

Keywords: kidney fibrosis, Klotho, peritubular capillary rarefaction, phosphotoxicity, vascular endothelial growth factor receptor 2
Abstract
Vascular endothelial growth factor (VEGF) and its cognate receptor (VEGFR2) system are crucial for cell functions associated with angiogenesis and vasculogenesis. Klotho contributes to vascular health maintenance in the kidney and other organs in mammals, but it is unknown whether renoprotection by Klotho is dependent on VEGF/VEGFR2 signaling. We used heterozygous VEGFR2-haploinsufficient (VEGFR2+/−) mice resulting from heterozygous knockin of green fluorescent protein in the locus of fetal liver kinase 1 encoding VEGFR2 to test the interplay of Klotho, phosphate, and VEGFR2 in kidney function, the vasculature, and fibrosis. VEGFR2+/− mice displayed downregulated VEGF/VEGFR2 signaling in the kidney, lower density of peritubular capillaries, and accelerated kidney fibrosis, all of which were also found in the homozygous Klotho hypomorphic mice. High dietary phosphate induced higher plasma phosphate, greater peritubular capillary rarefaction, and more kidney fibrosis in VEGFR2+/− mice compared with wild-type mice. Genetic overexpression of Klotho significantly attenuated the elevated plasma phosphate, kidney dysfunction, peritubular capillary rarefaction, and kidney fibrosis induced by a high-phosphate diet in wild-type mice but only modestly ameliorated these changes in the VEGFR2+/− background. In cultured endothelial cells, VEGFR2 inhibition reduced free VEGFR2 but enhanced its costaining of an endothelial marker (CD31) and exacerbated phosphotoxicity. Klotho protein maintained VEGFR2 expression and attenuated high phosphate-induced cell injury, which was reduced by VEGFR2 inhibition. In conclusion, normal VEGFR2 function is required for vascular integrity and for Klotho to exert vascular protective and antifibrotic actions in the kidney partially through the regulation of VEGFR2 function.
NEW & NOTEWORTHY This research paper studied the interplay of vascular endothelial growth factor receptor type 2 (VEGFR2), high dietary phosphate, and Klotho, an antiaging protein, in peritubular structure and kidney fibrosis. Klotho protein was shown to maintain VEGFR2 expression in the kidney and reduce high phosphate-induced cell injury. However, Klotho cytoprotection was attenuated by VEGFR2 inhibition. Thus, normal VEGFR2 function is required for vascular integrity and Klotho to exert vascular protective and antifibrotic actions in the kidney.
INTRODUCTION
Normal function and structure of the vasculature are highly dependent on appropriate intracellular and intercellular signaling by vascular endothelial growth factors (VEGFs) and their cognate receptors (VEGFRs) (1, 2). Normal cross talk between renal tubule-derived VEGF and endothelium-located VEGFR2 is crucial for the maintenance of peritubular capillaries in the kidney (3). The VEGF superfamily has three subtypes, among which VEGFA plays a crucial role in angiogenesis and vasculogenesis (1, 2). There are three VEGFRs that are activated upon binding to different VEGF isoforms that transduce intracellular signaling through VEGFR phosphorylation and internalization. VEGFR2, also called a kinase insert domain receptor (KDR) (4) or fetal liver kinase 1 (flk1) (5), plays a critical role in the maintenance of normal vasculature in the kidney (2, 6). Aberrant VEGFR2 is associated with kidney damage during chemotherapy with VEGFR inhibitors (7). Although the role of VEGFR2 in glomerular capillaries is well demonstrated (8, 9), its role in the maintenance of peritubular vasculature and the development of kidney fibrosis remains unknown.
Klotho, a type I single-pass transmembrane glycoprotein, was serendipitously discovered by Kuro-o and colleagues. Klotho is best described as a universal guardian of cell health (10). Klotho deficiency causes premature degeneration and increases susceptibility to disease in multiple organs including the kidney (11). Membrane Klotho is predominantly expressed in the kidney and brain (10). The extracellular domain of membrane Klotho can be shed from Klotho-expressing cells by secretases and released into the circulation as soluble Klotho (12). Membrane Klotho functions as a coreceptor for fibroblast growth factor (FGF)-23 (13), which plays multiple roles including the regulation of phosphate and other mineral metabolism (14–17). Soluble Klotho can function in both FGF23-dependent and FGF23-independnet modes (18) and more diverse biological actions including antioxidation (19), antisenescence (11), regulation of VEGFR2 signaling (20), and maintenance of vascular health (21). Extensive animal data support the protective role of Klotho in the vasculature, despite the fact that it is undetermined whether Klotho is expressed in vascular smooth muscle and endothelial cells (22, 23). Klotho-deficient mice have severe vasculopathy including endothelial dysfunction (24), vessel stiffness (25, 26), impaired angiogenesis and vasculogenesis (27), and massive vascular calcification (10, 21, 28). Kusaba et al. (20) showed that Klotho maintains endothelial integrity through modulation of VEGFR2 trafficking. However, it is unclear whether the vascular protection conferred by Klotho is mediated by the modulation of VEGFR2 in the kidney. Furthermore, overwhelming data support that Klotho reduces serum phosphate, which consequently decreases phosphotoxicity as a means of vascular protection (29).
Although phosphate is essential for cell structure and function, excessive accumulation of phosphate in the body causes cell dysfunction and injury and results in functional and morphological changes in many organs/tissues including the vasculature, collectively termed phosphotoxicity (11, 29–32). High serum phosphate is independently associated with cardiovascular morbidity and mortality in patients with chronic kidney disease (CKD) and also in subjects free of kidney disease (33, 34). We showed that long-term dietary phosphate loading contributes to kidney fibrosis in normal animals and promotes the progression of CKD (11, 35, 36). Phosphotoxicity is attributable at least in part to phosphate-induced cell apoptosis, decreased autophagy, and activated cellular senescence (11, 29). Phosphate is proposed to be a vascular toxin resulting in vasculopathy (32), but the molecular mechanism of phosphate-induced vasculopathy is not fully known.
To study the interplay between high phosphate, VEGFR2 activity, and Klotho in kidney fibrosis and vasculopathy, we used VEGFR2-haploinsufficient mice (VEGFR2+/−) generated by heterozygous green fluorescent protein (GFP) knockin in the flk1 locus (37) as an in vivo model to examine the effect of VEGFR2 deficiency on kidney function and histology. We used heterozygous Klotho hypomorphic mice (kl/+) (10) and transgenic mice with Klotho overexpression (Tg-kl) (38) to examine if Klotho under- and overexpression modifies kidney phenotypes in VEGFR2+/− mice after high dietary phosphate loading. We found that reduced VEGFR2 signaling activity induced abnormalities in kidney structure and function at baseline, suggesting that aberrant VEGFR2 signaling is a novel trigger for kidney fibrosis. Furthermore, we found that kidney fibrosis in VEGFR2+/− mice was exacerbated in the kl/+ background and partially attenuated in the Tg-kl background. Klotho is required to maintain normal VEGFA/VEGFR2 signaling in the kidney. The cytoprotective action of Klotho is partially dependent on intact VEGFR2 signaling, and the VEGFA/VEFR2-independent effects of Klotho are not sufficient to fully rescue kidney phenotypes in VEGFR2-deficient mice.
MATERIALS AND METHODS
Animal Lines and Animal Experiments
All animal experiments were conducted strictly following the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center. Animals were given ad libitum access to tap water and standard rodent chow containing 0.7% phosphate (wt/wt) (Teklad 2016, Harlan, Madison, WI) unless stated otherwise. High-phosphate rodent chow [2.0% (wt/wt), Harlan Teklad 08020, Harlan] fed to experimental animals for 12 wk unless otherwise specified. Ten mice per group (equal numbers of male and female mice) were used at 10–12 wk old unless otherwise specified.
WT 129 S1/SVlmJ (129 SV) mice, heterozygous knock-in-GFP-flk1, also called GFP-VEGFR2+/− and simply called VEGFR2+/− hereafter) (JAX Stock No. 017006) (Supplemental Table S1) (37), transgenic Tek-Cre (Tie2-Cre) (JAX Stock No. 008863) (39), and Rosa26 tdTomato (tdTomato) mouse lines (JAX Stock No. 007909) (40) were purchased from Jackson Laboratory (Bar Harbor, ME). Heterozygous Klotho hypomorphic (kl/+) mice (Supplemental Table S1) (10) and transgenic mice with overexpression of Klotho (Tg-kl) (Supplemental Table S1) (10, 38) were originally developed by Dr. Makoto Kuro-o and have been bred in our laboratory for years. tdTomato reporter mice were cross-mated with Tie2-Cre mice (Supplemental Table S1) and Tg-kl mice to generate one new mouse line harboring triple genes: tdTomato;Tie2-Cre;Tg-kl. All the genetically manipulated mouse lines have been crossbred into the WT 129 SV strain for more than 10 generations. The genotyping of these mouse lines was conducted with established methods (10, 37–40). To examine the effect of Klotho on the vasculature in the kidney of kl/kl mice, recombinant Klotho protein (0.3 mg/kg body wt/mo) was intraperitoneally administered using osmotic minipumps with normal saline as a vehicle for the control (No. 1004, Alzet, Durect, Cupertino, CA) starting at 4 wk old (29). Because homozygous GFP-VEGFR2 knockin mice were suffering embryonic lethality and homozygous Klotho hypomorphic (kl/kl) mice are too fragile to tolerate high phosphate challenge, VEGFR2+/- mice and kl/+ mice were used in this study.
Blood, Urine, and Kidney Collection
Twenty-four-hour urine was collected in individual metabolic cages. In terminal experiments, mice were anesthetized with isoflurane, blood was collected in heparinized tubes and centrifuged (3,000 g for 5 min at 4°C), and the plasma was separated and stored at −80°C until analysis. The kidneys were isolated, and one slice was fixed with 4% paraformaldehyde and embedded in Tissue Tek or paraffin for histological and immunohistologic experiments; the remaining parts were snap-frozen in liquid N2 and stored −80°C until analysis.
Plasma chemistries of animals were analyzed using a Vitros Chemistry Analyzer (Ortho-Clinical Diagnosis, Rochester, NY). Plasma and urine creatinine was measured using a P/ACE MDQ Capillary Electrophoresis System and photodiode detector (Beckman-Coulter, Fullerton, CA) (41).
Antibodies
Primary antibodies used for immunoblot analysis included mouse monoclonal β-actin antibody and mouse monoclonal α-smooth muscle actin (α-SMA) antibody (No. A5228, Sigma-Aldrich, St. Louis, MO); rabbit polyclonal Akt antibody (No. 9272), rabbit monoclonal phosphorylated (p-)Akt (Ser473) antibody (No. 4060), rabbit monoclonal p44/42 MAPK (Erk1/2) antibody (No. 4695), and mouse monoclonal p-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody (No. 92016) (Cell Signaling Technology, Danvers, MA); mouse monoclonal platelet-endothelial cell adhesion molecule (Pecam)-1/CD31 antibody (No. ab24590) and mouse polyclonal connective tissue growth factor (CTGF) antibody (No. ab51704) (Abcam, Cambridge, MA); and rabbit polyclonal VEGFA antibody (No. sc-152, Santa Cruz Biotechnology, Dallas, TX), respectively.
Primary antibodies used for immunohistochemistry were as follows: rat monoclonal Pecam-1/CD31 antibody (No. ab2550274, BD Biosciences, San Jose, CA); rat monoclonal endomucin antibody (No. sc-53941) and goat polyclonal VEGFA antibody (No. sc-152-G) (Santa Cruz Biotechnology); rat monoclonal Klotho antibody (No. KO603) (Trans Genics, Kobe, Japan); mouse monoclonal α-SMA antibody (No. MAB1420, R&D Systems, Minneapolis, MN); and rabbit monoclonal VEGFR2 (55B11) antibody (No. 2479, Cell Signaling Technology).
Secondary antibodies coupled to horseradish peroxidase for immunoblot analysis or to FITC, Alexa Fluor (647, 568, 488, and 405), or Cy5 as well as Syto 61 red fluorescent nuclear acid stain (Molecular Probes/Invitrogen Molecular Probes, Eugene, OR) were used for immunohistochemistry. Other reagents for immunohistochemistry included fluorescein-coupled Lotus tetragonolobus lectin for labeling proximal tubules and DAPI for labeling nuclei (Vector Laboratories, Burlingame, CA) and rhodamine phalloidin (R415) for labeling F-actin (Life Technologies, Carlsbad, CA).
Kidney Histology
Kidney tissues were fixed in 4% paraformaldehyde, and 4-µm sections of paraffin-embedded kidney tissues were stained with trichrome and examined and photographed by a researcher blinded to the experimental protocol. Fibrotic area and fibrosis intensity were quantified with ImageJ software (National Institutes of Health, Bethesda, MD) with previously published methods (41, 42).
Immunohistochemistry and Immunoblot Analysis
Four-micrometer sections of paraffin-embedded kidneys were subjected to immunohistochemistry using standard protocols (41, 42). We used a cocktail containing equal amounts of primary antibodies against Pecam-1/CD31 and endomucin (PE) to better image the vasculature in the kidney using a previously described protocol (43). To quantify vessel density in the kidney, PE staining or tdTomato signal density and area on kidney sections were obtained with ImageJ software similar to the quantification of fibrosis intensity in the kidney (41, 42).
Total kidney lysate covering all kidney zones was prepared as previously described (41, 42). Fifty micrograms protein of kidney lysate were solubilized in Laemmli sample buffer, fractionated by SDS-PAGE, transferred to PVDF membranes, and immunoblotted with different primary antibodies and β-actin as a loading control. The signal was visualized using an ECL kit (Perkin-Elmer LAS, Boston, MA) or with ChemiDoc Imaging Systems (Bio-Rad Laboratories, Hercules, CA).
Transmission Electron Microscopy
Kidney slices were fixed overnight with 2.5% glutaraldehyde and 2% paraformaldehyde in cacodylate buffer (0.1 M, pH 7.4), and samples were prepared as previously described (35). Ultrathin sections were cut on an ultracryomicrotome (Ultramicrotome Reichert Ultracut E, Leica Microsystems, Wetzlar, Germany) and visualized with a Jeol 1200 EX transmission electron microscope (Jeol, Akishima, Japan).
Quantitative PCR
Total RNA was extracted using the RNAeasy kit (Qiagen, Germantown, MD) from mouse kidneys. cDNA was generated with oligo-dT primers using the SuperScript III First Strand Synthesis System (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Primers used for quantitative PCR are provided in Supplemental Table S2 with conditions as previously described (41, 42). The reaction was performed in triplicate for each sample. Data are expressed at an amplification number of 2-ΔΔCt (where Ct is threshold cycle) by normalization of cyclophilin and comparison of controls.
Cell Culture
The mouse endothelial cell line (MS1, Stock No. CRL-2460) originally derived from the pancreas was purchased from the American Type Culture Collection and cultured at 37°C in a 95% air and 5% CO2 atmosphere. MS1 has been used for pancreatic vascular (44) and even commonly for nonpancreatic vascular studies (45) and successfully for the study of endothelial injury, regeneration, and endothelial to mesenchymal transition in acute kidney injury (46, 47). Cells were passed in high-glucose (450 mg/dL) DMEM supplemented with FBS (10%), sodium pyruvate (100 mM), penicillin (100 U/mL), and streptomycin (100 mg/mL) following the manufacturer’s protocol.
To examine for cellular phosphotoxicity, 1.0 or 2.0 mM phosphate culture media were added to MS1 cells for 24 or 48 h. To examine the effect of VEGFR2 inhibition on phosphotoxicity, Ki8751 (48) (Selleck Chemicals, Houston, TX) was added into culture media containing 1.0 or 2.0 mM phosphate to reach a final concentration of 10 or 20 nM. To assess Klotho cytoprotection, recombinant Klotho protein was prepared and purified from human embryonic kidney-293 cells as previously described (12) and added onto MS1 cells with different phosphate concentrations in culture media with or without Ki8751.
For the lactate dehydrogenase (LDH) assay, culture media were collected and immediately centrifuged at 4°C (1,400 rpm for 5 min) to remove cells and cellular debris. The supernatants were used for measurement of LDH release with the LDH cytotoxicity detection kit purchased from Clontech Laboratories (Mountain View, CA), according to the manufacturer’s instructions (49). For the immunoblot analysis and immunocytochemistry, MS1 cells were seeded on six-well plates or glass-cover slides, respectively.
Sex as a Biological Factor Determining Kidney Outcomes
Sex is a biological determinant of the severity of kidney damage and outcome based on our experiences and previously published data (50, 51). To present unbiased results, quantitative data are expressed as individual plots with open circles indicating male mice and pink circles indicating female mice. However, representative figures or immunoblots are only from male kidney tissues.
Statistical Analyses
Quantitative data are expressed as means ± SD. Individual values were plotted. Analysis was performed with SigmaPlot software (Systat Software, San Jose, CA). As appropriate, statistical analysis was performed using an unpaired Student’s t test or one-way or two-way ANOVA followed by a Student–Newman–Keuls post hoc test when applicable. Values of P ≤ 0.05 were considered statistically significant.
RESULTS
Homozygous Disruption of Flk1 Loci Leads to Embryonic Lethality
Given that homozygous global knockout of VEGFR2 (flk1−/−) is embryonic lethal at embryonic day 8.5 due to lack of vasculogenesis/angiogenesis (52), we examined whether knockin of GFP into the flk1 locus interferes with VEGFR2 protein function. We genotyped neonatal mice after weaning and found no homozygous mutant offspring from intercross of heterozygous VEGFR2+/− mice in more than 100 pups genotyped at weaning (Fig. 1, A and B), indicating that homozygous knockin of GFP into flk1 loci phenocopies global conventional knockout of VEGFR2−/− mice and death in utero. This finding confirms that normal VEGFR2 signaling is required for normal embryonic vasculogenesis and blood island formation (37, 43, 52). Because the study of embryonic vasculogenesis is beyond this research project, we did not carry out timed pregnant experiments in VEGFR2+/− female mice and were not able to pinpoint the exact time of embryonic lethality in our experimental animal.
Figure 1.

Knockin of green fluorescent protein (GFP) into one allele of fetal liver kinase 1 (Flk1), a gene encoding vascular endothelial growth factor (VEGF) receptor type 2 (VEGFR2), caused VEGFR2 haploinsufficiency and peritubular capillary rarefaction in the kidney. A: schema for heterozygous knockin of GFP into the VEGFR2 locus. B: homozygous knockin of GFP into the VEGFR2 locus led to embryonic lethality. PCR genotyping was carried out at weaning. The kidneys were harvested from wild-type (WT) mice (without knockin of GFP into the flk1 locus, i.e., VEGFR2+/+) and mice with heterozygous knockin of GFP into the flk1 locus (VEGFR2+/−). Mice were fed with normal phosphate chow at 6 wk of age. C: immunofluorescent images for endothelial marker (PE; red), collagen type IV (Col IV; blue), and exogenous inserted gene (GFP; green) in kidney sections. C, left: representative immunofluorescent images. Scale bar = 50 µm. C, right: quantitative analysis of PE staining density on kidney sections with ImageJ software. D: immunoblot analysis for endothelial marker (CD31), VEGFR2, VEGFA, GFP, and β-actin in total kidney lysates. D, left: representative immunoblots. D, right: quantitative analysis of all experiments from two groups. E: quantitative analysis of mRNA of CD31, VEGFR2, and VEGFA in the kidneys with quantitative PCR. F: immunoblot analysis for phosphorylated (p-)Erk and p-Akt as well as total (T-)Erk and Akt in total kidney lysates. F, left: representative blots. F, right: quantitative analysis of all immunoblots from two groups. C–F: quantitative data are expressed with scatterplots of individual data points (open circles indicate male mice and pink circles indicate female mice) and means ± SD (bars and errors) of all mice from each group. Statistical significance was evaluated by an unpaired Student’s t test, and significance was accepted when **P < 0.01 between two groups. The sample number in each group is presented in parentheses underneath each corresponding bar. PE, a cocktail of primary antibodies against platelet-endothelial cell adhesion molecule-1 and endomucin; VR2, VEGFR2.
Haploinsufficient VEGFR2 Mice Have Low VEGF/VEGFR Signal Activity and Peritubular Capillary Loss
We studied heterozygous VEGFR2+/− mice with an endothelial antibody cocktail (PE) to map the vessels in the kidney and to compare with the GFP signal. As shown in Fig. 1C, VEGFR2+/− mice had colocalization of GFP with the PE signal in the kidney, confirming endothelial expression of VEGFR2. Of note, the PE signal in VEGFR2+/− mice was much lower compared with WT mice. The signal of collagen type IV, a component of the basement membrane, was increased in VEGFR2+/− mice compared with WT mice. Its pathological implication in kidney fibrosis remains to be defined. Immunoblot analysis showed lower CD31 and VEGFR2 in kidney lysates of VEGFR2+/- mice than in WT mice (Fig. 1D). Lower VEGFA expression in kidney lysates was also found in VEGFR2+/− mice compared with WT mice (Fig. 1D), likely due to the kidney tubules affected by tubulointerstitial fibrosis. The detailed mechanism remains to be defined. Lower levels of CD31, VEGFA, and VEGFR2 mRNA were also found in the kidneys of VEGFR2+/− mice (Fig. 1E). Phosphorylation of Akt and Erk (p-Akt and p-Erk) is required for transduction of VEGF/VEGFR2 signaling (2, 53). We found lower p-Akt-to-total Akt and p-Erk-to-total Erk ratios in kidney lysates of VEGFR2+/− mice (Fig. 1F), indicating that VEGFR2 haploinsufficiency caused low VEGF/VEGFR signal activity.
To explore if there are perturbation of glomerular capillary and secondary damage of podocytes contributing to the kidney phenotype in VEGFR2+/− mice, we examined glomerular morphology and glomerular markers (nephrin, a glomerular podocyte protein). VEGFR2+/− mice had a normal ultrastructure of glomerular capillary structures (normal foot process of podocytes and glomerular basement membrane) (Supplemental Fig. S1A) and normal expression of nephrin and endothelial markers in glomeruli (Supplemental Fig. S1, B and C). Immunoblot analysis of nephrin in kidney lysates further confirmed the similar expression between VEGFR2+/− and WT mice (Supplemental Fig. S1D). In addition, the urinary protein excretion rate was similar between VEGFR2+/− and WT mice (Supplemental Fig. S1E), implying that VEGFR2+/− mice have normal glomerular capillary development and glomerular permeability at least in early life up to 3 mo old.
Haploinsufficient VEGFR2 Activity Accelerates Kidney Structural Abnormalities
One important feature of kidney injury is vascular loss (54). Impaired angiogenesis is an integral part of kidney disease, which debilitates tubular regeneration, induces and accelerates kidney fibrosis, and promotes CKD progression (54–56). We found higher levels of plasma creatinine and more kidney fibrosis in VEGFR2+/− mice at 12 wk of age, which were exacerbated with aging up to 1 yr (Fig. 2, A–C). There was a trend of higher plasma creatinine and Pi, higher kidney fibrotic scores, and higher CD31 mRNA in male mice compared with female mice, but without statistically significant differences between the two sexes (data not shown). Therefore, we presented the changes in plasma creatinine and Pi, kidney fibrotic scores, and CD31 mRNA in the kidneys in mixed-sex groups (Fig. 2, A, C, and D). VEGFR2+/− mice had higher plasma phosphate by 1 yr (Fig. 2A), more likely secondary to kidney dysfunction rather than a direct effect of impaired VEGFR signaling. Interestingly, VEGFR2+/− mice had lower levels of CD31 mRNA and protein in the kidney at 6 wk old (Fig. 2, D and E). Immunoblot analysis showed lower expression of Klotho and higher expression of fibrotic markers in the kidney at 12 wk old in VEGFR2+/− mice compared with WT mice at the same age (Fig. 2E). These findings suggest that Klotho deficiency and fibrosis may be the outcomes of impaired VEGFR2 signaling in the kidney.
Figure 2.

Spontaneous development of kidney fibrosis in heterozygous vascular endothelial growth factor receptor type 2 (VEGFR2)+/− mice. Wild-type (WT) and VEGFR2+/− mice were fed with normal rodent chow, and 48 mice were enrolled and randomly assigned into each group. Six mice per group were euthanized at the age of 6, 12, 24, and 52 wk. The plasma and kidneys were collected for further experiments. A: plasma creatinine (Cr) and phosphate (Pi). B: kidney fibrosis was assessed with trichrome staining. Representative microscopic images of mice from each group from 6 to 52 wk old are shown. Scale bar = 50 µm. C: quantitative analysis of kidney fibrosis based on trichrome-stained kidney sections with ImageJ software. D: quantitative analysis of CD31 mRNA in the kidneys with quantitative PCR. Data in A, C, and D are expressed as means ± SD of at least four mice from each group at the age of 6–52 wk. Statistical analysis was evaluated by an unpaired Student’s t test, and significance was accepted when *P < 0.05 and **P < 0.01 between two groups at same age. The results shown in A, C, and D are presented as mixed sexes. E: immunoblot analysis for endothelial marker (CD31), Klotho, α-smooth muscle actin (α-SMA), connective tissue growth factor (CTGF), and β-actin in total kidney lysates from two genotyped mice at 6 and 12 wk old. E, left: representative immunoblots. E, right: quantitative analysis of all immunoblots from each group. E: quantitative data are expressed with scatterplots of individual data points (open circles indicate male mice and pink circles indicate female mice) and means ± SD (bars and errors). Statistical significance was evaluated by two-way ANOVA followed by a Student–Newman–Keuls test. Statistical significance was accepted when *P < 0.05 and **P < 0.01 between two groups. The sample number in each group is presented in parentheses underneath each corresponding bar. VR2, VEGFR2.
Klotho Protein Is Required for Normal Peritubular Capillary Integrity
Klotho deficiency is associated with defective angiogenesis and vasculogenesis in skeletal muscle (27), but the Klotho effect on kidney vasculature has not been studied. To test for causality, we tested whether Klotho supplementation rescues peritubular capillary integrity in the kidneys of homozygous Klotho hypomorphic (kl/kl) mice. Klotho protein was not detectable in the kidney, and levels of endothelium-related markers, such as Pecam-1/CD31 and VEGFR2 protein, were lower in the kidneys of kl/kl mice (Fig. 3, A and B). Importantly, a 4-wk course of recombinant Klotho administration significantly improved vascular morphology and biomarkers of vessels, proving the causality between Klotho and vascular integrity.
Figure 3.

Klotho improved peritubular capillaries in Klotho-deficient mice. Twelve wild-type (WT) or kl/kl mice at 4 wk of age were fed with normal rodent chow and randomly assigned into two treatments (vehicle and recombinant Klotho protein) through intraperitoneal implantation of osmotic minipumps for a total duration of 4 wk. Mice were euthanized, and the kidneys were harvested for further experiments (A and B) after 4 wk of implantation. A: immunoblot analysis for endothelial marker (CD31), Klotho, vascular endothelial growth factor receptor type 2 (VEGFR2), and β-actin in total kidney lysates. A, left: representative immunoblots. A, right: quantitative analysis of all immunoblots from the four groups. B: immunofluorescent images for endothelial marker (PE; red) in kidney sections. B, left: representative immunofluorescent images. Scale bar = 100 µm. B, right: quantitative analysis of PE staining density on kidney sections with ImageJ software. C: WT and Tg-kl mice harboring tdTomato and Tie2-Cre genes were euthanized at 12 wk, and the kidneys were harvested to image the vasculature. C, top: representative immunofluorescent images for tdTomato signal (red) and Lotus tetragonolobus lectin (proximal tubular marker; green). Scale bar = 100 µm. C, bottom: quantitative analysis of tdTomato staining density on kidney sections with ImageJ software. A–C: quantitative data are expressed with scatterplots of individual data points (open circles indicate male mice and pink circles indicate female mice) and means ± SD (bars and errors) from each group. Statistical significance was evaluated by two-way ANOVA followed by a Student–Newman–Keuls test. Significant differences were accepted when *P < 0.05 and **P < 0.01 between two groups. The sample number in each group is presented in parentheses underneath each corresponding bar. G, glomerulus; PE, a cocktail of primary antibodies against platelet-endothelial cell adhesion molecule-1 and endomucin.
Similar to our previous findings (16), glomerular morphology was normal in kl/kl mice compared with WT mice based on both conventional light microscopy (Fig. 3B) and ultrastructural electron microscopy (Supplemental Fig. S2), although kl/kl mice had a low density of peritubular capillaries and low VEGFR2 expression (Fig. 3, A and B).
Next, we used Tg-kl mice with higher levels of circulating soluble Klotho to examine the Klotho effect on peritubular capillaries. Based on findings in endothelial reporter mice (tdTomoato;Tie2-Cre), peritubular capillary density was higher in Tg-kl mice than in WT mice (Fig. 3C). Thus, Klotho modulates the peritubular capillary more prominently than the glomerular capillary.
Klotho Deficiency Exacerbates Kidney Fibrosis and Vascular Loss in VEGFR2+/− Mice
Since VEGFR2+/− mice had low Klotho (Figs. 1 and 2) and kl/kl mice had low VEGFR2 in the kidney (Fig. 3A), we sought to see whether there is a reciprocal interplay between VEGFR2 and Klotho in peritubular capillaries by cross-breeding VEGFR2+/− and kl/+ mice to generate a new mouse line with haploinsufficiency for both Klotho and VEGFR2 (kl/kl;VEGFR2+/−). kl/kl;VEGFR2+/− mice did not have higher levels of plasma creatinine compared with the other three genotypes (WT, VEGFR2+/−, and kl/kl) at baseline (6 wk of age; Fig. 4A). However, kl/kl;VEGFR2+/− mice had higher levels of plasma phosphate than the other three genotypes (Fig. 4B). Trichrome stain showed that kl/kl mice had more kidney fibrosis than WT mice, and the fibrosis was further exacerbated in kl/kl mice when superimposed with haploinsufficient VEGFR2 (kl/kl;VEGFR2+/−; Fig. 4C). Immunoblot analysis demonstrated reduced vascular markers and higher kidney fibrotic markers in the kl/kl;VEGFR2+/− line compared with any of the other three lines (Fig. 4D).
Figure 4.

Vascular endothelial growth factor receptor type 2 (VEGFR2) haploinsufficiency exacerbated kidney dysfunction, fibrosis, and peritubular capillary rarefaction in Klotho-deficient mice. Six mice per line from four different genotypes [wild-type (WT), VEGFR2+/−, kl/kl, and kl/kl;VEGFR2+/−] were euthanized at 6 wk of age. The plasma and kidneys were collected from each mouse for further experiments. A: plasma creatinine (Cr). B: plasma Pi. C: kidney fibrosis assessed with trichrome staining. C, left: representative microscopic images. Scale bar = 200 µm. C, right: quantitative analysis of fibrotic scores on trichrome-stained kidney sections with ImageJ software. D: immunoblot analysis for endothelial marker (CD31), Klotho, α-smooth muscle actin (α-SMA), connective tissue growth factor (CTGF), and β-actin in total kidney lysates. D, left: representative blots. D, right: quantitative analysis of all immunoblots from the four groups. A–D: quantitative data are expressed with scatterplots of individual data points (open circles indicate male mice and pink circles indicate female mice) and means ± SD (bars and errors) of all mice from each group. Statistical significance was evaluated by two-way ANOVA followed by a Student–Newman–Keuls test, and significance was accepted when *P < 0.05 and **P < 0.01 between two groups. The sample number in each group is presented in parentheses underneath each corresponding bar. VR2, VEGFR2.
Because the severe hyperphosphatemia in kl/kl mice may contribute at least in part to the ill effects of Klotho deficiency (29), we examined the effect of high dietary phosphate on peritubular capillaries in mice with normal Klotho at baseline. Similar to previous findings (11), plasma phosphate was elevated in mice after 1 wk and maintained at higher levels compared with mice on the normal diet (Supplemental Table S3). VEGFA, CD31, and VEGFR2 expression in the kidney did not start to decline until 2 wk after high dietary phosphate feeding. Interestingly, Klotho protein was also reduced at 2 wk, along with an elevation of α-SMA (Supplemental Fig. 3, A and B). These results indicate that higher plasma phosphate may be a trigger to reduce VEGFR2 independent of Klotho reduction.
Hyperphosphatemia Enhances Peritubular Capillary Loss in the Kidney in Both Klotho-Deficient Mice and VEGFR2 Haploinsufficient Mice
We examined whether high dietary phosphate exacerbates abnormal peritubular capillaries in the background of Klotho deficiency and/or VEGFR2 haploinsufficiency (Fig. 5A). Because kl/kl mice are too fragile to tolerate dietary phosphate loading (29), heterozygous Klotho-deficient mice (kl/+) mice had to be used instead.
Figure 5.

Vascular endothelial growth factor receptor type 2 (VEGFR2) haploinsufficiency and Klotho deficiency synergistically impaired peritubular capillaries. A: mice with four different genotypes [wild-type (WT), VEGFR2+/−, kl/+, and kl/+;VEGFR2+/−] at 3 mo of age were fed normal- or high-phosphate diet for 12 wk. Each treatment per genotype had 10 mice. B: plasma creatinine (Cr). C: plasma Pi. D: total kidney lysates were subjected to immunoblot analysis for endothelial marker (CD31), Klotho, VEGFR2, and β-actin. D, top: representative blots. D, bottom: quantitative analysis of all immunoblots from four groups. B–D: quantitative data are expressed with scatterplots of individual data points (open circles indicate male mice and pink circles indicate female mice) and means ± SD (bars and errors) from each group. Statistical significance was evaluated by two-way ANOVA followed by a Student–Newman–Keuls test, and significance was accepted when *P < 0.05 and **P < 0.01 between two groups within same phosphate diet treatment and $P < 0.05 and $$P < 0.01 between normal- and high-phosphate diet within same genotype. The sample number in each group is presented in parentheses underneath each corresponding bar.
In the normal dietary phosphate cohort, kl/+;VEGFR2+/− mice at 6 mo old had higher plasma creatinine than the other three groups of mice (WT, VEGFR2+/−, and kl/+ mice; Fig. 5B). However, after high dietary phosphate loading, all four genotypes had higher plasma creatinine compared with mice fed normal dietary phosphate; among the groups, kl/+;VEGFR2+/− mice had the highest plasma creatinine (Fig. 5B). Similar to the change in plasma creatinine, plasma phosphate levels were marginally increased in kl/+ mice compared with WT mice under normal phosphate feeding. Note that high dietary phosphate increased plasma phosphate in all four groups of mice, among which kl/+;VEGFR2+/− mice had the highest plasma phosphate (Fig. 5C).
On a normal-phosphate diet, kl/+;VEGFR2+/− mice had much lower CD31, VEGFA, and VEGFR2 expression in the kidney than the other three genotypes (Fig. 5D). High dietary phosphate not only increased plasma creatinine and phosphate in kl/+;VEGFR2+/− mice but also further exacerbated vessel loss compared with WT, VEGFR2+/−, and kl/+ mice (Fig. 5D). Immunohistochemistry showed more peritubular capillary rarefaction in kl/+;VEGFR2+/− mice compared with the other three genotypes (Supplemental Fig. S4). As expected, Klotho was reduced by dietary phosphate loading in all four mouse lines, and kl/+;VEGFR2+/− mice had the lowest Klotho protein expression in the kidney (Fig. 5D).
Consistent with more severe changes in plasma creatinine, phosphate, and the vasculature in the kidney, kidney fibrosis was also more severe in kl/+;VEGFR2+/− mice and further exacerbated by high dietary phosphate (Fig. 6, A–C). Double staining showed more peritubular capillary rarefaction and higher α-SMA expression in the kidney after the high dietary phosphate challenge (Fig. 6B). High phosphate increased α-SMA and CTGF abundance in the kidney of each genotype (Fig. 6C). All changes induced by high phosphate diet were exacerbated in kl/+;VEGFR2+/− mice.
Figure 6.

Vascular endothelial growth factor receptor type 2 (VEGFR2) haploinsufficiency promoted high dietary phosphate-induced kidney fibrosis in kl/+ mice. A: kidneys from four different genotyped mice [wild-type (WT), VEGFR2+/−, kl/+, and kl/+;VEGFR2+/−] that were fed normal-phosphate (N Pi) or high-phosphate (H Pi) diet for 12 wk, respectively. Kidney sections were subjected to trichome (TC) stain to evaluate kidney fibrosis. A, top: representative microscopic images. Scale bar = 200 µm. A, bottom: quantitative analysis of fibrotic scores on TC-stained kidney sections with ImageJ software. B: immunofluorescent images for endothelial marker (PE; white), α-smooth muscle actin (α-SMA; red), and exogenous inserted gene [green fluorescent protein (GFP); green] in kidney sections. B, top: representative immunofluorescent images. Scale bar = 100 µm. White arrows locate double staining with α-SMA and GFP. B, bottom: quantitative analysis of PE staining density on kidney sections with ImageJ software. C: immunoblot analysis for α-SMA, connective tissue growth factor (CTGF), and β-actin in total kidney lysates. C, top: representative blots. C, bottom: quantitative analysis of all immunoblots from each group. A−C: quantitative data are expressed with scatterplots of individual data point (open circles indicate male mice and pink circles indicate female mice) and means ± SD (bars and errors) of all mice from each group. Statistical significance was evaluated by two-way ANOVA followed by a Student–Newman–Keuls test. Significant differences were accepted when *P < 0.05 and **P < 0.01 between two groups within the same phosphate diet treatment and $P < 0.05 and $$P < 0.01 between normal- and high-phosphate diet within the same genotype. The sample number in each group is presented in parentheses underneath each corresponding bar. PE, a cocktail of primary antibodies against platelet-endothelial cell adhesion molecule-1 and endomucin.
The Beneficial Effect of High Klotho on Peritubular Capillaries Is Attenuated by VEGFR2 Haploinsufficiency
Since Klotho is vascular protective and also regulates VEGFR2 function to support vasculogenesis and angiogenesis (20, 21, 57), we generated another mouse line (Tg-kl;VEGFR2+/−) with higher Klotho in the background of VEGFR2 haploinsufficiency to explore its response to high dietary phosphate (Fig. 7A).
Figure 7.

Klotho more effectively ameliorated high dietary phosphate-induced than vascular endothelial growth factor receptor type 2 (VEGFR2) haploinsufficiency-associated peritubular capillary rarefaction. A: mice with four different genotypes [wild-type (WT), VEGFR2+/−, Tg-kl, and Tg-kl;VEGFR2+/−] at 3-mo old were fed normal- or high-phosphate diet for 12 wk, respectively. Each treatment per genotype had 10 mice. B: plasma creatinine (Cr). C: plasma Pi. D: immunoblot analysis for endothelial marker (CD31), Klotho, VEGFR2, and β-actin in total kidney lysates. D, top: representative blots. D, bottom: quantitative analysis of all immunoblots from the four groups. B–D: quantitative data are expressed with scatterplots of individual data points (open circles indicate male mice and pink circles indicate female mice) and means ± SD (bars and errors) of mice from each group. Statistical significance was evaluated by two-way ANOVA followed by a Student–Newman–Keuls test. Significant differences were accepted when *P < 0.05 and **P < 0.01 between two groups within the same phosphate diet treatment and $P < 0.05 and $$P < 0.01 between normal- and high-phosphate diet within the same genotype. The sample number in each group is presented in parentheses underneath each corresponding bar.
The higher plasma creatinine in VEGFR2+/− mice was further elevated by high dietary phosphate and only marginally improved by overexpression of Klotho in Tg-kl mice, as one may expect (Fig. 7B). The higher Klotho in Tg-kl mice more effectively ameliorated the elevated plasma phosphate induced by high phosphate in the WT background compared with the VEGFR2 deficiency background (Fig. 7C). High phosphate diet-induced vessel loss (lower CD31 expression) and reduced VEGFA and VEGFR2 expression in the kidneys of WT mice, which was ameliorated by higher Klotho in Tg-Kl mice. Interestingly, the low vessel density and low VEGFR2 expression in the kidneys of VEGFR2+/− mice under either normal or high dietary phosphate were not as significantly improved by high Klotho as in WT mice (Fig. 7D and Supplemental Fig. S5). As expected, Tg-kl mice had the highest Klotho expression in the kidney under a normal phosphate diet, and this was only modestly decreased by a high-phosphate diet (Fig. 7D). Furthermore, the reduced Klotho level found in VEGFR2+/− mice under normal- or high-phosphate diet was only partially rescued by high expression of Klotho in Tg-kl;VEGFR2+/− mice (Fig. 7D).
Kidney fibrosis in VEGFR2+/− mice at 6 mo of age was marginally improved in Tg-kl;VEGFR2+/− mice compared with the WT background under normal dietary phosphate (Fig. 8, A–C). Notably, high phosphate-induced kidney fibrosis and peritubular capillary loss were significantly abolished in Tg-kl mice but only modestly improved in Tg-kl;VEGFR2+/− mice (Fig. 8, A–C). Immunoblot analysis of fibrotic markers (α-SMA and CTGF; Fig. 8C) confirmed the findings with trichrome stain and immunohistochemistry (Fig. 8, A and 7B). Therefore, Klotho effectively protects kidney fibrosis and peritubular capillary loss from phosphotoxicity in WT mice, but the Klotho renoprotection was dramatically attenuated by VEGFR2 deficiency.
Figure 8.

Klotho more significantly reduced high dietary phosphate-induced kidney fibrosis than vascular endothelial growth factor receptor type 2 (VEGFR2) haploinsufficiency-associated kidney fibrosis. A: kidneys from four different genotypes [wild-type (WT), VEGFR2+/−, Tg-kl, and Tg-kl;VEGFR2+/−] fed normal-phosphate (N Pi) or high-phosphate (H Pi) diet for 12 wk, respectively, as shown in Fig. 7A. Kidney sections were subjected to trichome (TC) stain to evaluate kidney fibrosis. A, top: representative microscopic images. Scale bar = 200 µm. A, bottom: quantitative analysis of fibrotic scores on TC-stained kidney sections with ImageJ software. B: immunofluorescent images for endothelial marker (PE; white), α-smooth muscle actin (α-SMA; red), and exogenous inserted gene [green fluorescent protein (GFP); green] in kidney sections. B, top: representative immunofluorescent images. Scale bar = 100 µm. White arrows indicate double staining of α-SMA with GFP. B, bottom: quantitative analysis of PE signal density on kidney sections with ImageJ software. C: immunoblot analysis for α-SMA, connective tissue growth factor (CTGF), and β-actin in total kidney lysates. C, left: representative blots. C, right: quantitative analysis of all immunoblots of all mice from each group. A–C: quantitative data are expressed with scatterplots of individual data points (open circles indicate male mice and pink circles indicate female mice) and means ± SD (bars and errors) of all mice from each group. Statistical significance was evaluated by two-way ANOVA followed by a Student–Newman−Keuls test, and significance was accepted when *P < 0.05 and **P < 0.01 between two groups within the same phosphate diet treatment and $P < 0.05 and $$P < 0.01 between normal- and high-phosphate diet within the same genotype. The sample number in each group is presented in parentheses underneath each corresponding bar. PE, a cocktail of primary antibodies against platelet-endothelial cell adhesion molecule-1 and endomucin.
Effect of Klotho on Endothelial Damage Induced by Inhibition of VEGFR2 and High Phosphate In Vitro
To test for a direct effect of Klotho against phosphate-induced endothelial toxicity, we used MS1, an immortalized mouse endothelial cell line, as an in vitro model. High phosphate media (2.0 mM) induced more LDH release into culture media compared with normal phosphate media (1.0 mM; Fig. 9A). Endothelial marker (CD31) and VEGFR2 expression were reduced in cell lysates (Fig. 9B), suggesting that high phosphate may induce endothelial cell dedifferentiation in addition to cell injury. Recombinant Klotho protein added to normal or high phosphate media significantly reduced LDH release and restored CD31 and VEGFR2 expression (Fig. 9, A and B), indicating that Klotho keeps cell health and protects endothelial cells from phosphotoxicity in vitro.
Figure 9.

Klotho attenuated endothelial injury and mesenchymal transition induced by high phosphate in cultured media. A–C: MS1 cells were incubated with Klotho (0.4 nM) in the presence of normal phosphate (N Pi; 1.0 mM) or high phosphate (H Pi; 2.0 mM). Culture media were collected for lactate dehydrogenase (LDH) release assay and cells for immunoblot analysis and immunohistochemistry at 24 h. A: LDH release in culture media. B: immunoblot analysis for CD31, vascular endothelial growth factor receptor type 2 (VEGFR2), and β-actin in total cell lysates extracted from MS1 cells. B, left: representative immunoblots. B, right: quantitative analysis of all immunoblots from six independent experiments. C: representative immunofluorescent images for endothelial marker (CD31; red) and VEGFR2 (green) in MS1 cells from six independent experiments. Scale bar = 25 µm. White arrowheads indicate CD31 costained with VEGFR2. D: MS1 cells were incubated in the conditions described in A–C but with 48-h incubation. Immunoblot analysis for α-smooth muscle actin (α-SMA) and β-actin in total cell lysates. D, left: representative immunoblots. D, right: quantitative analysis of all immunoblots from six independent experiments. A, B, and D: quantitative data are expressed with scatterplots of individual data points and means ± SD (bars and errors) of six independent experiments. Statistical significance was evaluated by two-way ANOVA followed by a Student–Newman–Keuls test, and significance was accepted when *P < 0.05 and **P < 0.01 between two groups. The sample number in each group is presented in parentheses underneath each corresponding bar.
VEGFR2 downstream signaling is mediated by multiple mechanisms at transcriptional, translational, and posttranslational levels as well as endocytosis of VEGFR2 protein (58). Fluorescent immunocytochemistry showed lower CD31 and VEGFR2 expression. Interestingly, and surprisingly, both free VEGFR2 and costaining of VEGFR2 with CD31 were increased by Klotho protein coincubation (Fig. 9C), which may indicate Klotho endothelial protection against phosphotoxicity.
Endothelial-mesenchymal transition is one mechanism of kidney fibrosis (59). Our previous study (11) and the present study on animals (Fig. 8) showed that chronic dietary phosphate-induced kidney fibrosis was suppressed by Klotho. We used cultured endothelial cells to examine if Klotho-inhibited fibrosis is associated with blocking endothelial-mesenchymal transition. We found that high phosphate significantly increased α-SMA protein and that Klotho attenuated the elevation induced by high phosphate media (Fig. 9D). Taken together with the results that high phosphate-downregulated CD31 and VEGFR2 expression was attenuated by Klotho (Fig. 9, A and B), it is conceivable that Klotho may suppress fibrosis in part through blocking endothelial-mesenchymal transition.
To further explore the direct role of VEGFR2 in protection against phosphotoxicity, we treated MS1 cells with the VEGFR2 inhibitor Ki8751 (48). Ki8751 increased baseline and enhanced high phosphate-induced LDH release in a dose-dependent manner (Fig. 10, A and B), implying that normal VEGFR2 signaling is required for endothelial cell integrity. Ki8751 significantly reduced CD31 expression and VEGFR2 expression in cell lysates, which were further exacerbated by high phosphate media (Fig. 10C). Interestingly, and rather surprisingly, Ki8751 increased VEGFR2 abundance, which was colocalized with CD31 (Fig. 10D). Whether this elevation implies that Ki8751 inhibits VEGFR2 endocytosis, consequently blocking VEGFR2 signaling, remains to be explored. Ki8751 did not reduce mRNA levels of CD31 and VEGFR2 (Supplemental Fig. S6), indicating that Ki8751 acutely suppresses VEGFR2 probably through inhibition of its endocytosis, a well-known mechanism to regulate VEGFR2 activity instead of VEGFR2 transcription regulation.
Figure 10.

Klotho cytoprotection against phosphotoxicity in endothelial cells was attenuated by vascular endothelial growth factor receptor type 2 (VEGFR2) inhibitor. A: mouse endothelial cells (MS1) were incubated with 0–5 nM Ki8751, a VEGFR2 inhibitor, in the presence of normal phosphate (N Pi; 1.0 mmol/L) or high phosphate (H Pi; 2.0 mmol/L) and cotreated with Klotho (0.4 nM) or normal saline (NS) as vehicle for 24 h. Culture media were collected for lactate dehydrogenase (LDH) assay. Data are expressed as averages from six independent experiments under each condition, and statistical differences among four groups at 5 nmol/L Ki8751 were determined by two-way ANOVA followed by a Student–Newman–Keuls test. Statistical significance was accepted when *P < 0.05 and **P < 0.01 between two groups. B: comparison of LDH release in culture media between normal- or high-phosphate media with or without Ki8751 (2 nM) and cotreatment of Klotho after 24-h treatment. C: immunoblot analysis for CD31, VEGFR2, and β-actin in total cell lysates extracted from MS1 cells after 24-h treatment. C, left: representative blots. C, right: quantitative analysis of all immunoblots from independent experiments. D: representative immunofluorescent images for endothelial marker (CD31; red) and VEGFR2 (green) in MS1 cells from six independent experiments. Scale bar = 25 µm. White arrows indicate CD31 costained with VEGFR2. B and C: quantitative data are expressed with scatterplots of individual data points and means ± SD of six independent experiments. Statistical significance was evaluated by two-way ANOVA followed by a Student–Newman–Keuls test. Statistical significance was accepted when *P < 0.05 and **P < 0.01 between two groups and $P < 0.05 and $$P < 0.01 between vehicle and Klotho treatment within the same experimental condition. The sample number in each group is presented in parentheses underneath each corresponding bar.
We next tested the relationship between Klotho and VEGFR2 in endothelial protection. Consistent with findings shown in Fig. 9, Klotho protected cells from phosphotoxicity, but its protective action was significantly reduced by inhibition of VEGFR2 (Fig. 10, A and B). Moreover, CD31 and VEGFR2 protein expression in cell lysates was reduced in cells treated with either high phosphate media and/or Ki8751 (Fig. 10C). However, Klotho more significantly alleviated CD31 and VEGFR2 reduction in cells treated with high phosphate media than in cells treated with Ki8751 (Fig. 10C), indicating that Klotho prevents cells more efficiently from phosphotoxicity than from low VEGFR2-induced toxicity.
DISCUSSION
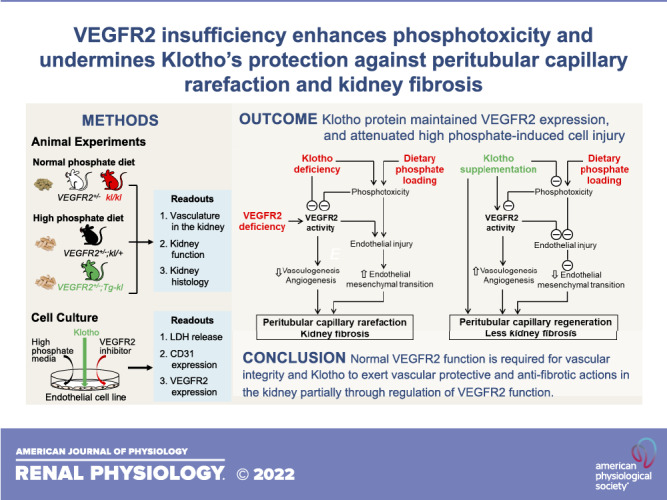
The role of VEGFR2 in angiogenesis and vasculogenesis in the kidney and extrarenal organs (37) is well known, but its role in peritubular capillary and kidney fibrosis has not been fully studied. Our in vivo and in vitro experiments collectively provide several salient findings. First, VEGFR2 deficiency in the kidney induces peritubular capillary rarefaction and kidney fibrosis. Second, VEGFR2 deficiency renders the kidney more vulnerable to phosphotoxicity. Third, VEGFR2 deficiency exacerbates structural and functional deterioration in the kidney seen in Klotho deficiency. Finally, VEGFR2 deficiency significantly attenuates the ability of Klotho to protect the kidney against phosphotoxicity. In summary, optimal VEGFR2 function is required to maintain kidney structure and function. Phosphate is an endothelial toxin, and VEGFR2 deficiency enhances phosphotoxicity and accelerates kidney damage. Klotho exerts vascular protection in part through maintaining VEGFR2 function in endothelial cells. A paradigm is shown in Fig. 11 to demonstrate the complex interplay between VEGFR2 signaling and Klotho in protecting against phosphotoxicity. The role of these factors in endothelial injury and protection is discussed below.
Figure 11.

Interplay of Klotho, phosphate, and vascular endothelial growth factor receptor type 2 (VEGFR2) in the peritubular vasculature and kidney fibrosis. A: high dietary phosphate loading induces cell damage. Damaged endothelial cells 1) dedifferentiate to fibroblasts through endothelial mesenchymal transition and 2) reduce VEGFR2 expression and consequently downregulate VEGFR2 signaling activity. On the other hand, VEGFR2 deficiency inhibits VEGFR2 signaling activity, which renders cells more vulnerable to phosphotoxicity. Low VEGFR2 signaling activity causes peritubular vascular loss. Peritubular capillary rarefaction induces kidney tubular ischemia and damage as well as tubulointerstitial fibrosis. Klotho deficiency suppresses VEGFR2 activity and exacerbates phosphotoxicity-induced endothelial injury. B: elevation of soluble Klotho through Klotho supplementation increases VEGFR2 signaling activity and attenuates phosphotoxicity on endothelial cells, thus improving vasculogenesis and angiogenesis and reducing endothelial-mesenchymal transition. Soluble Klotho also promotes peritubular capillary regeneration and reduces kidney fibrosis through a VEGFR2-independent pathway. Taken together, the peritubular capillary is preserved, and kidney fibrosis is attenuated.
Vascular Phosphotoxicity and VEGFR2 Protection
High phosphate is considered a vascular toxin (32). Vascular manifestations of phosphotoxicity include calcification (60), endothelium dysfunction, and hypertension (26). High phosphate challenge reduces VEGFR2 and CD31 expression in vivo and in vitro, and further blocking the VEGFR2 signal should enhance the CD31 and VEGFR2 reduction triggered by high phosphate, suggesting that VEGFR2 deficiency contributes to or accelerates phosphotoxicity. Therefore, VEGFR2 may be another target of vascular phosphotoxicity.
Antiangiogenic therapy, including VEGFA or VEGFR2 inhibitors, has been used to treat several types of cancers, but these treatments have been complicated by nephrotoxicity (7, 61). However, stimulation of VEGFR2 signaling could promote the progression of kidney damage in nitric oxide synthase-null mice (62, 63), and inhibition of VEGFR signaling reduced peritubular vascular rarefaction and fibrosis in the unilateral ureteral obstruction model (63) and improved diabetic nephropathy in diabetic mice (64). The present study, therefore, renders the role of VEGFR2 debatable in kidney physiology and pathophysiology. Sustainably and uncontrollably high VEGFR2 signaling activity (41, 62) could cause tubular injury and epithelial-mesenchymal transition, suggesting that either extremely low or high VEGFR2 signaling can exert deleterious effects. Our experiments in cultured endothelial cells showed that VEGFR2 inhibition may interfere with VEGFR2 trafficking, thus inducing cell injury and rendering the cells more vulnerable to phosphotoxicity. Therefore, normal VEGF/VEGFR2 signaling activity is required to protect cells from phosphotoxicity. To confirm whether upregulation of VEGFR2 signaling activity protects the kidney from phosphotoxicity would pave a new avenue to treat kidney fibrosis.
Interestingly, VEGFR2+/− mice have a defective peritubular vasculature but do not have detectable changes in glomerular vascular tuffs and glomerular podocyte structure. Our findings indicate that peritubular capillaries are more vulnerable to low Klotho- and high phosphate-induced vasculopathy than glomerular capillaries. Impaired angiogenesis in kl/kl mice is at least in part due to defects in VEGFA/VEGFR2 signaling activity (65), consequently inducing hypoxia, fibrosis, and chronic inflammation (65). Another fate of endothelial injury is endothelial-mesenchymal transition that contributes to kidney fibrosis, although this transition does not account for all forms of kidney fibrosis (59). Our data confirmed that high phosphate and VEGFR2 deficiency in endothelial cells induce endothelial-mesenchymal transition.
Vascular Protection by Klotho
Klotho is protective for the vasculature probably partly through upregulating oxidative scavengers (66) and binding to VEGFR2/endothelial transient receptor potential canonical Ca2+ channel 1 to promote their internalization (20). Klotho deficiency causes impaired angiogenesis and vasculogenesis (27) and amplifies vascular phosphotoxicity (26). We confirmed that Klotho upregulates VEGFR2 signaling possibly through regulation of VEGFR2 endocytosis, although we have not explored the molecular mechanisms of Klotho’s effect on VEGFR2 trafficking or on the fate of the endocytosed VEGFR2 protein. Nevertheless, we showed that Klotho protects endothelial cells against phosphotoxicity in part through a VEGFR2-dependent pathway. Klotho not only directly protects the vessel but also attenuates phosphotoxicity by improving systemic phosphate homeostasis (16). Klotho, as a renoprotective protein, may also protect peritubular capillaries and reduce kidney fibrosis through a VEGFR2-independent pathway including regulation of autophagy and senescence (Fig. 11; 11, 35).
Limitations
There are some limitations in our study. Mice with global homozygous deficiency for Klotho or VEGFR2 have early lethality, which impairs in vivo experiments in homozygous mouse lines. Conditional knockout of Klotho or VEGFR2 in kidney tubules is required. Finally, this study has not explored whether phosphate and Klotho regulates VEGFR2 endocytosis. The mechanism of VEGFR2 endocytosis by phosphate and Klotho and the fate of endocytosed VEGFR2 remain to be explored.
Conclusions
We conclude that perturbation of VEGFR2 signaling induces peritubular capillary loss and optimal VEGFR2 function is required for maintaining peritubular capillary structure and function, thereby reducing kidney fibrosis after kidney damage. Phosphate is an endothelial toxin, and VEGFR2 deficiency enhances phosphotoxicity and accelerates kidney damage. We propose that Klotho maintains normal peritubular capillary structure and function in part through the regulation of VEGFR2 signaling in endothelial cells (Fig. 11).
SUPPLEMENTAL DATA
Supplemental Figs. S1−S6 and Supplemental Tables S1 and S2: https://doi.org/10.6084/m9.figshare.20113385.v1.
GRANTS
This work was supported by National Institutes of Health Grants R01DK091392 (to M.C.H. and O.W.M.), R01DK092461 (to O.C., O.W.M., and M.C.H.), and R01HL126518 (to O.C.), Leducq Research Foundation Grant 21CVD03 (to O.C.), The George M. O’Brien Kidney Research Center/UT Southwestern Medical Center Grant P30DK07938 (to O.W.M.), and Charles and Jane Pak Foundation Endowed Professor Collaborative Research Support (to M.C.H. and O.W.M.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
O.W.M. and M.C.H. conceived and designed research; M.S. and J.O.M. performed experiments; M.S., J.O.M., and M.C.H. analyzed data; M.S., J.O.M., O.C., O.W.M., and M.C.H. interpreted results of experiments; M.S., J.O.M., and M.C.H. prepared figures; M.S., O.W.M., and M.C.H. drafted manuscript; O.C., O.W.M., and M.C.H. edited and revised manuscript; O.W.M. and M.C.H. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors are grateful to Dr. Shirley Yan for the technical help in transmission electron microscopic experiments and thank Brianna Flores, Nancy Gillings, and Sierra Shepard for technical assistance.
REFERENCES
- 1. Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol 7: 359–371, 2006. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 2. Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17: 611–625, 2016. doi: 10.1038/nrm.2016.87. [DOI] [PubMed] [Google Scholar]
- 3. Dimke H, Sparks MA, Thomson BR, Frische S, Coffman TM, Quaggin SE. Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J Am Soc Nephrol 26: 1027–1038, 2015. doi: 10.1681/ASN.2014010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Terman BI, Dougher-Vermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D, Böhlen P. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun 187: 1579–1586, 1992. doi: 10.1016/0006-291x(92)90483-2. [DOI] [PubMed] [Google Scholar]
- 5. Matthews W, Jordan CT, Gavin M, Jenkins NA, Copeland NG, Lemischka IR. A receptor tyrosine kinase cDNA isolated from a population of enriched primitive hematopoietic cells and exhibiting close genetic linkage to c-kit. Proc Natl Acad Sci USA 88: 9026–9030, 1991. doi: 10.1073/pnas.88.20.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tanabe K, Wada J, Sato Y. Targeting angiogenesis and lymphangiogenesis in kidney disease. Nat Rev Nephrol 16: 289–303, 2020. doi: 10.1038/s41581-020-0260-2. [DOI] [PubMed] [Google Scholar]
- 7. Estrada CC, Maldonado A, Mallipattu SK. Therapeutic inhibition of VEGF signaling and associated nephrotoxicities. J Am Soc Nephrol 30: 187–200, 2019. doi: 10.1681/ASN.2018080853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, Quaggin SE. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol 21: 1691–1701, 2010. [Erratum in J Am Soc Nephrol 22: 1390, 2011]. doi: 10.1681/ASN.2010030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bartlett CS, Jeansson M, Quaggin SE. Vascular growth factors and glomerular disease. Annu Rev Physiol 78: 437–461, 2016. doi: 10.1146/annurev-physiol-021115-105412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuro-O M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse Klotho gene leads to a syndrome resembling ageing. Nature 390: 45–51, 1997. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 11. Maique J, Flores B, Shi M, Shepard S, Zhou Z, Yan S, Moe OW, Hu MC. High phosphate induces and Klotho attenuates kidney epithelial senescence and fibrosis. Front Pharmacol 11: 1273, 2020. doi: 10.3389/fphar.2020.01273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL, Ravikumar P, Gillings N, Bian A, Sidhu SS, Kuro-O M, Moe OW. Renal production, uptake, and handling of circulating αKlotho. J Am Soc Nephrol 27: 79–90, 2016. doi: 10.1681/ASN.2014101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen G, Liu Y, Goetz R, Fu L, Jayaraman S, Hu MC, Moe OW, Liang G, Li X, Mohammadi M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 553: 461–466, 2018. doi: 10.1038/nature25451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakatani T, Sarraj B, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B, Razzaque MS. In vivo genetic evidence for Klotho-dependent, fibroblast growth factor 23 (Fgf23) -mediated regulation of systemic phosphate homeostasis. FASEB J 23: 433–441, 2009. doi: 10.1096/fj.08-114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andrukhova O, Bayer J, Schüler C, Zeitz U, Murali SK, Ada S, Alvarez-Pez JM, Smorodchenko A, Erben RG. Klotho lacks an FGF23-independent role in mineral homeostasis. J Bone Miner Res 32: 2049–2061, 2017. doi: 10.1002/jbmr.3195. [DOI] [PubMed] [Google Scholar]
- 16. Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-O M, Moe OW. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J 24: 3438–3450, 2010. doi: 10.1096/fj.10-154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wöhrle S, Bonny O, Beluch N, Gaulis S, Stamm C, Scheibler M, Müller M, Kinzel B, Thuery A, Brueggen J, Hynes NE, Sellers WR, Hofmann F, Graus-Porta D. FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J Bone Miner Res 26: 2486–2497, 2011. doi: 10.1002/jbmr.478. [DOI] [PubMed] [Google Scholar]
- 18. Dalton GD, Xie J, An SW, Huang CL. New insights into the mechanism of action of soluble Klotho. Front Endocrinol (Lausanne) 8: 323, 2017. doi: 10.3389/fendo.2017.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ravikumar P, Ye J, Zhang J, Pinch SN, Hu MC, Kuro-O M, Hsia CC, Moe OW. α-Klotho protects against oxidative damage in pulmonary epithelia. Am J Physiol Lung Cell Mol Physiol 307: L566–L575, 2014. doi: 10.1152/ajplung.00306.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kusaba T, Okigaki M, Matui A, Murakami M, Ishikawa K, Kimura T, Sonomura K, Adachi Y, Shibuya M, Shirayama T, Tanda S, Hatta T, Sasaki S, Mori Y, Matsubara H. Klotho is associated with VEGF receptor-2 and the transient receptor potential canonical-1 Ca2+ channel to maintain endothelial integrity. Proc Natl Acad Sci USA 107: 19308–19313, 2010. doi: 10.1073/pnas.1008544107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu MC, Kuro-O M, Moe OW. αKlotho and vascular calcification: an evolving paradigm. Curr Opin Nephrol Hypertens 23: 331–339, 2014. doi: 10.1097/01.mnh.0000447024.97464.a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lim K, Lu TS, Molostvov G, Lee C, Lam FT, Zehnder D, Hsiao LL. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 125: 2243–2255, 2012. doi: 10.1161/CIRCULATIONAHA.111.053405. [DOI] [PubMed] [Google Scholar]
- 23. Lindberg K, Olauson H, Amin R, Ponnusamy A, Goetz R, Taylor RF, Mohammadi M, Canfield A, Kublickiene K, Larsson TE. Arterial Klotho expression and FGF23 effects on vascular calcification and function. PLoS One 8: e60658, 2013. doi: 10.1371/journal.pone.0060658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nagai R, Saito Y, Ohyama Y, Aizawa H, Suga T, Nakamura T, Kurabayashi M, Kuroo M. Endothelial dysfunction in the Klotho mouse and downregulation of Klotho gene expression in various animal models of vascular and metabolic diseases. Cell Mol Life Sci 57: 738–746, 2000. doi: 10.1007/s000180050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen K, Zhou X, Sun Z. Haplodeficiency of Klotho gene causes arterial stiffening via upregulation of scleraxis expression and induction of autophagy. Hypertension 66: 1006–1013, 2015. doi: 10.1161/HYPERTENSIONAHA.115.06033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Six I, Okazaki H, Gross P, Cagnard J, Boudot C, Maizel J, Drueke TB, Massy ZA. Direct, acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PLoS One 9: e93423, 2014. doi: 10.1371/journal.pone.0093423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shimada T, Takeshita Y, Murohara T, Sasaki K, Egami K, Shintani S, Katsuda Y, Ikeda H, Nabeshima Y, Imaizumi T. Angiogenesis and vasculogenesis are impaired in the precocious-aging Klotho mouse. Circulation 110: 1148–1155, 2004. doi: 10.1161/01.CIR.0000139854.74847.99. [DOI] [PubMed] [Google Scholar]
- 28. Hu MC, Kuro-O M, Moe OW. Renal and extrarenal actions of Klotho. Semin Nephrol 33: 118–129, 2013. doi: 10.1016/j.semnephrol.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shi M, Maique J, Shaffer J, Davidson T, Sebti S, Fernández ÁF, Zou Z, Yan S, Levine B, Moe OW, Hu MC. The tripartite interaction of phosphate, autophagy, and αKlotho in health maintenance. FASEB J 34: 3129–3150, 2020. doi: 10.1096/fj.201902127R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Komaba H, Fukagawa M. Phosphate-a poison for humans? Kidney Int 90: 753–763, 2016. doi: 10.1016/j.kint.2016.03.039. [DOI] [PubMed] [Google Scholar]
- 31. Peri-Okonny P, Baskin KK, Iwamoto G, Mitchell JH, Smith SA, Kim HK, Szweda LI, Bassel-Duby R, Fujikawa T, Castorena CM, Richardson J, Shelton JM, Ayers C, Berry JD, Malladi VS, Hu MC, Moe OW, Scherer PE, Vongpatanasin W. High-phosphate diet induces exercise intolerance and impairs fatty acid metabolism in mice. Circulation 139: 1422–1434, 2019. doi: 10.1161/CIRCULATIONAHA.118.037550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shroff R. Phosphate is a vascular toxin. Pediatr Nephrol 28: 583–593, 2013. doi: 10.1007/s00467-012-2347-x. [DOI] [PubMed] [Google Scholar]
- 33. Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 11: 1088–1100, 2016. doi: 10.2215/CJN.11901115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Foley RN. Phosphate levels and cardiovascular disease in the general population. Clin J Am Soc Nephrol 4: 1136–1139, 2009. doi: 10.2215/CJN.01660309. [DOI] [PubMed] [Google Scholar]
- 35. Shi M, Flores B, Gillings N, Bian A, Cho HJ, Yan S, Liu Y, Levine B, Moe OW, Hu MC. αKlotho mitigates progression of AKI to CKD through activation of autophagy. J Am Soc Nephrol 27: 2331–2345, 2016. doi: 10.1681/ASN.2015060613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu MC, Shi M, Gillings N, Flores B, Takahashi M, Kuro OM, Moe OW. Recombinant α-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int 91: 1104–1114, 2017. doi: 10.1016/j.kint.2016.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ema M, Takahashi S, Rossant J. Deletion of the selection cassette, but not cis-acting elements, in targeted Flk1-lacZ allele reveals Flk1 expression in multipotent mesodermal progenitors. Blood 107: 111–117, 2006. doi: 10.1182/blood-2005-05-1970. [DOI] [PubMed] [Google Scholar]
- 38. Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-O M. Suppression of aging in mice by the hormone Klotho. Science 309: 1829–1833, 2005. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol 230: 230–242, 2001. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 40. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140, 2010. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shi M, Flores B, Li P, Gillings N, McMillan KL, Ye J, Huang LJ, Sidhu SS, Zhong YP, Grompe MT, Streeter PR, Moe OW, Hu MC. Effects of erythropoietin receptor activity on angiogenesis, tubular injury, and fibrosis in acute kidney injury: a “U-shaped” relationship. Am J Physiol Renal Physiol 314: F501–F516, 2018. doi: 10.1152/ajprenal.00306.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shi M, Maique J, Shepard S, Li P, Seli O, Moe OW, Mc H. In vivo evidence for therapeutic applications of beclin 1 to promote recovery and inhibit fibrosis after acute kidney injury. Kidney Int 101: 63–78, 2021. doi: 10.1016/j.kint.2021.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Daniel E, Azizoglu DB, Ryan AR, Walji TA, Chaney CP, Sutton GI, Carroll TJ, Marciano DK, Cleaver O. Spatiotemporal heterogeneity and patterning of developing renal blood vessels. Angiogenesis 21: 617–634, 2018. doi: 10.1007/s10456-018-9612-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Franco M, Roswall P, Cortez E, Hanahan D, Pietras K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood 118: 2906–2917, 2011. doi: 10.1182/blood-2011-01-331694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perry B, Banyard J, McLaughlin ER, Watnick R, Sohn A, Brindley DN, Obata T, Cantley LC, Cohen C, Arbiser JL. AKT1 overexpression in endothelial cells leads to the development of cutaneous vascular malformations in vivo. Arch Dermatol 143: 504–506, 2007. doi: 10.1001/archderm.143.4.504. [DOI] [PubMed] [Google Scholar]
- 46. Chen J, Matzuk MM, Zhou XJ, Lu CY. Endothelial pentraxin 3 contributes to murine ischemic acute kidney injury. Kidney Int 82: 1195–1207, 2012. doi: 10.1038/ki.2012.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lu LH, Oh DJ, Dursun B, He Z, Hoke TS, Faubel S, Edelstein CL. Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. J Pharmacol Exp Ther 324: 111–117, 2008. doi: 10.1124/jpet.107.130161. [DOI] [PubMed] [Google Scholar]
- 48. Kubo K, Shimizu T, Ohyama S, Murooka H, Iwai A, Nakamura K, Hasegawa K, Kobayashi Y, Takahashi N, Takahashi K, Kato S, Izawa T, Isoe T. Novel potent orally active selective VEGFR-2 tyrosine kinase inhibitors: synthesis, structure-activity relationships, and antitumor activities of N-phenyl-N'-{4-(4-quinolyloxy)phenyl}ureas. J Med Chem 48: 1359–1366, 2005. doi: 10.1021/jm030427r. [DOI] [PubMed] [Google Scholar]
- 49. Li P, Shi M, Maique J, Shaffer J, Yan S, Moe OW, Hu MC. Beclin 1/Bcl-2 complex-dependent autophagy activity modulates renal susceptibility to ischemia-reperfusion injury and mediates renoprotection by Klotho. Am J Physiol Renal Physiol 318: F772–F792, 2020. doi: 10.1152/ajprenal.00504.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kang KP, Lee JE, Lee AS, Jung YJ, Kim D, Lee S, Hwang HP, Kim W, Park SK. Effect of gender differences on the regulation of renal ischemia-reperfusion-induced inflammation in mice. Mol Med Rep 9: 2061–2068, 2014. doi: 10.3892/mmr.2014.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shi M, McMillan KL, Wu J, Gillings N, Flores B, Moe OW, Hu MC. Cisplatin nephrotoxicity as a model of chronic kidney disease. Lab Invest 98: 1105–1121, 2018. doi: 10.1038/s41374-018-0063-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376: 62–66, 1995. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 53. Walker AMN, Warmke N, Mercer B, Watt NT, Mughal R, Smith J, Galloway S, Haywood NJ, Soomro T, Griffin KJ, Wheatcroft SB, Yuldasheva NY, Beech DJ, Carmeliet P, Kearney MT, Cubbon RM. Endothelial insulin receptors promote VEGF-A signaling via ERK1/2 and sprouting angiogenesis. Endocrinology 162: bqab104, 2021. doi: 10.1210/endocr/bqab104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Basile DP. Rarefaction of peritubular capillaries following ischemic acute renal failure: a potential factor predisposing to progressive nephropathy. Curr Opin Nephrol Hypertens 13: 1–7, 2004. doi: 10.1097/00041552-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 55. Menshikh A, Scarfe L, Delgado R, Finney C, Zhu Y, Yang H, de Caestecker MP. Capillary rarefaction is more closely associated with CKD progression after cisplatin, rhabdomyolysis, and ischemia-reperfusion-induced AKI than renal fibrosis. Am J Physiol Renal Physiol 317: F1383–F1397, 2019. doi: 10.1152/ajprenal.00366.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tanaka T, Nangaku M. Angiogenesis and hypoxia in the kidney. Nat Rev Nephrol 9: 211–222, 2013. doi: 10.1038/nrneph.2013.35. [DOI] [PubMed] [Google Scholar]
- 57. Chen L, Liu H, Liu J, Zhu Y, Xu L, He H, Zhang H, Wang S, Wu Q, Liu W, Liu Y, Pan D, Ren S, Xu J, Gu J. Klotho endows hepatoma cells with resistance to anoikis via VEGFR2/PAK1 activation in hepatocellular carcinoma. PLoS One 8: e58413, 2013. doi: 10.1371/journal.pone.0058413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Simons M. An inside view: VEGF receptor trafficking and signaling. Physiology (Bethesda) 27: 213–222, 2012. doi: 10.1152/physiol.00016.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 19: 2282–2287, 2008. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yamada S, Giachelli CM. Vascular calcification in CKD-MBD: roles for phosphate, FGF23, and Klotho. Bone 100: 87–93, 2017. doi: 10.1016/j.bone.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gu X, Zhang S, Zhang T. Abnormal crosstalk between endothelial cells and podocytes mediates tyrosine kinase inhibitor (TKI)-induced nephrotoxicity. Cells 10: 869, 2021. doi: 10.3390/cells10040869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sato W, Tanabe K, Kosugi T, Hudkins K, Lanaspa MA, Zhang L, Campbell-Thompson M, Li Q, Long DA, Alpers CE, Nakagawa T. Selective stimulation of VEGFR2 accelerates progressive renal disease. Am J Pathol 179: 155–166, 2011. [Erratum in Am J Pathol 179: 2674–2675, 2011]. doi: 10.1016/j.ajpath.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin SL, Chang FC, Schrimpf C, Chen YT, Wu CF, Wu VC, Chiang WC, Kuhnert F, Kuo CJ, Chen YM, Wu KD, Tsai TJ, Duffield JS. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol 178: 911–923, 2011. doi: 10.1016/j.ajpath.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lavoz C, Rodrigues-Diez RR, Plaza A, Carpio D, Egido J, Ruiz-Ortega M, Mezzano S. VEGFR2 blockade improves renal damage in an experimental model of type 2 diabetic nephropathy. JCM 9: 302, 2020. doi: 10.3390/jcm9020302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kang DH, Anderson S, Kim YG, Mazzalli M, Suga S, Jefferson JA, Gordon KL, Oyama TT, Hughes J, Hugo C, Kerjaschki D, Schreiner GF, Johnson RJ. Impaired angiogenesis in the aging kidney: vascular endothelial growth factor and thrombospondin-1 in renal disease. Am J Kidney Dis 37: 601–611, 2001. doi: 10.1053/ajkd.2001.22087. [DOI] [PubMed] [Google Scholar]
- 66. Mencke R, Hillebrands JL; NIGRAM consortium. The role of the anti-ageing protein Klotho in vascular physiology and pathophysiology. Ageing Res Rev 35: 124–146, 2017. doi: 10.1016/j.arr.2016.09.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1−S6 and Supplemental Tables S1 and S2: https://doi.org/10.6084/m9.figshare.20113385.v1.