

Abstract
Rationale
Patients with chronic obstructive pulmonary disease (COPD) develop more severe coronavirus disease (COVID-19); however, it is unclear whether they are more susceptible to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and what mechanisms are responsible for severe disease.
Objectives
To determine whether SARS-CoV-2 inoculated primary bronchial epithelial cells (pBECs) from patients with COPD support greater infection and elucidate the effects and mechanisms involved.
Methods
We performed single-cell RNA sequencing analysis on differentiated pBECs from healthy subjects and patients with COPD 7 days after SARS-CoV-2 inoculation. We correlated changes with viral titers, proinflammatory responses, and IFN production.
Measurements and Main Results
Single-cell RNA sequencing revealed that COPD pBECs had 24-fold greater infection than healthy cells, which was supported by plaque assays. Club/goblet and basal cells were the predominant populations infected and expressed mRNAs involved in viral replication. Proteases involved in SARS-CoV-2 entry/infection (TMPRSS2 and CTSB) were increased, and protease inhibitors (serpins) were downregulated more so in COPD. Inflammatory cytokines linked to COPD exacerbations and severe COVID-19 were increased, whereas IFN responses were blunted. Coexpression analysis revealed a prominent population of club/goblet cells with high type 1/2 IFN responses that were important drivers of immune responses to infection in both healthy and COPD pBECs. Therapeutic inhibition of proteases and inflammatory imbalances reduced viral titers and cytokine responses, particularly in COPD pBECs.
Conclusions
COPD pBECs are more susceptible to SARS-CoV-2 infection because of increases in coreceptor expression and protease imbalances and have greater inflammatory responses. A prominent cluster of IFN-responsive club/goblet cells emerges during infection, which may be important drivers of immunity. Therapeutic interventions suppress SARS-CoV-2 replication and consequent inflammation.
Keywords: COVID-19, COPD, single-cell RNA sequencing, protease, interferon
At a Glance Commentary
Scientific Knowledge on the Subject
To date, most studies examining the susceptibility of patients with chronic obstructive pulmonary disease to COVID-19 are on the basis of clinical observations; however, there is no experimental evidence to support these observations.
What This Study Adds to the Field
This study shows that patients with chronic obstructive pulmonary disease have increased severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, greater inflammatory responses to infection, and examine therapeutic interventions to reduce susceptibility to infection.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is causing a major global pandemic of coronavirus disease (COVID-19). Mortality rates are approximately 1%, and age, sex, demographics, and variants of concern are predictors of death (1). Disease manifestations are variable, with 80% of patients presenting with asymptomatic or mild disease features including fever, cough, fatigue, and loss of taste or smell (2). However, approximately 20% of patients experience severe disease, characterized by diffuse pneumonia, acute lung injury, and acute respiratory distress syndrome, often requiring mechanical ventilation.
SARS-CoV-2 is predominantly transmitted via inhalation of respiratory droplets from infected individuals. On entering the airways, the viral spike protein binds to the host viral entry receptor ACE2 (angiotensin converting-enzyme-2), which is highly expressed in the epithelium of the nasal cavities, trachea, and large and small airways (3, 4). ACE2 is highly expressed on differentiated primary bronchial epithelial cells (pBECs) with minimal expression in undifferentiated or submerged ex vivo cultures (5). ACE2 expression increases with age and in both alveolar and pBECs after cigarette smoke exposure, indicating a potential mechanism of increased risk of SARS-CoV-2 infection in the elderly, current smokers, and those with underlying chronic lung diseases (3, 6–8).
Cellular (e.g., TMPRSS2 [transmembrane serine protease-2]) and endosomal cysteine (e.g., CTSB [cathepsin-B] and CTSL [cathepsin-L]) proteases prime and cleave the spike protein from the receptor-binding domain, enabling SARS-CoV-2 internalization (2, 9, 10). In the lungs of patients with chronic obstructive pulmonary disease (COPD), there is a protease imbalance with greater activity of proteases and lower expression of protease inhibitors (e.g., serpins), which contribute to emphysema (11, 12). Whether this imbalance increases susceptibility to SARS-CoV-2 in patients with COPD is unknown.
COPD is a widely reported risk factor for hospitalization, mechanical ventilation, severe disease, and mortality in COVID-19 (2, 13–15). Patients with COPD generally have increased susceptibility to respiratory viral infections, such as influenza and virally induced exacerbations (16–19). Most clinical studies fail to adequately assess the infectivity rate of patients with COPD compared with the general population because of confounding factors including age, sex, comorbidities, unknown viral loads, and variants of concern with increased infectivity. Comparatively, infection of ex vivo pBECs differentiated at the air–liquid interface (ALI) under controlled conditions provides a true comparison of infectivity and disease severity while recapitulating important physiological tissue-specific conditions of human disease. However, no study has directly examined the responses of COPD pBECs cultured at ALI to SARS-CoV-2 infection.
Here, we infected pBECs from COPD and healthy donors with SARS-CoV-2 and examined viral load and host responses using single-cell RNA sequencing (scRNAseq). We also performed therapeutic interventions to elucidate the mechanisms of increased susceptibility.
Methods
Study approvals/patient recruitment, collection/culture of human pBECs, SARS-CoV-2 infection, therapeutic interventions, preparation of single-cell suspensions, background correction of ambient RNA, scRNAseq, single-cell transcriptomics, genotype identification, infected cell comparisons, disease state with infection comparisons, trajectory analysis, and in vivo comparisons are described in the online supplement and/or as described previously (3, 16, 17, 20–26).
Results
A Prominent Club/Goblet Cell Population Expressing IFN Responses Emerges in SARS-CoV-2–infected COPD and healthy pBECs
pBECs from COPD (n = 4) and healthy (n = 3) donors were differentiated at the ALI for 42 days and then infected with SARS-CoV-2 (Multiplicity of Infection 0.1) or sham (Figure 1A, Table 1). Samples were collected on Day 7 postinfection (dpi), and mRNA expression was assessed using scRNAseq (BD Rhapsody). Overall, 16,597 pBECs passed quality control across all samples (Figures 1B and E1A in the online supplement). scRNAseq revealed nine different cell clusters on the basis of transcriptomic profiles (Figure 1C), which encompassed three major cell types: club/goblet, basal, and ciliated cells (Figure 1D). Clusters were identified using known markers of club/goblet (MUC5AC, SCGB1A1, MSMB, and CEACAM6), basal (KRT5, TP63, and C1orf74), cycling basal (MKI67), and ciliated (FOXJ1 and C10orf95) cells (Figure 1B–1E).
Figure 1.
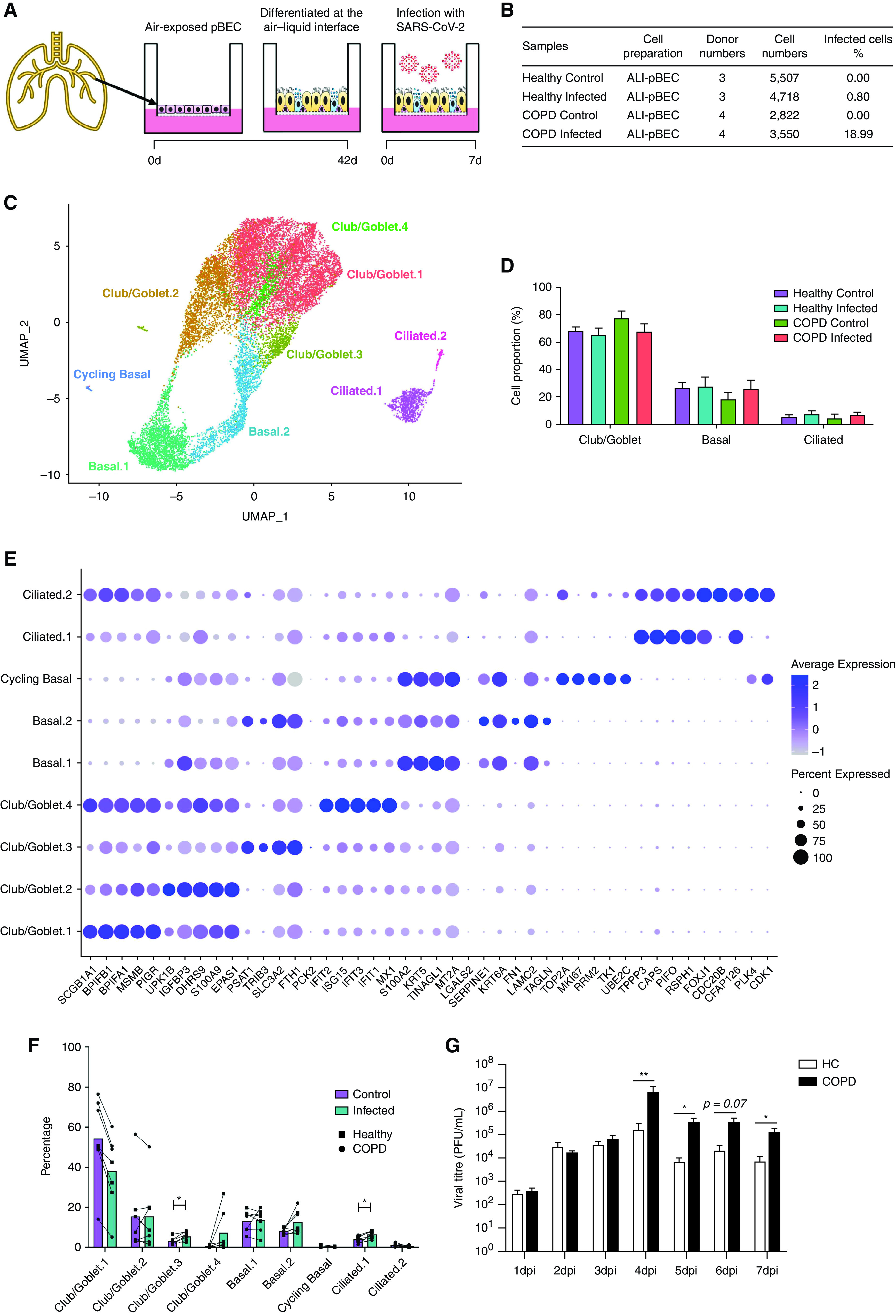
scRNAseq analysis of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-infected human primary bronchial epithelial cells (pBECs) differentiated at the air–liquid interface (ALI). (A) Experimental design of the SARS-CoV-2 infection of human pBECs differentiated ALI ex vivo. (B) Baseline characteristics of the samples showing cell numbers sequenced. (C) UMAP plot of the scRNAseq dataset identifying cell clusters. (D) Bar chart of proportions (in percentage) of major cell types (club/goblet, basal, and ciliated). (E) Cell proportions (in percentage) of control and infected healthy and chronic obstructive pulmonary disease (COPD) pBECs. (F) Average expression of the top five markers for each cluster. (G) Viral titers recovered from daily apical washes from COPD and healthy pBECs at ALI. Statistical differences between the groups are indicated whereby *P ⩽ 0.05 and **P ⩽ 0.01. HC = healthy control; scRNAseq = single-cell RNA sequencing; UMAP = uniform manifold approximation and projection.
Table 1.
Characteristics of the Study Participant Donors of Cells for Single-Cell RNA-Sequencing Analysis
| Sex | Age, yr | Smoker Status | Pack-years | GOLD stage | FEV1%p | FVC%p | FEV1/FVC | |
|---|---|---|---|---|---|---|---|---|
| Healthy | ||||||||
| HC234 | Female | 77 | Never | — | — | 70 | 80 | 70 |
| HC241 | Male | 55 | Never | — | — | 106 | 103 | 80 |
| HC243 | Female | 75 | Never | — | — | 94 | 102 | 73 |
| COPD | ||||||||
| CA299 | Female | 85 | Ex-smoker | 71 | 2B | 67 | 85 | 21 |
| CA340 | Male | 67 | Current | 52 | 2B | 72 | 104 | 12 |
| CA342 | Female | 76 | Ex-smoker | 53 | 2A | 54 | 66 | 4 |
| CA381 | Male | 78 | Current | 58 | 2B | 61 | 73 | 19 |
Definition of abbreviations: COPD = chronic obstructive pulmonary disease; FEV1%p = FEV1 percentage predicted (the volume of air that can be exhaled from the lungs in 1 second); FVC%p = FVC percentage predicted (the largest volume of air that can be exhaled after inhaling as deep as possible); GOLD = Global Initiative for Chronic Obstructive Lung Disease; Pack-years = packs smoked per day × years as a smoker.
At baseline, COPD control pBECs had significantly greater percentages of club/goblet cells compared with healthy control pBECs (78.1 vs. 68.19%, P = 0.0397) (Figure 1D). Goblet cell hyperplasia in pBECs from patients with COPD is well-known but was lost with infection (27). Further stratification into subclusters showed decreased proportions of Club/Goblet.1 in infected compared with control samples (Figures 1F and E1G). A prominent club/goblet population, Club/Goblet.4, was characterized by high expression of IFN response genes (IRGs) (Figure 1E). This cluster was found almost exclusively in infected compared with control samples, suggesting that the decreased proportion of cells in Club/Goblet.1 with infection may be attributed to a shift into an IFN-responsive population (Figures 1E and 1F).
COPD pBECs Have Increased SARS-CoV-2 Susceptibility
Strikingly, scRNAseq showed that COPD pBECs had approximately 24-times the number of infected cells compared with healthy pBECs (18.99% vs. 0.80%) (Figure 1A). This was further confirmed with elevated viral titers in plaque assays of apical washes from SARS-CoV-2–infected COPD pBECs from 4 dpi, which was significantly higher by 7 dpi (Figure 1G).
To further define the increased susceptibility of COPD pBECs, we examined the SARS-CoV-2 transcriptional profile in single cells using well-characterized viral markers ORF1ab/3a/6/7a/7b/8/10, N, E, M, and S (Figure 2A). This enabled us to explore transcriptional changes in infected versus bystander cells and identify response signatures associated with infection. Again, COPD pBECs had substantially increased numbers of infected cells compared with healthy pBECs (18.99% vs. 0.80%) (Figures 2B and 2C). This was further reflected in the negative strand mapping, indicative of an actively replicating virus (Figure E4). SARS-CoV-2 markers were primarily detected in club/goblet and basal cell populations, with some ciliated cells. There were increased proportions of infected Club/Goblet.1, 2, and 4 clusters, as well as basal.1 and 2 clusters in COPD versus healthy pBECs. Cycling basal cells occurred in COPD but not healthy pBECs, although this may be influenced by low cell numbers identified in this cluster.
Figure 2.
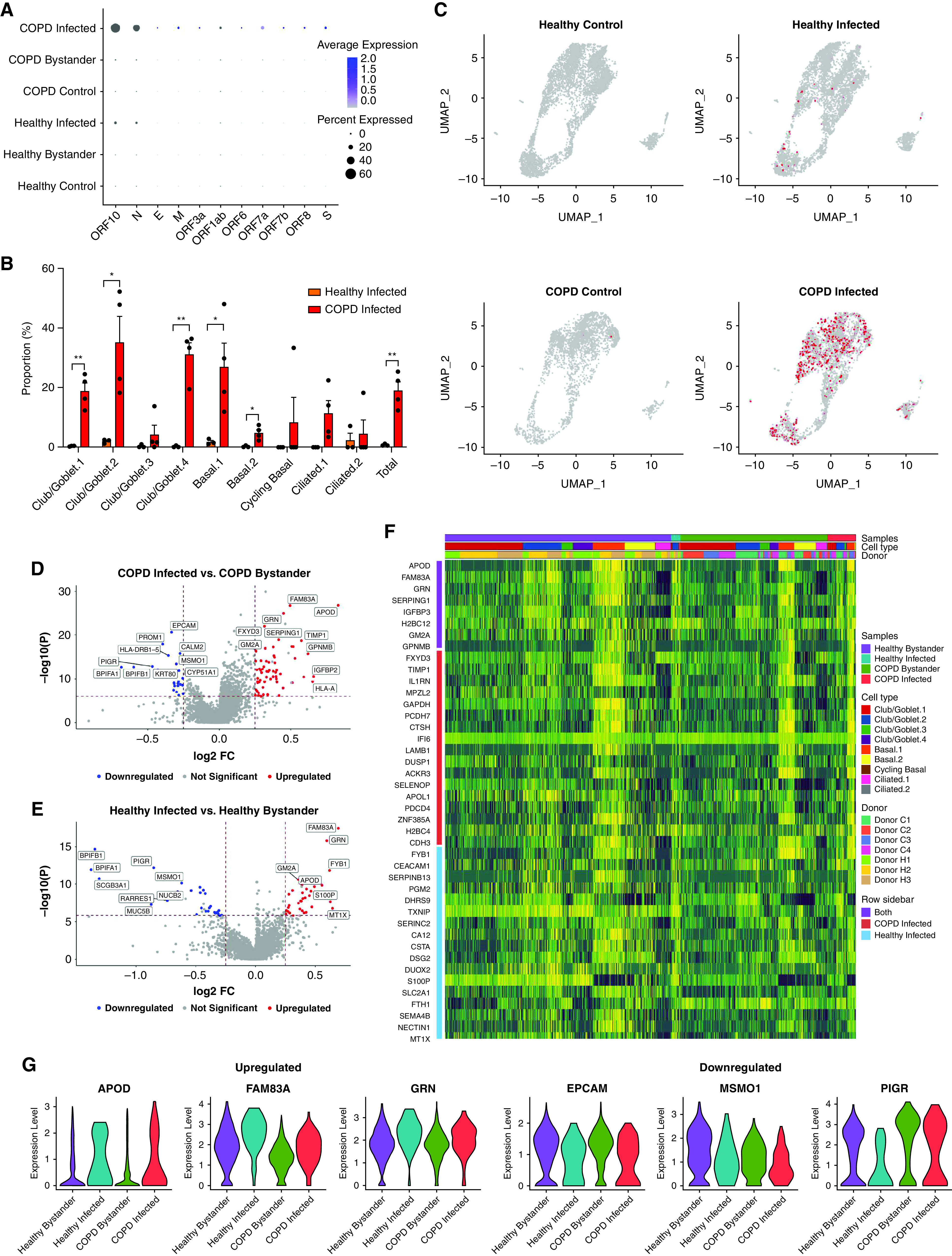
Differential gene expression analysis between infected and bystander cells in infected chronic obstructive pulmonary disease (COPD) and healthy primary bronchial epithelial cells. (A) Signature plots of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) markers. (B) Proportion of infected healthy (orange) and COPD (red) cells in each cell cluster. (C) Expression of SARS-CoV-2 markers in each of the different groups. (D) Volcano plot comparison of infected and bystander COPD cells. (E) Volcano plot comparison of infected and bystander healthy cells. (F) Heatmap of the top 25 genes in infected compared with bystander healthy and COPD cells. (G) Violin plots of three common upregulated and downregulated genes in infected healthy and COPD cells compared with their respective bystander cells. Statistical differences between the groups are indicated whereby *P ⩽ 0.05 and **P ⩽ 0.01. FC = fold change; UMAP = uniform manifold approximation and projection.
Differential gene expression analysis between infected and bystander cells showed that infected COPD and healthy cells had similar gene expression profiles compared with bystander cells (Figures 2D and 2G). APOD, FAM83A, and GRN were upregulated, whereas EPCAM, MSMO1, and PIGR were downregulated in infected cells. This shows that there are commonly used pathways in SARS-CoV-2 pathogenesis that are independent of preexisting disease status.
Protease Imbalances in COPD May Underpin Increased Infection Susceptibility
We next examined factors that may predispose patients with COPD to SARS-CoV-2 infection (Figure 3). We compared pseudobulk-infected and control differential gene expression of COPD with healthy datasets (Figures 3A–3C). Many of the most highly differentiated factors are linked to a broad range of different functions and do not form pathways. However, sham-infected COPD pBECs had increased expression of CCL2 and HMOX1 and downregulation of LIME1 and HLA-DRA-2, indicating a skewed proinflammatory state favoring monocyte/macrophage recruitment and decreased antigen presentation (Figure 3B). Infected COPD cells had greater expression of PIM1 and CLN3 supporting lysosomal function, elevated proinflammatory responses, and may contribute to enhanced monocyte and granulocyte signaling compared with infected healthy cells (Figure 3C). We also observed increased expression of cell adhesion-associated molecules like NEDD9, which may promote immune cell recruitment, signal transduction, and inflammation reported in COPD (Figure 3C).
Figure 3.
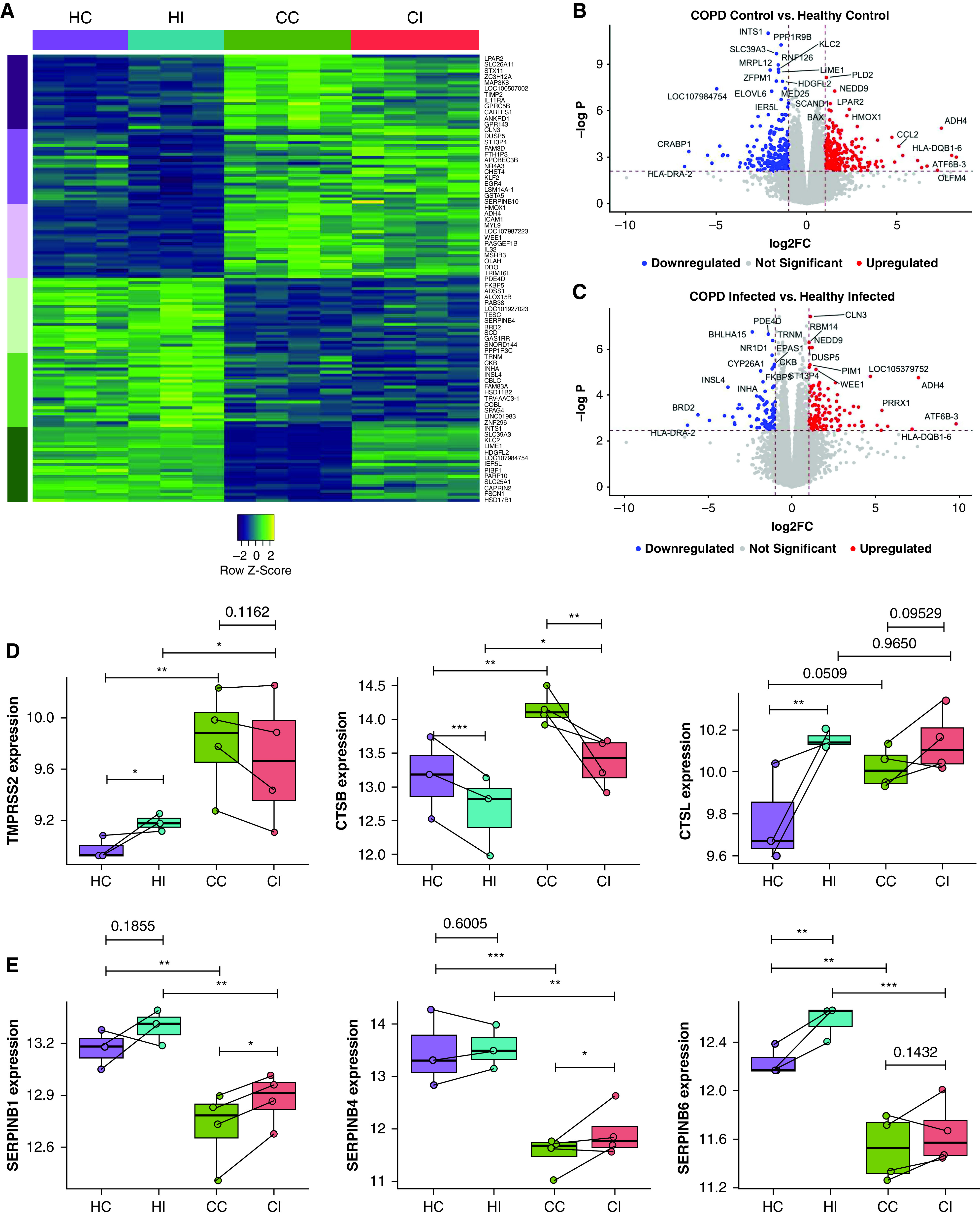
Differential gene expression analysis between infected chronic obstructive pulmonary disease (COPD) and healthy primary bronchial epithelial cells. (A) Heatmap of the top 50 differentially upregulated genes in infected COPD compared with healthy cells. Columns are grouped by samples, and rows represent genes grouped by the top upregulated (purple) and downregulated (green) in each or both COPD groups. (B) Volcano plot of differentially expressed genes in sham-infected COPD and healthy cells. (C) Volcano plot of differentially expressed genes in infected COPD and healthy cells. Average expression of (D) protease genes involved in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) entry (TMPRSS2, CTSB, and CTSL) and (E) protease inhibitor genes (SERPINB1, SERPINB4, and SERPINB6). Each dot represents a donor. Statistical differences between the groups are indicated by asterisks, in which *P ⩽ 0.05, **P ⩽ 0.01, and ***P ⩽ 0.001. CC = COPD control; CI = COPD infected; FC = fold change; HC = healthy control; HI = healthy infected.
Strikingly, pseudobulk analysis also showed increased expression of TMPRSS2, the major serine protease involved in S protein cleavage from SARS-CoV-2 (10), in COPD compared with healthy cells, which may increase infection (Figure 3D). CTSB, a lysosomal cysteine protease that promotes SARS-CoV-2 infection, was also upregulated. We also found elevated baseline expression of CTSL in COPD donors. In contrast, serine protease inhibitors SERPINB1/4/6 were consistently reduced with or without infection in COPD pBECs (Figure 3E). These findings highlight a protease imbalance in COPD pBECs that may be critical for increased SARS-CoV-2 infectivity and severe disease.
COPD pBECs Have Elevated Proinflammatory Responses and Disease Severity Markers
There is currently no explanation for increased disease severity and exacerbations in SARS-CoV-2–infected patients with COPD (14, 15). Pseudobulk analysis showed that COPD pBECs had increased expression of inflammatory genes IL1A/B/6/32, CCL20, CSF1, and NFKBIA/Z associated with COVID-19 at baseline and with infection (Figure 4A). Thus, higher expression of proinflammatory genes may drive greater inflammatory responses in SARS-CoV-2–infected patients with COPD.
Figure 4.
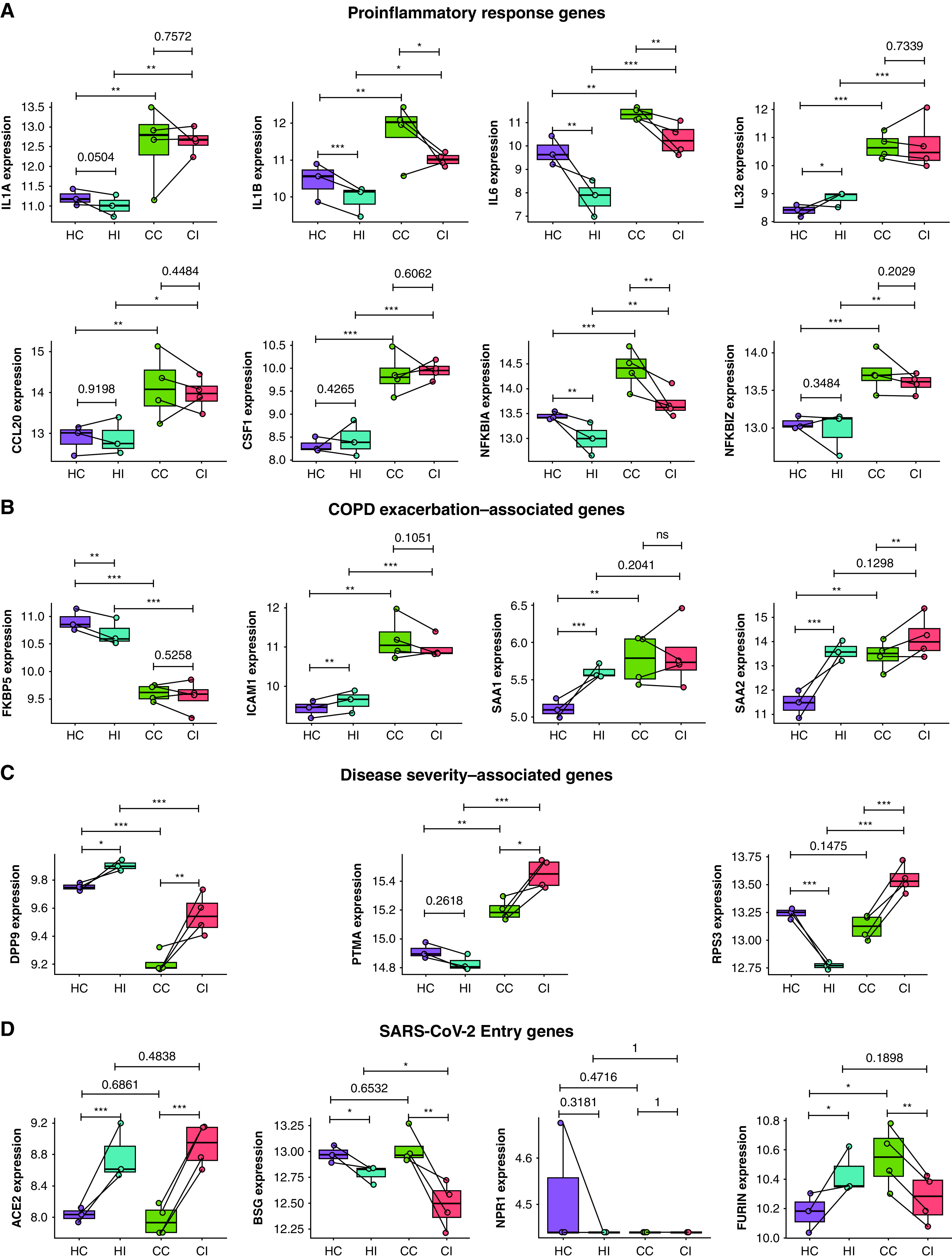
Elevated proinflammatory, exacerbation, and disease severity genes and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) entry genes in chronic obstructive pulmonary disease (COPD) compared with healthy primary bronchial epithelial cells. Average expression of (A) proinflammatory response genes (IL1A, IL1B, IL6, IL32, CCL20, CSF1, NFKBIA, and NFKBIZ), (B) COPD exacerbation–associated genes (FKBP5, ICAM1, SAA1, and SAA2), (C) disease severity–associated genes (DPP9, PTMA, and RPS3), and (D) known SARS-CoV-2 entry genes (ACE2, BSG, NPR1, and FURIN). Each dot represents a donor. Statistical differences between groups are indicated whereby *P ⩽ 0.05, **P ⩽ 0.01, and ***P ⩽ 0.001. CC = COPD control; CI = COPD infected; HC = healthy control; HI = healthy infected; ns = not significant.
We then examined the expression of COPD exacerbation- and disease severity-associated gene signatures that may account for increased mortality. FKBP5, a modulator of corticosteroid responsiveness, antiinflammatory function, and antiinfluenza virus factor (28), was reduced in COPD independently of infection (Figure 4B). ICAM1, the rhinovirus entry receptor, was upregulated with and without infection, reflecting increases in viral-induced COPD exacerbations. SAA1 and SAA2, which contribute to acute exacerbations of severe COPD (29), were also elevated in COPD pBECs at baseline and further with infection.
DPP9, which predisposes to acute inflammatory responses and is a risk factor for severe COVID-19 (30), was lower in COPD with and without infection. PTMA, elevated in severe COVID-19 (31), was increased in infected versus sham-infected COPD and infected healthy pBECs, contributing to more severe COVID-19 in patients with COPD. RPS3, a ribosomal protein involved in DNA repair and oxidative stress and triggers caspase-driven apoptosis, was increased in infected COPD pBECs.
We also investigated the expression of all other SARS-CoV-2 entry factors. ACE2 was upregulated with infection, regardless of COPD status. BSG was downregulated with infection, more so in COPD. NPR1 showed no significant differences. Importantly, FURIN was increased in COPD pBECs at baseline; however, on infection, it was upregulated in healthy but downregulated in COPD pBECs (Figure 4D).
Thus, COPD pBECs display critical gene expression signatures of increased inflammatory responses at baseline, which, coupled with elevated exacerbation and disease-severity predictors, may contribute to increased severity and mortality in COPD.
Coexpression Network Analysis Reveals COPD-infected pBECs Have Elevated Inflammatory Response Modules
We next used coexpression network analysis with the CoCeNa2 pipeline to assess genes associated with infection and disease status. Unlike classical differential gene expression, this identifies similarly regulated genes and groups them into modules on the basis of each sample, cell type, or other variables. We analyzed the four sample types independently for each major cell type. This revealed group-specific enrichment of coexpressed modules for each (Figure 5A). Modules are colored on the basis of group fold change, which is the mean of each module. This analysis showed three cell types, club/goblet, basal, and ciliated cells, predominated.
Figure 5.
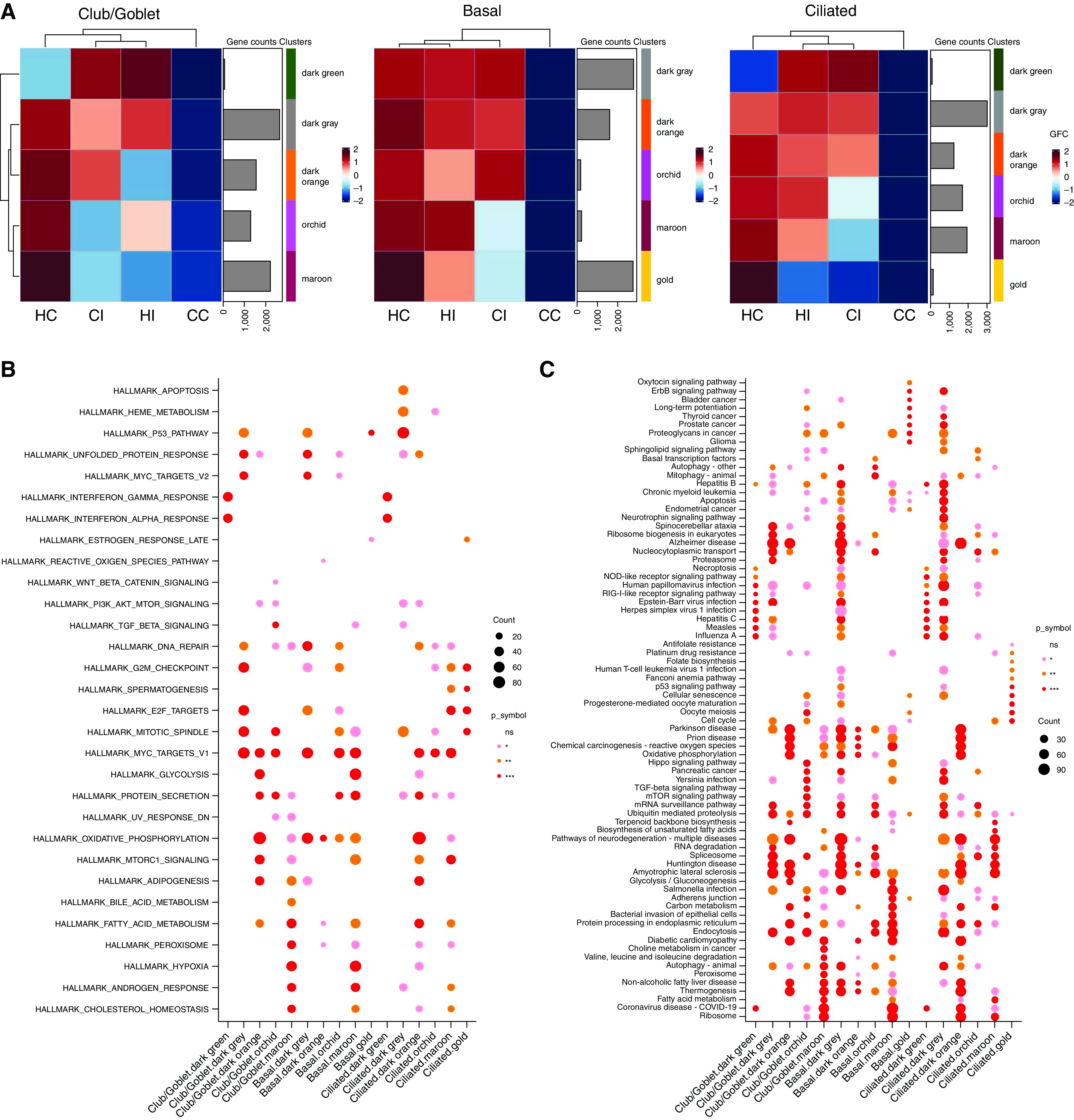
Coexpression network analysis using the CoCeNa2 pipeline. (A) Coexpression network analysis was performed on the basis of club/goblet, basal, and ciliated cells. Heatmaps for the samples are colored on the basis of the group fold change, which is the mean expression of each gene in the module. (B) Hallmark and (C) KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis using the CoCeNa2 pipeline for each module. Top pathways for each module are shown. Statistical differences between groups are indicated by different colored symbols, in which pink dots represent P ⩽ 0.05, orange dots represent P ⩽ 0.01, red dots represent P ⩽ 0.001, and the absence of a dot represents nonsignificant pathways. The size of dots is proportional to the number of gene counts that are associated with the respective pathway. CC = COPD control; CI = COPD infected; COPD = chronic obstructive pulmonary disease; GFC = group fold change; HC = healthy control; HI = healthy infected; ns = not significant.
Club/goblet and basal cells were clustered into five and ciliated cells into six modules (Figure 5A). The dark green module in club/goblet and ciliated cells showed a similar pattern in which infected COPD and healthy pBECs had a higher group fold change relative to control samples and were higher in infected COPD pBECs. This module is not present in the basal cell cluster. Clearly, this gene module most represents responses to SARS-CoV-2 infection. Pathway analysis in this module revealed activation of IFN-α and -γ and inflammatory responses and pathways related to viral infection and COVID-19 (Figures 5B and 5C). Regardless of cell type, COPD-infected pBECs had higher expression of all modules.
We also assessed Hallmark (Figure 5B) and KEGG (Kyoto Encyclopedia of Genes and Genomes) (Figure 5C) pathways for each module, and those in maroon for each cell type identify common pathways. In Hallmark pathway analysis, IFN-γ and -α and inflammatory responses (Nf-κB and p53) and cell death pathways were the top common pathways. Several KEGG pathways related to viral infection, including COVID-19, were associated with each cell type. The maroon module of each cell type also had overlapping pathways related to hypoxia, oxidative stress, oxidative phosphorylation, and mTORC1 (mammalian target of rapamycin complex 1) signaling, the latter being associated with lung cell senescence and stable growth arrest in COPD (32).
Club/Goblet.4 Cells Are Major Drivers of IFN Responses to SARS-CoV-2 Infection
The Club/Goblet.4 cluster was predominantly found in COPD and healthy infected populations, characterized by high expression of IRGs. We investigated the emergence of club/goblet cell populations through trajectory analysis (Figure 6A). This analysis explores the concept that cells are not stationary but transition from one biological state to another, characterized by unique gene expression signatures. Trajectory analysis creates pseudotime to measure the progress of single cells in biological processes. We identified seven different states of club/goblet cells (Figures 6A and 6B). Trajectory maps are influenced by SARS-CoV-2 infection, with control samples predominantly on the left and infected samples clustering on the right. This indicates that cell trajectories are moving to mucus hypersecretion, inflammation, and metabolism states with infection across pseudotime.
Figure 6.
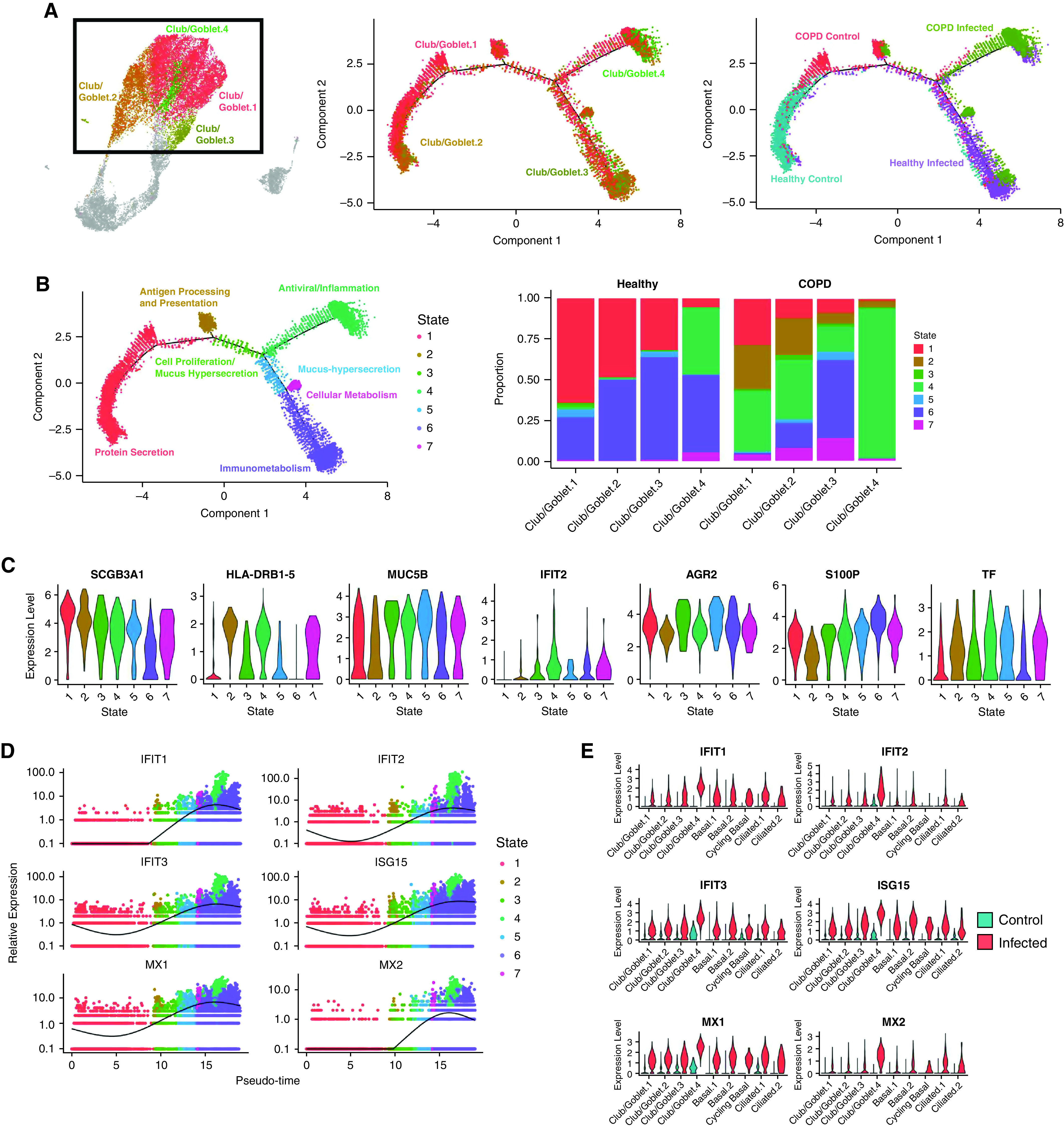
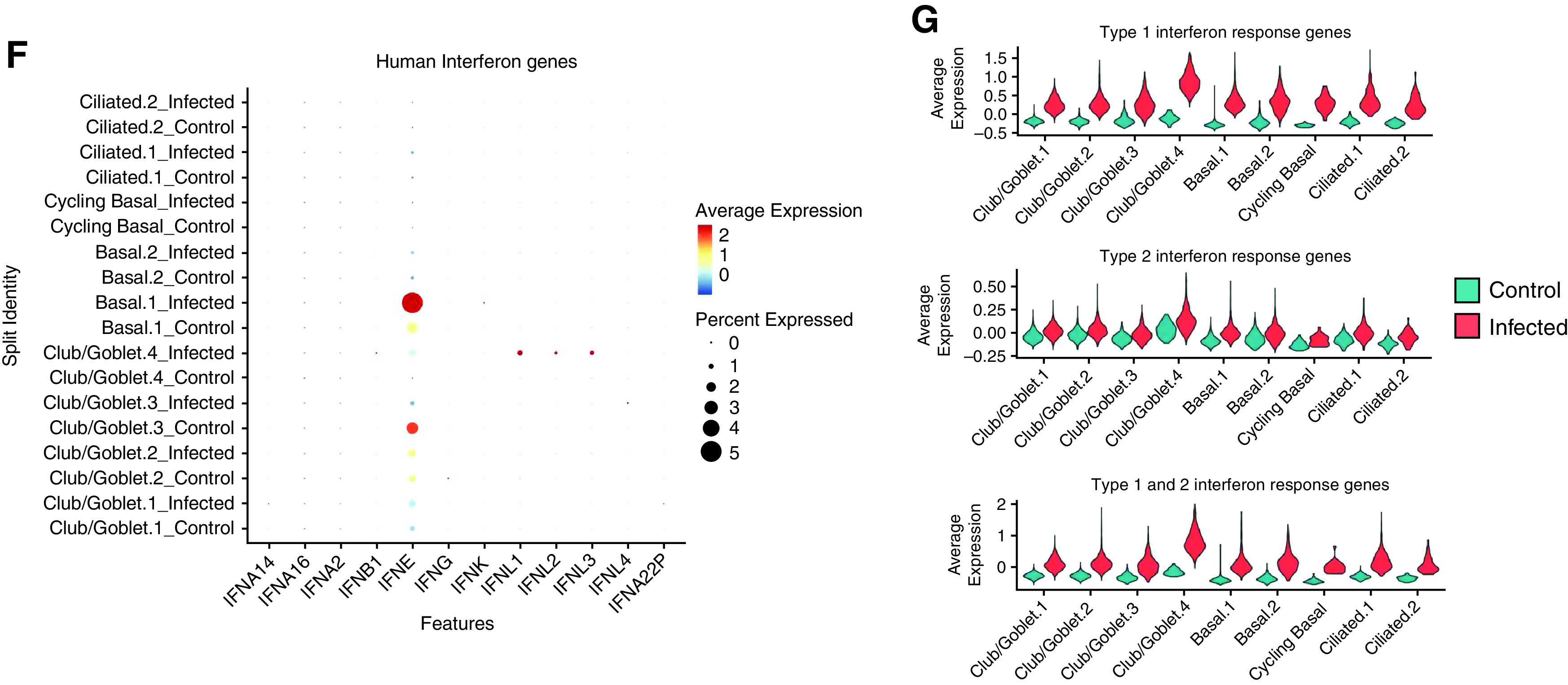
Emergence of the Club/Goblet.4 cluster is driven by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. (A) Trajectory analysis in club/goblet cells colored by the four identified clusters, samples, and (B) cell states. The proportion of cell states in different cell types is shown in the barplot. (C) Violin plots of differentially expressed genes among cell states. (D) Pseudotime trajectory of IFN response genes (IRGs) colored by cell states. (E) Violin plots of IRGs in control and infected cells. (F) Average expression of human IFN genes. (G) Violin plots of type 1, type 2, and both type 1 and type 2 IRGs. COPD = chronic obstructive pulmonary disease.
Of seven club/goblet cell states, four (States 4–7) were predominantly present in infected pBECs. State 4 is an “antiviral/inflammation” state because of the high expression of proinflammatory (CXCL6) and IRGs (IFIT1/2/3, ISG15, and MX1/2), whereas State 5 is a “mucus-hypersecretion” state with increased AGR2. State 6 is an “immunometabolism” state with a high expression of secretory genes (S100P), and State 7 is a “cellular metabolism” state with increased TF (Figure 6C). The contributing proportions of cell states were vastly different between COPD and healthy pBECs (Figure 6B). In infected COPD pBECs, the inflammation state occurred in all four club/goblet cell clusters, whereas in infected healthy cells, it predominated in the Club/Goblet.4 cluster. Infected COPD cells had a much higher proportion of the State 4 antiviral/inflammation state in the Club/Goblet.4 cluster. Infected healthy cells had a higher proportion of the State 6 immunometabolism state.
By plotting cell types across pseudotime, we showed that the expression pattern of IRGs (IFIT1/2/3, ISG15, and MX1/2) increased as cells progressed from sham infected in State 1 (red) to States 4 and 6 (green and purple) with infection across pseudotime (Figure 6D). This was largely controlled by Club/Goblet.4, as control samples expressed low concentrations of IRGs (Figure 6E).
We also examined IRGs, which showed that IFN-e expression was substantially higher in infected pBECs (Figure 6F). We then assessed Type 1/2 IRGs and found that infection induced markedly higher expression of type 1 and type 2 IRGs in all clusters, especially in Club/Goblet.4 cells (Figure 6G). The same pattern was observed in genes that respond to both type 1 and 2 IFNs. We then assessed IFN receptor expression in these IFN-responsive cells but found extremely low expression across all samples (Figure E3). Surprisingly, there was no overrepresentation of IFN receptors in Club/Goblet.4 cells, indicating another mechanism is responsible for IRG induction independent of direct receptor–ligand interactions.
Validation of Gene Expression Signatures in In Vivo Human Cohorts
We next validated our findings in publicly available datasets from patients with COVID-19. Gene set variation analysis (GSVA) calculates the expression of a given gene set for each individual. We performed GSVA of our data with differentially expressed genes from a large cohort study of nasal swabs from SARS-CoV-2 (n = 138) and healthy (n = 93) donors (GSE163151) (33). The upregulated genes in our COPD and healthy datasets mirrored those in the nasal swab cohort (Figure 7A). Similarly, gene expression that was downregulated in COPD and healthy samples in our study was significantly associated with those downregulated in SARS-CoV-2–positive and –negative samples in the nasal swab cohort. GSVA was also performed on cluster-specific genes of the Club/Goblet.4 population in our study (Figure 7B). Up- and downregulated genes in our Club/Goblet.4 population were in agreement with the infected nasal swab cohort and were significantly different compared with uninfected control samples (Figures 7A and 7B).
Figure 7.
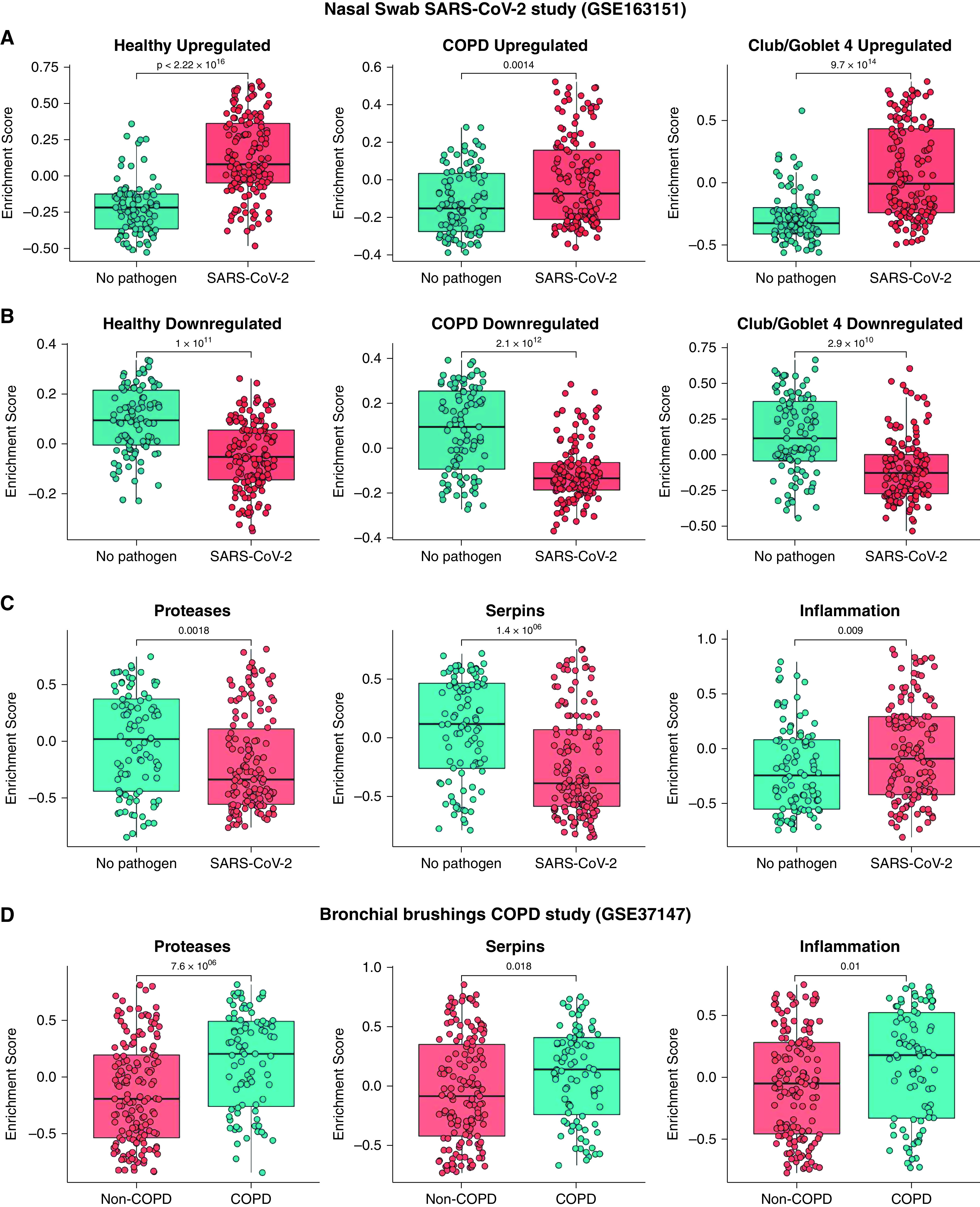
Comparing ex vivo with publicly available severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in vivo data. Gene set variation analysis (GSVA) was performed with bulk RNA sequencing dataset of in vivo SARS-CoV-2 nasal swabs (n = 231, GSE163151). GSVA of (A) up- and (B) downregulated genes in infected healthy and chronic obstructive pulmonary disease (COPD) samples compared with their control cells and Club/Goblet.4 cells compared with other cell types. (C) GSVA was performed for protease, serpin, and inflammatory genes with a nasal swab in vivo data. (D) GSVA of proteases, serpin, and inflammatory genes with bronchial brushing in vivo COPD data (n = 238, GSE37147). Statistical differences between the groups were accepted at P ⩽ 0.05.
We assessed whether our findings of protease and inflammatory imbalances driving COPD susceptibility (Figures 3 and 4) were also clinically relevant. We examined the nasal swab cohort (GSE163151) and another publicly available dataset from bronchial brushings from COPD (n = 87) and healthy (n = 151) donors (GSE37147) (23). The nasal swab cohort had decreases in protease and serpin-associated gene signatures and increases in inflammatory signatures after SARS-CoV-2 infection (Figure 7C). Comparatively, the bronchial brushing COPD dataset also had increases in protease, serpin, and inflammatory-associated signatures associated with SARS-CoV-2 infection (Figure 7D).
Validation of these publicly available datasets demonstrates the clinical relevance of our findings.
Therapeutic Interventions Targeting Protease Imbalances and Elevated Proinflammatory Responses Reduce SARS-Cov-2 Replication in pBECs
To further characterize biological responses to SARS-CoV-2 infection in COPD and healthy pBECs, we examined proinflammatory and IFN protein concentrations (Figure 8A). Strikingly, we observed an almost complete absence of IFN-β in infected COPD compared with infected healthy pBECs from 4 dpi, providing a strong mechanistic basis for elevated viral titers in COPD pBECs (Figure 1F). Furthermore, there was a significant elevation of IL-6 in infected COPD pBECs from 3 dpi, likely as a consequence of increased viral titers and absent IFN-β (Figure 8A). There were also lower concentrations of IP-10, which agrees with previous findings of the protective role of IP-10 in SARS-CoV-2 infection (34) and provides further evidence that patients with COPD are predisposed to more severe COVID-19.
Figure 8.
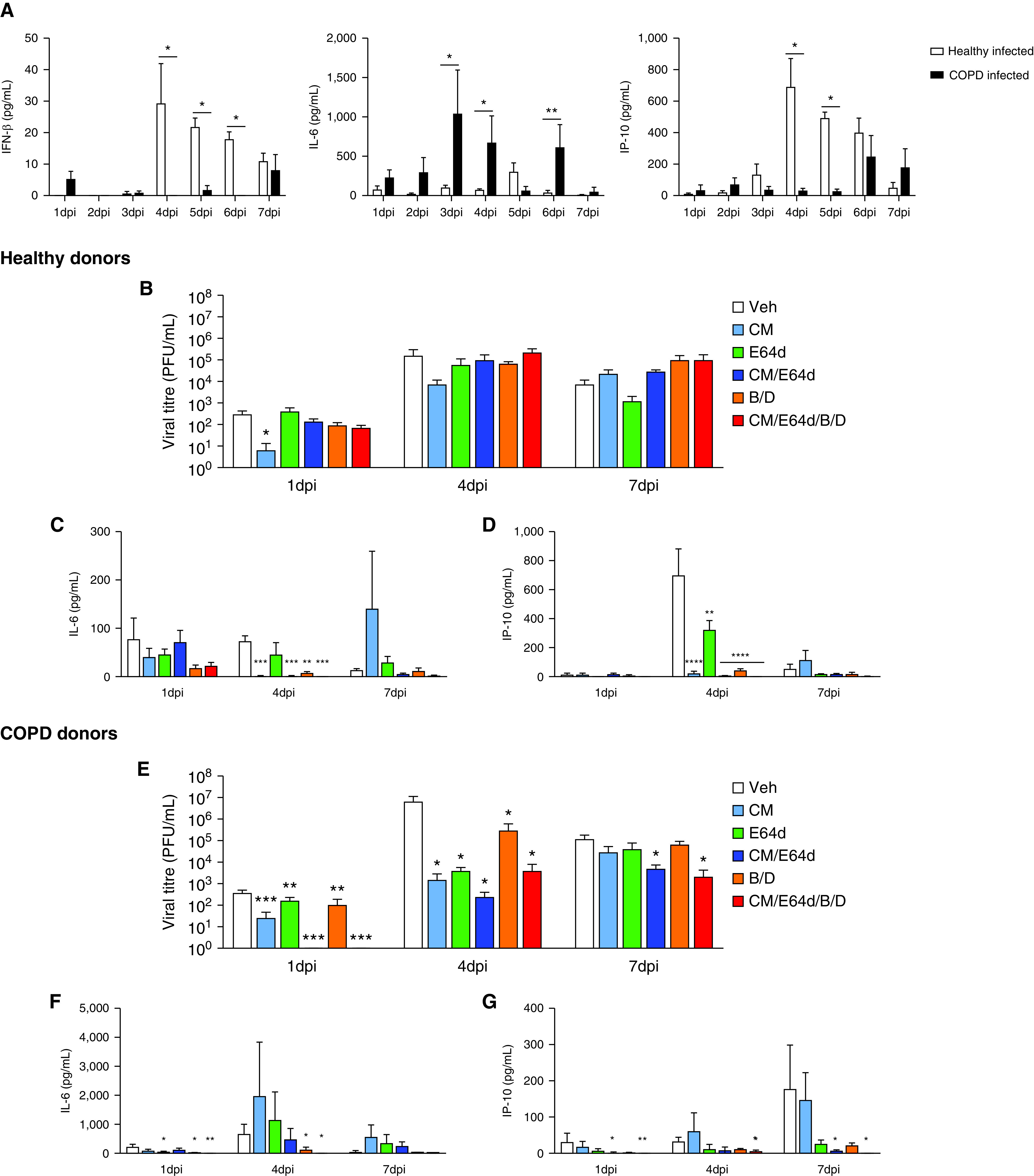
Therapeutic interventions targeting protease imbalances and excessive proinflammatory responses limit severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) replication and blunt virally induced hyperinflammation. (A) Proinflammatory cytokines and IFN concentrations were quantified at the protein level using the LEGENDplex Human Anti-virus Response Panel in chronic obstructive pulmonary disease (COPD) and healthy infected samples across time. (B) Viral titers were quantified in daily apical washes from healthy infected samples after daily treatments with Camostat Mesylate (CM), E64d, CM/E64d, baricitinib/dexamethasone (B/D), or a combined therapy (CM/E64d/B/D). (C) IL-6 and IP-10 (CXCL10) were quantified in apical washes from healthy infected samples using the LEGENDplex Human Anti-virus Response Panel. (D) Viral titers were quantified in daily apical washes from COPD-infected samples after daily treatments. (E) IL-6 and IP-10 were quantified in the apical washes from COPD-infected samples. Statistical differences between the groups are indicated, whereby *P ⩽ 0.05, **P ⩽ 0.01, ***P ⩽ 0.001, and ****P ⩽ 0.0001. In B–G, statistical differences shown are relative to their respective vehicle control at each time point examined. PFU = plaque-forming unit; Veh = vehicle.
We identified protease imbalances in COPD pBECs with elevated TMPRSS2 and CTSB, together with increased proinflammatory, exacerbation, and disease severity responses (Figures 3D, 3E, and 4). Consequently, we confirmed their involvement in increased infection of COPD pBECs by therapeutically targeting these pathways (Table 2). We treated infected COPD and healthy pBECS with Camostat Mesylate (CM, TMPRSS2 inhibitor), E64d (CTSB inhibitor), CM/E64d (dual TMPRSS2 and E64d inhibition), baricitinib/dexamethasone (B/D, anti-inflammatory, JAK/STAT [Janus kinase signal transducer and activator of transcription] inhibitor, and glucocorticoid), or all therapies combined (CM/E64d/B/D).
Table 2.
Characteristics of the Study Participant Donors of Cells for Therapeutic Intervention Studies
| Sex | Age, yr | Smoker Status | Pack-years | GOLD Stage | FEV1%p | FVC%p | FEV1/FVC | |
|---|---|---|---|---|---|---|---|---|
| Healthy | ||||||||
| HC181 | Male | 53 | Never | — | — | 100 | 112 | 77 |
| HC243 | Female | 75 | Never | — | — | 94 | 112 | 75 |
| HC250 | Female | 77 | Never | — | — | 110 | 102 | 73 |
| COPD | ||||||||
| CA303 | Male | 70 | Ex-smoker | 40 | 2B | 70 | 74 | 77 |
| CA319 | Male | 77 | Ex-smoker | 44 | 2C | 79 | 101 | 58 |
| CA321 | Female | 76 | Ex-smoker | 36 | 2C | 64 | 92 | 52 |
For definition of abbreviations, see Table 1.
In healthy pBECs, only CM reduced viral titers at 1 dpi (Figures 8B and E5A). CM, CM/E64d, B/D, or a combination of all therapies reduced IL-6 and IP-10 concentrations at 2–5 dpi (Figures 8C, 8D, E6A, and E6B). In stark contrast, in COPD pBECs, all therapies individually or in combination significantly reduced viral titers at all time points from 1 dpi to 7 dpi (Figures 8E and E5B). Moreover, CM/E64d, B/D, or a combination of all therapies significantly reduced IL-6 and IP-10 concentrations from 1 dpi to 7 dpi (Figures 8F, 8G, E6C, and E6D).
Collectively, these findings show that interventions targeting proteases and proinflammatory responses in SARS-CoV-2 infection have excellent therapeutic potential in reducing SARS-CoV-2 replication and associated hyperinflammation, particularly in patients with COPD.
Discussion
We define for the first time the transcriptomic dynamics of SARS-CoV-2 infection in differentiated pBECs from COPD and healthy donors using scRNAseq. It is also the first study to show biological evidence that COPD pBECs are substantially more permissive to SARS-CoV-2 infection compared with healthy pBECs. Moreover, we demonstrated that COPD pBECs have an inherent protease imbalance that increases the production of the key SARS-CoV-2 entry cofactors TMPRSS2 and CTSB. COPD pBECs also had higher mRNA and protein concentrations of proinflammatory cytokines, signatures of acute COPD exacerbations, and disease severity genes associated with poor COVID-19 outcomes. We identified a unique Club/Goblet.4 cell population that develops almost exclusively in SARS-CoV-2–infected cells, characterized by IRG signatures despite the low or absent expression of IFN receptors. Trajectory analysis of club/goblet cell populations revealed that they transition to this IFN-specific cluster, ultimately driving transcriptomic responses. We used publicly available transcriptomic datasets to validate our findings in in vivo SARS-CoV-2 datasets. Therapeutic interventions targeting protease imbalances and elevated proinflammatory responses identified potential therapies that reduce SARS-CoV-2 viral titers and virally induced hyperinflammation in healthy and especially COPD pBECs. This further validates our single-cell findings and can be used to inform future therapeutics for COVID-19, particularly in more susceptible patients with COPD.
ALI cultures recapitulate important physiological features of respiratory epithelia and are composed of important cell populations like club, goblet, basal, and ciliated cells. Infected COPD pBECs expressed higher proinflammatory cytokines and COPD exacerbation genes (35). We identified that infected COPD cells had almost all Club/Goblet.4 cells assigned to the antiviral/inflammation state, which had high expression of MUC5B. Comparatively, all healthy infected populations had generally less MUC5B-associated states, which highlights that the predisposed disease status of pBECs in vivo is recapitulated in ALI cultures. These findings reconfirm important features of COPD pathogenesis and provide strong evidence that the findings of this study are relevant to patients.
We demonstrated that club/goblet and basal cells were the predominant populations infected with SARS-CoV-2. This differs from other studies in which ciliated cells were reported as the primary infected cell type in pBECs. However, clinical reports have suggested that ciliated cells are diminished during COVID-19 (36). Airway epithelial cells cultured at ALI for longer durations are more susceptible to SARS-CoV-2 owing to greater expression of viral entry factors like ACE2 (37). Our pBECs were maintained at ALI for more than 6 weeks before infection, which may promote greater infectivity of different cell subpopulations not observed in other studies. Importantly, pBECs from patients with COPD cultured for up to 10 weeks at ALI show marked changes associated with previous disease status and chronic cigarette smoke exposure, likely because of modifications in progenitor cells (38).
We did not observe significantly elevated expression of ACE2 between COPD and healthy pBECs at baseline. Although it was upregulated with infection in both settings, there were no significant differences between them. This suggests that other mechanisms likely contribute to increased infectivity after expansion at ALI (increased TMPRSS2 and CTSB, reduced serpins). Furthermore, we performed scRNAseq at 7 dpi, representing a well-established infection that has likely progressed past ciliated cells, which are the first line of airway defense. Our data at 7 dpi suggest that club/goblet cells are predominant infected populations during later stages, whereas previous studies identified that ciliated cells may be the primary infected cells at earlier stages. Clinical observations report the median SARS-CoV-2 incubation period as 5.1 days before the onset of disease symptoms; however, some develop symptoms as late as 12 days after exposure (39, 40). Thus, our scRNA-seq data at 7 dpi represents a clinically relevant time point in SARS-CoV-2 infection. Our findings provide an in-depth understanding of the transcriptomic profile during later-stage infection and provide important insights into the significance of different cell populations at different stages of SARS-CoV-2 infection in COPD.
A major finding was that COPD pBECs were substantially more susceptible to SARS-CoV-2 infection compared with healthy pBECs. This has been postulated from clinical studies and informed by other viral infections (7, 41). We show the first experimental evidence to support these concepts. This was reflected at the single-cell level with viral transcripts and supported by negative-strand mapping representing replicating virus that was significantly elevated in COPD pBECs (Figure E4). This was supported by elevated viral titers in COPD pBECs recovered from apical washes from as early as 4 dpi and was maintained until 7 dpi (Figure 1G). Our previous work has shown that patients with COPD express higher concentrations of ICAM1, which increases susceptibility to rhinovirus infection (42). We also demonstrate that COPD pBECs have elevated concentrations of SARS-CoV-2 entry factors, TMPRSS2 and CTSB, and downregulated protease inhibitors such as serpin genes (10). TMPRSS2 and CTSB independently facilitate SARS-CoV-2 entry and likely contribute to increased infectivity of COPD pBECs. We, therefore, propose that protease imbalances likely confer increased susceptibility of COPD pBECs to infection. Using therapeutic interventions targeting these protease imbalances, we demonstrated proof of concept in reducing SARS-CoV-2 titers, which may be used as potential therapies in patients with COPD to reduce susceptibility to infection. This is supported by reports of high lung protease activity in patients with COPD (43) and elevated TMPRSS2 expression in bronchial epithelial brushings from current compared with never smokers, suggesting that smoking is partially responsible for the increased susceptibility of patients with COPD (44, 45).
Another study found that TMPRSS2 was not increased in patients with COPD compared with ex-smokers, although this does not reflect the true comparison of COPD versus healthy status observed in our study (46). Comparing our scRNAseq data to publicly available datasets, we also observed striking upregulation of proteases in bronchial brushings from patients with COPD, independent of SARS-CoV-2 infection. Furthermore, SARS-CoV-2–positive nasal swab datasets correlate with our data, with downregulation of CTSB in infected COPD and healthy pBECs. Although these datasets did not support our observations regarding the downregulation of serpin-associated genes, this could be attributed to many confounding factors like age, sex, smoking status, and comorbidities, which were not made publicly available and may contribute to protease imbalances. In addition, differences between nasal and bronchial samples result in some differences in gene expression because of different tissue tropisms and biological roles (47). SARS-CoV-2 does infect and replicate in nasal epithelial cells, which is the initial exposure site. However, the major site for SARS-CoV-2 tropism is the lower airways, bronchioles, and alveoli (48). Thus, our findings define the significance of protease imbalances in driving COPD susceptibility to SARS-CoV-2 infection and highlight the importance of understanding the complex interactions of different tissues in interpreting infection outcomes.
Patients with COPD are at greater risk of hospitalization and death from COVID-19, likely because of disease exacerbations; however, there is little evidence to substantiate these concepts (49). Our data demonstrate that COPD pBECs express higher mRNA and protein concentrations of proinflammatory cytokines, likely contributing to excessive inflammation in COVID-19 in these patients. This matches clinical observations that IL-6 is a key component of viral-induced hyperinflammation and that patients with higher baseline IL-6 concentrations have poor COVID-19 outcomes (50, 51). Nf-κB regulatory genes NFKBIA and NFKBIZ were also significantly elevated in COPD pBECs, providing a plausible mechanism for the heightened proinflammatory responses we observed. Furthermore, gene expression of predictors of COVID-19 severity, DPP9 and PTMA, were dysregulated in COPD compared with healthy cells. There is emerging evidence that decreased lung expression of DPP9 correlates with excessive inflammatory responses and more severe COVID-19 (30), suggesting decreased concentrations in COPD pBECs are indicative of severe infection outcomes. PTMA is significantly elevated in patients with severe COVID-19, matching our observations in COPD pBECs (31). Thus, our findings reflect clinical observations in patients with severe COVID-19 and define biological mechanisms responsible for exacerbations and severe COVID-19 in COPD. Future studies should further investigate the molecular pathways and the relevance of our candidate gene signatures.
IFN responses are critical host immune defense mechanisms against invading viral infections (16, 52, 53). There is conflicting evidence on their roles in SARS-CoV-2 infection, with some suggesting that IFN signatures are diminished or absent and are also dependent on location along the respiratory tract (52, 54, 55). Furthermore, recent studies have shown that nasal IFN responses were significantly delayed relative to viral replication as compared with other respiratory viruses such as the influenza A virus (56). In line with these reports, we detected substantially reduced IFN-β concentrations in infected COPD pBECs compared with healthy cells, which we postulate is a key driver for increased susceptibility to elevated inflammatory responses and viral titers. This is further supported by nasopharyngeal scRNAseq datasets, which demonstrated that patients with severe COVID-19 had muted antiviral immune signatures (57). Moreover, we identified a prominent cluster of club/goblet cells with high IRG signatures almost exclusively in infected samples (Figure 6). Pseudotime analysis predicted that these cells emerged from Club/Goblet.1 and led to the emergence of Club/Goblet.2, 3, and 4 populations on the basis of gene expression profiles. Club/Goblet.4 populations almost exclusively occurred in infected samples, suggesting that club/goblet cell differentiation may be a response to IFN responses. However, we did not identify gene expression patterns attributable to increased IFN responses and no enhanced IFN receptor expression. These data suggest that this population of club/goblet cells is stimulated through alternative pathways independently of IFN. There is emerging evidence that previous immune activity can influence cell state and drive long-term inflammatory memory in barrier tissues such as differentiated epithelium (58), which may provide an explanation for club/goblet cell differentiation in our ex vivo ALI cultures. In support of this, previous work has shown that rhinovirus infection before SARS-CoV-2 exposure led to accelerated IRG signatures, which prevented SARS-CoV-2 replication (59). Moreover, SARS-CoV-2 can directly induce IRGs without receptor engagement, although this warrants further investigation (52). The diminished IFN responses at the single-cell level at 7 dpi are not entirely unexpected, as IFN responses are rapidly switched on and off to potentiate and control immune responses (60). This IFN-responsive population was not a major cell type that was infected with SARS-CoV-2 in COPD or healthy cells, suggesting that they are hyperresponsive subpopulations that make major contributions to excessive IRG signatures. It is likely that the heightened IFN responses reflected the increases in SARS-CoV-2–infected cells in COPD compared with healthy cells. Importantly, bioinformatics and pathway analyses found no induction of type 2 IRGs; however, there was a strong type 1/2 IRG signature, although they were unable to be separated in our analyses. On the basis of our findings, it is very likely that these are type 1 IRG signatures, which is also supported by previous studies that have shown type 2 IFN responses are almost exclusively produced by immune cells (61). Few studies have profiled the dynamics of IFN responses in respiratory epithelial cells after SARS-CoV-2 infection, which should be a focus of future investigations. Future studies should also explore the contribution of different pBEC subpopulations to IFN responses to decipher the primary IFN producers triggering the differentiation of club/goblet cells.
There are some limitations. We selected cells with a higher percentage (35%) of mitochondrial genome reads. Ideally, mitochondrial genome reads should be 10–15%, as higher mitochondrial contamination represents low-quality or dying cells. We selected 35%, as infected COPD and healthy pBECs had more than 90% of cells with greater than 20% of mitochondrial genome reads. This is unavoidable and is an expected risk in our study. We also could not differentiate club and goblet cells because of biological similarities in transcriptional profiles, which may influence our interpretation of the role of different cell subpopulations in driving host immune responses. Finally, because of the expense of scRNAseq, it was not feasible to explore the transcriptomic profile at multiple time points after infection.
Conclusions
We show that SARS-CoV-2 infection is increased in COPD compared with healthy pBECs, which is likely because of protease imbalances resulting from increased concentrations of SARS-CoV-2 entry proteases TMPRSS2 and CTSB and downregulation of serpin protease inhibitors. COPD cells have heightened baseline proinflammatory responses (mRNA and protein) and increased markers of exacerbation and severe disease outcomes, providing mechanistic insights into severe COVID-19 outcomes in patients with COPD. Therapeutic suppression of viral coreceptor proteases, addressing the protease imbalance, and/or treatment with antiinflammatories or type 1 and 2 IFN responses may be effective therapies for severe COVID-19 in patients with COPD.
Acknowledgments
Acknowledgment
The authors thank Melbourne Health and VIDRL at the Peter Doherty Institute for Infection and Immunity for providing the SARS-CoV-2 isolate. We also thank Dr. Carole Ford for helpful discussions in establishing the BD Rhapsody scRNAseq platform and the Garvan Kinghorn Cancer Centre for their Illumina next-generation sequencing platform. We would also like to thank Fia Boedijono for bioinformatics assistance.
Footnotes
Supported by the Rainbow Foundation, National Health and Medical Research Council (2011467 and 117534), University of Technology Sydney, Medical Research Future Fund (2007221) of Australia, NSW RNA Production Network, and Maridulu Budyari Gumal-Sydney Partnership for Health, Education, Research and Enterprise (SPHERE).
Author Contributions: M.D.J., R.M.M., S.I., D.H.N., S.M., P.P., K.N., N.G.H., W.J.B., B.M.S., P.A.W., A.F., and P.M.H. designed and performed experiments and analyzed data. L.J.G., P.J.H., and D.G.-O. provided bioinformatics assistance and assisted with data interpretation. M.D.J., P.A.W., A.F., and P.M.H. conceptualized the study and wrote the draft manuscript. All authors contributed to the editing of the first draft manuscript. P.M.H. funded the study.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202108-1901OC on May 12, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. O’Driscoll M, Ribeiro Dos Santos G, Wang L, Cummings DAT, Azman AS, Paireau J, et al. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature . 2021;590:140–145. doi: 10.1038/s41586-020-2918-0. [DOI] [PubMed] [Google Scholar]
- 2. Johansen MD, Irving A, Montagutelli X, Tate MD, Rudloff I, Nold MF, et al. Animal and translational models of SARS-CoV-2 infection and COVID-19. Mucosal Immunol . 2020;13:877–891. doi: 10.1038/s41385-020-00340-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wark PAB, Pathinayake PS, Kaiko G, Nichol K, Ali A, Chen L, et al. ACE2 expression is elevated in airway epithelial cells from older and male healthy individuals but reduced in asthma. Respirology . 2021;26:442–451. doi: 10.1111/resp.14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med . 2020;46:586–590. doi: 10.1007/s00134-020-05985-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jia HP, Look DC, Shi L, Hickey M, Pewe L, Netland J, et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol . 2005;79:14614–14621. doi: 10.1128/JVI.79.23.14614-14621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baker SA, Kwok S, Berry GJ, Montine TJ. Angiotensin-converting enzyme 2 (ACE2) expression increases with age in patients requiring mechanical ventilation. PLoS One . 2021;16:e0247060. doi: 10.1371/journal.pone.0247060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leung JM, Yang CX, Tam A, Shaipanich T, Hackett T-L, Singhera GK, et al. ACE-2 expression in the small airway epithelia of smokers and COPD patients: implications for COVID-19. Eur Respir J . 2020;55:2000688. doi: 10.1183/13993003.00688-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bui LT, Winters NI, Chung MI, Joseph C, Gutierrez AJ, Habermann AC, et al. Human Cell Atlas Lung Biological Network Chronic lung diseases are associated with gene expression programs favoring SARS-CoV-2 entry and severity. Nat Commun . 2021;12:4314. doi: 10.1038/s41467-021-24467-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Christie MJ, Irving AT, Forster SC, Marsland BJ, Hansbro PM, Hertzog PJ, et al. Of bats and men: Immunomodulatory treatment options for COVID-19 guided by the immunopathology of SARS-CoV-2 infection. Sci Immunol . 2021;6:eabd0205. doi: 10.1126/sciimmunol.abd0205. [DOI] [PubMed] [Google Scholar]
- 10. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell . 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chillappagari S, Preuss J, Licht S, Müller C, Mahavadi P, Sarode G, et al. Altered protease and antiprotease balance during a COPD exacerbation contributes to mucus obstruction. Respir Res . 2015;16:85–85. doi: 10.1186/s12931-015-0247-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kelly-Robinson GA, Reihill JA, Lundy FT, McGarvey LP, Lockhart JC, Litherland GJ, et al. The serpin superfamily and their role in the regulation and dysfunction of serine protease activity in COPD and other chronic lung diseases. Int J Mol Sci . 2021;22:6351. doi: 10.3390/ijms22126351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alqahtani JS, Oyelade T, Aldhahir AM, Alghamdi SM, Almehmadi M, Alqahtani AS, et al. Prevalence, severity and mortality associated with COPD and smoking in patients with COVID-19: a rapid systematic review and meta-analysis. PLoS One . 2020;15:e0233147. doi: 10.1371/journal.pone.0233147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aveyard P, Gao M, Lindson N, Hartmann-Boyce J, Watkinson P, Young D, et al. Association between pre-existing respiratory disease and its treatment, and severe COVID-19: a population cohort study. Lancet Respir Med . 2021;9:909–923. doi: 10.1016/S2213-2600(21)00095-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gerayeli FV, Milne S, Cheung C, Li X, Yang CWT, Tam A, et al. COPD and the risk of poor outcomes in COVID-19: a systematic review and meta-analysis. EClinicalMedicine . 2021;33:100789. doi: 10.1016/j.eclinm.2021.100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsu AC, Parsons K, Barr I, Lowther S, Middleton D, Hansbro PM, et al. Critical role of constitutive type I interferon response in bronchial epithelial cell to influenza infection. PLoS One . 2012;7:e32947. doi: 10.1371/journal.pone.0032947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hsu ACY, Dua K, Starkey MR, Haw T-J, Nair PM, Nichol K, et al. MicroRNA-125a and -b inhibit A20 and MAVS to promote inflammation and impair antiviral response in COPD. JCI Insight . 2017;2:e90443. doi: 10.1172/jci.insight.90443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mallia P, Message SD, Gielen V, Contoli M, Gray K, Kebadze T, et al. Experimental rhinovirus infection as a human model of chronic obstructive pulmonary disease exacerbation. Am J Respir Crit Care Med . 2011;183:734–742. doi: 10.1164/rccm.201006-0833OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Papi A, Bellettato CM, Braccioni F, Romagnoli M, Casolari P, Caramori G, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med . 2006;173:1114–1121. doi: 10.1164/rccm.200506-859OC. [DOI] [PubMed] [Google Scholar]
- 20. Hsu AC, Starkey MR, Hanish I, Parsons K, Haw TJ, Howland LJ, et al. Targeting PI3K-p110α suppresses influenza virus infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2015;191:1012–1023. doi: 10.1164/rccm.201501-0188OC. [DOI] [PubMed] [Google Scholar]
- 21. Hao Y, Hao S, Andersen-Nissen E, Mauck WM, III, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell . 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods . 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Steiling K, van den Berge M, Hijazi K, Florido R, Campbell J, Liu G, et al. A dynamic bronchial airway gene expression signature of chronic obstructive pulmonary disease and lung function impairment. Am J Respir Crit Care Med . 2013;187:933–942. doi: 10.1164/rccm.201208-1449OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol . 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burnett DL, Jackson KJL, Langley DB, Aggrawal A, Stella AO, Johansen MD, et al. Immunizations with diverse sarbecovirus receptor-binding domains elicit SARS-CoV-2 neutralizing antibodies against a conserved site of vulnerability. Immunity . 2021;54:2908–2921.e6. doi: 10.1016/j.immuni.2021.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Counoupas C, Johansen MD, Stella AO, Nguyen DH, Ferguson AL, Aggarwal A, et al. A single dose, BCG-adjuvanted COVID-19 vaccine provides sterilizing immunity against SARS-CoV-2 infection. NPJ Vaccines . 2021;6:143. doi: 10.1038/s41541-021-00406-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gohy S, Carlier FM, Fregimilicka C, Detry B, Lecocq M, Ladjemi MZ, et al. Altered generation of ciliated cells in chronic obstructive pulmonary disease. Sci Rep . 2019;9:17963. doi: 10.1038/s41598-019-54292-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hao W, Wang L, Li S. FKBP5 regulates RIG-I-mediated NF-κB activation and influenza A virus infection. Viruses . 2020;12:672. doi: 10.3390/v12060672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao D, Abbasi A, Rossiter HB, Su X, Liu H, Pi Y, et al. Serum amyloid A in stable COPD patients is associated with the frequent exacerbator phenotype. Int J Chron Obstruct Pulmon Dis . 2020;15:2379–2388. doi: 10.2147/COPD.S266844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, et al. GenOMICC Investigators ISARIC4C Investigators; COVID-19 Human Genetics Initiative; 23andMe Investigators; BRACOVID Investigators; Gen-COVID Investigators. Genetic mechanisms of critical illness in COVID-19. Nature . 2021;591:92–98. doi: 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- 31. Yu K, He J, Wu Y, Xie B, Liu X, Wei B, et al. Dysregulated adaptive immune response contributes to severe COVID-19. Cell Res . 2020;30:814–816. doi: 10.1038/s41422-020-0391-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Houssaini A, Breau M, Kebe K, Abid S, Marcos E, Lipskaia L, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight . 2018;3:e93203. doi: 10.1172/jci.insight.93203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ng DL, Granados AC, Santos YA, Servellita V, Goldgof GM, Meydan C, et al. A diagnostic host response biosignature for COVID-19 from RNA profiling of nasal swabs and blood. Sci Adv . 2021;7:eabe5984. doi: 10.1126/sciadv.abe5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bergamaschi C, Terpos E, Rosati M, Angel M, Bear J, Stellas D, et al. Systemic IL-15, IFN-γ, and IP-10/CXCL10 signature associated with effective immune response to SARS-CoV-2 in BNT162b2 mRNA vaccine recipients. Cell Rep . 2021;36:109504. doi: 10.1016/j.celrep.2021.109504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Comer DM, Kidney JC, Ennis M, Elborn JS. Airway epithelial cell apoptosis and inflammation in COPD, smokers, and nonsmokers. Eur Respir J . 2013;41:1058–1067. doi: 10.1183/09031936.00063112. [DOI] [PubMed] [Google Scholar]
- 36. Ravindra NG, Alfajaro MM, Gasque V, Huston NC, Wan H, Szigeti-Buck K, et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human airway epithelium identifies target cells, alterations in gene expression, and cell state changes. PLoS Biol . 2021;19:e3001143. doi: 10.1371/journal.pbio.3001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang H, Rostami MR, Leopold PL, Mezey JG, O’Beirne SL, Strulovici-Barel Y, et al. Expression of the SARS-CoV-2 ACE2 receptor in the human airway epithelium. Am J Respir Crit Care Med . 2020;202:219–229. doi: 10.1164/rccm.202003-0541OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carlier FM, Detry B, Lecocq M, Collin AM, Planté-Bordeneuve T, Verleden SE, et al. The memory of airway epithelium damage in smokers and COPD patients. bioRxiv . 2021;8:113. doi: 10.26508/lsa.202302341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lauer SA, Grantz KH, Bi Q, Jones FK, Zheng Q, Meredith HR, et al. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann Intern Med . 2020;172:577–582. doi: 10.7326/M20-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med . 2020;382:1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leung JM, Niikura M, Yang CWT, Sin DD. COVID-19 and COPD. Eur Respir J . 2020;56:2002108. doi: 10.1183/13993003.02108-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shukla SD, Mahmood MQ, Weston S, Latham R, Muller HK, Sohal SS, et al. The main rhinovirus respiratory tract adhesion site (ICAM-1) is upregulated in smokers and patients with chronic airflow limitation (CAL) Respir Res . 2017;18:6. doi: 10.1186/s12931-016-0483-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Turino GM. The origins of a concept: the protease-antiprotease imbalance hypothesis. Chest . 2002;122:1058–1060. doi: 10.1378/chest.122.3.1058. [DOI] [PubMed] [Google Scholar]
- 44. Aliee H, Massip F, Qi C, Stella de Biase M, van Nijnatten J, Kersten ETG, et al. U-BIOPRED study group Cambridge Lung Cancer Early Detection Programme; INER-Ciencias Mexican Lung Program. Determinants of expression of SARS-CoV-2 entry-related genes in upper and lower airways. Allergy . 2022;77:690–694. doi: 10.1111/all.15152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cai G, Bossé Y, Xiao F, Kheradmand F, Amos CI. Tobacco smoking increases the lung gene expression of ACE2, the receptor of SARS-CoV-2. Am J Respir Crit Care Med . 2020;201:1557–1559. doi: 10.1164/rccm.202003-0693LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Watson A, Öberg L, Angermann B, Spalluto CM, Hühn M, Burke H, et al. MICA II Studygroup Dysregulation of COVID-19–related gene expression in the COPD lung. Respir Res . 2021;22:164. doi: 10.1186/s12931-021-01755-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Imkamp K, Berg M, Vermeulen CJ, Heijink IH, Guryev V, Kerstjens HAM, et al. Nasal epithelium as a proxy for bronchial epithelium for smoking-induced gene expression and expression quantitative trait loci. J Allergy Clin Immunol . 2018;142:314–317.e15. doi: 10.1016/j.jaci.2018.01.047. [DOI] [PubMed] [Google Scholar]
- 48. Hu B, Guo H, Zhou P, Shi Z-L. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol . 2021;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gómez Antúnez M, Muiño Míguez A, Bendala Estrada AD, Maestro de la Calle G, Monge Monge D, Boixeda R, et al. SEMI-COVID-19 Network Clinical characteristics and prognosis of COPD patients hospitalized with SARS-CoV-2. Int J Chron Obstruct Pulmon Dis . 2021;15:3433–3445. doi: 10.2147/COPD.S276692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe . 2020;27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sabaka P, Koščálová A, Straka I, Hodosy J, Lipták R, Kmotorková B, et al. Role of interleukin 6 as a predictive factor for a severe course of Covid-19: retrospective data analysis of patients from a long-term care facility during Covid-19 outbreak. BMC Infect Dis . 2021;21:308. doi: 10.1186/s12879-021-05945-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim Y-M, Shin E-C. Type I and III interferon responses in SARS-CoV-2 infection. Exp Mol Med . 2021;53:750–760. doi: 10.1038/s12276-021-00592-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schroeder S, Pott F, Niemeyer D, Veith T, Richter A, Muth D, et al. Interferon antagonism by SARS-CoV-2: a functional study using reverse genetics. Lancet Microbe . 2021;2:e210–e218. doi: 10.1016/S2666-5247(21)00027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sa Ribero M, Jouvenet N, Dreux M, Nisole S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog . 2020;16:e1008737. doi: 10.1371/journal.ppat.1008737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sposito B, Broggi A, Pandolfi L, Crotta S, Clementi N, Ferrarese R, et al. The interferon landscape along the respiratory tract impacts the severity of COVID-19. Cell . 2021;184:4953–4968.e16. doi: 10.1016/j.cell.2021.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hatton CF, Botting RA, Dueñas ME, Haq IJ, Verdon B, Thompson BJ, et al. Delayed induction of type I and III interferons mediates nasal epithelial cell permissiveness to SARS-CoV-2. Nat Commun . 2021;12:7092. doi: 10.1038/s41467-021-27318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ziegler CGK, Miao VN, Owings AH, Navia AW, Tang Y, Bromley JD, et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell . 2021;184:4713–4733.e22. doi: 10.1016/j.cell.2021.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ordovas-Montanes J, Beyaz S, Rakoff-Nahoum S, Shalek AK. Distribution and storage of inflammatory memory in barrier tissues. Nat Rev Immunol . 2020;20:308–320. doi: 10.1038/s41577-019-0263-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cheemarla NR, Watkins TA, Mihaylova VT, Wang B, Zhao D, Wang G, et al. Dynamic innate immune response determines susceptibility to SARS-CoV-2 infection and early replication kinetics. J Exp Med . 2021;218:e20210583. doi: 10.1084/jem.20210583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol . 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tau G, Rothman P. Biologic functions of the IFN-gamma receptors. Allergy . 1999;54:1233–1251. doi: 10.1034/j.1398-9995.1999.00099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kedzierski L, Tate MD, Hsu AC, Kolesnik TB, Linossi EM, Dagley L, et al. Suppressor of cytokine signaling (SOCS)5 ameliorates influenza infection via inhibition of EGFR signaling. eLife . 2017;6:e20444. doi: 10.7554/eLife.20444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Veerati PC, Troy NM, Reid AT, Li NF, Nichol KS, Kaur P, et al. Airway epithelial cell immunity is delayed during rhinovirus infection in asthma and COPD. Front Immunol . 2020;11:974. doi: 10.3389/fimmu.2020.00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fleming SJ, Marioni JC, Babadi M. CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. bioRxiv . 2019:791699.
- 65. Heaton H, Talman AM, Knights A, Imaz M, Gaffney DJ, Durbin R, et al. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat Methods . 2020;17:615–620. doi: 10.1038/s41592-020-0820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]