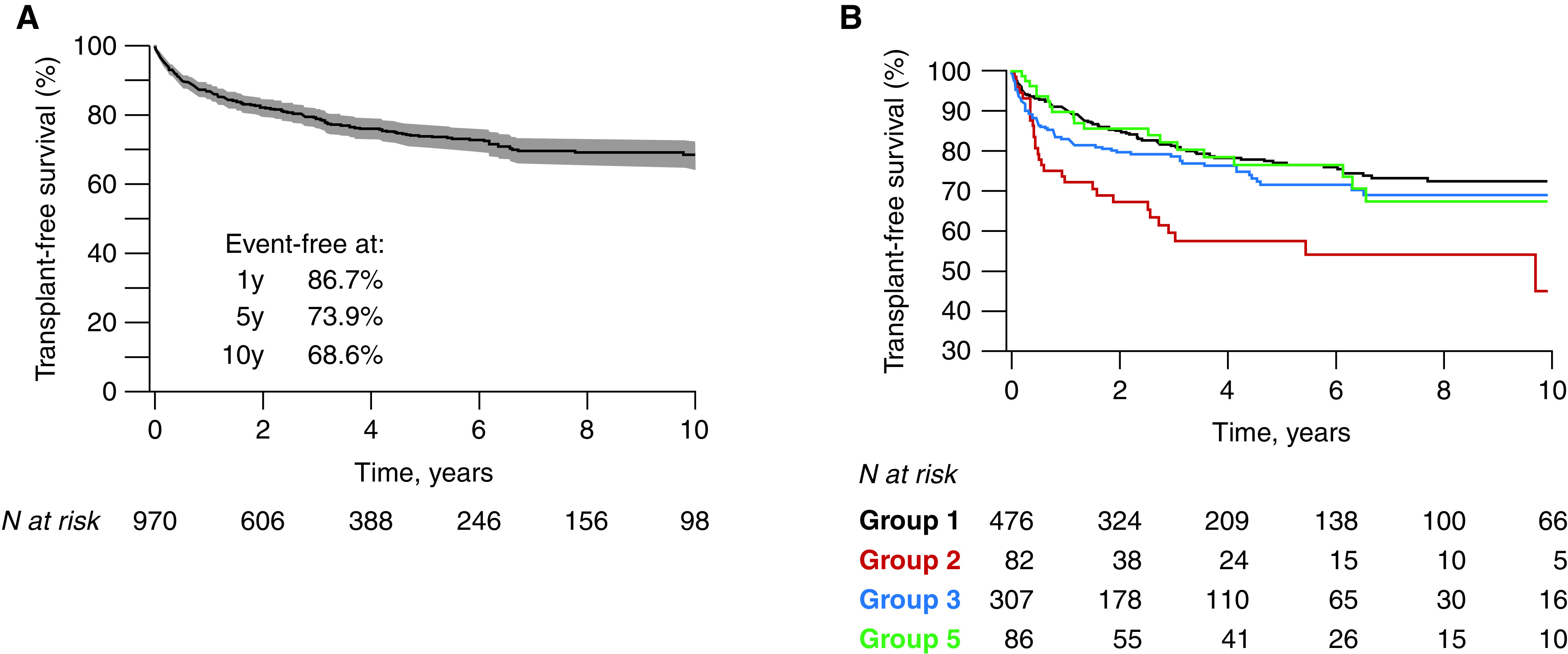
Kaplan-Meier survival curves in children with pulmonary hypertension, (A) in the overall cohort and (B) for the different pulmonary hypertension groups. The numbers below the horizontal axis represent the number of patients at risk.
Discussion
This paper describes the epidemiology, clinical characteristics, and outcomes of the largest national cohort of consecutive incident patients with pediatric PH, assessed over a 20-year period within a universal healthcare system. We found a substantial peak in the incidence of pediatric PH in infancy, as well as changes in subgroup distribution and sex ratio by age. Multiple contributory causes of PH were common, as was resolution of disease, especially in group 3 patients. Short- and longer-term outcomes were poor for all types of pediatric PH, despite significant usage of PAH therapies. The unique features of childhood PH described in this report challenge current disease paradigms and should inform future systems of disease classification.
Our study reports incidence and prevalence by pediatric PH group, as well as incidence by age. We found that PH incidence was highest in infancy for all groups and fell sharply thereafter, indicative of the dominance of developmental conditions related to pediatric disease. The incidence and prevalence of PH in childhood were similar to those reported in other clinical registries (2, 12, 13) and predictably lower than in studies that used administrative or claims data, given the different methodologies used (4, 14, 15). Indeed, registry design, and especially inclusion and exclusion criteria, affect the clinical phenotypes of the patients enrolled.
Our study included all five World Symposium on PH groups and did not require cardiac catheterization for the diagnosis of PH. PH due to lung disease was the predominant group in infancy, and these patients undergo cardiac catheterization less commonly than patients with other types of PH. Our cohort had a higher proportion of group 3 PH (32.3%) and a lower median age (2.6 yr) than the Tracking Outcome and Practice in Pediatric PH and the Spanish Registry for Pediatric PH (REHIPED), both of which required cardiac catheterization for registry inclusion. Similar to our cohort, the PPHNet registry did not require cardiac catheterization and had an even younger population (median age, 0.5 yr) with a higher proportion of group 3 PH (3, 16–18). Although group 1 PH has been the focus of research in pediatric PH, our results support the inclusion of patients with PH due to lung disease in pediatric PH registries and the development of initiatives seeking to improve diagnosis and management in this subset of patients.
One in six children in our study had PH related to more than one diagnostic group. The most common association was between bronchopulmonary dysplasia (group 3) and PAH related to CHD (group 1). The frequent cooccurrence of diverse causes of PH highlights the need for systematic phenotyping and care provided by multispecialty, multidisciplinary teams with PH clinicians working alongside pediatric cardiologists, respiratory pediatricians, neonatologists, and clinical geneticists, among others (6). Understanding the relative contribution of each cause of PH can be extremely challenging, especially as different conditions can evolve in different ways. For example, bronchopulmonary dysplasia may resolve, whereas PAH typically progresses. Therefore, the presence of multiple causes of PH may influence prognosis and the response to treatment.
PAH therapies are increasingly used in pediatrics and were used in a significant proportion of our patients (6). The use of PAH therapy in our population was similar to other registries in terms of the proportion of patients with PAH treated and the use of combination therapy. However, a higher proportion of patients received prostacyclin in the North American registry than our cohort, possibly pointing to differences in prescription practices and/or treatment incentives. In the past, the use of PAH therapies in children has been extrapolated almost exclusively from studies in adults given the common etiology between pediatric and adult group 1 disease. More recently, pediatric PH programs have enabled clinical trials of therapy in the childhood PAH population (19, 20). As expected, group 1 patients, and particularly those with idiopathic or heritable PAH, were far more likely to be treated with combination PAH therapy than other forms of PH. However, a sizeable proportion of patients outside of group 1 PH were treated with PAH therapy, including patients with PH associated with left heart disease and lung disease, who are rarely treated in adult centers. It should be noted that the etiology of pediatric PH outside of group 1 differs substantially from adult disease; hence, the role of PAH therapies in these children needs to be evaluated separately. For example, pediatric group 3 PH was caused primarily by developmental lung disease related to bronchopulmonary dysplasia or congenital diaphragmatic hernia. These involve completely different pathophysiological processes compared with adult group 3 PH, which is most commonly seen in the context of chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis (21). In group 2 PH, the main pediatric etiologies were pulmonary vein stenosis and childhood cardiomyopathies (22), and patients referred to the pediatric PH service are typically those with advanced disease, in whom experts may use PAH therapies as an adjunct to conventional heart failure therapy. Finally, group 5 PH in childhood was related to either complex congenital heart disease, or to rare systemic or metabolic diseases, for which an evidence base for PAH therapy is largely absent. While awaiting clinical trials in these groups, PAH therapies should only be considered and managed in specialist centers on an individual-patient basis, with careful monitoring of side effects and therapeutic response.
This cohort covers an extended period of 20 years. To avoid immortal time bias, we restricted our survival analysis to consecutive, incident patients (16, 23). As such, the results likely provide an accurate indication of expected outcome in newly diagnosed individuals. Almost one-third of patients had undergone transplantation or died within 10 years of diagnosis, and patients with group 2 PH had the worst outcome. Our outcomes were broadly similar to those reported by REHIPED and intermediate between other European and North American registries (2, 23). Overall transplant-free survival in the PPHNet registry was 84% at 5 years, compared with 73.9% in the present study. Clinical characteristics of the study population potentially explain small differences in outcomes. Apart from being younger at enrollment, with a higher proportion of group 3 PH, children in the PPHNet registry had more favorable hemodynamics (e.g., PVRI 7.1 vs. 10 WU.m2) and a higher rate of disease resolution than our cohort. This was especially striking in patients with PAH, for whom resolution at 5 years in the North American registry was 42% versus 10.7% in our study. Indeed, patients with transient forms of PH were present in greater numbers in PPHNet: PPHN was the primary diagnosis in 7.6% versus 0.7% of patients in our cohort. Our overall outcomes were broadly similar to those reported by REHIPED, where survival at 3 years was 80%. However, the Spanish registry reported superior outcomes in patients with PAH compared with the remainder, which neither our study nor the North American registry was able to demonstrate. Pediatric PH of all types still carries significant morbidity and mortality despite major improvements in diagnosis, supportive care, and pharmacological therapies that have improved the natural history of the disease. There remains an urgent need for therapeutic innovation for these patients (24, 25).
Resolution of PH is a unique feature of pediatric disease, occurring in one in seven patients diagnosed in our center, and was far more common in group 3 PH. Resolution occurred at a median 2 years from study inclusion, indicating that the majority of these patients do not have “transient PH” as previously defined (4). Currently, there are no data to predict which patients will experience resolution of their disease. Further study of the process of PH resolution may provide novel mechanistic insights, with the potential to drive therapeutic advances aimed at developing a cure for PH in children and adults.
The main limitation of our study was its retrospective design, with data collected from clinical records and databases. As with all retrospective cohort studies, our analysis may be subject to selection bias owing to referral patterns and inclusion criteria. For example, we did not have access to data, including outcomes, of patients who were not referred to the PH service. As a national referral center, however, the influence of geographic coverage and competing referral pathways is minimized, especially for patients with significant or progressive PH. Although right heart catheterization is required to confirm the diagnosis of PH, we also enrolled a smaller proportion of patients without a cardiac catheter after thorough noninvasive assessment, which permitted the inclusion of individuals who were too unwell or not suitable for catheterization. Moreover, our cohort included very few patients with transient forms of PH, such as PPHN, reflecting referral practices in the United Kingdom. Such transient forms of PH are not fully captured in our study, and our estimates of incidence and prevalence focus mainly on more persistent types of PH. Finally, reporting changes in survival over time and the impact of PAH therapy/therapeutic strategy on hemodynamics and clinical outcomes was beyond the scope of the current study.
Despite these limitations, this study represents the largest reported national cohort of consecutive children with PH. The size of the population, the completeness and granularity of the clinical data, and the extensive follow-up allowed an accurate description of the epidemiology, clinical phenotypes, and long-term outcomes of the overall PH group and diagnostic subgroups.
In conclusion, pediatric patients with PH managed in a tertiary referral center are a vastly heterogeneous group, with a high burden of disease in infancy and early childhood, and significantly adverse outcomes despite widespread use of PAH therapies. Structured clinical programs are necessary to ensure high quality of care but also facilitate research and accelerate the transition of therapeutic innovations from adult PH to children (26).
Acknowledgments
Acknowledgment
The authors thank the Dinosaur trust and Great Ormond Street Hospital children’s charity for supporting research capacity within the pediatric pulmonary hypertension unit. All research at Great Ormond Street Hospital National Health Service Foundation Trust and University College London Great Ormond Street Institute of Child Health is made possible by the National Institute for Health and Care Research Great Ormond Street Hospital Biomedical Research Center. The views expressed are those of the author(s) and not necessarily those of the National Health Service, the National Institute for Health and Care Research, or the Department of Health.
Footnotes
Author Contributions: S.M., S.G.H., and A.C. conceptualized the study. A.C. performed data curation. A.C., K.D., and S.M. performed formal analysis and data visualization. Supervision was performed by S.M. and K.D. A.C. and S.M. wrote the original draft, which was critically reviewed and edited by K.D. and V.M.
Data sharing statement: To minimize the possibility of unintentionally sharing information that can be used to reidentify private information, and in line with the conditions of the ethics approval, patient-level data are not available for use outside of this study.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202110-2428OC on May 17, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Rosenzweig EB, Abman SH, Adatia I, Beghetti M, Bonnet D, Haworth S, et al. Paediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. Eur Respir J . 2019;53:1801916. doi: 10.1183/13993003.01916-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. del Cerro Marín MJ, Sabaté Rotés A, Rodriguez Ogando A, Mendoza Soto A, Quero Jiménez M, Gavilán Camacho JL, et al. REHIPED Investigators Assessing pulmonary hypertensive vascular disease in childhood. Data from the Spanish registry. Am J Respir Crit Care Med . 2014;190:1421–1429. doi: 10.1164/rccm.201406-1052OC. [DOI] [PubMed] [Google Scholar]
- 3. Berger RM, Beghetti M, Humpl T, Raskob GE, Ivy DD, Jing Z-C, et al. Clinical features of paediatric pulmonary hypertension: a registry study. Lancet . 2012;379:537–546. doi: 10.1016/S0140-6736(11)61621-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Loon RLE, Roofthooft MTR, Hillege HL, ten Harkel ADJ, van Osch-Gevers M, Delhaas T, et al. Pediatric pulmonary hypertension in the Netherlands: epidemiology and characterization during the period 1991 to 2005. Circulation . 2011;124:1755–1764. doi: 10.1161/CIRCULATIONAHA.110.969584. [DOI] [PubMed] [Google Scholar]
- 5.Constantine A, Dimopoulos K, Muthurangu V, Haworth S, Moledina S.Twenty-year experience and outcomes in a national paediatric pulmonary hypertension service Eur Heart J 202142XXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, et al. American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Surgery and Anesthesia; and the American Thoracic Society Pediatric pulmonary hypertension: guidelines from the American Heart Association and American Thoracic Society. Circulation . 2015;132:2037–2099. doi: 10.1161/CIR.0000000000000329. [DOI] [PubMed] [Google Scholar]
- 7. Flegal KM, Cole TJ. Construction of LMS parameters for the Centers for Disease Control and Prevention 2000 growth charts. Natl Health Stat Rep . 2013;63:1–3. [PubMed] [Google Scholar]
- 8. Zemel BS, Pipan M, Stallings VA, Hall W, Schadt K, Freedman DS, et al. Growth charts for children with Down Syndrome in the United States. Pediatrics . 2015;136:e1204–e1211. doi: 10.1542/peds.2015-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fenton TR, Kim JH. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr . 2013;13:59. doi: 10.1186/1471-2431-13-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Galiè N, Humbert M, Vachiery J-L, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT) Eur Respir J . 2015;46:903–975. doi: 10.1183/13993003.01032-2015. [DOI] [PubMed] [Google Scholar]
- 11.Estimates of the population for the UK https://www.ons.gov.uk/peoplepopulationandcommunity/populationandmigration/populationestimates/datasets/populationestimatesforukenglandandwalesscotlandandnorthernireland.
- 12. Kwiatkowska J, Zuk M, Migdal A, Kusa J, Skiba E, Zygielo K, et al. Children and adolescents with pulmonary arterial hypertension: baseline and follow-up data from the Polish Registry of Pulmonary Hypertension (BNP-PL) J Clin Med . 2020;9:E1717. doi: 10.3390/jcm9061717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moledina S, Hislop AA, Foster H, Schulze-Neick I, Haworth SG. Childhood idiopathic pulmonary arterial hypertension: a national cohort study. Heart . 2010;96:1401–1406. doi: 10.1136/hrt.2009.182378. [DOI] [PubMed] [Google Scholar]
- 14. Li L, Jick S, Breitenstein S, Hernandez G, Michel A, Vizcaya D. Pulmonary arterial hypertension in the USA: an epidemiological study in a large insured pediatric population. Pulm Circ . 2017;7:126–136. doi: 10.1086/690007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leber L, Beaudet A, Muller A. Epidemiology of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: identification of the most accurate estimates from a systematic literature review. Pulm Circ . 2021;11:2045894020977300. doi: 10.1177/2045894020977300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abman SH, Mullen MP, Sleeper LA, Austin ED, Rosenzweig EB, Kinsella JP, et al. Pediatric Pulmonary Hypertension Network Characterisation of paediatric pulmonary hypertensive vascular disease from the PPHNet Registry. Eur Respir J . 2021;59:2003337. doi: 10.1183/13993003.03337-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest . 2010;137:376–387. doi: 10.1378/chest.09-1140. [DOI] [PubMed] [Google Scholar]
- 18. Marín MJ del C, Rotés AS, Ogando AR, Soto AM, Jiménez MQ, Camacho JLG, et al. Assessing pulmonary hypertensive vascular disease in childhood. Data from the Spanish registry. Am J Respir Crit Care Med . 2014;190:1421–1429. doi: 10.1164/rccm.201406-1052OC. [DOI] [PubMed] [Google Scholar]
- 19. Barst RJ, Ivy DD, Gaitan G, Szatmari A, Rudzinski A, Garcia AE, et al. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension. Circulation . 2012;125:324–334. doi: 10.1161/CIRCULATIONAHA.110.016667. [DOI] [PubMed] [Google Scholar]
- 20. Barst RJ, Beghetti M, Pulido T, Layton G, Konourina I, Zhang M, et al. STARTS-2 Investigators STARTS-2: long-term survival with oral sildenafil monotherapy in treatment-naive pediatric pulmonary arterial hypertension. Circulation . 2014;129:1914–1923. doi: 10.1161/CIRCULATIONAHA.113.005698. [DOI] [PubMed] [Google Scholar]
- 21. Vachiéry J-L, Adir Y, Barberà JA, Champion H, Coghlan JG, Cottin V, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol . 2013;62:D100–D108. doi: 10.1016/j.jacc.2013.10.033. [DOI] [PubMed] [Google Scholar]
- 22. Kalfa D, Belli E, Bacha E, Lambert V, di Carlo D, Kostolny M, et al. European Congenital Heart Surgeons Association Primary pulmonary vein stenosis: outcomes, risk factors, and severity score in a multicentric study. Ann Thorac Surg . 2017;104:182–189. doi: 10.1016/j.athoracsur.2017.03.022. [DOI] [PubMed] [Google Scholar]
- 23. Zijlstra WMH, Douwes JM, Rosenzweig EB, Schokker S, Krishnan U, Roofthooft MTR, et al. Survival differences in pediatric pulmonary arterial hypertension: clues to a better understanding of outcome and optimal treatment strategies. J Am Coll Cardiol . 2014;63:2159–2169. doi: 10.1016/j.jacc.2014.02.575. [DOI] [PubMed] [Google Scholar]
- 24. Barst RJ. Children deserve the same rights we do: the need for paediatric pulmonary arterial hypertension clinical drug development. Heart . 2010;96:1337–1338. doi: 10.1136/hrt.2010.195248. [DOI] [PubMed] [Google Scholar]
- 25. Newman JH, Rich S, Abman SH, Alexander JH, Barnard J, Beck GJ, et al. Enhancing insights into pulmonary vascular disease through a precision medicine approach. A joint NHLBI-Cardiovascular Medical Research and Education Fund workshop report. Am J Respir Crit Care Med . 2017;195:1661–1670. doi: 10.1164/rccm.201701-0150WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ollivier C, Sun H, Amchin W, Beghetti M, Berger RMF, Breitenstein S, et al. New strategies for the conduct of clinical trials in pediatric pulmonary arterial hypertension: outcome of a multistakeholder meeting with patients, academia, industry, and regulators, held at the European Medicines Agency on Monday, June 12, 2017. J Am Heart Assoc . 2019;8:e011306. doi: 10.1161/JAHA.118.011306. [DOI] [PMC free article] [PubMed] [Google Scholar]