

Keywords: bile acids, cholehepatic shunt, gut-liver axis, primary sclerosing cholangitis
Abstract
Primary sclerosing cholangitis (PSC) is characterized by increased ductular reaction (DR), liver fibrosis, hepatic total bile acid (TBA) levels, and mast cell (MC) infiltration. Apical sodium BA transporter (ASBT) expression increases in cholestasis, and ileal inhibition reduces PSC phenotypes. FVB/NJ and multidrug-resistant 2 knockout (Mdr2−/−) mice were treated with control or ASBT Vivo-Morpholino (VM). We measured 1) ASBT expression and MC presence in liver/ileum; 2) liver damage/DR; 3) hepatic fibrosis/inflammation; 4) biliary inflammation/histamine serum content; and 5) gut barrier integrity/hepatic bacterial translocation. TBA/BA composition was measured in cholangiocyte/hepatocyte supernatants, intestine, liver, serum, and feces. Shotgun analysis was performed to ascertain microbiome changes. In vitro, cholangiocytes were treated with BAs ± ASBT VM, and histamine content and farnesoid X receptor (FXR) signaling were determined. Treated cholangiocytes were cocultured with MCs, and FXR signaling, inflammation, and MC activation were measured. Human patients were evaluated for ASBT/MC expression and histamine/TBA content in bile. Control patient- and PSC patient-derived three-dimensional (3-D) organoids were generated; ASBT, chymase, histamine, and fibroblast growth factor-19 (FGF19) were evaluated. ASBT VM in Mdr2−/− mice decreased 1) biliary ASBT expression, 2) PSC phenotypes, 3) hepatic TBA, and 4) gut barrier integrity compared with control. We found alterations between wild-type (WT) and Mdr2−/− mouse microbiome, and ASBT/MC and bile histamine content increased in cholestatic patients. BA-stimulated cholangiocytes increased MC activation/FXR signaling via ASBT, and human PSC-derived 3-D organoids secrete histamine/FGF19. Inhibition of hepatic ASBT ameliorates cholestatic phenotypes by reducing cholehepatic BA signaling, biliary inflammation, and histamine levels. ASBT regulation of hepatic BA signaling offers a therapeutic avenue for PSC.
NEW & NOTEWORTHY We evaluated knockdown of the apical sodium bile acid transporter (ASBT) using Vivo-Morpholino in Mdr2KO mice. ASBT inhibition decreases primary sclerosing cholangitis (PSC) pathogenesis by reducing hepatic mast cell infiltration, altering bile acid species/cholehepatic shunt, and regulating gut inflammation/dysbiosis. Since a large cohort of PSC patients present with IBD, this study is clinically important. We validated findings in human PSC and PSC-IBD along with studies in novel human 3-D organoids formed from human PSC livers.
INTRODUCTION
Primary sclerosing cholangitis (PSC) is a cholestatic liver disease that mainly targets cholangiocytes, the epithelial cells lining bile ducts (1). PSC is characterized by hepatic fibrosis, inflammation, ductular reaction (DR), and impaired bile acid (BA) circulation (1). Up to 70% of PSC patients present with irritable bowel disease (IBD) comorbidity (2), implicating the gut-liver axis in the progression of PSC and PSC-IBD damage. Damaged cholangiocytes exhibit proliferative and/or senescent phenotypes and secrete proinflammatory and profibrotic factors (3–5). Biliary damage, in conjunction with intestinal inflammation in PSC-IBD patients, suggests a role for bile duct and intestinal cross talk signaling during cholestasis.
We have demonstrated the damaging role of mast cells (MCs) in the progression of cholestatic liver diseases through various factors including stem cell factor (SCF), transforming growth factor β1 (TGF-β1), and histamine/histamine receptor (HR) signaling (3, 5–7). Furthermore, MCs, injected via tail vein, infiltrate the liver and intestine, triggering both cholestatic liver injury and intestinal inflammation in MC-deficient mice (4). MCs have long been implicated to induce intestinal inflammation in IBD patients (8, 9); however, the role for MC regulation of the gut-liver axis during PSC and PSC-IBD remains elusive.
BA synthesis is regulated by nuclear receptor farnesoid X receptor (FXR) and its downstream effector fibroblast growth factor 15 (FGF15) in mice (10). The primary BAs synthesized include cholic acid (CA) and muricholic acids (MCAs), which are converted from chenodeoxycholic acid (CDCA), with CDCA serving as the natural agonist for FXR activation in humans (11). FXR is expressed in numerous organs of the body including the liver and intestine (12). After activation, FXR nuclear translocation induces the expression of target genes including short heterodimer partner (SHP) and FGF15 (13). Although intestinal FGF15 is the main BA synthesis regulator, cholangiocyte FGF15 may also regulate biliary and hepatic BA biosynthesis (13, 14). Cholangiocytes alter bile composition via the secretion of bicarbonate, water, and signaling hormones, which can alter bile flow (15). Cholangiocytes and enterocytes both express the apical sodium bile acid transporter (ASBT), the transporter responsible for enterohepatic and cholehepatic conjugated BA circulation. Cholangiocytes play an important role in modifying bile content; therefore, it is reasonable to assume that the role of cholangiocytes in BA circulation may be more complex than previously speculated (16).
We have demonstrated that PSC patients have increased MC infiltration, biliary FXR expression, and elevated serum FGF19 and histamine levels (4). Because of the role of MCs in the regulation of biliary FXR signaling, we aimed to assess how biliary ASBT inhibition alters cholestatic damage and MC activation in multidrug-resistant 2 knockout (Mdr2−/−) mice.
MATERIALS AND METHODS
Materials
All reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless stated otherwise. All authors have access to the study data and have reviewed and approved the final manuscript.
In Vivo Model
We used 12-wk-old male Mdr2−/− mice, a mouse model of PSC (17), raised on an FVB/NJ background (wild type, WT). To assess the effect of ASBT knockdown, WT and Mdr2−/− mice were treated with Control or ASBT Vivo-Morpholino (VM; 12.5 mg oligo/kg body wt) via two tail vein injections 7 and 3 days before euthanasia as described previously (5). Control (5′- CCTCTTACCTCAGTTACAATTTATA-3′) and ASBT VM (5′- GGAGTTATCCATCACTGCTTGTGCT-3′) were purchased from Gene-Tools Inc (Philomath, OR). Mice were kept in standard housing with a 12:12-h day-night lighting cycle and free access to standard chow and water. All housing and procedures adhered to Institutional Animal Care and Use Committee (IACUC) protocols approved by Indiana University–Purdue University Indianapolis. Serum, liver (snap frozen and formalin fixed/paraffin embedded), isolated cholangiocytes and hepatocytes, and respective supernatants were collected from all mice as described previously (6, 18). Briefly, livers were perfused with 1× HEPES-buffered saline (HBS) containing 0.02% (wt/vol) EGTA until blood clearance, and liver was then digested with 1× HBS containing 0.01% (wt/vol) MgSO4, 0.02% (wt/vol) collagenase, and 3.4 mM CaCl2. Livers were then broken down in HBS over ice with forceps. Hepatocytes were pelleted, and supernatants were collected and stored at −80°C before analysis. Cholangiocytes were isolated with antibody-bound magnetic beads for antigen expressed by cholangiocytes (19). Isolated cholangiocyte and hepatocyte number and viability were determined by trypan blue exclusion with light microscopy. Isolated cell supernatant was obtained from a portion of cells (1 × 106 cells per 1 mL) incubated in HBS containing 0.01% (wt/vol) MgSO4 and 3.4 mM CaCl2 for 4 h in a 37°C shaking water bath. Since ASBT is responsible for enterohepatic BA circulation (20), whole small intestine (with content, snap frozen, n = 3–5), ileum (snap frozen and paraffin embedded, n = 4 or 5), colon (paraffin embedded, n = 7–10), and feces (from colon, frozen, n = 7–10) were collected from all groups.
Hepatic Damage Evaluation
Alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) were measured with the IDEXX Catalyst One Serum Chemistry Analyzer (IDEXX, Westbrook, ME). Serum from 7–10 mice was combined and run in the IDEXX Catalyst One Serum Chemistry Analyzer in triplicate. The data are presented as means ± SD of technical replicates. Hepatic damage was assessed on hematoxylin and eosin Y (H&E)-stained tissue sections and assessed by a semiquantitative scoring system: lobular necrosis and inflammation scoring: 0 = none, 1 = one focus or less per ×10 objective, 2 = two to four foci per ×10 objective, 3 = more than five foci per ×10 objective; portal inflammation: 0 = none, 1 = mild, some or all portal areas, 2 = moderate, some or all portal areas, 3 = moderate/marked, all portal areas, 4 = marked, all portal areas.
PCR, Immunohistochemical Staining, and Immunofluorescence
Total RNA was isolated with TRI Reagent (Sigma Life Science, St. Louis, MO) and reverse transcribed with the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) and the Ready First Strand cDNA Synthesis Kit (Qiagen, Valencia, CA), following manufacturers’ protocols. Quantitative PCR (qPCR) was performed with RT2 Primary Assay from Qiagen (Valencia, CA). Immunoreactivity was detected with ImmPRESS horseradish peroxidase and alkaline phosphatase detection kits obtained from Vector Laboratories (Olean, NY). After immunohistochemical staining was completed, hematoxylin and eosin Y (H&E) and Fast Green-Sirius Red (FG-SR) staining, slides were scanned and imaged with Leica Aperio ImageScope 12.1 (Wetzlar, Germany). Immunofluorescence imaging was performed by confocal microscopy (LEICA TCS SP5 X system; Leica Microsystems, Inc., Buffalo Grove, IL). Percent positive staining was calculated from positive-stained area over the whole field area for each image with Image Pro software from Media Cybernetics (Rockville, MD).
ASBT Expression
To determine knockdown of ASBT via VM, ASBT expression was determined in ileum sections (4-μm thickness) from all mice by immunofluorescent staining with cytokeratin-19 (CK-19, epithelial marker) (21). Percent positive staining in the ileum was calculated from positive-stained apical membrane over the whole field area for each image with Image Pro software from Media Cybernetics (Rockville, MD). To confirm hepatic ASBT knockdown, ASBT expression was imaged in liver sections (4-μm thickness) from all mice via immunohistochemistry (3). Percent positive staining was calculated from positive-stained area over the whole field area for each image with Image Pro software from Media Cybernetics (Rockville, MD). For small (<15 μm) and large (>15 μm) ASBT bile duct quantification, we first performed morphometric analysis (22) and selected bile ducts by size to measure positive-stained areas over whole field area for each image with Image Pro software from Media Cybernetics (Rockville, MD).
Evaluation of Hepatic Damage and Intrahepatic Bile Duct Mass
Elevated ALP serum levels and hepatocyte damage are hallmarks of PSC patients, accompanied by increased intrahepatic bile duct mass (IBDM) (1); therefore, we assessed these phenotypes in our mice. Small (<15 μm) and large (>15 μm) IBDMs (22) were assessed by immunohistochemistry for CK-19, a marker for cholangiocytes in the liver (6). Stained slides were scanned by a digital scanner (Aperio Scanscope CS System; Leica Biosystems, Milan, Italy) and processed by ImageScope. The number of small and large bile ducts was quantified by morphometric analysis (23, 24).
Evaluation of Hepatic Fibrosis, Inflammation, Biliary Senescence-Associated Secretory Phenotype, and MC Infiltration/Activation
Patients with PSC often present with bridging and portal hepatic fibrosis (3, 4). We assessed hepatic fibrosis in all mice by FG-SR staining to detect collagen deposition, followed by semiquantification of positive collagen staining. Hepatic fibrosis was confirmed with Masson’s trichrome staining, following the manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA), and qPCR of fibronectin (Fn1) in total liver (3, 4).
Increased hepatic inflammation is often detected in PSC patients, resulting in cholangiocyte damage (1). To determine hepatic inflammation, we measured F4/80 (a marker of Kupffer cells) via immunohistochemistry (4), with semiquantification of positive staining. Biliary inflammation was confirmed with macrophage inflammatory protein-1α/C-C motif ligand 3 (MIP-1α/CCL3) secretion on Mouse Cytokine ELISA Plate Array I (Signosis, Inc., Santa Clara, CA) in cholangiocyte supernatant.
Serum histamine levels were measured in all mice via commercially available enzyme immunoassay (EIA) kits, following the manufacturer’s protocol (Cayman Chemical Co., Ann Arbor, MI) (25). Murine MC infiltration was detected with immunohistochemistry for tryptase β-2 in liver and ileum sections from all groups of mice (26, 27). Semiquantification was performed as described above.
BA Signaling and Composition
Patients with PSC have dysregulated BA signaling and elevated hepatic total bile acid (TBA) content (1). TBA levels (Crystal Chem, Inc., Downers Grove, IL) were measured in serum, liver, hepatocyte and cholangiocyte supernatants, feces, and whole small intestinal tract contents from all mice (28). BA species, including conjugated and unconjugated, were determined in liver via liquid chromatography tandem mass spectrometry (LC-MC/MS; 8600 system at McGuire Research Institute, Richmond, VA) (29). Hydrophobic primary BAs, CA, and CDCA were measured by EIA (Cell Bio Labs, Inc., San Diego, CA), following the manufacturer’s protocol, in serum and fecal lysate (10 µL per mg of sample). To assess hepatocyte response to altered BA composition, BA transporters NTCP (Slc10a1) and BSEP (Abcb11) and BA efflux transporter (MRP3, Abcc3) were measured in isolated hepatocytes by qPCR. To understand the role of ASBT VM in enterohepatic BA signaling of Mdr2−/− mice, we determined hepatic and ileal FXR expression via immunofluorescence, costained with CK-19 to mark epithelial cells, including cholangiocytes. We performed semiquantification as described above. The downstream FXR target effector FGF15 was measured in isolated cholangiocyte supernatants by EIA.
Biliary ASBT and MC Activation in Human Patients
Healthy, nondiseased liver tissues were purchased from Sekisui XenoTech, LLC (Kansas City, KS, n = 2) or provided by B. Ekser (n = 4) from deceased donors. Liver tissues from patients with PSC (n = 4), PSC with IBD comorbidity (PSC-IBD) (n = 5), primary biliary cholangitis (PBC, n = 3), biliary atresia (n = 1), alcoholic steatohepatitis (ASH, n = 1), and nonalcoholic steatohepatitis (NASH, n = 2) were collected as explants during liver transplantation, deidentified, and provided by B. Ekser. Liver sections (formalin fixed, 5-μm thickness, n = 2–4 per group) from healthy human control subjects and patients with cholestatic liver diseases were used for immunohistochemistry for ASBT and tryptase (MC marker). Percent positive staining of bile duct ASBT was calculated as bile duct area positive staining over the whole field area for each image with Image Pro software from Media Cybernetics (Rockville, MD). MC number was counted and averaged for each image. Bile from healthy nondiseased and cholestatic [biliary atresia (n = 1), PBC (n = 2), PSC (n = 2), and PSC-IBD (n = 3)] patients was deidentified and provided by B. Ekser. Patient demographics are detailed in Supplemental Table S1. Samples were received under a protocol approved by Indiana University Health and the Indiana University Institutional Review Board.
Histamine levels (Cayman Chemicals, Inc., Ann Arbor, MI) and TBA content (Crystal Chem, Inc., Downers Grove, IL) were measured in bile from control and cholestatic patients by EIA, following manufacturer protocols. For all patient samples written informed consent was obtained, and the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as described and approved by the Indiana University Institutional Review Board. No donor organs or bile were obtained from executed prisoners or other institutionalized persons.
In Vitro Studies
Immortalized intrahepatic murine cholangiocyte line cells (IMCLs) were cultured as described previously (28), and 4 × 105 cells were seeded on Transwell membranes with 0.4-μm pores (Corning, Corning, NY). IMCLs were serum starved overnight and treated with vehicle (H2O) or ASBT VM (2 μM) for 4 h, for enhanced inhibition and cell viability (30), before stimulation with taurocholic acid (TCA, 100 μM) (31) for 24 h at 37°C. Basolateral supernatant and cells were collected for EIA and qPCR analysis, respectively. BA signaling was assessed by qPCR for FXR (Nr1h4), SHP (Nr0b2), and FGF15 (Fgf15). Inflammation was assessed by qPCR for IL-1β. Basolateral CA (Cell Bio Labs, Inc., San Diego, CA) and histamine (Cayman Chemical Co, Ann Arbor, MI) were measured by EIA.
Fetal murine-derived hepatic MCs (MC/9, ATCC CRL8306) were cultured as described previously (6). MCs were seeded at 1 × 106 cells/mL density and starved of serum overnight. MCs were treated with vehicle (H2O), TCA (100 μm), or basolateral cholangiocyte supernatants for 24 h at 37°C. MC activation was determined by qPCR for markers of activation (FcRε1α and tryptase) and inflammation (IL-1β and TGF-1β). Furthermore, MC BA response was determined by qPCR for ASBT (Slc10a2), FXR (Nr1h4), and FGF15 (Fgf15). The in vitro experimental design is outlined in Supplemental Fig. S5A.
Separately, IMCLs, MCs, and the hepatocyte cell line AML12 (ATCC CRL2254) were cultured and seeded at 5 × 105 cells/well density and allowed to grow overnight. RNA was isolated, and mRNA expression of FXR (Nr1h4), SHP (Nr0b2), and FGF15 (Fgf15) was determined by qPCR.
Patient-Derived Cholangiocyte Isolation and Immortalization
Liver samples obtained from explanted livers (n = 1 for each group) were minced with scissors and rinsed three times with 1× phosphate-buffered saline (PBS). Liver tissues were digested in 1.66 mg/mL collagenase type XI (Sigma-Aldrich) diluted in sterile Dulbecco’s modified Eagle’s medium-F12 (DMEM-F12; Lonza, Walkersville, MD) treated with 10% antibiotic-antimycotic (anti/anti) in a 37°C shaking water bath for 30 min. Digested liver tissue was filtered with sterile gauze, and lysate was further filtered with a 100-mm cell strainer. Remaining lysate was centrifuged at 100 g for 4 min at 4°C, and the supernatant was collected and centrifuged at 700 g for 5 min at 4°C. The resulting pellet was washed in DMEM-F12 treated with 10% anti/anti before being centrifuged at 700 g for 5 min at 4°C. The final pellet was resuspended in H69 medium until confluent. The cells were isolated and purified with cholangiocyte marker EpCAM (HEA125; Progen, Wayne, PA) tagging and selection by fluorescence-activated cell sorting (FACS). Primary cholangiocytes were immortalized by lentivirus containing HPV E6/E7 (Applied Biological Materials, Inc., Richmond, BC, Canada) at second passage, following the manufacturer’s protocol.
Organoid Generation with Immortalized MCs, Hepatic Stellate Cells, and Patient-Derived Cholangiocyte
Normal and PSC cholangiocytes (CHOL) were isolated from explants of normal donor (female, 54 yr old) and PSC patient (female, 56 yr) liver tissue, respectively. At eighth passage, immortalized normal and PSC patient-derived cholangiocytes were cocultured for up to 72 h and combined with MCs purchased from Kerafast (Boston, MA) and hepatic stellate cells (HSCs) purchased from ScienCell (Carlsbad, CA). To generate multicellular organoids (see Fig. 7A), a total of 25,000 cells (CHOL:HSC:MC, 2.5:1:0.1) were mixed and suspended in a mixture composed of respective culture media in the same ratio as the cell composition [CHOL = H69 medium, HSC = Stellate Cell medium (ScienCell, Carlsbad, CA), and MC = StemProTM-34 SFM (Thermo Fisher, Waltham, MA)]. The cell mixture was then seeded in PrimeSurface Ultra-Low Attachment U-bottom 96-well plates (S-BIO, Hudson, NH) and centrifuged at 400 g for 10 min to pellet cells for organoid formation. The organoids were maintained and allowed to grow in the same medium composition for 3 days, with medium changes every other day. Basal organoids were collected and fixed in 2% paraformaldehyde in 1× PBS for immunological staining, and supernatant was collected for EIA analysis.
Figure 7.

Generation of human 3-dimensional (3-D) organoids and assessment of apical sodium bile acid (BA) transporter (ASBT)/chymase/histamine/fibroblast growth factor (FGF) signaling. Control and primary sclerosing cholangitis (PSC) cholangiocytes (from n = 1 patient per group) were isolated, immortalized, and used for organoid formation as detailed in the schematic created with bioRender.com (A). Human PSC organoids have enhanced ASBT (red) immunoreactivity, costained with cytokeratin-19 (CK-19, green) and desmin (yellow) to mark cholangiocytes and hepatic stellate cells (HSCs), respectively, compared with control organoids (B). Furthermore, histamine (C) and FGF19 (D) secretion were measured in PSC organoids compared with human control organoids. Chymase (red) immunoreactivity (E) was detected in control and PSC organoids. Data are presented as mean ± SD of 4 and 5 experiments for histamine and FGF19 enzyme immunoassay (EIA), respectively, from 25 organoids created from 1 human PSC patient and 1 human control patient. Representative images are presented at ×10 with ×40 zoom from 1 organoid per group.
Organoids and supernatant were collected for further analysis. Histamine (Cayman Chemicals, Ann Arbor, MI) and FGF19 (Abcam, Cambridge, MS) secretion were determined by EIA on organoid supernatant (collected from n = 25 organoids per patient). ASBT expression was determined by immunofluorescence with desmin and CK-19 costaining to mark HSCs and cholangiocytes, respectively. Chymase immunoreactivity was also determined by immunofluorescence costained with CK-19.
Microbiome Composition and Bacterial Translocation
Microbiome dysbiosis and bacterial translocation have been implicated in PSC development and progression (32). Fecal microbiome composition was determined with shallow shotgun whole genome sequencing by Transnetyx (Cordova, TN) (33). Fecal samples were collected at time of euthanasia from four to six mice per treatment group, snap frozen, and stored at −80°C until time of analysis. Samples were placed in individual tubes containing DNA stabilization buffer to ensure stability and traceability and shipped for DNA extraction, library preparation, and sequencing by Transnetyx protocol. DNA extraction was fully automated and optimized with the DNeasy 96 PowerSoil Pro QIAcube HT extraction kit (Qiagen, Germantown, MD), following the manufacturer’s protocol, for reproducible extraction of inhibitor-free, high-molecular weight genomic DNA that captures true microbial diversity of fecal samples. After DNA extraction and quality control (QC), genomic DNA was converted into sequencing libraries with unique dual indexed (UDI) adapters, to ensure that reads and/or organisms are not misassigned, with the KAPA HyperPlus Kit (Roche, Indianapolis, IN), following the manufacturer’s protocol. After QC, the libraries were sequenced by the shotgun sequencing method (a depth of 2 million 2 × 150 bp read pairs), which enables species- and strain-level taxonomic resolution, with NovaSeq (Illumina, San Diego, CA), following the manufacturer’s protocol. Sequencing data, in the form of FASTQ files, were uploaded automatically onto One Codex analysis software and analyzed against the One Codex database consisting of >115,000 whole microbial reference genomes. The classification results were filtered through several statistical postprocessing steps designed to eliminate false positive results caused by contamination, host reads, or sequencing artifacts. Abundance analyses were determined by One Codex microbiome analysis software based on Shannon index (alpha diversity) and principal coordinate analysis (PCoA, beta diversity) based on Bray–Curtis dissimilarity matrix. Comparisons with statistical significance for family and genera are detailed in Supplemental Tables S5 and S6, respectively.
To assess intestinal leakage, which is associated with gut dysbiosis, we stained colon from our mice for tight junction protein zonula occludens-1 (ZO-1) on formalin-fixed paraffin-embedded sections (4–6 μm thick). Percent positive staining of ZO-1 in the colon was calculated from positive-stained apical membrane area over the whole field area for each image with Image Pro software from Media Cybernetics (Rockville, MD). The mRNA expressions of tricellulin, cingulin, and ZO-1 were determined in the total colon RNA from all groups of mice by qPCR. Bacterial translocation was determined through colony-forming units (CFUs) from liver homogenates. Frozen liver samples from individual mice were handled aseptically and homogenized in sterile filtered 1× PBS (10 µL per 1 mg tissue, 10 mg minimum) with sterile ceramic bead tubes on Bead Blaster 24 (Benchmark, Tempe, AZ); program run: 4,260 revolutions per minute for 30 s, repeated thrice, at 4°C. Homogenates were centrifuged, and liver lysate (100 µL) was sterilely plated and spread on nonselective brain heart infusion agar plates (BD, Franklin Lakes, NJ) by aseptic technique and allowed to absorb for 15 min at room temperature. Plates were then inverted and incubated for 96 h at 37°C, and colonies were manually counted. CFU per milligram of tissue was determined by dividing total colony number per mouse by 10.
Statistical Analysis
Data are presented as means ± SD. Student’s unpaired t test was used when two groups were analyzed. When more than two groups were analyzed, with one independent variable, Welch one-way ANOVA was used, followed by appropriate post hoc test with GraphPad Prism 9 (San Diego, CA). When more than two groups were analyzed, with two independent variables, two-way ANOVA was used, followed by appropriate post hoc test with GraphPad Prism 9. We considered P < 0.05 as significant.
RESULTS
ASBT VM Treatment Reduces Hepatic (but Not Ileal) ASBT Immunoreactivity Compared with Control
We that found ileal ASBT immunoreactivity (green) was unchanged between wild-type (WT) and Mdr2−/− mice treated with either Control or ASBT VM (Supplemental Fig. S1A). Mdr2−/− Control mice have significantly increased hepatic ASBT immunoreactivity in large (red arrowheads) but not small (yellow arrowheads) bile ducts compared with WT Control, and, in both WT and Mdr2−/− mice, ASBT VM treatment significantly reduced hepatic large (but not small) bile duct ASBT immunoreactivity (Supplemental Fig. S1B).
ASBT VM Treatment Reduces Hepatic Damage and Large Intrahepatic Bile Duct Mass in Mdr2−/− Mice
Compared with WT Control, ALT and ALP levels increased in Mdr2−/− Control mice, and treatment with ASBT VM decreased both ALT and ALP in both WT and Mdr2−/− mice (Fig. 1A). AST serum levels increased in Mdr2−/− Control compared with WT Control and were decreased by ASBT VM treatment in Mdr2−/− mice. WT ASBT VM mice displayed increased AST serum levels compared with WT Control mice (Fig. 1A). Mdr2−/− Control mice had increased lobular necrosis compared with WT ASBT VM, but this was unchanged compared with WT Control and Mdr2−/− ASBT VM mice (Fig. 1B). Portal inflammation and lobular inflammation significantly increased in Mdr2−/− Control mice compared with WT mice, and Mdr2−/− ASBT VM mice had a significant reduction of portal inflammation compared with Mdr2−/− Control mice (Fig. 1B). Lobular inflammation was unchanged between Mdr2−/− Control and ASBT VM mice and was increased in Mdr2−/− ASBT VM-treated mice compared with WT. Mdr2−/− Control mice had increased large (Fig. 1C) and pooled (Supplemental Fig. S2A) IBDM compared with WT mice, and ASBT VM treatment reduced large and pooled IBDM (Fig. 1C and Supplemental Fig. S2A), but not small IBDM (Supplemental Fig. S2B), in Mdr2−/− mice compared with Control-treated Mdr2−/− mice.
Figure 1.

Apical sodium bile acid (BA) transporter (ASBT) Vivo-Morpholino (VM) treatment reduces hepatic damage and large intrahepatic bile duct mass (IBDM) in multidrug-resistant 2 knockout (Mdr2−/−) mice. Alanine aminotransferase (ALT) and alkaline phosphatase (ALP) levels increased in Mdr2−/− Control mice compared with wild-type (WT) Control mice and were significantly decreased in both WT and Mdr2−/− mice treated with ASBT VM (A). Aspartate aminotransferase (AST) serum levels significantly increased in WT ASBT VM and Mdr2−/− Control mice but were reduced in Mdr2−/− mice after ASBT VM treatment (A). There was increased portal inflammation in Mdr2−/− mice treated with Control that was significantly reduced after ASBT VM treatment, and no differences in lobular inflammation were found between WT or Mdr2−/− mice treated with Control or ASBT VM (B). Large IBDM increased in Mdr2−/− Control mice compared with WT mice. ASBT VM reduced large IBDM in Mdr2−/− mice but had no effect in WT mice (C). Serum from 7–10 mice was combined and run in triplicates. The data are presented as mean ± SD of our technical replicates for serum chemistry. Data are presented as mean ± SD of whole tissue scanning for cytokeratin-19 (CK-19) and hematoxylin and eosin Y (H&E) analysis from 4 or 5 slides containing liver samples from 7 to 10 mice per group. Representative images are presented as ×10. *P < 0.05, ***P < 0.001, ****P < 0.0001.
ASBT VM Treatment Reduces Hepatic Fibrosis and Inflammation in Mdr2−/− Mice
There was no visible hepatic fibrosis in WT mice treated with either Control or ASBT VM (Fig. 2, A and B). Mdr2−/− Control mice had increased collagen deposition (Fig. 2, A and B) and fibronectin-1 (Fn1) expression (Fig. 2C) compared with WT Control mice, and these parameters were reduced in Mdr2−/− mice after ASBT VM treatment (Fig. 2, A–C).
Figure 2.

Hepatic fibrosis is ameliorated in multidrug-resistant 2 knockout (Mdr2−/−) mice after apical sodium bile acid (BA) transporter (ASBT) Vivo-Morpholino (VM). We found increased collagen deposition shown by Fast Green-Sirius Red (FG-SR) staining and semiquantification (A) and confirmed with Masson’s trichrome (B) in Mdr2−/− Control mice compared with wild-type (WT) mice. ASBT VM treatment reduced collagen deposition in Mdr2−/− mice (A and B). Hepatic Fn1 expression increased in Mdr2−/− Control mice but was unchanged in WT mice and was reduced after ASBT VM (C). Representative images are presented as ×10. Data are presented as mean ± SD of 5 images for FG-SR semiquantification and 4 experiments for qPCR from 7 to 10 mice per group. **P < 0.01, ***P < 0.001, ****P < 0.0001.
There was elevated Kupffer cell/macrophage presence (F4/80) in Mdr2−/− Control mice compared with both WT Control and WT ASBT VM mice (Fig. 3A). ASBT VM treatment reduced Kupffer cell/macrophage presence compared with Control in Mdr2−/− mice (Fig. 3A). There was elevated biliary secretion of macrophage inflammatory protein-1α/C-C motif ligand 3 (MIP-1α/CCL3) in isolated cholangiocyte supernatants from WT ASBT VM, Mdr2−/− Control, and Mdr2−/− ASBT VM compared with WT Control mice. ASBT VM treatment reduced biliary MIP-1α/CCL3 secretion in isolated cholangiocyte supernatants from Mdr2−/− mice compared with Control mice (Fig. 3B).
Figure 3.

Apical sodium bile acid (BA) transporter (ASBT) Vivo-Morpholino (VM) reduces hepatic inflammation, biliary senescence-associated secretory phenotype (SASP) secretion, and histamine serum content. There was increased Kupffer cell presence (F4/80) (A) with semiquantification and macrophage inflammatory protein-1α/C-C motif ligand 3 (MIP1-α/CCL3) secretion from isolated cholangiocyte supernatant (B) in multidrug-resistant 2 knockout (Mdr2−/−) Control mice compared with wild-type (WT) mice. ASBT VM treatment reduced hepatic inflammation and biliary MIP1-α/CCL3 in Mdr2−/− mice (A and B). Serum histamine content increased in Mdr2−/− Control compared with WT mice but was reduced after ASBT VM treatment (C). Representative images are presented as ×10 for F4/80. Data are presented as mean ± SD of n = 5 images for F4/80 quantification from 7–10 mice per group. Data are presented as mean ± SD of 4 technical replicates for histamine enzyme immunoassay (EIA) and 3 technical replicates for biliary MIP1-α/CCL3 secretion pooled from 7 to 10 mice per group. *P < 0.05, **P < 0.01, ****P < 0.0001.
In Mdr2−/− Mice, Histamine Serum Content Decreases with ASBT VM Treatment and MC Presence Increases in Both Liver and Intestine
Serum histamine levels increased in Mdr2−/− mice treated with Control compared with WT mice treated with Control (Fig. 3C). ASBT VM treatment significantly reduced serum histamine levels in WT and Mdr2−/− mice (Fig. 3C). We found minimal hepatic MC infiltration in WT mice treated with Control or ASBT VM; however, a significant increase in hepatic MC infiltration (red arrows in Supplemental Fig. S2C) was found in the portal area of Mdr2−/− Control mice. Treatment with ASBT VM did not change MC infiltration in WT or Mdr2−/− mice compared with respective controls (Supplemental Fig. S2C). Mdr2−/− Control mice had increased ileal MC infiltration compared with WT mice that was unchanged in Mdr2−/− mice treated with ASBT VM (Supplemental Fig. S2D). These data demonstrate that MCs infiltrate the liver and intestine during cholestatic liver injury, and, whereas MC presence was not altered by ASBT VM treatment, histamine serum content was significantly reduced when ASBT was inhibited. Since the majority of histamine is derived from degranulated MCs (34, 35), we speculate that ASBT VM treatment may act on some, but not all, MCs to stabilize and reduce histamine release; however, this requires further evaluation to confirm.
ASBT VM Alters TBA and BA Species in Serum, Liver, and Feces and Decreases Cholehepatic TBA
We found increased TBA levels in serum, liver, and feces of Mdr2−/− Control mice compared with WT Control and WT ASBT VM mice (Fig. 4, A–C). ASBT VM treatment reduced serum TBA in Mdr2−/− mice compared with Mdr2−/− Control; however, liver TBA content remained unchanged (Fig. 4, A and B, respectively). ASBT VM treatment increased fecal TBA content in WT mice compared with Control-treated WT mice. Surprisingly, Mdr2−/− mice treated with ASBT VM had reduced fecal TBA excretion compared with Control-treated Mdr2−/− mice (Fig. 4C). Whole small intestine TBA content was unchanged between WT and Mdr2−/− mice treated with or without ASBT VM (Fig. 4D). Next, we evaluated cholehepatic TBA content and found that supernatants from both hepatocytes and cholangiocytes had increased TBA levels in Mdr2−/− mice treated with Control compared with WT mice. ASBT VM treatment significantly reduced both hepatocyte and cholangiocyte supernatant TBA levels in Mdr2−/− mice compared with Control treatment; however, in WT mice TBA levels remained unchanged (Fig. 4, E and F).
Figure 4.
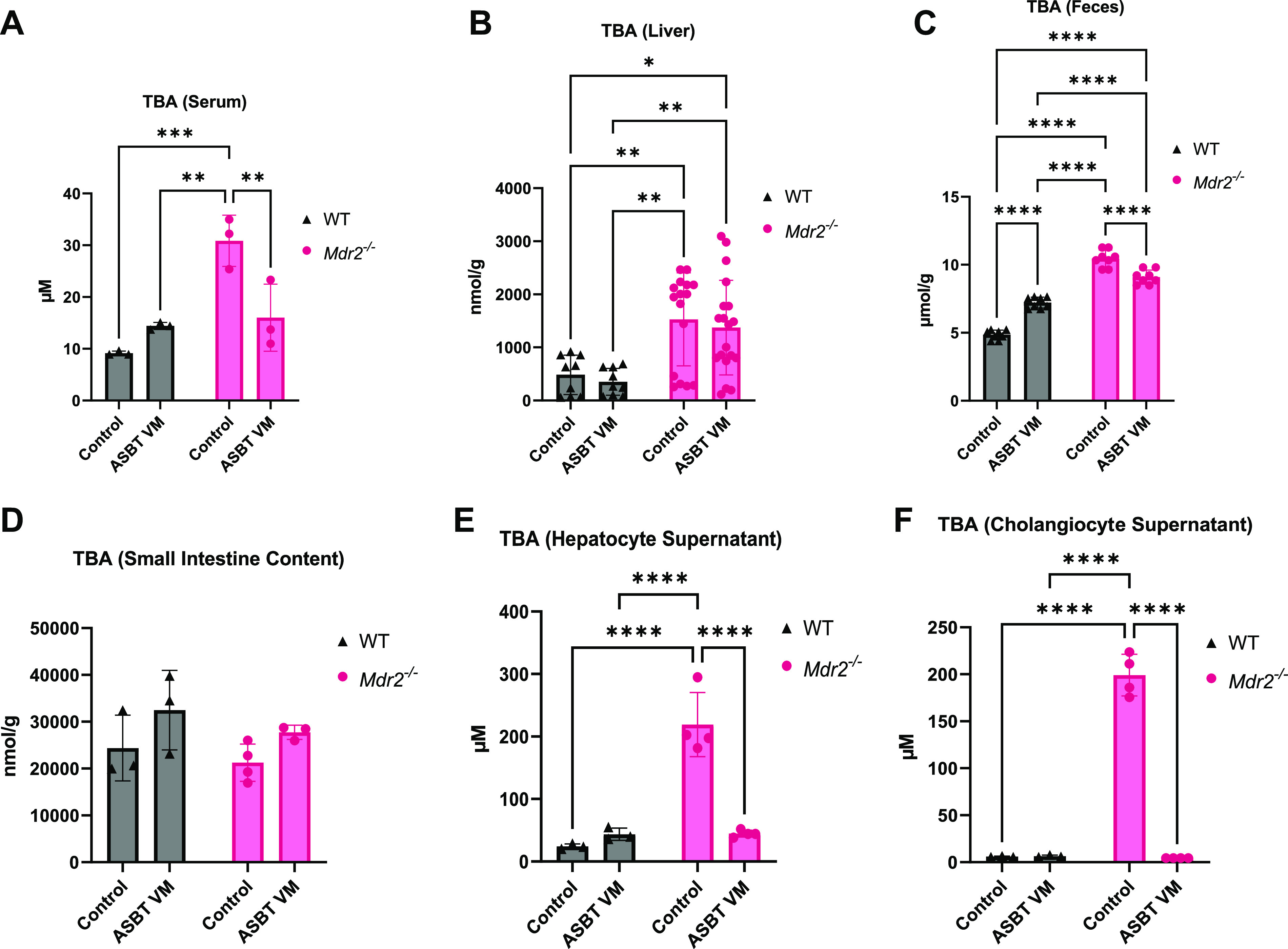
Apical sodium bile acid (BA) transporter (ASBT) ASBT Vivo-Morpholino (VM) treatment alters enterohepatic and decreases cholehepatic total bile acid (TBA) levels. Multidrug-resistant 2 knockout (Mdr2−/−) Control mice had increased TBA levels in serum (A), liver (B), feces (C) compared with all wild-type (WT) mice. ASBT VM treatment reduced serum (A) and feces (C) TBA in Mdr2−/− mice and increased serum (A) TBA in WT mice. Intestinal TBA levels were unchanged between WT and Mdr2−/− mice treated with Control or ASBT VM (D). Isolated hepatocyte supernatant (E) and cholangiocyte supernatant (F) TBA content increased in Mdr2−/− Control mice compared with WT mice, and ASBT VM treatment reduced TBA content in both hepatocyte and cholangiocyte supernatants [collected from isolated cells pooled from 7 to 10 mice per group and incubated in HEPES-buffered saline (HBS) with 0.1 M CaCl2 in a 37°C shaking water bath for 4 h]. Data are presented as mean ± SD of 4–7 experiments from 7–10 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Pharmacological ileal ASBT inhibition increases fecal BA levels (36), and our results indicate that hepatic ASBT inhibition reduces fecal TBA. Therefore, we measured fecal and serum CA and CDCA in our mice. Fecal CA and CDCA were unchanged in both WT and Mdr2−/− mice treated with Control or ASBT VM (Supplemental Fig. S3, A and B). Serum CA was also unchanged among all groups, whereas CDCA was significantly increased in WT ASBT VM mice compared with WT Control mice (Supplemental Fig. S3, C and D). Serum CDCA was significantly reduced in Mdr2−/− Control mice compared with WT Control but was increased after ASBT VM treatment in Mdr2−/− mice (Supplemental Fig. S3D). The proportion of hepatic total conjugated and unconjugated BAs was unchanged in both WT and Mdr2−/− mice treated with either Control or ASBT VM (Supplemental Fig. S3E), indicating that BA modification pathways are not affected by ASBT inhibition. Together, these findings suggest that inhibition of biliary ASBT alters serum CDCA levels in both WT and Mdr2−/− mice.
We further evaluated BA species in the liver (Supplemental Fig. S3F and Supplemental Table S2) and found that tauro-β-muricholic acid (TβMCA) is significantly increased in Mdr2−/− mice compared with WT mice and is unchanged after ASBT VM treatment (Supplemental Fig. S3G). With regard to fecal BA species (Supplemental Fig. S3H), we found that fecal secondary BAs, glycol-ursodeoxycholic acid (GUDCA) (Supplemental Fig. S3I), and lithocholic acid (LCA) (Supplemental Fig. S3J) increased in Mdr2−/− Control compared with WT Control mice, and ASBT VM treatment decreased these parameters in Mdr2−/− mice compared with Control (Supplemental Fig. S3, I and J, and Supplemental Table S3).
ASBT VM Regulates Hepatic and Ileum BA Transporters
Since we identified changes in BA pool and species, we examined BA transporters in both the liver and ileum. Hepatocyte gene expression of the BA efflux transporters multidrug resistance-associated protein 3 (MRP3) and bile salt export pump (BSEP) increased in Mdr2−/− Control mice compared with Control and ASBT VM WT mice (Fig. 5, A and B). Although these findings appear contradictory, it has been shown that mRNA expression of BSEP remains unchanged whereas MRP3 increases in Mdr2−/− mice; however, the protein levels of these transporters decrease (37), suggesting that transport alteration occurs at the translational level. ASBT VM treatment in Mdr2−/− mice reduced hepatocyte expression of MRP3 and BSEP compared with Control-treated Mdr2−/− mice, respectively (Fig. 5, A and B). Next, we found reduced hepatocyte expression of the BA uptake transporter sodium-dependent uptake transporter (NTCP) in Mdr2−/− mice treated with Control compared with Control- and ASBT VM-treated WT mice (Fig. 5C). NTCP expression was partially reversed by ASBT VM treatment in Mdr2−/− mice (Fig. 5C).
Figure 5.
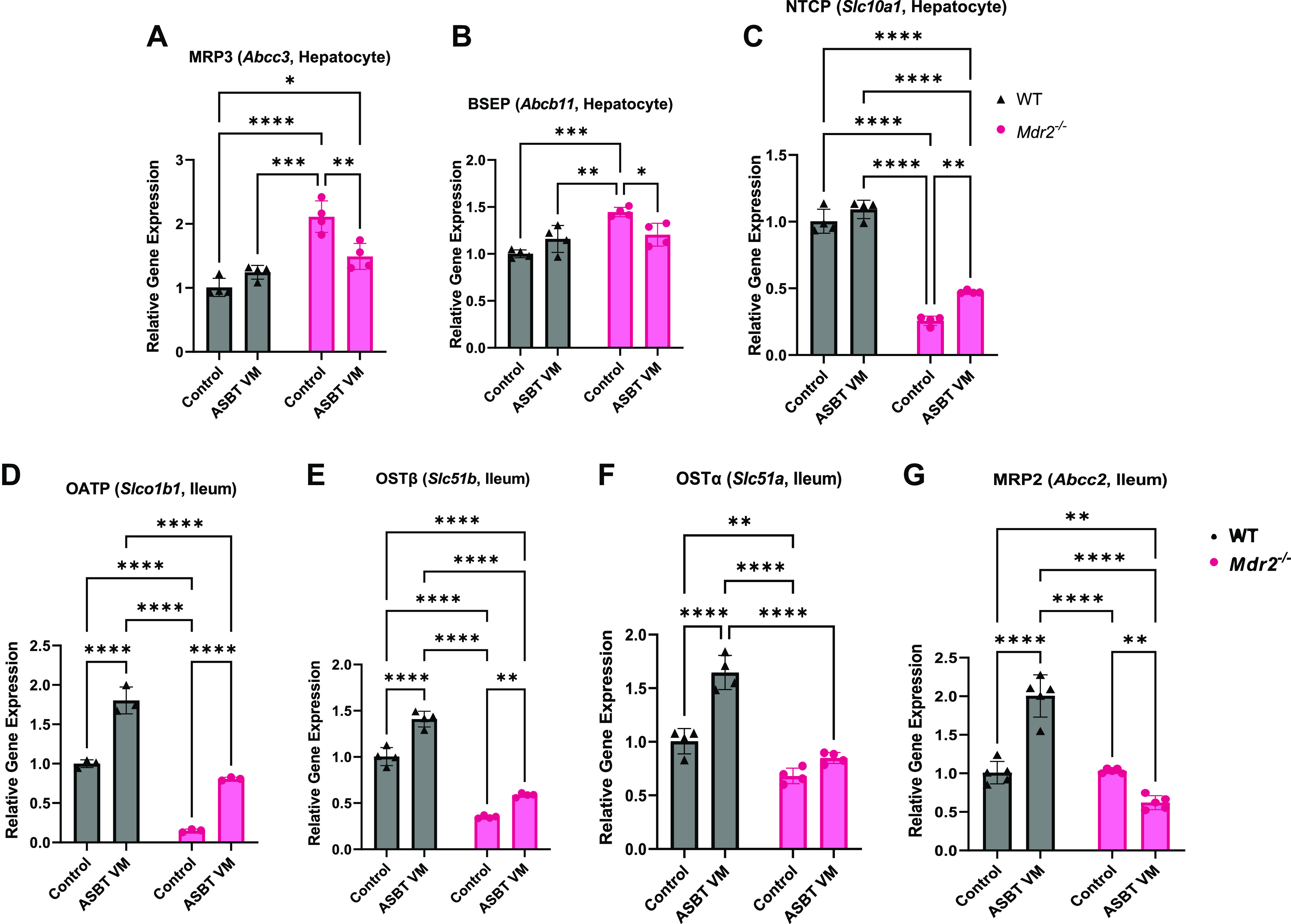
Apical sodium bile acid (BA) transporter (ASBT) inhibition alters hepatic and ileal BA transporter expression. Hepatocyte mRNA expression of BA transporters multidrug resistance-associated protein 3 (MRP3, A) and bile salt export pump (BSEP, B) significantly increased, while NTCP (C) significantly decreased, in multidrug-resistant 2 knockout (Mdr2−/−) Control mice compared with wild-type (WT) mice. ASBT Vivo-Morpholino (VM) treatment decreased MRP3 (A) and BSEP (B) while increasing NTCP (C) expression in Mdr2−/− mice compared with Control. ASBT VM treatment increased OATP (Slco1b1) (D), OSTβ (Slc51b) (E), OSTα (Slc5a1) (F), and MRP2 (Abcc2) (G) in WT mice compared with Control; however, OATP (D) and OSTβ (E) were increased, OSTα (F) unchanged, and MRP2 (G) decreased in Mdr2−/− after ASBT VM treatment compared with Control. Data are presented as mean ± SD of 4 experiments for qPCR from 7 to 10 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To further understand intestinal BA signaling, we measured the expression of BA transporters in the ileum. OATP and OSTβ expression significantly decreased in Mdr2−/− mice treated with Control compared with Control WT mice, whereas expression increased in both WT and Mdr2−/− mice after ASBT VM treatment (Fig. 5, D and E). Interestingly, OSTα and MRP2 expression increased in WT mice after ASBT VM treatment and expression was unchanged (OSTα) or significantly reduced (MRP2) in Mdr2−/− mice treated with ASBT VM compared with Control-treated mice (Fig. 5, F and G). Although these findings indicate cholestasis-dependent alterations in BA transporter expression, the role of ileal BA transporters in ASBT-regulated fecal TBA excretion remains unclear.
Hepatic and Ileal FXR and Biliary FGF15 Are Dysregulated in Mdr2−/− Mice after ASBT VM Treatment
We have demonstrated that biliary FXR and FGF15 expression increase after MC-induced cholestasis in MC-deficient mice (4), and it has been shown that ASBT expression is regulated by FXR activation in the ileum and bile ducts (38). We found that hepatocyte and cholangiocyte FXR immunoreactivity was present in both WT and Mdr2−/− mice treated with Control but was significantly reduced in Mdr2−/− mice compared with WT mice (Supplemental Fig. S4A). In the ileum, FXR immunoreactivity increased in WT mice treated with ASBT VM and in Mdr2−/− Control compared with WT Control mice, whereas FXR immunoreactivity decreased in Mdr2−/− ASBT VM-treated mice compared with Mdr2−/− Control (Supplemental Fig. S4B). Downstream of FXR, we found that biliary FGF15 secretion increased in Control Mdr2−/− mice compared with WT Control but was reduced in Mdr2−/− mice after ASBT VM treatment (Supplemental Fig. S4C), suggesting that biliary FXR/FGF15 may have a positive relationship with ASBT expression/activation. There were no significant differences in FGF15 secretion between WT groups (Supplemental Fig. S4C).
Biliary ASBT, MC Presence, and Bile Histamine Content Are Elevated in Patients with Liver Disease
Based on histology and semiquantification, there is a significant increase in ASBT immunoreactivity primarily in bile ducts of patients with cholestatic liver disease (PSC, PSC-IBD, biliary atresia, and PBC) and in alcohol-induced steatohepatitis (ASH) and nonalcoholic steatohepatitis (NASH) accompanied by a significant elevation of MC number compared with healthy, nondiseased control liver (Fig. 6A). There was no difference in TBA levels (Fig. 6B) in bile from healthy, nondiseased control subjects and cholestatic patients (PSC n = 2, PSC-IBD n = 3, biliary atresia n = 2, and PBC n = 1); however, cholestatic patients had increased bile histamine content compared with healthy, nondiseased control subjects (Fig. 6C). Since bile is secreted to the intestine to aid in lipid absorption, and MCs are known to participate in the liver and intestinal health, histamine may serve as a mediator of gut-liver communication, using bile as a conduit.
Figure 6.

Biliary apical sodium bile acid (BA) transporter (ASBT) and mast cell (MC) presence increase in patients with liver disease. Patients with primary sclerosing cholangitis (PSC) or PSC with irritable bowel disease (IBD) have significant (assessed by semiquantification) increased biliary ASBT expression (brown) accompanied by significant elevated MC number (tryptase, red) (A) compared with nondiseased control patients. We also found that cholestatic patients [primary biliary cholangitis (PBC), biliary atresia] and patients with fatty liver diseases [alcoholic steatohepatitis (ASH), nonalcoholic steatohepatitis (NASH)] display ASBT-positive bile ducts (brown) accompanied by MC presence (tryptase, red) compared with control subjects (A). Bile total bile acids (TBA) (B) were unchanged whereas histamine levels (C) increased in cholestatic patients compared with nondiseased control patients. Representative images are presented at ×40. Data are presented as mean ± SD of 3 experiments per patient for TBA and histamine enzyme immunoassay (EIA) from 7 cholestatic patients and 5 control patients and n = 5 nonoverlapping images for ASBT and MC quantification. *P < 0.05, ****P < 0.0001.
Human PSC-Derived Cholangiocyte Organoids Display ASBT and Chymase Immunoreactivity and Histamine and FGF19 Secretion
Cholangiocytes were isolated and immortalized from human control (n = 1) and PSC (n = 1) liver and utilized to form multicellular three-dimensional (3-D) organoids with cultured human HSCs and MCs, as described in Fig. 7A. We found that control and PSC organoids expressed CK-19, desmin, and ASBT, with enhanced immunoreactivity of ASBT in the PSC organoid compared with control (Fig. 7B). Furthermore, 3-D human PSC organoids displayed increased histamine (Fig. 7C) and FGF19 (Fig. 7D) secretion compared with healthy control organoids. Both PSC and control organoids display chymase immunoreactivity, and MCs were found near cholangiocytes (Fig. 7E). Our data demonstrate the feasibility of generating multicellular organoids with a combination of cells derived from human liver explants and cultured cell lines, including MCs. Although our data suggest that PSC organoids have enhanced ASBT, histamine, and FGF19, limitations of these findings include the number of patients organoids were derived from (n = 1), and further experiments are warranted to increase the rigor and robustness of these findings.
ASBT VM Reduces Biliary BA Transport and Subsequent MC Activation in Vitro
The murine in vitro experimental design is outlined in Supplemental Fig. S5A. Before assessing biliary BA transport and MC activation, we evaluated ASBT immunoreactivity by IF and semiquantification after treatment with ASBT VM. We found that treatment with ASBT VM did not induce any significant decrease in IMCL ASBT immunoreactivity compared with basal and water treatment (data not shown). We suspect this is due to the short in vitro treatment time; however, the following data demonstrate that treatment with ASBT VM regulates biliary BA signaling in vitro. We found increased biliary basolateral total CA levels in IMCLs treated with TCA after apical treatment compared with vehicle treatment, which was reduced with ASBT VM treatment (Supplemental Fig. S5B), confirming ASBT inhibition of conjugated BA transport. Interestingly, basolateral IMCL histamine secretion levels were unchanged between all groups, indicating that biliary histamine may not respond to direct BA stimulation (Supplemental Fig. S5C). We found that TCA treatment led to an increase of l1b in IMCL that was reduced when ASBT VM treatment was used before TCA treatment (Supplemental Fig. S5D). Finally, we found that biliary FXR signaling [FXR (Nr1h4), SHP (Nr0b2), and FGF15 (Fgf15)] was increased after TCA treatment and SHP and FGF15 were reduced after ASBT VM treatment (Supplemental Fig. S5, E–G), indicating that cholangiocytes respond to BAs through endogenous FXR signaling.
To understand the relationship between cholangiocyte ASBT-dependent BA transport and MC activation, we treated MCs with TCA along with conditioned media from our IMCLs treated with TCA and ASBT VM/TCA cotreatment (Supplemental Fig. S5A). MC expression of FXR (Nr1h4), FGF15 (Fgf15), and ASBT (Slc10a2) increased after TCA treatment or stimulation with basolateral media from IMCLs treated with TCA compared with controls; however, MC expression of these BA signaling genes was reduced after stimulation with basolateral media from IMCLs cotreated with ASBT VM and TCA (Supplemental Fig. S5, H–J). Next, we found increased MC expression of inflammatory (Il1b and Tgfb1) and activation (Fcer1a and Tpsb2) markers after treatment with basolateral media from IMCLs treated with TCA compared with controls, but this was reduced when MCs were stimulated with basolateral media from IMCLs cotreated with ASBT VM and TCA (Supplemental Fig. S5, K–N). TCA treatment did not alter MC inflammatory or activation expression compared with vehicle control.
To understand the expression of FXR in a cell-specific manner, we measured FXR (Nr1h4), SHP (Nr0b2), and FGF15 (Fgf15) in MCs and IMCLs. Since hepatocytes are involved in BA synthesis and cholehepatic shunt, we also included AML12, a hepatocyte line. We found that FXR (Nr1h4) is minimally expressed in MCs and IMCLs compared with AML12, whereas SHP (Nr0b2) is unchanged between all cell types (Supplemental Fig. S5O). Furthermore, MCs have a significant increase of basal FGF15 (Fgf15) mRNA expression compared with AML12 and IMCLs (Supplemental Fig. S5O), which may be due to the partial activation induced by growth media. These data show that, in vitro, FXR/FGF15 signaling is mediated by multiple cell types including contributions from hepatocytes, cholangiocytes, and MCs.
Fecal Microbiome Composition is Altered in Mdr2−/− Mice, and ASBT VM Challenges Hepatic Bacteria Translocation
Overall, we found no changes in total bacterial reads (Supplemental Fig. S6A) and family alpha diversity (Supplemental Fig. S6F) between any groups treated with or without ASBT VM; however, there were changes within family taxa relative abundance (Supplemental Fig. S6B). Rikenellaceae abundance (Supplemental Fig. S6C) significantly decreased whereas Muribaculaceae (Supplemental Fig. S6D) abundance significantly increased in Mdr2−/− mice compared with WT mice in both Control and ASBT VM treatment (Supplemental Table S4). Furthermore, Prophyromondaceae significantly increased in Mdr2−/− mice after ASBT VM treatment compared with Mdr2−/− Control and WT mice (Supplemental Fig. S6E). Principal coordinate analysis (PCoA), based on Bray–Curtis distance matrix, demonstrated a separation of family taxa abundance between WT and Mdr2−/− mice but was not altered by ASBT VM treatment (Supplemental Fig. S9A).
Relative abundance analysis demonstrated genera differences between WT and Mdr2−/− mice, which were not affected by ASBT VM treatment (Supplemental Fig. S7A). Akkermansia decreased in Mdr2−/− mice compared with WT mice treated with ASBT VM (Supplemental Fig. S7B). Alistipes genus was reduced in Mdr2−/− and WT ASBT VM mice compared with WT Control mice (Supplemental Fig. S7C). Furthermore, both Mdr2−/− Control and ASBT VM-treated mice displayed a significantly increased abundance of Bacteroides compared with corresponding WT groups (Supplemental Fig. S7D). Lactobacillus genus decreased in Mdr2−/− Control and ASBT VM groups compared with WT ASBT VM (Supplemental Fig. S7E). Shannon diversity index and PCoA analysis demonstrated increased diversity within Mdr2−/− samples compared with WT mice; however, ASBT VM treatment did not affect either analysis (Supplemental Figs. S7F and S9B).
Relative abundance was altered in species taxa level between groups of mice (Supplemental Fig. S8A). Alistipes sp. UBA6068 was significantly decreased in Mdr2−/− compared with WT mice regardless of treatment (Supplemental Fig. S8B). Mdr2−/− mice displayed increased Shannon diversity index compared with WT mice, whereas ASBT VM treatment did not affect species taxa abundance in either WT or Mdr2−/− mice (Supplemental Fig. S8C). Species beta diversity displayed separation between WT and Mdr2−/− Control mice that was unchanged after ASBT VM treatment (Supplemental Fig. S9C). Together these data indicate that microbiome composition and diversity are altered in cholestatic mice, which may be due to BA retention and signaling; however, downregulation of hepatic ASBT did not affect the microbiome composition.
To evaluate the potential role of alteration in gut barrier integrity, we performed semiquantification of ZO-1 immunofluorescence and found no change between all groups of mice (Fig. 8A); however, colonic tricellulin mRNA expression significantly increased after ASBT VM treatment in both WT and Mdr2−/− mice (Fig. 8B), whereas cingulin and ZO-1 mRNA expression increased after ASBT VM treatment in WT mice only (Fig. 8, C and D). Furthermore, there were increased CFUs in the liver lysate of Mdr2−/− Control mice compared with WT mice, which was significantly reduced after ASBT VM treatment (Fig. 8E). Together, these data suggest that disruption of biliary ASBT expression may influence gut barrier integrity and allow bacterial translocation to the liver, perpetuating hepatic damage.
Figure 8.

Apical sodium bile acid (BA) transporter (ASBT) Vivo-Morpholino (VM) treatment reduces gut barrier integrity and bacterial translocation. Intestinal tight junction protein zonula occludens-1 (ZO-1) immunofluorescence and semiquantification was unchanged between wild-type (WT) and multidrug-resistant 2 knockout (Mdr2−/−) groups treated with either Control or ASBT VM (A). In the ileum, tight junction tricellulin expression decreased in Mdr2−/− control mice compared with WT but was increased in both WT and Mdr2−/− ASBT VM-treated mice compared with respective controls (B). Similarly, Mdr2−/− Control mice displayed decreased cingulin and ZO-1 expression compared with WT mice, whereas ASBT VM increased cingulin and ZO-1 expression in WT mice but they were unchanged in Mdr2−/− mice (C and D). Hepatic bacterial translocation was increased in hepatic tissue lysate of Mdr2−/− Control compared with WT mice but was reduced in Mdr2−/− mice treated with ASBT VM compared with Control-treated mice (E). Representative images presented at ×40 magnification; white arrows denote ZO-1-positive area. Data are presented as mean ± SD of 3 experiments for colony-forming units (CFU) from 3 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
DISCUSSION
We have identified cross talk between MC/histamine signaling and ASBT-induced BA transport in the liver of cholestatic Mdr2−/− male mice. Our data indicate that elevated ASBT expression, and suspectedly more active cholehepatic shunt, assists in increased histamine release, thus perturbing the BA pool, leading to increased ileal MC infiltration, colonic permeability, increased fecal TBA, and microbiome dysbiosis in Mdr2−/− mice (Fig. 9). MCs express FXR and ASBT (39), and mucosal MCs are activated by BAs (40, 41); therefore, we suspect that increased cholehepatic shunting of BAs within the portal tract can activate MCs and contribute to PSC liver damage. Although MCs and cholangiocytes are unlikely to synthesize BAs, cholangiocytes and MCs express Cyp27a1 (14), which catabolizes cholesterol into oxysterols, a BA precursor, suggesting a cross talk mechanism between MCs and cholangiocytes. Furthermore, inhibition of ASBT expression by VM may not affect microbiome composition but may reduce hepatic bacterial translocation, which has been speculated to initiate liver cirrhosis; however, we were unable to explain unexpected results regarding fecal TBA excretion in Mdr2−/− mice (42). It is unclear whether Mdr2−/− fecal TBA excretion is expected to increase or decrease compared with WT (36, 43); however, it is surprising that ASBT VM reduced fecal TBA excretion in our cholestatic mice. Based on these findings, there is a need to investigate the relationship between ASBT-dependent BA circulation, MC activation, and their collective effects on the gut-liver axis in PSC and PSC-IBD.
Figure 9.

Working model: increased apical sodium bile acid (BA) transporter (ASBT) expression, and suspected increased cholehepatic shunt, assists in increased histamine release perturbing the BA pool, leading to increased ileal mast cell (MC) activation, colonic permeability, increased fecal total bile acids (TBA), and microbiome dysbiosis in multidrug-resistant 2 knockout (Mdr2−/−) mice. FGF, fibroblast growth factor; FXR, farnesoid X receptor; ZO-1, zonula occludens-1. Figure made with Bio-Render.com, with permission.
The function of ileal ASBT is well established in rodent models of cholestasis and nonalcoholic fatty liver disease (NAFLD) (36, 44, 45); however, there is limited understanding of biliary ASBT regarding regulation of BA signaling. The cholehepatic shunt is a widely accepted, but understudied, feature of cholestatic liver injury (46), largely because of the limited capability to visualize biliary transport, in vivo. ASBT expression in mouse models is mostly unknown, although ASBT expression has been confirmed in large cholangiocytes in the rat (47, 48). In our study, we found that, in mice, both small and large cholangiocytes express ASBT; however, ASBT expression was significantly increased in large (but not small) bile ducts of Mdr2−/− mice, which was reduced by ASBT VM treatment. In vitro, we found that ASBT inhibition with VM treatment reduced TCA transport through the cholangiocyte monolayer and reduced TCA-induced biliary inflammation. Based on our in vitro data, biliary ASBT may play an important role in mediating cholangiocyte inflammatory response through increased IL-1β expression following BA insult, which may, in turn, activate MCs within the portal tract.
Enterohepatic ASBT-mediated BA transport regulates the gut-liver axis, an important component regulating overall health. We found that inhibition of ASBT reduced serum histamine levels, and it is well known that MCs are the prime source of histamine (34, 35). These data suggest that ASBT may mediate MC activation, although further analysis is warranted. Furthermore, we found that PSC and PSC-IBD patients have increased hepatic MC infiltration accompanied by increased bile histamine levels, implying that histamine can be secreted into bile before its secretion into the small intestine. In turn, histamine, which is mainly secreted from mucosal MCs in the intestine (49), is a key mediator of gut-related disorders, including IBD, Crohn’s disease, and ulcerative colitis (UC) (50). In patients with Crohn’s disease and UC, both histamine content and secretion are upregulated in the gut mucosal tissue compared with healthy patients (51). These studies provide a potential link with our findings in PSC-IBD patients and indicate that histamine may be a key signaling molecule that may compromise the balance of the gut-liver axis. Further evaluation of intestinal histamine secretion from enterocytes and MC presence in the gut of PSC and PSC-IBD patients is necessary to clarify this signaling mechanism.
Our previous work demonstrated that inhibition of MC FXR signaling reduced MC-induced PSC phenotypes and decreased ASBT expression (4). In the present study we evaluated whether manipulation of ASBT would, in turn, alter FXR. We found that FXR was significantly reduced in ileum of ASBT VM Mdr2−/− mice compared with controls. Although our data present conflicting evidence of FXR activation, it is unclear whether this phenotype is a result of compensation for increased hepatic TBA content in Mdr2−/− mice (52, 53), dampened microbiome-dependent BA deconjugation (54), or increased hepatic activity of Cyp2c70, the enzyme responsible for MCA conversion (55). In contrast, FXR immunoreactivity significantly increased in the ileum of Control Mdr2−/− mice and ASBT VM decreased ileum FXR immunoreactivity. Furthermore, ASBT VM reduced biliary FGF15 secretion, suggesting that although FXR may not be directly altered by ASBT, the downstream effectors are modulated, including FGF15.
Histamine can be synthesized by the gut microbiome and, primarily, MCs but is readily degraded by enzymes including diamine oxidase (DAO) and monoamine oxidase B (56), leading to limited time of bioavailability. PSC patients have increased serum histamine and biliary H1HR and H2HR expression (6), demonstrating a deleterious role in disease progression. Conversely, histamine exerts positive effects in a murine model of IBD after dextran sodium sulfate treatment, where introduction of histamine-producing bacteria (Lactobacillus reuteri) reduced colonic tumor formation compared with control mice (57). Patients with DAO deficiency present with microbiome dysbiosis, specifically increased presence of Proteobacteria, which has been implicated as damaging in progression of metabolic disorders and IBD (58, 59). Furthermore, Control-treated Mdr2−/− mice displayed increased fecal abundance of Bacteroides, increased intestinal MC infiltration, gut barrier integrity, and hepatic bacterial translocation compared with WT mice. The intestinal microbiome regulates ASBT-dependent BA cycling under physiological conditions (60), and mucosal MCs impair the intestinal barrier integrity, leading to inflammation and microbiome dysbiosis in IBD patients (61); however, the relationship between MCs, the microbiome, and ASBT in PSC is not yet defined.
Our data indicate that MC histamine signaling responds to biliary ASBT-dependent BA transport; however, functional ASBT expression in PSC and fatty liver disease remains controversial (36, 44). Ileal ASBT expression is canonically regulated by FXR activation postprandially (62), and its expression is expected to reduce in diseases with elevated BA circulation like PSC; however, ASBT inhibition has been utilized in clinical trials to reduce BA enterohepatic circulation (36, 63). In these studies, ASBT inhibition has been isolated to the ileum, allowing for continued cholehepatic circulation of BAs within the liver. It has been previously shown that TCA or taurolithocholic acid (TLCA) feeding increases cholangiocyte proliferation, in vivo and ex vivo, in an ASBT-dependent manner in rats (64). Furthermore, inhibition of ileal ASBT improves liver fibrosis and NAFLD activity scores in choline-deficient and high-fat diet-fed mice, respectively, compared with control animals (44, 65). The present study demonstrates that inhibition of biliary ASBT reduces hepatic damage and fibrosis deposition typically exhibited in Mdr2−/− mice, adding to current knowledge on hepatic and enterohepatic ASBT in PSC.
Limitations of our study include the artificial exploration of the cholehepatic shunt using isolated cholangiocyte and hepatocyte supernatants and IMCLs on Transwell membranes. Since genetic mouse models include global deletion of Slc10a2 (ASBT) and pharmacological inhibition is limited to the ileum, there are limited options for the inhibition of biliary ASBT. Development of biliary-specific ASBT inhibition or deletion is necessary to fully grasp the importance of the cholehepatic shunt in the initiation and progression of cholestatic damage in murine models of PSC. Furthermore, visualization and analysis of BA transport from the bile duct lumen would help confirm the cholehepatic shunt.
In conclusion, we have demonstrated that MC infiltration and activation in Mdr2−/− mice may be dependent on ABST-mediated BA transport. Human patients exhibit elevated biliary ASBT expression accompanied by increased MC number compared with healthy control subjects. Our study supports the growing body of evidence demonstrating MCs as pivotal targets in PSC progression. MCs infiltrate enterohepatic organs, so their influence likely extends to the gut-liver axis. Although our results are pathological in nature, they provide a strong rationale for targeting the MC/ASBT signaling and the cholehepatic shunt as a therapeutic strategy during PSC.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Tables S1–S6 and Supplemental Figures S1–S9: https://doi.org/10.6084/m9.figshare.21441195.v1.
GRANTS
This work was supported by the Hickam Endowed Chair, Gastroenterology, Medicine, Indiana University, the Indiana University Health–Indiana University School of Medicine Strategic Research Initiative, the United States Department of Veterans Affairs (VA) Senior Career Scientist Award (IK6 BX004601) and the VA Merit award (5I01BX000574) to G.A. and the VA Career Scientist Award (IK6BX005226) and the VA Merit award (1I01BX003031) to H.F., and Career Development Award-2 to L.K. (1IK2BX005306); Biomedical Laboratory Research and Development Service; by National Institutes of Health (NIH) Grants DK108959 and DK119421 (H.F.), DK054811, DK115184, DK076898, DK107310, DK110035, DK062975, and AA028711 (to G.A.); and by the PSC Partners Seeking a Cure (to G.A.). Portions of these studies were supported by resources at the Richard L. Roudebush VA Medical Center, Indianapolis, IN.
DISCLAIMERS
The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
V.M. and H.F. conceived and designed research; V.M., C.M., B.E., D.K., T.Z., K.K., L.P., L. Chen, L.K., L. Ceci, N.W., G.C., W.Z., A.I., A.M., T.O., E.G., P.O., and H.F. performed experiments; V.M., C.M., B.E., D.K., T.Z., K.K., L.P., L. Ceci, L. Chen, N.W., G.C., A.M., T.O., E.G., P.O., G.A., and H.F. analyzed data; V.M., N.W., G.C., T.O., E.G., P.O., G.A., and H.F. interpreted results of experiments; V.M. and H.F. prepared figures; V.M. and H.F. drafted manuscript; V.M., C.M., B.E., D.K., T.Z., K.K., L.P., L. Chen, L.K., A.M., T.O., P.O., G.A., and H.F. edited and revised manuscript; V.M., C.M., B.E., D.K., T.Z., K.K., L.P., L. Chen, L.K., L. Ceci, N.W., G.C., W.Z., A.I., A.M., T.O., E.G., P.O., G.A., and H.F. approved final version of manuscript.
ACKNOWLEDGMENTS
Figures 7 and 9 were created with BioRender.com.
Present addresses: V. Meadows, Dept. of Pharmacology & Toxicology, Rutgers University, Piscataway, NJ; L. Pham, Dept. of Science and Mathematics, Texas A&M University-Central Texas, Killeen, TX.
REFERENCES
- 1. Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary sclerosing cholangitis—a comprehensive review. J Hepatol 67: 1298–1323, 2017. doi: 10.1016/j.jhep.2017.07.022. [DOI] [PubMed] [Google Scholar]
- 2. Mertz A, Nguyen NA, Katsanos KH, Kwok RM. Primary sclerosing cholangitis and inflammatory bowel disease comorbidity: an update of the evidence. Ann Gastroenterol 32: 124–133, 2019. doi: 10.20524/aog.2019.0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kyritsi K, Kennedy L, Meadows V, Hargrove L, Demieville J, Pham L, Sybenga A, Kundu D, Cerritos K, Meng F, Alpini G, Francis H. Mast cells induce ductular reaction mimicking liver injury in mice through mast cell-derived transforming growth factor beta 1 signaling. Hepatology 73: 2397–2410, 2021. doi: 10.1002/hep.31497. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4. Meadows V, Kennedy L, Ekser B, Kyritsi K, Kundu D, Zhou T, Chen L, Pham L, Wu N, Demieville J, Hargrove L, Glaser S, Alpini G, Francis H. Mast cells regulate ductular reaction and intestinal inflammation in cholestasis through farnesoid X receptor signaling. Hepatology 74: 2684–2698, 2021. doi: 10.1002/hep.32028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meadows V, Kennedy L, Hargrove L, Demieville J, Meng F, Virani S, Reinhart E, Kyritsi K, Invernizzi P, Yang Z, Wu N, Liangpunsakul S, Alpini G, Francis H. Downregulation of hepatic stem cell factor by Vivo-Morpholino treatment inhibits mast cell migration and decreases biliary damage/senescence and liver fibrosis in Mdr2-/- mice. Biochim Biophys Acta Mol Basis Dis 1865: 165557, 2019. doi: 10.1016/j.bbadis.2019.165557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kennedy L, Hargrove L, Demieville J, Karstens W, Jones H, DeMorrow S, Meng F, Invernizzi P, Bernuzzi F, Alpini G, Smith S, Akers A, Meadows V, Francis H. Blocking H1/H2 histamine receptors inhibits damage/fibrosis in Mdr2-/- mice and human cholangiocarcinoma tumorigenesis. Hepatology 68: 1042–1056, 2018. [Erratum in Hepatology 76: 1898, 2022] doi: 10.1002/hep.29898. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7. Kennedy L, Meadows V, Demieville J, Hargrove L, Virani S, Glaser S, Zhou T, Rinehart E, Jaeger V, Kyritsi K, Pham L, Alpini G, Francis H. Biliary damage and liver fibrosis are ameliorated in a novel mouse model lacking l-histidine decarboxylase/histamine signaling. Lab Invest 100: 837–848, 2020. doi: 10.1038/s41374-020-0405-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chi Z, Xu J, Saxena R. increased mast cell counts and degranulation in microscopic colitis. Gastroenterol Res Pract 2020: 9089027, 2020. doi: 10.1155/2020/9089027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crowe SE, Luthra GK, Perdue MH. Mast cell mediated ion transport in intestine from patients with and without inflammatory bowel disease. Gut 41: 785–792, 1997. doi: 10.1136/gut.41.6.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tu H, Okamoto AY, Shan B. FXR, a bile acid receptor and biological sensor. Trends Cardiovasc Med 10: 30–35, 2000. doi: 10.1016/s1050-1738(00)00043-8. [DOI] [PubMed] [Google Scholar]
- 11. Fiorucci S, Distrutti E. The pharmacology of bile acids and their receptors. Handb Exp Pharmacol 256: 3–18, 2019. doi: 10.1007/164_2019_238. [DOI] [PubMed] [Google Scholar]
- 12. Han CY. Update on FXR biology: promising therapeutic target? Int J Mol Sci 19: 2069, 2018. doi: 10.3390/ijms19072069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ji S, Liu Q, Zhang S, Chen Q, Wang C, Zhang W, Xiao C, Li Y, Nian C, Li J, Li J, Geng J, Hong L, Xie C, He Y, Chen X, Li X, Yin Z-Y, You H, Lin K-H, Wu Q, Yu C, Johnson RL, Wang L, Chen L, Wang F, Zhou D. FGF15 activates Hippo signaling to suppress bile acid metabolism and liver tumorigenesis. Dev Cell 48: 460–474.e9, 2019. doi: 10.1016/j.devcel.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 14. Jung D, York JP, Wang L, Yang C, Zhang A, Francis HL, Webb P, McKeehan WL, Alpini G, Lesage GD, Moore DD, Xia X. FXR-induced secretion of FGF15/19 inhibits CYP27 expression in cholangiocytes through p38 kinase pathway. Pflugers Arch 466: 1011–1019, 2014. doi: 10.1007/s00424-013-1364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tabibian JH, Masyuk AI, Masyuk TV, O’Hara SP, LaRusso NF. Physiology of cholangiocytes. Compr Physiol 3: 541–565, 2013. doi: 10.1002/cphy.c120019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dawson PA. Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb Exp Pharmacol 201: 169–203, 2011. doi: 10.1007/978-3-642-14541-4_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)-/- mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol 43: 1045–1054, 2005. doi: 10.1016/j.jhep.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 18. Glaser S, Lam IP, Franchitto A, Gaudio E, Onori P, Chow BK, Wise C, Kopriva S, Venter J, White M, Ueno Y, Dostal D, Carpino G, Mancinelli R, Butler W, Chiasson V, DeMorrow S, Francis H, Alpini G. Knockout of secretin receptor reduces large cholangiocyte hyperplasia in mice with extrahepatic cholestasis induced by bile duct ligation. Hepatology 52: 204–214, 2010. doi: 10.1002/hep.23657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Glaser SS, Gaudio E, Rao A, Pierce LM, Onori P, Franchitto A, Francis HL, Dostal DE, Venter JK, DeMorrow S, Mancinelli R, Carpino G, Alvaro D, Kopriva SE, Savage JM, Alpini GD. Morphological and functional heterogeneity of the mouse intrahepatic biliary epithelium. Lab Invest 89: 456–469, 2009. doi: 10.1038/labinvest.2009.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shneider BL, Dawson PA, Christie DM, Hardikar W, Wong MH, Suchy FJ. Cloning and molecular characterization of the ontogeny of a rat ileal sodium-dependent bile acid transporter. J Clin Invest 95: 745–754, 1995. doi: 10.1172/JCI117722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pastuszak M, Groszewski K, Pastuszak M, Dyrla P, Wojtuń S, Gil J. Cytokeratins in gastroenterology. Prz Gastroenterol 10: 61–70, 2015. doi: 10.5114/pg.2015.51182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alpini G, Roberts S, Kuntz SM, Ueno Y, Gubba S, Podila PV, LeSage G, LaRusso NF. Morphological, molecular, and functional heterogeneity of cholangiocytes from normal rat liver. Gastroenterology 110: 1636–1643, 1996. doi: 10.1053/gast.1996.v110.pm8613073. [DOI] [PubMed] [Google Scholar]
- 23. de Jong IE, Overi D, Carpino G, Gouw AS, van den Heuvel MC, van Kempen LC, Mancone C, Onori P, Cardinale V, Casadei L, Alvaro D, Porte RJ, Gaudio E. Persistent biliary hypoxia and lack of regeneration are key mechanisms in the pathogenesis of posttransplant nonanastomotic strictures. Hepatology 75: 814–830, 2022. doi: 10.1002/hep.32166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Francis HL, Demorrow S, Franchitto A, Venter JK, Mancinelli RA, White MA, Meng F, Ueno Y, Carpino G, Renzi A, Baker KK, Shine HE, Francis TC, Gaudio E, Alpini GD, Onori P. Histamine stimulates the proliferation of small and large cholangiocytes by activation of both IP3/Ca2+ and cAMP-dependent signaling mechanisms. Lab Invest 92: 282–294, 2012. doi: 10.1038/labinvest.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kennedy L, Meadows V, Kyritsi K, Pham L, Kundu D, Kulkarni R, Cerritos K, Demieville J, Hargrove L, Glaser S, Zhou T, Jaeger V, Alpini G, Francis H. Amelioration of large bile duct damage by histamine-2 receptor Vivo-Morpholino treatment. Am J Pathol 190: 1018–1029, 2020. [Erratum in Am J Pathol 190: 1580, 2020]. doi: 10.1016/j.ajpath.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Payne V, Kam PC. Mast cell tryptase: a review of its physiology and clinical significance. Anaesthesia 59: 695–703, 2004. doi: 10.1111/j.1365-2044.2004.03757.x. [DOI] [PubMed] [Google Scholar]
- 27. Rychter J, Ortega O, Berdun S, Arenas C, Lopez I, Espin F, Vergara P, Clavé P. Mast cell degranulation inhibits motor patterns of human ileum and sigmoid colon in vitro: relevance for postoperative ileus. Neurogastroenterol Motil 27: 1098–1109, 2015. doi: 10.1111/nmo.12589. [DOI] [PubMed] [Google Scholar]
- 28. Jones H, Hargrove L, Kennedy L, Meng F, Graf-Eaton A, Owens J, Alpini G, Johnson C, Bernuzzi F, Demieville J, DeMorrow S, Invernizzi P, Francis H. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2-/- mice. Hepatology 64: 1202–1216, 2016. doi: 10.1002/hep.28704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Puri P, Daita K, Joyce A, Mirshahi F, Santhekadur PK, Cazanave S, Luketic VA, Siddiqui MS, Boyett S, Min H-K, Kumar DP, Kohli R, Zhou H, Hylemon PB, Contos MJ, Idowu M, Sanyal AJ. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 67: 534–548, 2018. doi: 10.1002/hep.29359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Phumesin P, Junking M, Panya A, Yongpitakwattana P, Noisakran S, Limjindaporn T, Yenchitsomanus PT. Inhibition of dengue virus replication in monocyte-derived dendritic cells by vivo-morpholino oligomers. Virus Res 260: 123–128, 2019. doi: 10.1016/j.virusres.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 31. Cai SY, Ouyang X, Chen Y, Soroka CJ, Wang J, Mennone A, Wang Y, Mehal WZ, Jain D, Boyer JL. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2: e90780, 2017. doi: 10.1172/jci.insight.90780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sabino J, Vieira-Silva S, Machiels K, Joossens M, Falony G, Ballet V, Ferrante M, Van Assche G, Van der Merwe S, Vermeire S, Raes J. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 65: 1681–1689, 2016. doi: 10.1136/gutjnl-2015-311004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hillmann B, Al-Ghalith GA, Shields-Cutler RR, Zhu Q, Gohl DM, Beckman KB, Knight R, Knights D. Evaluating the information content of shallow shotgun metagenomics. mSystems 3: e00069-18, 2018. doi: 10.1128/mSystems.00069-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hill SJ. Multiple histamine receptors: properties and functional characteristics. Biochem Soc Trans 20: 122–125, 1992. doi: 10.1042/bst0200122. [DOI] [PubMed] [Google Scholar]
- 35. Thangam EB, Jemima EA, Singh H, Baig MS, Khan M, Mathias CB, Church MK, Saluja R. The role of histamine and histamine receptors in mast cell-mediated allergy and inflammation: the hunt for new therapeutic targets. Front Immunol 9: 1873, 2018. doi: 10.3389/fimmu.2018.01873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baghdasaryan A, Fuchs CD, Österreicher CH, Lemberger UJ, Halilbasic E, Påhlman I, Graffner H, Krones E, Fickert P, Wahlström A, Ståhlman M, Paumgartner G, Marschall HU, Trauner M. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J Hepatol 64: 674–681, 2016. doi: 10.1016/j.jhep.2015.10.024. [DOI] [PubMed] [Google Scholar]
- 37. Cai SY, Mennone A, Soroka CJ, Boyer JL. Altered expression and function of canalicular transporters during early development of cholestatic liver injury in Abcb4-deficient mice. Am J Physiol Gastrointest Liver Physiol 306: G670–G676, 2014. doi: 10.1152/ajpgi.00334.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sinha J, Chen F, Miloh T, Burns RC, Yu Z, Shneider BL. beta-Klotho and FGF-15/19 inhibit the apical sodium-dependent bile acid transporter in enterocytes and cholangiocytes. Am J Physiol Gastrointest Liver Physiol 295: G996–G1003, 2008. doi: 10.1152/ajpgi.90343.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meng F, Kennedy L, Hargrove L, Demieville J, Jones H, Madeka T, Karstens A, Chappell K, Alpini G, Sybenga A, Invernizzi P, Bernuzzi F, DeMorrow S, Francis H. Ursodeoxycholate inhibits mast cell activation and reverses biliary injury and fibrosis in Mdr2-/- mice and human primary sclerosing cholangitis. Lab Invest 98: 1465–1477, 2018. doi: 10.1038/s41374-018-0101-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gelbmann CM, Schteingart CD, Thompson SM, Hofmann AF, Barrett KE. Mast cells and histamine contribute to bile acid-stimulated secretion in the mouse colon. J Clin Invest 95: 2831–2839, 1995. doi: 10.1172/JCI117988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Quist RG, Ton-Nu HT, Lillienau J, Hofmann AF, Barrett KE. Activation of mast cells by bile acids. Gastroenterology 101: 446–456, 1991. doi: 10.1016/0016-5085(91)90024-F. [DOI] [PubMed] [Google Scholar]
- 42. Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol 60: 197–209, 2014. doi: 10.1016/j.jhep.2013.07.044. [DOI] [PubMed] [Google Scholar]
- 43. Fuchs CD, Paumgartner G, Mlitz V, Kunczer V, Halilbasic E, Leditznig N, Wahlström A, Ståhlman M, Thüringer A, Kashofer K, Stojakovic T, Marschall HU, Trauner M. Colesevelam attenuates cholestatic liver and bile duct injury in Mdr2-/- mice by modulating composition, signalling and excretion of faecal bile acids. Gut 67: 1683–1691, 2018. doi: 10.1136/gutjnl-2017-314553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rao A, Kosters A, Mells JE, Zhang W, Setchell KD, Amanso AM, Wynn GM, Xu T, Keller BT, Yin H, Banton S, Jones DP, Wu H, Dawson PA, Karpen SJ. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci Transl Med 8: 357ra122, 2016. doi: 10.1126/scitranslmed.aaf4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. van de Peppel IP, Rao A, Dommerholt MB, Bongiovanni L, Thomas R, de Bruin A, Karpen SJ, Dawson PA, Verkade HJ, Jonker JW. The beneficial effects of apical sodium-dependent bile acid transporter inactivation depend on dietary fat composition. Mol Nutr Food Res 64: e2000750, 2020. doi: 10.1002/mnfr.202000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hofmann AF. Current concepts of biliary secretion. Dig Dis Sci 34: 16S–20S, 1989. doi: 10.1007/BF01536657. [DOI] [PubMed] [Google Scholar]
- 47. Alpini G, Glaser SS, Rodgers R, Phinizy JL, Robertson WE, Lasater J, Caligiuri A, Tretjak Z, LeSage GD. Functional expression of the apical Na+-dependent bile acid transporter in large but not small rat cholangiocytes. Gastroenterology 113: 1734–40, 1997. doi: 10.1053/gast.1997.v113.pm9352879. [DOI] [PubMed] [Google Scholar]
- 48. Luo ZL, Cheng L, Wang T, Tang LJ, Tian FZ, Xiang K, Cui L. Bile acid transporters are expressed and heterogeneously distributed in rat bile ducts. Gut Liver 13: 569–575, 2019. doi: 10.5009/gnl18265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Z, Kurashima Y. Two sides of the coin: mast cells as a key regulator of allergy and acute/chronic inflammation. Cells 10: 1615, 2021. doi: 10.3390/cells10071615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schnedl WJ, Enko D. Histamine intolerance originates in the gut. Nutrients 13: 1262, 2021. doi: 10.3390/nu13041262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Raithel M, Matek M, Baenkler HW, Jorde W, Hahn EG. Mucosal histamine content and histamine secretion in Crohn’s disease, ulcerative colitis and allergic enteropathy. Int Arch Allergy Immunol 108: 127–133, 1995. doi: 10.1159/000237129. [DOI] [PubMed] [Google Scholar]
- 52. Wu G, Wen M, Sun L, Li H, Liu Y, Li R, Wu F, Yang R, Lin Y. Mechanistic insights into geniposide regulation of bile salt export pump (BSEP) expression. RSC Adv 8: 37117–37128, 2018. doi: 10.1039/c8ra06345a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu T, Zhang Q, Li J, Chen H, Wu J, Song H. Up-regulation of BSEP and MRP2 by Calculus Bovis administration in 17alpha-ethynylestradiol-induced cholestasis: Involvement of PI3K/Akt signaling pathway. J Ethnopharmacol 190: 22–32, 2016. doi: 10.1016/j.jep.2016.05.056. [DOI] [PubMed] [Google Scholar]
- 54. Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17: 225–35, 2013. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 55. de Boer JF, Verkade E, Mulder NL, de Vries HD, Huijkman N, Koehorst M, Boer T, Wolters JC, Bloks VW, van de Sluis B, Kuipers F. A human-like bile acid pool induced by deletion of hepatic Cyp2c70 modulates effects of FXR activation in mice. J Lipid Res 61: 291–305, 2020. doi: 10.1194/jlr.RA119000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Elieh Ali Komi D, Wöhrl S, Bielory L. Mast cell biology at molecular level: a comprehensive review. Clin Rev Allergy Immunol 58: 342–365, 2020. doi: 10.1007/s12016-019-08769-2. [DOI] [PubMed] [Google Scholar]
- 57. Shi Z, Fultz RS, Engevik MA, Gao C, Hall A, Major A, Mori-Akiyama Y, Versalovic J. Distinct roles of histamine H1- and H2-receptor signaling pathways in inflammation-associated colonic tumorigenesis. Am J Physiol Gastrointest Liver Physiol 316: G205–G216, 2019. doi: 10.1152/ajpgi.00212.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rizzatti G, Lopetuso LR, Gibiino G, Binda C, Gasbarrini A. Proteobacteria: a common factor in human diseases. Biomed Res Int 2017: 9351507, 2017. doi: 10.1155/2017/9351507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schink M, Konturek PC, Tietz E, Dieterich W, Pinzer TC, Wirtz S, Neurath MF, Zopf Y. Microbial patterns in patients with histamine intolerance. J Physiol Pharmacol 69: 579–593, 2018. doi: 10.26402/jpp.2018.4.09. [DOI] [PubMed] [Google Scholar]
- 60. Out C, Patankar JV, Doktorova M, Boesjes M, Bos T, de Boer S, Havinga R, Wolters H, Boverhof R, van Dijk TH, Smoczek A, Bleich A, Sachdev V, Kratky D, Kuipers F, Verkade HJ, Groen AK. Gut microbiota inhibit Asbt-dependent intestinal bile acid reabsorption via Gata4. J Hepatol 63: 697–704, 2015. doi: 10.1016/j.jhep.2015.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. De Zuani M, Dal Secco C, Frossi B. Mast cells at the crossroads of microbiota and IBD. Eur J Immunol 48: 1929–1937, 2018. doi: 10.1002/eji.201847504. [DOI] [PubMed] [Google Scholar]
- 62. Neimark E, Chen F, Li X, Shneider BL. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology 40: 149–56, 2004. doi: 10.1002/hep.20295. [DOI] [PubMed] [Google Scholar]
- 63. Al-Dury S, Wahlström A, Wahlin S, Langedijk J, Elferink RO, Ståhlman M, Marschall HU. Pilot study with IBAT inhibitor A4250 for the treatment of cholestatic pruritus in primary biliary cholangitis. Sci Rep 8: 6658, 2018. doi: 10.1038/s41598-018-25214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alpini G, Ueno Y, Glaser SS, Marzioni M, Phinizy JL, Francis H, Lesage G. Bile acid feeding increased proliferative activity and apical bile acid transporter expression in both small and large rat cholangiocytes. Hepatology 34: 868–76, 2001. doi: 10.1053/jhep.2001.28884. [DOI] [PubMed] [Google Scholar]
- 65. Rao A, van de Peppel IP, Gumber S, Karpen SJ, Dawson PA. Attenuation of the hepatoprotective effects of ileal apical sodium dependent bile acid transporter (ASBT) inhibition in choline-deficient L-amino acid-defined (CDAA) diet-fed mice. Front Med (Lausanne) 7: 60, 2020. doi: 10.3389/fmed.2020.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables S1–S6 and Supplemental Figures S1–S9: https://doi.org/10.6084/m9.figshare.21441195.v1.
Data Availability Statement
Data will be made available upon reasonable request.