

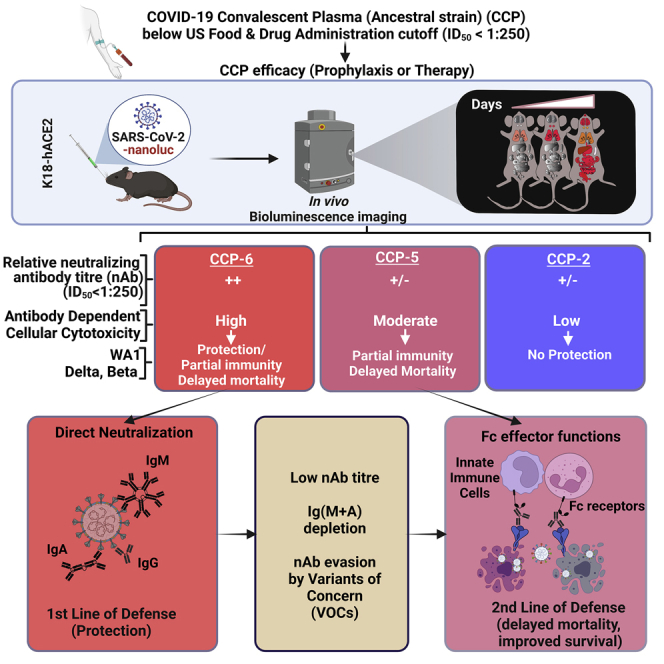
Summary
COVID-19 convalescent plasmas (CCPs) are chosen for plasma therapy based on neutralizing titers and anti-Spike immunoglobulin levels. However, CCP characteristics that promote SARS-CoV-2 control are complex and incompletely defined. Using an in vivo imaging approach, we demonstrate that CCPs with low neutralizing (ID50 ≤ 1:250), but moderate to high Fc-effector activity, in contrast to those with poor Fc function, delay mortality and/or improve survival of SARS-CoV-2-challenged K18-hACE2 mice. The impact of innate immune cells on CCP efficacy depended on their residual neutralizing activity. Fractionation of a selected CCP revealed that IgG and Ig(M + A) were required during therapy, but the IgG fraction alone sufficed during prophylaxis. Finally, despite reduced neutralization, ancestral SARS-CoV-2-elicited CCPs significantly delayed Delta and Beta-induced mortality suggesting that Fc-effector functions contribute to immunity against VOCs. Thus, Fc activity of CCPs provide a second line of defense when neutralization is compromised and can serve as an important criterion for CCP selection.
Keywords: Fc-effector, convalescent plasma, ADCC, COVID-19, neutrophils, macrophages, SARS-CoV-2, IgG, IgM, IgA
Graphical abstract
Highlights
-
•
COVID-19 convalescent plasma (CCP) therapy with robust Fc function can protect mice
-
•
Fc activity of CCPs can serve as secondary defense when neutralization is compromised
-
•
Fc functions facilitate cross-reactive immunity against SARS-CoV-2 variants of concern
-
•
Fc functions can serve as one of the key profiles when selecting CCPs for therapy
SARS-CoV-2 control by COVID-19 convalescent plasmas (CCPs) is complex and poorly understood. Here Ullah et al. show that CCPs with low neutralizing activity but robust Fc function can delay SARS-CoV-2-induced disease progression and promote cross-reactive immunity against variants of concern, suggesting that Fc functions can demarcate CCPs for therapeutic use.
Introduction
Convalescent plasma (CP) therapy is a first line of treatment when the human population lacks pathogen-specific immunity and treatment options are limited.1,2,3,4 CP therapy may be of particular interest for the aged, immune-suppressed cancer or transplant patients where vaccination fails to elicit protective antibody responses as well as in co-morbid populations where vaccination cannot be used.5,6,7,8 Unlike vaccines and monoclonal antibodies (mAbs), CP therapy requires limited development and a standard infrastructure for blood collection. This makes CP therapy rapidly deployable even under low resource settings, especially in developing nations, which constitute most of the world population where the alternative-mAb based therapies are prohibitively expensive.9 CP therapy is adaptable to emerging SARS-CoV-2 variants of concern (VOCs) when the plasma is sourced from convalescent human subjects infected with homologous variants. In addition, the polyclonal nature of CPs makes them relatively effective against heterologous variants. In contrast, targeted immune therapies need development from scratch to be specific, as is currently the case with like mRNA vaccines and neutralizing antibody (nAb) cocktails requiring reengineering to target newly arising variants such as the SARS-CoV-2 Omicron and its sublineages.10,11,12,13,14,15 Therefore, CP therapy remains a go-to countermeasure for emerging and future pathogens with pandemic potential.
Currently, the choice of COVID-19 convalescent plasma (CCP) for therapy is driven by high titers of anti-SARS-CoV-2 Spike immunoglobulin (Ig)G (Median titer: 1:3,200) and neutralization titer (inhibitory dilution [ID50 > 1:250]).16 Neutralizing antibodies (nAbs) in CCPs can inactivate virus and reduce inflammation to help mitigate SARS-CoV-2-induced acute respiratory disorder syndrome.17 SARS-CoV-2 neutralizing titers are therefore a critical criterion for selection of CCPs for therapy.18,19 The emerging consensus from large randomized clinical trials is that CCPs should contain the highest neutralizing titers possible and be transfused early in the disease course before patients require greater supportive therapies to increase the likelihood of benefit.19,20,21
Due to their inherent complexity, the plasma milieu may not necessarily produce the antiviral activities required to protect recipients even when beneficial nAbs are present. In the CONCOR-1 trial, higher levels of IgG specific for the membrane-bound Spike with disproportionally low neutralizing and Fc-effector functions were associated with worse outcomes.21 Therefore, additional signatures of CCP that track with positive outcomes are required to better characterize the clinical utility and choice of CCP for plasma therapy.22 In addition to direct neutralization, antibodies (Abs) can use their Fc domain for mediating effector functions by interacting with Fc receptors (FcRs) expressed on innate immune cells.23,24,25,26 FcR engagement on neutrophils, monocytes, and natural killer (NK) cells can elicit multiple activities including the clearance of viral particles through phagocytosis (antibody-dependent phagocytosis; ADP) and cytotoxic killing of virus-infected cells (antibody-dependent cellular cytotoxicity; ADCC).27,28,29,30 Several studies have now shown that purified monoclonal SARS-CoV-2 nAbs rely on Fc-effector functions for improved in vivo efficacy especially during therapy.24,26,31,32 Moreover, introducing Fc-FcγR binding enhancer mutations (GASDALIE) have improved in vivo nAb efficacy and reduced dosage.23,31,33,34,35 Fc-effector functions of Abs elicited by prior infection or vaccination were suggested as a correlate for continued immunity against emerging VOCs despite compromised neutralization.35,36,37,38 Given that the predominant proportion of Spike-binding antibodies in plasma elicited during natural infection are non-nAbs (nnAbs),34 their contribution to the overall Fc- mediated targeting of SARS-CoV-2 virions and virus-infected cells is expected to be significant. Moreover, nnAbs, through Fc function, may synergize with nAbs through Fc-effector functions to improve overall efficacies especially in the context of polyclonal plasma.23,35 Thus, given the emerging evidence of Fc-mediated Ab effector functions in both protection and disease caused by SARS-CoV-2, the presence of robust Fc-effector activities may serve as an additional criterion to select CCPs for therapeutic applications. However, while the presence of SARS-CoV-2-specific Abs in CCPs that elicit Fc-mediated effector activity have independently correlated with therapeutic benefits,21 or continued protection against VOCs,37,38 direct in vivo evidence beyond correlation is lacking.
Here we used the K18-hACE2 mouse model with bioluminescence imaging (BLI) to track SARS-CoV-2 infection for screening CCPs with neutralizing titers below the US Food and Drug Administration (FDA) cutoff (ID50 <1:250) but varying Fc-effector activity. In contrast to CCPs with poor Fc-effector activity that did not confer any protection, CCPs with moderate to robust Fc function delayed mortality and/or improved survival during lethal challenge with homologous WA1 strain under both prophylaxis and therapy. As neutralizing activity among the analyzed CCPs decreased, Fc-effector functions became more important for in vivo efficacy. Depletion of Ab classes from a selected CCP revealed the importance of IgG as well as Ig(M + A) fractions for maximal in vivo efficacy during therapy while the IgG fraction alone sufficed for prophylaxis. However, IgG-driven Fc-effector functions were crucial for prophylaxis in the absence of Ig(M + A). Furthermore, ancestral SARS-CoV-2-elicited CCPs with moderate to robust Fc function delayed disease progression and/or improved survival during Delta and Beta VOCs infection despite low neutralizing activity, demonstrating the importance of polyclonal Fc-effector functions as a second line of defense in cross-immunity against VOCs. These data make a compelling case for the relevance of Fc-effector activities when assessing CCP therapeutic potency and suggest that it could potentially serve as an additional criterion for selection.
Results
SARS-CoV-2-challenged K18-hACE2 mice allow investigation of CCPs with beneficial Fc-effector activities during prophylaxis and therapy
To evaluate if Fc-effector functions constitute a beneficial CCP profile, we selected CCPs collected during the first wave of COVID-19 with low neutralizing inhibitory dilutions (ID50 ≤ 1:250; below the FDA cutoff)16,39 and analyzed their ADCC activity using an in vitro assay (Figure 1A). CCPs were arbitrarily graded (represented in blue to red color scale) into low (<5%), moderate (15%–25%) and high (>30%) based on their ADCC activity. Since variation in the relative neutralizing titer of CCPs can influence outcomes despite being below FDA cutoff, we selected CCP-2 (low ADCC, nAb ID50 titer = 1:25 [+/−]), CCP-3 (moderate ADCC and nAb ID50 titer = 1:70 [+]), CCP-5 (moderate ADCC, nAb ID50 titer = 1:25 [+/−]), and CCP-6 (high ADCC, nAb ID50 titer = 1:160 [++]) for comparative in vivo efficacy analyses using BLI.24 We first prophylactically treated K18-hACE2 mice with CCPs before challenge with homologous SARS-CoV-2 WA1 expressing the nanoluc luciferase (nLuc) (Figure 1B). Temporal BLI imaging and quantification of nLuc signals to monitor virus replication in whole body and brain revealed that prophylaxis with CCP-2 did not prevent SARS-CoV-2 WA1 nLuc infection and subsequent virus spread compared with isotype hIgG1-admnistered mice (mock) (Figures 1C–1E). CCP-3 and CCP-5 prophylaxis allowed 50% of the animals in the cohort to control infection with significant delay in virus replication kinetics and neuroinvasion (9–10 dpi versus 4–6 dpi) in the remaining mice that succumbed to infection. In contrast, CCP-6-prophylaxis controlled virus infection, as nLuc signals were undetectable in challenged mice. In accordance with the BLI, mice subjected to isotype and CCP-2 prophylaxes suffered gradual weight loss and succumbed to infection by 6 dpi, whereas those under CCP-6 prophylaxis did not lose any weight and demonstrated 100% survival (Figures 1F and 1G). Further, 50% of the mice that survived during CCP-3- or 5-prophylaxis began regaining lost body weight by 9 dpi, while the remaining mice showed a 3- to 4-day delay in SARS-CoV-2-induced mortality. nLuc signals measured after necropsy in isolated target organs (lung, brain, and nose) corresponded to viral loads (N mRNA expression, nLuc activity) with decreases seen in organs of surviving mice under CCP-3-, 5-, or 6-prophylaxis compared with CCP-2 or hIgG1-administered cohorts (Figures S1A–S1C and 1H). Analyses of inflammatory cytokine mRNA expression (Il-6, Ccl2, Cxcl10, and Ifng) in target organs revealed a 10- to 1,000-fold induction in mice under CCP-2 prophylaxis compared with uninfected mice (Figures 1I and 1J). Although cytokine mRNA levels in organs of mice under CCP-3- or 5-prophylaxis were overall not significantly lower than those pretreated with hIgG1 or CCP-2, the surviving mice in the cohort showed a distinctively diminished induction profile. In contrast, cytokine mRNA expression in CCP-6 pretreated animals were at basal levels, indicating protection from SARS-CoV-2 infection. Our analyses based on CCP-6 suggested that both neutralization and Fc function likely contribute to protection. In addition, comparative efficacy of CCP-2 with CCP-5 (0% versus 50% survival) with similar low neutralizing activity (ID50 = 1:25, +/−) but differing in ADCC activities (1.3% versus 22.66%) suggested an important contribution of Fc activity even during prophylactic interventions.
Figure 1.

In vivo efficacies of selected CCPs in K18-hACE2 mice against lethal SARS-CoV-2 challenge during prophylaxis
(A) WA1-neutralizing activity (left y axis) of indicated CCPs plotted as inverse of plasma inhibitory dilution (ID50) that reduces FFUs by 50% using Vero E6 cells as targets. The right y axis shows %ADCC (low to high color-coded in blue to red scale) in the presence of CCP using a 1:1 ratio of parental CEM.NKr cells and CEM.NKr.Spike cells as target cells while PBMCs from uninfected donors were used as effector cells.
(B) Experimental design for screening in vivo efficacy of indicated CCPs delivered intraperitoneally (i.p.) under prophylaxis (−1 dpi) in K18-hACE2 mice intranasally (i.n.) challenged with 1 × 105 FFU WA1 SARS-CoV-2-nLuc. hIgG1-treated mice were used as control (Mock).
(C) Representative BLI images of SARS-CoV-2-nLuc-infected mice in ventral (v) and dorsal (d) positions for an experiment as in (B). Scale bars denote radiance (photons/s/cm2/steradian).
(D and E) Temporal quantification of nLuc signal as flux (photons/s) computed non-invasively in indicated tissues.
(F) Temporal changes in mouse body weight with initial body weight set to 100%. Cross symbol, death.
(G) Kaplan-Meier survival curves of mice (n = 4–7 per group) statistically compared by log-rank (Mantel-Cox) test.
(H) Viral loads (nLuc activity/mg) in indicated tissue measured on Vero E6 cells as targets. Undetectable virus amounts were set to 1.
(I and J) Fold change in indicated cytokine mRNA expression in brain and lung tissues. The data were normalized to Gapdh mRNA expression in the same sample and that in uninfected mice after necropsy. CCP classification for associated %ADCC (Fc) are shown as low (L), Moderate (M), and High (H). Relative nAb titer of CCPs (ID50 < 1:250) are shown as ++, +, and +/−. Each curve in (D–F) represents an individual mouse. Data in (D–J) are from two independent experiments and n = 2–4 mice per group. Grouped data in (D–F) and (H–J) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for group comparisons to mock controls are shown in black, with convalescent plasma CCP-2 are shown in blue, with CCP-3 are shown in purple, CCP-5 are shown light red. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; Mean values ± SD are depicted. See also Figure S1
We next investigated the ability of selected CCPs to clear established infection (therapeutic mode) by treating mice with SARS-CoV-2-WA1 nLuc at 2 dpi (Figure 2A). Quantification of nLuc signals after temporal BLI revealed that therapy with the CCP-2 or hIgG1 did not control the expanding SARS-CoV-2 replication in the lungs and allowed virus dissemination into the brain in K18-hACE2 mice (Figures 2B–2D). Though CCP-3 or -5 treatment reduced virus replication in mice compared with control cohorts, SARS-CoV-2 eventually invaded the brain with delayed kinetics at 8 dpi. In contrast, mice treated with CCP-6 cleared pre-established infection in the lungs by 8 dpi. Remarkably, despite detectable neuroinvasion at 6 dpi, CCP-6 treatment controlled and subsequently cleared virus in the brain of infected animals by 10 dpi (Figures 2B and 2D). Accordingly, body weight analyses and survival experiments showed that CCP-2 and mock-treated mice lost ∼20% of their starting body weight and succumbed to infection by 6 dpi while all CCP-6-treated mice regained body weight and survived (Figures 2E and 2F). Body weight loss and subsequent mortality were delayed by 2 or 2–6 days, respectively, in 75% of mice treated with CCP-3 or -5. The relatively better prognosis with CCP-5 compared with CCP-3 despite the lower neutralizing activity may be due to a better nnAb-mediated Fc function (22.66% versus 16.17% ADCC; p < 0.0045, non-parametric Mann-Whitney test). An overall decrease in nLuc flux, viral loads (N mRNA expression, nLuc activity) and inflammatory cytokine mRNA expression was also seen in individual target tissues (nose, lung brain) in CCP-3, -5, or -6-treated animals, especially in the surviving mice when compared with mock or CCP-2-treated cohorts of mice (Figures 2G–2I and S1D–S1F). Thus, in addition to robust polyclonal Fc-effector function, a threshold level of neutralizing activity such in CCP-6 was needed to completely clear established infections in mice during therapy. However, it is possible to significantly delay viral replication kinetics and extend survival using a low neutralizing CCP such as CCP-5 with moderate Fc-effector functions. Thus, our data showcases the utility of K18-hACE2 mice in rapidly screening CCPs to identify those with net protective profiles for an optimal therapeutic outcome.
Figure 2.

In vivo efficacies of selected CCPs in K18-hACE2 mice against lethal SARS-CoV-2 challenge during therapy
(A) Experimental design for screening in vivo efficacy of indicated CCPs delivered under therapy (+2 dpi, i.p.) in K18-hACE2 mice challenged with 1 × 105 FFU WA1 SARS-CoV-2-nLuc (i.n.). hIgG1-treated mice were used as control (Mock).
(B) Representative BLI images of SARS-CoV-2-nLuc-infected mice in ventral (v) and dorsal (d) positions for experiment as in (A). Scale bars denote radiance (photons/s/cm2/steradian).
(C and D) Temporal quantification of nLuc signal as flux (photons/s) computed non-invasively in indicated tissues.
(E) Temporal changes in mouse body weight with initial body weight set to 100% for experiment. Cross symbol, death.
(F) Kaplan-Meier survival curves of mice (n = 4 per group) statistically compared by log-rank (Mantel-Cox) test.
(G) Viral loads (nLuc activity/mg) in indicated tissue measured after necropsy on Vero E6 cells as targets. Undetectable virus amounts were set to 1.
(H and I) Fold change in indicated cytokine mRNA expression in brain and lung tissues. The data were normalized to Gapdh mRNA expression in the same sample and that in non-infected mice after necropsy. CCP classification for associated %ADCC (Fc) are shown as low (L), Moderate (M), and High (H). Relative nAb titer of CCPs (ID50 < 1:250) are shown as ++, +, and +/−. Each curve in (C–E) represents an individual mouse. Data in (C–I) are from two independent experiments and n = 2 mice per group. Grouped data in (C–E) and (G–I) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for group comparisons to mock controls are shown in black, with convalescent plasma CCP-2 shown in blue, with CCP-3 shown in purple, and CCP-5 are shown in light red. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; mean values ± SD are depicted. See also Figure S1
Innate effector cells contribute to CCP-mediated protection during prophylaxis
CCP potency against SARS-CoV-2 is a combination of its neutralizing and Fc-effector activities. Fc domains of Abs recruit innate immune cells to eliminate infected producer cells and free virus particles. Therefore, we evaluated the contribution of Fc-effector function first during prophylaxis with CCP-3, -5, and -6 by immuno-depleting neutrophils (anti-Ly6G) or macrophages (anti-CSF1R; for CCP-6 alone). Flow cytometry confirmed that ∼98% neutrophils (CD45+CD11b+Ly6G+) in blood or ∼75% of lung-resident macrophages (CD45+CD11b+ Ly6G−Ly6C−CD68+) were depleted following treatment with depleting Abs (Figures S2A–S2D). Depletion of these innate immune cell types on their own did not alter the susceptibility of K18-hACE2 mice to SARS-CoV-2 infection (Figures S2E–S2J).
BLI (non-invasive and post necropsy), body weight, and survival analyses revealed that neutrophil depletion severely compromised virologic control and partial protection offered by CCP-3- or 5-prophylaxis with 100% of mice succumbing to infection by 6–7 dpi (Figures 3A–3F). Neutrophil-depleted CCP-3 and -5 pretreated cohorts were also unable to diminish inflammatory cytokine induction as well as viral loads (in target organs (Figures 3G and 3H and S3A–S3D). In contrast, BLI analyses of mice pretreated with CCP-6 with relatively high neutralizing activity revealed only a transient and weak SARS-CoV-2 replication in the lungs at 4 and 6 dpi that cleared by 10 dpi with immune cell depletion (Figures 3A–3C). In addition, CCP-6 could still prevent virus dissemination to the brain (Figure 3D). However, a transient body weight loss (up to 10%) in K18-hACE2 mice occurred before complete recovery in contrast to mice that were not depleted of these innate cell types (Figure 3E). Post-necropsy analyses (organ flux, tissue viral loads, and inflammatory cytokine mRNA expression) also confirmed virological control at the experimental endpoint (Figures 3G, 3H, and S3A–S3D). However, inflammatory cytokines were significantly higher in the lungs compared with undepleted cohorts. Overall, our data suggested that below a certain amount of neutralizing activity, Fc-mediated innate immune cell engagement played a significant role in partial protection seen during CCP-3- or -5-prophylaxis. In the presence of higher neutralizing activity, such as during CCP-6 prophylaxis, Fc-effectors played a marginal, yet distinctive role in reducing residual infection and inflammation.
Figure 3.

Innate immune cell depletion compromises CCP-mediated immunity against SARS-CoV-2 during prophylaxis in K18-hACE2 mice
(A) Experimental design to test the contribution of macrophages (CD45+Ly6G−Ly6C−CD11b+CD68+) and neutrophils CD45+CD11b+Ly6G+) in K18-hACE2 mice challenged with WA1 SARS-CoV-2-nLuc (1 × 105 FFU, i.n.) and treated prophylactically (i.p.; −1 dpi, 1 mL/20–25 g body weight) with indicated CCPs. αCSF1R or αLy6G mAbs (i.p., 20 mg/kg body weight) were used to deplete macrophages and neutrophils respectively every 48 h starting 2 days before infection. Human and rat isotype mAb-treated cohorts served as controls (Isotype). Animals were followed by BLI every 2 days as indicated.
(B) Representative BLI images of SARS-CoV-2-nLuc-infected mice in ventral (v) and dorsal (d) positions. Scale bars denote radiance (photons/s/cm2/steradian).
(C and D) Temporal quantification of nLuc signal as flux (photons/s) computed non-invasively.
(E) Temporal changes in mouse body weight with initial body weight set to 100%. Cross symbol, death.
(F) Kaplan-Meier survival curves of mice (n = 4 per group) statistically compared by log-rank (Mantel-Cox) test.
(G and H) Fold change in cytokine mRNA expression in brain and lung tissues at the time of death after necropsy. The data were normalized to Gapdh mRNA expression in the same sample and that in uninfected mice after necropsy. Each curve in (C–E) and each data point in (G–H) represents an individual mouse. Data in (C–H) are from two independent experiments and n = 2 mouse per group. Grouped data in (C–E) and (G–H) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for group comparisons to isotype control are shown in black, with CCP-3 to CCP-3+αLy6G shown in purple, with CCP5 to CCP-5+αLy6G shown in light red, CCP-6+αCSF1R shown in green, and with CCP-6 αLy6G shown in red. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; mean values ± SD are depicted. See also Figures S2 and S3
Innate effector cells contribute significantly to CCP potency during therapy
Next, we depleted neutrophils (anti-Ly6G for CCP-3, -5, -6) or macrophages (anti-CSF1R for CCP-6) to analyze the role of innate effector cells during CCP therapy (Figure 4A). Longitudinal BLI analyses and nLuc flux revealed that depletion of either neutrophils or macrophages (for CCP-6-treated mice) significantly compromised CCP-mediated virologic control (Figures 4B–4D and S3E–S3H). Cohorts that underwent innate immune cell depletion during CCP therapy lost 20%–30% of their body weight like isotype-treated mice and succumbed to infection, in some cases with a 1-day delay in death (Figures 4E and 4F). One hundred percent of the mice in CCP-3 or -5-treated cohorts showed virus neuroinvasion when neutrophils were depleted. However, CCP-6 with highest neutralizing activity was able to prevent virus neuroinvasion even in 50% of mice depleted for macrophages or 75% of mice depleted for neutrophils compared with isotype Ab-treated mice (Figures 4B and 4D). These data suggested a higher contribution of macrophages compared with neutrophils in CCP-6-mediated Fc-effector functions during therapy. Innate immune cell depletion compromised all CCP-mediated virologic control, resulting in higher viral loads in the brain and lungs like control cohorts (hIgG1- and rat IgG2A-treated) at experimental endpoints (Figures 4G and 4H). The ability of CCPs to diminish exacerbated expression of inflammatory cytokine mRNAs in the lungs was also significantly compromised when neutrophils or macrophages were depleted (Figure 4H). However, inflammatory cytokines (Ccl2, Cxcl10) in the brain remained under control in CCP-6-treated cohorts reflecting the delay in neuroinvasion compared with isotype Ab-treated cohorts (Figure 4G). These data show that Fc-effector functions mediated by innate immune effector cells significantly contributed to CCP-mediated protection during therapy and were also required to dampen inflammation, especially in the lungs where SARS-CoV-2 established infection.
Figure 4.

Innate immune cells are required to eliminate established SARS-CoV-2 infection during CCP therapy in K18-hACE2 mice
(A) Experimental design to test the contribution of macrophages (CD45+CD11b+CD68+) and neutrophils (CD45+CD11b+Ly6G+) in K18-hACE2 mice therapeutically treated at 2 dpi with indicated CCPs (i.p., 1 mL/20–25 g body weight) after challenge with WA1 SARS-CoV-2-nLuc (i.n., 1 × 105 FFU). αCSF1R or αLy6G mAbs (i.p., 20 mg/kg body weight) were used to deplete macrophages and neutrophils, respectively, every 48 h starting at 0 dpi. Human and/or rat isotype mAb-treated cohorts served as controls (Isotype). The mice were followed by non-invasive BLI every 2 days from the start of infection.
(B) Representative BLI images of SARS-CoV-2-nLuc-infected mice in ventral (v) and dorsal (d) positions. Scale bars denote radiance (photons/s/cm2/steradian).
(C and D) Temporal quantification of nLuc signal as flux (photons/s) computed non-invasively.
(E) Temporal changes in mouse body weight with starting weight set to 100%. Cross symbol, death.
(F) Kaplan-Meier survival curves of mice (n = 4 per group) statistically compared by log-rank (Mantel-Cox) test for experiment as in (A).
(G and H) Fold change in cytokine mRNA expression in brain and lung tissues after necropsy at the time of death. The data were normalized to Gapdh mRNA expression in the same sample and that in non-infected mice after necropsy. Each curve in (C–E) and each data point in (G–H) represents an individual mouse. Data in (C–H) are from two independent experiments and n = 2–3 mice per group. Grouped data in (C–E) and (G–H) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for group comparisons to isotype control are shown in black, with CCP-3 to CCP-3+αLy6G shown in purple, with CCP5 to CCP-5+αLy6G shown in light red, CCP-6+αCSF1R shown in green, and with CCP-6 αLy6G shown in red. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; mean values ± SD are depicted. See also Figure S3
Polyclonal IgGs contribute to protection during CCP-6 prophylaxis
IgM and IgA are mucosal Abs that function as the first line of defense against mucosal pathogens.40 Although not as potent as IgG, multivalent Abs like IgM (pentamer: decavalent) and IgA (dimer: tetravalent) can exhibit enhanced neutralization due to their avidity.41,42,43 To evaluate the contribution of specific Ab classes toward protection, we focused on CCP-6 due its excellent protective profile. We depleted IgG (designated as CCP-6/IgM+A fraction) or Ig(M + A) (designated as CCP-6/IgG fraction) from CCP-6 and confirmed successful depletion of antibody class-subsets by immunoglobulin class-specific ELISA (<99% of IgG or 90%–95% of Ig[M + A]) (Figures S4A and S4B) and flow-cytometric evaluation of class-specific anti-Spike content using Spike-expressing HEK293 cells (Figure S4C). ADCC analyses of the undepleted and depleted CCP-6 fractions revealed that in vitro Fc activities predominantly tracked with CCP6/IgG fraction (Figure S4D). While both fractions displayed SARS-CoV-2 neutralizing activity (Figure S4E), the CCP6/IgG fraction demonstrated ∼2.3-fold higher neutralizing activity than the CCP-6/Ig(M + A) fraction.
We next investigated the anti-SARS-CoV-2 in vivo efficacy of class-depleted plasma fractions during prophylaxis (Figure 5A). Unfractionated CCP-6 was diluted before use to account for the loss in IgG (Equalized IgG) in the CCP-6/IgG fraction incurred during the depletion procedure. Longitudinal BLI revealed that CCP-6/IgG fraction displayed virologic control like undepleted CCP-6 (Equalized IgG) with 100% survival efficacy (Figure 5F) despite a small reduction (<10%) in body weight compared with undepleted plasma (Figures 5E and 5F). In contrast, CCP-6/Ig(M + A) fraction exhibited near-complete loss in CCP-6-mediated protection with uncontrolled virus replication, neuroinvasion, 15%–20% body weight loss, and 100% mortality (Figures 5B–5F and S5A and S5B). Significantly higher viral loads and inflammatory cytokine mRNA expression in target organs reflected the loss of virologic control in mice treated with CCP-6/Ig(M + A) compared with mice treated with unfractionated and CCP-6/IgG plasma (Figures 5G–5I). Thus, polyclonal IgGs predominantly contributed to virologic control and protection during CCP-6 prophylaxis.
Figure 5.

Polyclonal IgGs in CCP-6 predominantly contribute to protection during prophylaxis in SARS-CoV-2-infected K18-hACE2 mice
(A) Experimental design to test in vivo efficacies of CCP-6, CCP-6/Ig(M + A), and CCP-6/IgG fraction (1 mL × 2 i.p. injections, 4 h apart) in SARS-CoV-2-nLuc infected K18-hACE2 mice (i.n., 1 × 105 FFU) under prophylaxis (−1 dpi). For CCP-6 treatment, plasma was diluted to equalize IgG content of IgG fractionated plasma. αLy6G mAb (i.p., 20 mg/kg body weight) was used to deplete neutrophils respectively every 48 h starting 2 days before infection. Mice treated with hIgG1 served as controls (Isotype). The mice were followed by non-invasive BLI every 2 days from the start of infection.
(B) Representative BLI images of SARS-CoV-2-nLuc-infected mice in ventral (v) and dorsal (d) positions. Scale bars denote radiance (photons/s/cm2/steradian).
(C and D) Temporal quantification of nLuc signal as flux (photons/s) computed non-invasively.
(E) Temporal changes in mouse body weight with starting weight set to 100%. Cross symbol, death.
(F) Kaplan-Meier survival curves of mice (n = 4 per group) statistically compared by log-rank (Mantel-Cox) test for experiment as in (A).
(G) Viral loads (nLuc activity/mg) from indicated tissues using Vero E6 cells as targets. Undetectable virus amounts were set to 1.
(H and I) Fold change in cytokine mRNA expression in brain and lung tissues. The data were normalized to Gapdh mRNA expression in the same sample and that in non-infected mice after necropsy. Viral loads (G) and inflammatory cytokine profile (H, I) were determined at the time of death at 6 dpi or 10 dpi for surviving mice after necropsy. Each curve in (C–E) and each data point in (G–I) represents an individual mouse. Data in (C–I) are from are from two independent experiments and n = 2 mice per group. Grouped data in (C–E), (G–I) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for group comparisons with isotype control are shown in black, with IgG equalized CCP-6 shown in cyan, with CCP-6/Ig(M + A) fraction shown in red, with CCP-6/IgG fraction shown in green, and with CCP-6/IgG fractionated under neutrophil depletion shown in orange. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; mean values ± SD are depicted. See also Figures S4 and S5.
To decipher if direct neutralization and/or Fc-mediated innate cell-recruitment contributed to protection during prophylaxis with CCP-6/IgG fraction, we immuno-depleted neutrophils (anti-Ly6G). Compared with the undepleted plasma where innate cells contributed marginally during prophylaxis, neutrophil depletion had a significant impact on protection conferred by CCP-6/IgG fraction (Figures 5B–5F). BLI analyses revealed loss of virologic control with visible infection at 2–4 dpi and dissemination of virus into the brain at 8 dpi (Figures 5B, 5D, S5A, and S5B) with all the mice in the neutrophil-depleted cohort losing weight and succumbing to SARS-CoV-2 challenge, albeit with a delay of 1–3 days (Figures 5E and 5F). These data correlated with increased viral loads in tissues and enhanced inflammatory cytokine mRNA expression in neutrophil-depleted cohorts prophylactically treated with CCP-6/IgG (Figures 5G–5I and S5C). Together, these data suggest a functional interplay between Ig(M + A) and IgG to promote virus neutralization. When Ig(M + A) was depleted, the reliance on Fc functions mediated by effector immune cells over direct neutralization by the IgG fraction was significantly increased for effective virological control. Thus, when neutralization by CCP-6/IgG was insufficient to prevent virus infection, IgG Fc-driven recruitment of innate immune cells acted as a second line of defense to promote infected-cell clearance and control virus replication during prophylaxis.
Polyclonal IgG and Ig(M + A) Fc-effector activities are required for in vivo CCP-6 efficacy during therapy
Longitudinal BLI analyses revealed that the in vivo efficacy of both CCP-6/IgG and CCP-6/Ig(M + A) fractions against SARS-CoV-2 were severely compromised compared with undepleted CCP-6 during therapy (Figures 6A–6D). SARS-CoV-2-nLuc replicated and disseminated to the brain in six out of seven mice in both cohorts that received Ig class-depleted plasma (Figures 6B–6D, 6G, and S6). Although 14% of the mice (one out of seven) in both cohorts survived, body weight and survival analyses showed that mice that received the CCP-6/IgG fraction exhibited decelerated body weight loss and delayed mortality compared with those that received the CCP-6/Ig(M + A) fraction (8 dpi versus 6 dpi) (Figures 6E and 6F). Thus, IgG played a larger role than Ig(M + A) in CCP-6-mediated protection. The capacity of CCP-6-depleted fractions to inhibit tissue virus replication and inflammation was also significantly compromised compared with mice treated with undepleted plasma (Figures 6G–6I). Interestingly, cytokine mRNA expression (Ccl2 and Cxcl10) in the lungs of mice that received depleted plasma fractions were significantly higher than the unfractionated CCP-6-treated or the isotype IgG1-treated cohorts (Figure 6I). These data reveal the contribution of both Ig fractions in dampening inflammatory immune responses. Thus, as with prophylaxis, Ig class-depletion analyses suggest a functional interplay between IgG and Ig(M + A) for optimal in vivo efficacy of CCP-6.
Figure 6.

Antibody classes collaborate to achieve maximal in vivo protection during CCP-6 therapy in SARS-CoV-2-infected K18-hACE2 mice
(A) Experimental design to test in vivo efficacies of CCP-6, CCP-6/Ig(M + A), CCP-6/IgG fraction (1 mL × 2 i.p. injections, 4 h apart) in SARS-CoV-2-nLuc infected mice K18-hACE2 mice (i.n., 1 × 105 FFU) under therapy (+2 dpi). For CCP-6 treatment, plasma was diluted to equalize IgG content of Ig(M + A)-depleted plasma. Mice treated with hIgG1 served as controls (Isotype). The mice were followed by non-invasive BLI every 2 days from the start of infection.
(B) Representative BLI images of SARS-CoV-2-nLuc-infected mice in ventral (v) and dorsal (d) positions. Scale bars denote radiance (photons/s/cm2/steradian).
(C and D) Temporal quantification of nLuc signal as flux (photons/s) computed non-invasively.
(E) Temporal changes in mouse body weight with starting weight set to 100%. Cross symbol, death.
(F) Kaplan-Meier survival curves of mice (n = 7 per group) statistically compared by log-rank (Mantel-Cox) test for experiment as in (A).
(G) Viral loads (FFUs/mg) from indicated tissue using Vero E6 cells as targets. Undetectable virus amounts were set to 1.
(H and I) Fold change in cytokine mRNA expression in brain and lung tissues. The data were normalized to Gapdh mRNA expression in the same sample and that in non-infected mice after necropsy. Viral loads (G) and inflammatory cytokine profile (H, I) were determined at the time of death for mice that succumbed to infection (F) and at 18 dpi for surviving mice. Each curve in (C–E) and each data point in (G–I) represents an individual mouse. Data in (C–I) are from two to three independent experiments n = 2–3 mouse per group. Grouped data in (C–E), (G–I) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for group comparisons to isotype control are shown in black, with IgG-equated CCP-6 shown in cyan, with CCP-6/Ig(M + A) fraction shown in red, and with CCP-6/IgG fraction shown in green. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; mean values ± SD are depicted. See also Figure S6.
Cross-reactive ADCC activity can contribute to immunity against VOCs
Recent in vitro studies suggest that broad Fc-effector functions elicited by prior infection or vaccination may offer continued protection against VOCs despite loss in neutralization.37,38,44 However, if cross-reactive Fc-effector functions can provide in vivo protective efficacy when neutralization is diminished remains unexplored. We sought to extend these observations to in vivo studies using ancestral strain-elicited CCPs against heterologous SARS-CoV-2 VOCs B.1.617.2 (Delta) and B.1.352 (Beta). The neutralizing half maximal inhibitory concentration (IC50) values against Delta compared with WA1 for all other CCPs were not statistically different except for CCP-5, although a trend toward loss in activity was observed (Figure 7A). In contrast, the ability to neutralize Beta VOC compared with WA1 was significantly diminished for all the CCPs tested. ADCC assays to evaluate cross-VOC activity of ancestral SARS-CoV-2-elicited CCPs using human PBMCs showed that all CCPs were significantly less active against Beta VOC, whereas CCP-6 also showed significantly reduced activity against Delta VOC compared with CCP-3 or -5 (Figure 7B).
Figure 7.

Fc-mediated cross-protective efficacy profiles of WA1-elicited CCPs against Delta and Beta VOCs in K18-hACE2 mice
(A) A graph depicting WA1, Delta, and Beta-neutralizing activity of indicated CCPs expressed as 1/inhibitory concentration of plasma (IC50). IC50 was defined as the plasma amount (μL) that reduces FFUs by 50% using Vero E6 cells as targets.
(B) A graph depicting %ADCC activities in the presence of CCP using a 1:1 ratio of parental CEM.NKr cells and CEM.NKr.Spike (WA1, Delta or Beta) cells as target cells, while PBMCs from uninfected donors were used as effector cells.
(C) Experimental design for screening in vivo efficacy of indicated CCPs delivered 1 mL per 20–25 g body weight of mouse intraperitoneally (i.p.) under prophylaxis (−1dpi) and therapeutically (+2 dpi) in K18-hACE2 mice intranasally (i.n.) challenged with 1 × 105 FFU of B.1.617.2 (Delta VOC) or B.1.351 (Beta VOC). PBS-treated mice were used as control (Mock).
(D–G) Temporal changes in mouse body weight with initial body weight set to 100% during CCP prophylaxis (−1 dpi) and therapy (+2 dpi) for experiment as in (C) in mice challenged with Delta and Beta VOC. Cross symbol, death.
(H–K) Kaplan-Meier survival curves of mice (n = 4 per group) statistically compared by log-rank (Mantel-Cox) test.
(L–O) Fold change in SARS-CoV-2 nucleocapsid (N gene) expression in indicated tissue at the time of death or 16 dpi for surviving mice during CCP prophylaxis and therapy for experiment shown in (C). The data were normalized to Gapdh mRNA expression in the same sample and that in non-infected mice after necropsy. Grouped data in (A and B) were analyzed by one-way ANOVA with Dunnett’s multiple comparisons test to determine if Delta and Beta VOC-neutralizing titers or %ADCC in CCPs differed significantly from WA1. Each curve in (D–G) and each data point in (M−P) represent an individual mouse. Data in these from two independent experiments and n = 2–3 mice per group. Grouped data in (D–G), (M−P) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for group comparisons to isotype control are shown in black, with CCP-2-treated cohorts shown as blue, with CCP-2-treated cohorts shown as purple, with CCP-5-treated cohorts shown as light red, and CCP-6-treated cohorts shown in red. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; mean values ± SD are depicted.
We next examined the in vivo efficacy of CCPs in K18-hACE2 mice challenged with Delta and Beta VOCs under prophylaxis (−1 dpi) and therapy (+2 dpi) (Figure 7C). Prophylaxis using CCP-2 with low Fc-effector activities failed to protect against both VOCs, and the mice exhibited body weight loss and death at 6 dpi like mock-treated control animals (Figures 7C, 7D, 7F, 7H, and 7J). In contrast, prophylaxis with CCP-6, that retained considerable Delta VOC-neutralizing activity protected 50% of the mice, delayed mortality by 3–4 days in mice that succumbed to infection, and reduced N mRNA expression significantly in target organs during challenge with Delta VOC (Figures 7C, 7D, 7H, and 7M). CCP-3, with modest neutralizing and Fc activities against Delta, did not improve survival but significantly delayed mortality by 2–5 days and reduced N mRNA expression in the lungs as well as the brain in pretreated animals (Figures 7C, 7D, 7H, and 7M). Notably, CCP-5, with significantly diminished Delta neutralizing potency (IC50WA1 3.5 versus IC50Delta 12.59) but with better ADCC activity than CCP-3 (%ADCCDelta 17.48 versus 12.86; p < 0.0022; non-parametric Mann-Whitney test), demonstrated 25% survival, significantly delayed mortality by 5–7 days, and reduced N mRNA expression in lungs and brain (Figures 7C, 7D, 7H, and 7M). Accordingly, neutrophil depletion abolished the partial immunity offered by CCP-5 and confirmed the contribution of Fc-effector functions in CCP-5 efficacy against Delta VOC during prophylaxis (Figure 7H). CCPs 3, 5, or 6 when administered therapeutically in K18-hACE2 mice were unable to improve survival but significantly delayed body weight loss and mortality compared with mock or CCP-2 treatment (Figures 7E and 7I). Although CCP-6 maintained neutralizing activities, it showed a significant drop in ADCC activity against Delta compared with WA1, which likely resulted in compromised in vivo efficacy. While neutralizing activity was vital, our efficacy data with CCP-5 and -6 indicate a distinct contribution of Fc functions toward Delta VOC immunity during prophylaxis and therapy.
CCP prophylaxis failed to prevent Beta VOC-induced mortality in mice, consistent with all CCPs suffering substantial declines in neutralizing and ADCC activities (Figures 7J and 7L). However, prophylaxis with CCP-3, -5, or -6 significantly delayed weight loss and Beta VOC-induced death by 1–2 days compared with mock or CCP-2 pretreated animals (Figures 7F and 7J). CCP-5 or -6 prophylaxis also reduced N mRNA expression in the lungs of infected mice (Figure 7O). Given that Beta-neutralizing activity was similar in CCP-2 and -5, higher Fc functions likely contributed to delayed weight loss and mortality during CCP-5 prophylaxis (Figures 7A, 7B, 7E, 7J, and 7O). Under therapy, only CCP-6 treatment, which had the highest neutralizing and ADCC activities against Beta, resulted in a significant delay in weight loss and diminished N mRNA expression in mice (Figures 7A, 7B, 7G, and 7O). However, none of the CCPs were effective in improving survival in mice against the highly evasive Beta VOC during therapy (Figure 7L). Thus, while cross-VOC Fc-effector functions distinctly played a role in immunity against VOC by delaying disease progression, contribution from VOC cross-reactive neutralizing activity in CCPs was vital for protection against VOCs.
Discussion
The constituents of CCPs are complex, and it is difficult to predict their in vivo efficacies based solely on neutralizing titers or Spike-specific immunoglobulin content. To navigate the intricacies of CCPs, additional measures of selection that track with in vivo protection are required and important to guide best practices in future infectious disease outbreaks. Furthermore, in vivo models that allow testing CPs with protective profiles can help identify properties suitable for incorporating into and complementing high-throughput screening assays in vitro. Here we combined the highly susceptible K18-hACE2 mouse model of SARS-CoV-2 with BLI to track virus replication for studying efficacies and characteristics of CCPs that contribute to in vivo protection. CCPs with low, moderate, and high ADCC activities were selected with neutralizing activities below the FDA cutoff (ID50 ≤ 1:250) together with innate immune cell depletion to evaluate the contribution of associated Fc-effector activities toward in vivo efficacy (summarized in Table S1). Our data reveal that the extent to which CCP relies on Fc-effector functions for in vivo efficacy is determined by its neutralizing activity during prophylaxis. However, Fc-effector functions played a crucial role during CCP therapy for both virologic control and protection. Exacerbated inflammatory response is one of the hallmarks of SARS-CoV-2-induced disease.45 In addition to virologic control, we found that recruitment of innate immune cells through polyclonal Fc-FcR interactions can dampen the SARS-CoV-2-induced inflammatory response. Thus, CCP-associated polyclonal Fc-effector functions have the potential to mitigate SARS-CoV-2-induced disease.
CCP-mediated Fc-effector function was rarely measured in COVID-19 clinical studies. Given that the CONCOR-1 trial reported only a partial correlation between neutralizing and Fc-effector functions, one can assume that even studies using stringent CCP selection criteria likely used plasma with variable degrees of Fc-effector function.21 Developing the capacity to adapt and disseminate Fc-effector function testing rapidly may be key to its wider use in future pandemics and a more optimal use of CCP, directing those with high neutralizing but low Fc-effector function toward prophylaxis trials while reserving those with both high neutralizing and high Fc-effector functions for the acutely ill.
Our in vivo efficacy analyses in mice under prophylaxis revealed that Fc-effector activities elicited by the ancestral SARS-CoV-2 can be effective in delaying disease progression and death during VOC challenge. These data mirrored our previous analyses where an Fc-enhanced nnAb did not offer complete protection but delayed mortality in mice.23 However, combining Fc-enhanced nnAb with Fc-compromised nAb completely protected mice despite each antibody failing to protect on its own. Thus, cross-reactive Fc-effector functions on their own are likely not enough for complete protection against VOCs. Polyclonal neutralizing activity, although diminished, forms a critical component of the mix with Fc-effector activities to engender protection against VOCs. The Fc-effector functions of CCPs likely augment protection afforded by nAbs and serve as a second line of defense against neutralization-resistant VOCs. Overall, our in vivo analyses endorse inclusion of Fc-effector activities in addition to neutralization as additional criteria to select CCPs for therapeutic applications. Several high-throughput in vitro assays, including those for measuring Fc-effector functions, exist that can examine multiple signatures of CCPs.21,22,46 A rapid in vivo efficacy analyses can complement these assays to navigate complex CCP characteristics for identifying those with net protective profiles. Demarcating plasma signatures that track with protective or detrimental effects will be key to the success of CP therapy for future infectious disease outbreaks and pandemics.
Limitations of the study
K18-hACE2 mice are highly susceptible to SARS-CoV-2 infection.47,48,49,50,51 This model provides a practical way to rapidly navigate through the various activities of CCP and identify those that contribute to protection, overcoming the limitations of in vitro assay-driven plasma analyses that cannot predict in vivo effects. While mice are suitable to evaluate Fc-effector functions of human Abs as mouse FcγRs bind with similar affinities to human IgG, specific innate immune Fc-effector cells differ between mice and humans.52 In mice, FcγRIV expressed on neutrophils and monocytes dominate IgG-driven Fc-effector functions, while in humans, FcγRIIIA expressing NK cells play a prominent role as effector cells.24,26,53,54,55 Our in vivo data in mice indicate that CCPs with low neutralizing activity can significantly delay mortality primarily due to associated Fc-effector functions. However only a clinical trial using CCPs selected based on ADCC activity can provide more insights into the relative importance of measuring neutralization and ADCC activities for optimal therapeutic efficacy.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Fc block anti mouse-CD16/CD32 (93) | BioLegend Inc | Cat # 101302; RRID: AB_312801 |
| PE anti-mouse CD11b (M1/70) | BioLegend Inc | Cat # 101207; RRID: AB_312790 |
| APC/Cy7 anti-mouse Ly-6C (HK1.4) | BioLegend Inc | Cat #128025; RRID: AB_10643867 |
| APC/Cy7 anti-mouse Ly-6G (Clone 1A8) | BioLegend Inc | Cat #127617; RRID: AB_1877262 |
| Alexa Fluor® 488 anti-mouse Ly-6G (Clone 1A8) | BioLegend Inc | Cat # 127626; RRID: AB_2561340 |
| APC Rat anti-mouse CD45 (30-F11) | BD-Pharmingen | Cat # 559864; RRID: AB_398672 |
| Alexa Fluor® 488 anti-mouse CD45 (Clone 30-F11) | BioLegend Inc | Cat # 103122; RRID: AB_493531 |
| InVivoMAb anti-mouse LY6G (clone: 1A8) for neutrophil depletion | Bio X Cell | Cat # BE0075-1; RRID: AB_1107721 |
| InVivoMAb anti-mouse CSF1R (CD115) for macrophage depletion | Bio X Cell | Cat # BE0213; RRID: AB_ 2687699 |
| InVivoMab rat IgG2a clone C1.18.4; Isotype controls for macrophage and neutrophil depletion | Bio X Cell | Cat # BE0085; RRID: AB_1107771 |
| InVivoMAb human IgG1 isotype control | Bio X Cell | Cat # BE0297; RRID: AB_2687817 |
| Goat anti-Human IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat # A-21445; RRID: AB_2535862 |
| Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat # A-21235; RRID: AB_2535804 |
| Goat anti-Human IgG Fc Cross-Adsorbed Secondary Antibody, HRP | Invitrogen | Cat # A-18823; RRID: AB_2535600 |
| Goat Anti-human IgM (μ-chain specific) 1 mg | Sigma-Aldrich | I01759-1MG |
| Goat anti-human IgA α chain spécific | Jackson ImmunoResearch | 109-005-011 |
| Human IgA subclasses standard serum | Cedarlane | NOR-04 |
| Peroxidase AffiniPure F(ab')₂ Fragment Goat Anti-Human Serum IgA, α chain specific | Jackson ImmunoResearch | 109-036-011 |
| Goat anti-Human IgG Fc specific | Jackson ImmunoResearch | 109-005-098 |
| Serum Human Standard à 9.5 mg/mL | Cedarlane | NOR-01 |
| Peroxidase AffiniPure Goat Anti-Human IgG (H + L) (min X Bov, Hrs, Ms Sr Prot) | Jackson ImmunoResearch | 109-035-088 |
| Goat anti-human IgM Fc specific | Jackson ImmunoResearch | 109-005-129 |
| Low control human serum | Cedarlane | 35-S4321H000-L4 |
| Peroxidase AffiniPure Goat Anti-Human IgA + IgG + IgM (H + L) | Jackson ImmunoResearch | 109-035-064 |
| IgM from human serum | Sigma Aldrich | I-8260 |
| Deposited data | ||
| Raw data used to generate all graphs shown in Figures | This manuscript | Mendeley Data, V1, https://doi.org/10.17632/xx7j82dv97.1 |
| Bacterial and Virus Strains | ||
| SARS-CoV-2-nLuc (strain 2019-nCoV/USA_WA1/2020) | Craig B Wilen (Yale University) | K. Plante and Pei-Yong Shi, World Reference Center for Emerging Viruses and Arboviruses, University of Texas Medical Branch) |
| SARS-CoV-2 USA-WA1/2020 | BEI resources | Cat # NR-52281 |
| SARS-CoV-2, Isolate hCoV-19/USA/PHC658/2021 (Delta Variant) | BEI resources | Cat # NR-55611 |
| SARS-CoV-2, Isolate hCoV-19/USA/MD-HP01542/2021 | BEI resources | Cat # NR-55282 |
| Biological samples | ||
| CCP-1 (Age:50, Male, AB Rh+, 50 dpi) | This manuscript | N/A |
| CCP-2 (Age:58, Male, A Rh+, 78 dpi) | This manuscript | N/A |
| CCP-3 (Age:49, Male, O Rh+, 77 dpi) | This manuscript | N/A |
| CCP-4 (Age:23, Female, O Rh+, unknown dpi) | This manuscript | N/A |
| CCP-5 (Age: 30, Male, O Rh-, 30 dpi) | This manuscript | N/A |
| CCP-6 (Age: 51, Male, A Rh+, 69 dpi) | This manuscript | N/A |
| Primary human peripheral blood mononuclear cells (PBMCs) | FRQS AIDS network | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Liberase TL Research Grade | Sigma-Aldrich | Cat# 5401020001 |
| Dispase (5U/mL) | STEMCELL technologies | Catalog # 07913 |
| DNAse I recombinant, RNAse-free | Roche | Ref # 04716728001 |
| Gibco™ RPMI 1640 medium | Thermo Fisher Scientific | Cat # 11875093 |
| Gibco™ Dulbecco’s modified Eagle’s medium (DMEM) | Thermo Fisher Scientific | Cat # 11965118 |
| Gibco™ MEM Non-essential amino acid (NEAA) solution | Thermo Fisher Scientific | Cat # 11140050 |
| Gibco™ Penicillin-streptomycin solution (10,000 U/mL) | Thermo Fisher Scientific | Cat # 15140122 |
| Gibco™ Dulbecco’s Phosphate Buffered Saline (DPBS) | Thermo Fisher Scientific | Cat # 14190144 |
| Gibco™ L-Glutamine (200mM) | Thermo Fisher Scientific | Cat # 25030081 |
| Gibco™ 0.05% Trypsin-EDTA, phenol red | Thermo Fisher Scientific | Cat # 25300054 |
| Fetal bovine serum | Atlanta Biologicals | Cat #S11550 |
| eBioscience™ RBC Lysis Buffer (1X) | Invitrogen | Cat # 00-4333-57 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat# A9647-100G CAS: 9048-46-8 |
| Accutase | BioLegend Inc | Cat # 423201 |
| 0.05% Trypsin-EDTA (1X) | Life Technologies | Cat # 25300–054 |
| K3 EDTA 15% Solution | Fisher Scientific | Cat # BD 366450 |
| Sodium pyruvate (100 mM) | Life technologies | Ref # 11360–070 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat # M3148 |
| L-Glutamine (200mM) | Life technologies | Ref # 25030–081 |
| Tris-buffered saline (TBS) | Thermo Fisher Scientific | Cat # BP24711 |
| BD Cytofix/Cytoperm™ | BD Biosciences | Cat # 554722 |
| BD Perm/Wash™ | BD Biosciences | Cat # 554723 |
| Passive lysis buffer | Promega | Cat #E1941 |
| Paraformaldehyde (16% PFA) | Electron Microscopy Sciences | Cat # 19200 CAS: 30525-89-4 |
| Rat serum | Stemcell Biotechnologies | Cat # 13551 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat #D2650-5X5ML CAS: 67-68-5 |
| Sodium azide | Sigma-Aldrich | Cat # S-8032 EC No: 247-852-1 |
| Glycine | American Bioanalytical | Cat # AB00730-01000 CAS: 56-40-6 |
| The PEG-it Virus precipitation solution (5X) | System Bioscience | Cat # LV810A-1 |
| Avicel® Pharma Grade | FMC Honko et. al., 202056 |
Cat # RC-581 NF |
| Puromycin dihydrochloride | Millipore Sigma | Cat #P8833 |
| D-Luciferin potassium salt | Thermo Fisher Scientific | Cat #L2916 |
| LIVE/DEAD Fixable AquaVivid Cell Stain | Thermo Fisher Scientific | Cat #L34957 |
| Cell proliferation dye eFluor670 | Thermo Fisher Scientific | Cat # 65-0840-85 |
| Cell proliferation dye eFluor450 | Thermo Fisher Scientific | Cat # 65-0842-85 |
| SARS-CoV-2 S2 ectodomain C-His tag protein | BEI Resources | NR-53799 |
| SIGMAFAST OPD | EMD Millipore | Cat #P9187 |
| Ethanolamine | Sigma Aldrich | E0135-100mL |
| Sodium chloride 5M | Sigma Aldrich | 59222C-500mL |
| Hydrochloric acid | Biolab | 351285–212 |
| Sodium acetate | Sigma Aldrich | S2889-250g |
| Carbonate, 0.5M buffer soln., pH 9.6 250mL | Fisher Scientific | AAJ62610AK |
| NHS Act Sepharose® 4 Fast Flow | Sigma Aldrich | GE17-0906-01 |
| Peptide M/Agarose 2 mL | Cedarlane | GEL-PDM-2 |
| Sodium bicarbonate | Sigma Aldrich | S6014 |
| Sodium carbonate | Sigma Aldrich | S2127 |
| Tween-20 | Fisher Scientific | BP337-500 |
| Casein, Hammarsten bovine | Sigma Aldrich | E0789-500g |
| TMB soluble reagent high sensitivity | ESBE Scientific | SCY-TM4999 |
| H2SO4 10N | Fisher Scientific | SA200-1 |
| Critical commercial assays | ||
| Nano-Glo Luciferase Assay System (nanoluc substrate) | Promega | Cat #N1120 |
| Pierce™ Gaussia Luciferase Glow Assay Kit | Thermo Fisher Scientific | Cat # 16160 |
| KAPA SYBR FAST qPCR Master Mix (2X) Kit | KAPA Biosystems | Cat # KK4600 and KK4601 |
| Ambion DNase I (RNase-free) | Thermo Fisher Scientific | Cat # AM2222 |
| RNeasy Mini Kit (50) | Qiagen | Cat #/ID 74104 |
| iScript advanced cDNA kit | Bio Rad | Cat #1725038 |
| iQ Multiplex Powermix | Bio Rad | Cat # 1725848 |
| iScript™ cDNA Synthesis Kit | Bio Rad | Cat # 95047–100 |
| Protein G HP SpinTrap | Cytiva | 28903134 |
| Experimental models: Cell lines | ||
| Vero E6 (female, Chlorocebus sabaeus) | ATCC | Cat # CRL-1586; RRID: CVCL_0574 |
| Vero E6-TMPRSS2 (female, Chlorocebus sabaeus) | Craig B. Wilen, Yale University | N/A |
| HEK293 (female, Homo sapiens) | ATCC | Cat # CRL-1573; RRID: CVCL_0045 |
| HEK293T (female, Homo sapiens) | ATCC | Cat # CRL-3216; RRID: CVCL_0063 |
| 293T-ACE2 (female, Homo sapiens) | Prevost et al., 2020 | N/A |
| Cf2Th (female, Canis lupus familiaris) | ATCC | Cat # CRL-1430; RRID: CVCL_3363 |
| CEM.NKr-CCR5+ (female, Homo sapiens) | NIH AIDS Reagent Program | Cat # 4376; RRID: CVCL_X623 |
| CEM.NKr-Spike (female, Homo sapiens) | Anand et al., 2021 | N/A |
| CEM.NKr-Delta-Spike (female, Homo sapiens) | This paper | N/A |
| CEM.NKr- Beta-Spike (female, Homo sapiens) | This paper | N/A |
| TZM-bl (female, Homo sapiens) | NIH AIDS Reagent Program | Cat # 8129; RRID: CVCL_B478 |
| THP-1 (male, Homo sapiens) | ATCC | Cat # TIB-202; RRID: CVCL_0006 |
| Experimental models: Organisms/strains | ||
| B6.Cg-Tg(K18-ACE2)2Prlmn/J (males and females); 6–12 weeks old | The Jackson Laboratory | Stock No: 034860 RRID:IMSR_JAX:034860 |
| Oligonucleotides | ||
| SARS-CoV-2 N F: 5′-ATGCTGCAATCGTGCTACAA-3′ | Yale School of Medicine, W. M. Keck Foundation, Oligo Synthesis Resource | N/A |
| SARS-CoV-2 N R: 5′-GACTGCCGCCTCTGCTC-3′ | Yale School of Medicine, W. M. Keck Foundation, Oligo Synthesis Resource | N/A |
| FAM-Gapdh | Bio Rad | Cat # 12001950 |
| HEX-IL6 | Bio Rad | Cat # 10031228 |
| TEX615-CCL2 | Bio Rad | Cat # 10031234 |
| Cy5-CXCL10 | Bio Rad | Cat # 10031231 |
| Cy5.5-IFNg | Bio Rad | Cat # 10031237 |
| Transgene Forward: GAC CCC TGA GGG TTT CAT ATA G | Yale School of Medicine, W. M. Keck Foundation, Oligo Synthesis Resource | #53437, Genotyping primers for K18-hACE2 mice. The Jackson Laboratory |
| Common: CAC CAA CAC AGT TTC CCA AC | Yale School of Medicine, W. M. Keck Foundation, Oligo Synthesis Resource | #53438, Genotyping primers for K18-hACE2 mice. The Jackson Laboratory |
| Wildtype forward: AAG TTG GAG AAG ATG CTG AAA GA | Yale School of Medicine, W. M. Keck Foundation, Oligo Synthesis Resource | #53439, Genotyping primers for K18-hACE2 mice. The Jackson Laboratory |
| Recombinant DNA | ||
| pNL4.3 R-E− Luc | NIH AIDS Reagent Program | Cat # 3418 |
| pSVCMV-IN-VSV-G | Lodge et al., 199757 | N/A |
| Lentiviral packaging plasmids (pLP1, pLP2) | Liu et al., 201358 | N/A |
| pLenti-C-mGFP-P2A-Puro-ACE2 | OriGene | Cat # RC208442L4 |
| pIRES2-eGFP vector | Clontech | Cat # 6029–1 |
| pLTR-Tat | Finzi et al., 201059 | N/A |
| SARS-CoV-2 Spike (B.1.617.2) Gene Lentiviral ORF cDNA clone expression plasmid (Codon Optimized), C-GFPSpark tag (Delta) | SinoBiological | Cat # VG40804-ACGLN |
| SARS-CoV-2 (2019-nCoV) Spike (B.1.351) Gene Lentiviral ORF cDNA clone expression plasmid (Codon Optimized), C-GFPSpark tag (Beta) | SinoBiological | Cat # VG40772-ACGLN |
| Software and algorithms | ||
| Accuri CSampler software | BD Biosciences | |
| FlowJo v10 | Treestar | RRID:SCR_008520 |
| Adobe Photoshop CC | Adobe Systems Inc | RRID:SCR_014199 |
| Adobe Illustrator CC | Adobe Systems Inc | RRID:SCR_010279 |
| BioRender (schematics in figures) | BioRender.com | RRID:SCR_018361 |
| CFX MaestroTM Software (qPCR analyses) | Bio-rad Inc | RRID:SCR_018064 |
| Graphpad Prism v9.4.1 | GraphPad Software | RRID:SCR_002798 |
| Living Image software | Perkin Elmer | http://www.perkinelmer.com/catalog/category/id/living%20image%20software RRID:SCR_014247 |
| Gen5 microplate reader and imager software | Biotek | RRID:SCR_017317 |
| Other | ||
| TriStar LB 941 Multimode Microplate Reader and Luminometer | BERTHOLD TECHNOLOGIES GmbH & Co. KG | Mothes and Finzi Lab |
| BD Biosciences C6 Accuri Flow Cytometer, | BD Biosciences | Yale, MMPATH, Central Facility RRID:SCR_019591 |
| BD LSR II Flow Cytometer | BD Biosciences | https://medicine.yale.edu/immuno/flowcore/ |
| C1000 Touch thermal cycler | Bio-Rad | RRID:SCR_019688 |
| CFX Connect™ Real-Time PCR Detection System | Bio-Rad | RRID:SCR_018064 |
| Nanodrop Spectrophotometer ND-1000 | Thermo Fisher Scientific | RRID:SCR_016517 |
| 27G × ½’’ insulin syringe with needle | TERUMO | Cat # SS∗05M2713 |
| 31G insulin syringe | BD Biosciences | Cat # 328468 |
| 70 μm Nylon cell strainer | FALCON | Cat # 352350 |
| Acrodisc 25 mm Syringe Filter w/0.45 μm HT Tuffryn Membrane | PALL Life Sciences | Cat # 4184 |
| 96-well white plates for luciferase assays | Costar | Cat # 3917 |
| Cryotubes | Thermo Fisher Scientific Nunc | Cat # 340711 |
| Polystyrene Round-bottom Tube | FALCON | Ref # 352058 |
| Optical Flat 8-Cap Strips for 0.2 mL tube stripes/plates | Bio-Rad | Cat # TCS0803 |
| Individual PCR tubes 8-tube Strip, clear | Bio-Rad | Cat # TLS0801 |
| ThermalGrid Rigid Strip PCR tubes | Denville Scientific INC | Ref #C18064 |
| 96 well U bottom plate | FALCON | Ref # 353077 |
| XIC-3 animal isolation chamber | PerkinElmer | N/A |
| Perkin Elmer IVIS Spectrum In-Vivo Imaging System | PerkinElmer | Yale University ABSL-3 facility. RRID:SCR_018621 |
| RAS-4 Rodent Anesthesia System | PerkinElmer | CLS146737 |
| Synergy LX multi-mode reader | Biotek | RRID:SCR_019763 |
| Millex-GV Filter, 0.22uM | Fisher Scientific | SLGV013SL |
| MicroSpin Columns, 50 Pieces | Cytiva | 27356501 |
| Immulon 2HB 96-Wells Flat bottom | VWR | 62402–972 |
| EBA 21 centrifuge | Hettich | 1004–31 |
| Legend 21R Micro centrifuge | Thermo Fisher Scientific | 75002446 |
| Micro plate reader Synergy | Biotek | H1 |
| Microplate washer 405 TS | Biotek | 405TSRSQ |
Resource availability
Lead contact
Requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Pradeep D. Uchil (pradeep.uchil@yale.edu).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Cell and viruses
Vero E6 (CRL-1586, American Type Culture Collection (ATCC), were cultured at 37°C in RPMI supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES pH 7.3, 1 mM sodium pyruvate, 1× non-essential amino acids, and 100 U/mL of penicillin–streptomycin. The SARS-CoV-2/USA_WA1/2019 isolate expressing nanoluc luciferase (nLuc) was obtained from Craig B Wilen, Yale University and generously provided by K. Plante and Pei-Yong Shi, World Reference Center for Emerging Viruses and Arboviruses, University of Texas Medical Branch).60 SARS-CoV-2 USA-WA1/2020, B.1.617.2 (Delta) and B.1.351 (Beta) isolates without reporters were obtained through BEI Resources. Viruses were propagated in Vero E6 TMPRSS2 by infecting them in T150 cm2 flasks at a MOI of 0.1. The culture supernatants were collected after 72 h when cytopathic effects were clearly visible. The cell debris was removed by sedimentation and filtered through 0.45-micron filter to generate virus stocks. Viruses were concentrated by adding one volume of cold (4°C) 4x PEG-it Virus Precipitation Solution [40% (w/v) PEG-8000 and 1.2 M NaCl; System Biosciences] to three volumes of virus-containing supernatant. The solution was mixed by inverting the tubes several times and then incubated at 4°C overnight. The precipitated virus was harvested by centrifugation at 1,500 × g for 60 minutes at 4°C. The concentrated virus was then resuspended in PBS then aliquoted for storage at −80°C. All work with infectious SARS-CoV-2 was performed in Institutional Biosafety Committee approved BSL3 and A-BSL3 facilities at Yale University School of Medicine using appropriate positive pressure air respirators and protective equipment. For the generation of CEM.NKr cells stably expressing the SARS-CoV-2 Spike glycoproteins of Delta and Beta VOCs, transgenic lentiviruses were produced in 293T using a third-generation lentiviral vector system. Briefly, 293T cells were co-transfected with two packaging plasmids (pLP1 and pLP2), an envelope plasmid (pSVCMV-IN-VSV-G) and a lentiviral transfer plasmid coding for a GFP-tagged SARS-CoV-2 Delta or Beta Spike (Sinobiological). Supernatant containing lentiviral particles was used to transduce CEM.NKr cells in presence of 5 μg/mL polybrene. CEM.NKr CCR5+ cells stably expressing SARS-CoV-2 Spike (GFP+) were sorted by flow cytometry. CEM.NKr, CEM.NKr-Spike, CEM.NKr-Delta-Spike, CEM.NKr-Beta-Spike and peripheral blood mononuclear cells (PBMCs) were maintained at 37°C under 5% CO2 in RPMI media, supplemented with 10% FBS and 100 U/mL penicillin/ streptomycin. 293T (or HEK293T), 293T-ACE261 cells were maintained at 37°C under 5% CO2 in DMEM media, supplemented with 5% FBS and 100 U/mL penicillin/streptomycin. CEM.NKr (NIH AIDS Reagent Program) is a T lymphocytic cell line resistant to NK cell-mediated lysis. CEM.NKr-Spike stably expressing ancestral, Delta and Beta VOC Spikes were used as target cells in ADCC assays.36,62 PBMCs were obtained from healthy donor through leukapheresis and were used as effector cells in ADCC assay.
Ethics statement
CCP was obtained from individuals who were infected during the first wave of the pandemic, after at least fourteen days of resolution of COVID-19 symptoms.63 All participants consented to the study (CER #2020–004). PBMCs from healthy individuals as a source of effector cells in our ADCC assay were obtained under CRCHUM institutional review board (protocol #19.381). Research adhered to the standards indicated by the Declaration of Helsinki. All participants were adults and provided informed written consent prior to enrollment in accordance with Institutional Review Board approval.
Plasma samples
Recovered COVID-19 patients who have received a COVID-19 diagnosis by the Québec Provincial Health Authority and met the donor selection criteria for plasma donation in use at Héma-Québec were recruited. They were allowed to donate plasma at least 14 days after complete resolution of COVID-19 symptoms. A volume of 500 mL to 750 mL of plasma was collected by plasmapheresis (TRIMA Accel®, Terumo BCT). Disease severity (date of symptoms onset, end of symptoms, type, and intensity of symptoms, need for hospitalization/ICU) was documented for each donor using a questionnaire administered at the time of recruitment. For additional details of CCPs (sex, age, blood group of the convalescent donor and day of collection post infection, please refer to key resource table.
Mouse experiments
All animals were maintained in the (SPF-free) barrier facility of the Yale University Animal Resource Centre (YARC) within a 14:10 light: dark cycle. Breeding population of mice and infected animals are maintained in separate rooms. All SARS-CoV-2-infected animals were housed in animal room under BSL3 containment. Cages, animal waste, bedding, and animal carcasses were disposed and decontaminated following the guidelines of Environmental Health Services at Yale. All replication competent virus-infected animals were handled under ABSL3 conditions with personnel’s donning pressurized air purified respirators (PAPR), double gloves, shoe covers, sleeve covers and disposable gowns. All experiments described here were approved by Institutional Animal Care and Use Committees (IACUC) as well as SOPs approved by Institutional Environmental Health and Biosafety committee. hACE2 transgenic B6 mice (heterozygous) were obtained from Jackson Laboratory. 6–8-week-old male and female mice were used for all the experiments. The heterozygous mice were crossed and genotyped to select heterozygous mice for experiments by using the primer sets recommended by Jackson Laboratory. Each cohort size was n = 4–8 to allow statistical testing and conducted as 2–3 biological replicates (n = 2–3 per replicate) to allow parallel evaluation of different CCPs and virus combinations. The number of animals (n = 4–8 per cohort) needed to achieve statistically significant results were calculated based on a priori power analysis. We calculated power and sample sizes required based on data from pilot experiments and previous studies.23,24,64 Animals with sex- and age-matched littermates were included randomly in the experiments. No animals were excluded due to illness after the experiments. At the time of experimentation, care was taken to include equal numbers of male and female mice whenever possible to ensure that sex of the animals does not constitute a biological variable during analysis.
Method details
SARS-CoV-2 infection and treatment conditions
For all in vivo experiments, the 6 to 8 weeks male and female mice were intranasally challenged with 1 × 105 FFU SARS-CoV-2_WA1_nLuc, WA1, Delta and Beta VOCs in 25–30 μL volume under anesthesia (0.5 - 5% isoflurane delivered using precision Dräger vaporizer with oxygen flow rate of 1 L/min). For human convalescent plasma treatment using prophylaxis regimen, mice were administered 1 mL of indicated plasma intraperitoneally (i.p.), 24 h prior to infection. For therapy, the same amount was administered two-day post infection (2 dpi). For IgG and Ig(M + A)-depletion, the plasma had to be diluted 1:1. Hence 2 mL of the Class-depleted plasma was administered intraperitoneally in two injections, 1 mL each and 1 h apart. The starting body weight was set to 100%. For survival experiments, mice were monitored every 8–12 h starting six days after virus challenge. Lethargic and moribund mice or mice that had lost more than 20% of their body weight were sacrificed and considered to have succumbed to infection for Kaplan-Meier survival plots. Mice were considered to have recovered if they gained back all the lost weight.
IgG and Ig(M + A) depletion of CCP-6
Selective depletion of IgM, IgA or IgG was done by adsorption on class-specific ligands immobilized on sepharose or agarose beads starting with a two-fold dilution of plasma in PBS. IgG and IgA antibodies were depleted from plasma obtained from one recovered COVID-19 patient (CCP-6) using Protein G HP Spintrap (GE Healthcare Life Sciences, Buckinghamshire, UK) and Peptide M/Agarose (InvivoGen, San Diego, CA), respectively, according to the manufacturer’s instructions with the exception that no elution step for the recovery of the targeted antibodies was done. For IgM depletion, anti-human IgM (μ-chain specific, Sigma, St.Louis, MO) was covalently coupled to NHS Activated Sepharose® 4 Fast Flow (GE Healthcare) at 815 μg/mL of matrix. Depletion was performed according to the manufacturer’s instructions with the exception that no elution step for the recovery of the targeted class was done. All non-depleted and class-depleted samples were filtered on a 0.22 μm Millex GV filter (SLGV013SL, Millipore, Burlington, MA) to ensure sterility for the virus capture and neutralization assays. For the preparation of Ig(M + A) depleted samples, plasmas were depleted sequentially in IgM and then in IgA as described above.
To assess the extent of IgM, IgG and IgA depletion, ELISA were performed on non-depleted as well as IgM/IgA- and IgG-depleted plasma samples. Wells of a 96-well microplate were filled with either goat anti-human IgM (μ-chain specific) at 5 μg/mL, goat anti-human serum IgA (a-chain specific) at 0.3 μg/mL or goat anti-human IgG (γ-chain specific) at 5 μg/mL (all from Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Microtiter plates were sealed and stored overnight at 2–8°C. After four (IgA) to six (IgM and IgG) washes with H2O-0.1% Tween 20 (Sigma), 200 μL of blocking solution (10 mmol/L phosphate buffer, pH 7.4, containing 0.85% NaCl, 0.25% Hammerstein casein (EMD Chemicals Inc., Gibbstown, NJ,) were added to each well to block any remaining binding sites. The blocking solution for the IgG and IgM ELISA also contained 0.05% Tween 20. After 0.5 h (IgA) to 1h (IgM and IgG) incubation at 37°C and washes, samples and the standard curves (prepared with human calibrated standard serum, Cedarlane, Burlington, Canada) were added to the plates in triplicates. Plates were incubated for 1h at 37°C. After washes, 100 μL of either goat anti-human IgA + G + M (H + L) HRP conjugate (1/30 000), goat anti-human IgG (H + L) HRP conjugate (1/30 000) or goat anti-human IgA (a-chain specific) HRP conjugate (1/5000) (all from Jackson ImmunoResearch Laboratories, Inc.) were added and samples were incubated at 37°C for 1h. Wells were washed and bound antibodies were detected by the addition of 100 μL of 3,3′,5,5′-tetramethylbenzimidine (TMB, ScyTek Laboratories, Logan, UT). The enzymatic reaction was stopped by the addition of 100 μL 1 N H2SO4 and the absorbance was measured at 450/630 nm within 5 minutes.
Bioluminescence imaging (BLI) of SARS-CoV-2 infection
All standard operating procedures and protocols for IVIS imaging of SARS-CoV-2 infected animals under ABSL-3 conditions were approved by IACUC, IBSCYU and YARC. All the imaging was carried out using IVIS Spectrum® (PerkinElmer) in XIC-3 animal isolation chamber (PerkinElmer) that provided biological isolation of anesthetized mice or individual organs during the imaging procedure. All mice were anesthetized via isoflurane inhalation (3–5% isoflurane, oxygen flow rate of 1.5 L/min) prior and during BLI using the XGI-8 Gas Anesthesia System. Prior to imaging, 100 μL of Nanoluc substrate, furimazine (NanoGlo™, Promega, Madison, WI) diluted 1:40 in endotoxin-free PBS was retroorbitally administered to mice under anesthesia. The mice were then placed into XIC-3 animal isolation chamber (PerkinElmer) pre-saturated with isothesia and oxygen mix. The mice were imaged in both dorsal and ventral position at indicated days post infection. The animals were then imaged again after euthanasia and necropsy by spreading additional 200 μL of substrate on to exposed intact organs. Infected areas identified by carrying out whole-body imaging after necropsy were isolated, washed in PBS to remove residual blood and placed onto a clear plastic plate. Additional droplets of furimazine in PBS (1:40) were added to organs and soaked in substrate for 1–2 min before BLI.
Images were acquired and analyzed with Living Image v4.7.3 in vivo software package (Perkin Elmer Inc). Image acquisition exposures were set to auto, with imaging parameter preferences set in order of exposure time, binning, and f/stop, respectively. Images were acquired with luminescent f/stop of 2, photographic f/stop of 8. Binning was set to medium. Comparative images were compiled and batch-processed using the image browser with collective luminescent scales. Photon flux was measured as luminescent radiance (p/sec/cm2/sr). During luminescent threshold selection for image display, luminescent signals were regarded as background when minimum threshold setting resulted in displayed radiance above non-tissue-containing or known uninfected regions.
Focus forming assay
Titers of virus stocks was determined by standard plaque assay. Briefly, the 4 × 105 Vero-E6 cells were seeded on 12-well plate. 24 h later, the cells were infected with 200 μL of serially diluted virus stock. After 1 hour, the cells were overlayed with 1mL of pre-warmed 0.6% Avicel (RC-581 FMC BioPolymer) made in complete RPMI medium. Plaques were resolved at 48 h post infection by fixing in 10 % paraformaldehyde for 15 min followed by staining for 20 min with 0.2% crystal violet made in 20% ethanol. Plates were rinsed in water to visualize plaques.
Measurement of viral burden
Indicated organs (nasal cavity, brain, lungs) from infected or uninfected mice were collected, weighed, and homogenized in 1 mL of serum free RPMI media containing penicillin-streptomycin and homogenized in 2 mL tube containing 1.5 mm Zirconium beads with BeadBug 6 homogenizer (Benchmark Scientific, TEquipment Inc). Virus titers were measured using three highly correlative methods.24 Frist, the total RNA was extracted from homogenized tissues using RNeasy plus Mini kit (Qiagen Cat # 74136), reverse transcribed with iScript advanced cDNA kit (Bio-Rad Cat #1725036) followed by a SYBR Green Real-time PCR assay for determining copies of SARS-CoV-2 N gene RNA using primers SARS-CoV-2 N F: 5′-ATGCTGCAATCGTGCTACAA-3′ and SARS-CoV-2 N R: 5′-GACTGCCGCCTCTGCTC-3′. All our real-time PCR assays based on SYBR Green had a built-in melt-curve that were checked to ensure estimation of only specific PCR products and not false-positives. Second, serially diluted clarified tissue homogenates were used to infect Vero-E6 cell culture monolayer. The titers per gram of tissue were quantified using standard plaque forming assay described above. Third, we used Nanoluc activity as a shorter surrogate for plaque assay. Infected cells were washed with PBS and then lysed using 1X Passive lysis buffer. The lysates transferred into a 96-well solid white plate (Costar Inc) and Nanoluc activity was measured using Tristar multiwell Luminometer (Berthold Technology, Bad Wildbad, Germany) for 2.5 seconds by adding 20 μL of Nano-Glo® substrate in nanoluc assay buffer (Promega Inc, WI, USA). Uninfected monolayer of Vero cells treated identically served as controls to determine basal luciferase activity to obtain normalized relative light units. The data were processed and plotted using GraphPad Prism 8 v8.4.3.
Analyses of signature inflammatory cytokines mRNA expression
Brain and lung samples were collected from mice at the time of necropsy. Approximately, 20 mg of tissue was suspended in 500 μL of RLT lysis buffer, and RNA was extracted using RNeasy plus Mini kit (Qiagen Cat # 74136), reverse transcribed with iScript advanced cDNA kit (Bio-Rad Cat #1725036). To determine mRNA copy numbers of signature inflammatory cytokines, multiplex qPCR was conducted using iQ Multiplex Powermix (Bio Rad Cat # 1725848) and PrimePCR Probe Assay mouse primers FAM-GAPDH, HEX-IL6, TEX615-CCL2, Cy5-CXCL10, and Cy5.5-IFNgamma. The reaction plate was analyzed using CFX96 touch real time PCR detection system. Scan mode was set to all channels. The PCR conditions were 95°C 2 min, 40 cycles of 95°C for 10 s and 60°C for 45 s, followed by a melting curve analysis to ensure that each primer pair resulted in amplification of a single PCR product. mRNA copy numbers of Il6, Ccl2, Cxcl10 and Ifng in the cDNA samples of infected mice were normalized to Gapdh mRNA with the formula ΔCt(target gene) = Ct(target gene)-Ct(Gapdh). The fold increase was determined using 2−ΔΔCt method comparing treated mice to uninfected controls.
Antibody depletion of immune cell subsets
Macrophages and neutrophils were depleted during using anti-CSF1R (BioXcell; clone AFS98; 20 mg/kg body weight)65 and anti-Ly6G (clone: 1A8; 20 mg/kg body weight)66 respectively. The mAbs were administered to mice by i.p injection every two days starting at −2 dpi for during CCP prophylaxis or 0 dpi for CCP therapy. Rat IgG2a mAb (BioXCell; clone C1.18.4; 20 mg/kg body weight) or human IgG1 mAb (BioXCell; 12.5 mg/kg body weight) was used as isotype control. The mice were sacrificed and bled 2–3 days after antibody administration or at necropsy to ascertain depletion of desired population.
Flow cytometric analyses for immune cell depletion
For analysis of neutrophil depletion, peripheral blood was collected 2–3 days after administration of depleting antibodies. Erythrocytes were lysed with eBioscience 1X RBC lysis buffer (Invitrogen), PBMCs fixed with 4% PFA and quenched with PBS containing 0.1M glycine. PFA-fixed cells PBMCs were resuspended and blocked in Cell Staining buffer (BioLegend Inc.) containing Fc blocking antibody against CD16/CD32 (BioLegend Inc) before staining with antibodies. Neutrophils were identified as CD45+CD11b+Ly6G+ cells using APC Rat anti-mouse CD45 (30-F11), PE anti-mouse CD11b (M1/70) APC/Cy7 and anti-mouse Ly-6G (1A8) antibodies.
For analyses of macrophage depletion, lung tissue was harvested 2 days after administration of antibodies. The tissue was minced and incubated in Hanks' Balanced Salt Solution containing Dispase (5 U/mL; STEMCELL technologies), Liberase TL (0.2 mg/mL, Sigma-Aldrich) and DNase I (100 mg/mL, Roche) at 37°C for 1 h and passed through a 70 μm cell strainer (Falcon, Cat # 352350). The single cell suspension was fixed in BD Cytofix/Cytoperm buffer and stained in BD Cytoperm buffer containing Fc blocking antibody against CD16/CD32 (BioLegend Inc). Macrophages were identified as CD45+CD11b+Ly6G−L6C−CD68+ population using Alexa 488 Rat anti-mouse CD45 (30-F11), PE anti-mouse CD11b (M1/70) APC/Cy7 and anti-mouse Ly-6G (1A8), APC/Cy7 anti-mouse Ly-6C (HK1.4) and Alexa 647 anti-mouse CD68 (FA-11) antibodies.
Data were acquired on an Accuri C6 (BD Biosciences) and were analyzed with Accuri C6 software. 100,000–200,000 viable cells were acquired for each sample. FlowJo software (Treestar) was used to generate FACS plots.
Antibody dependent cellular cytotoxicity (ADCC) assay
This assay was previously described.36,62 Briefly, for evaluation of anti-SARS-CoV-2 antibody-dependent cellular cytotoxicity (ADCC) activity, parental CEM.NKr CCR5+ cells were mixed at a 1:1 ratio with CEM.NKr cells stably expressing a GFP-tagged full length SARS-CoV-2 ancestral Spike (CEM.NKr.SARS-CoV-2.Spike cells) or their Delta and Beta Spike counterparts. These cells were stained for viability (Aqua fluorescent reactive dye, Invitrogen) and with a cellular dye (cell proliferation dye eFluor670; Thermo Fisher Scientific) and subsequently used as target cells. Overnight rested PBMCs were stained with another cellular marker (cell proliferation dye eFluor450; Thermo Fisher Scientific) and used as effector cells. Stained target and effector cells were mixed at a ratio of 1:10 in 96-well V-bottom plates. Plasma (1/500 dilution) was added to the appropriate wells. Monoclonal antibodies CR3022 and CV3-13 were also included (1 μg/mL) in each experiment as a positive control. The plates were subsequently centrifuged for 1 min at 300 x g, and incubated at 37°C, 5% CO2 for 5 hours and then fixed in a 2% PBS-formaldehyde solution. ADCC activity was calculated using the formula: [(% of GFP + cells in Targets plus Effectors) - (% of GFP + cells in Targets plus Effectors plus plasma/antibody)]/(% of GFP + cells in Targets) x 100 by gating on transduced live target cells. All samples were acquired on an LSRII cytometer (BD Biosciences) and data analysis was performed using FlowJo v10.7.1 (Tree Star).
Flow cytometry analysis of the different anti-Spike classes
For evaluation of the different antibody classes (IgG, IgM, IgA and Total Ig) targeting the SARS-CoV-2 Spike, CEM.NKr cells stably expressing a GFP-tagged full length SARS-CoV-2 Spike and CEM.NKr CCR5+ parental cells were stained for 45 minutes at 25°C with plasma CCP-6, plasma CCP-6 depleted in IgG and plasma CCP-6 depleted in IgA and IgM (1/500). Cells were then washed and further stained with a viability dye staining (Aqua fluorescent reactive dye, Invitrogen) and specific secondary antibodies targeting IgGs (Alexa Fluor® 647 anti-human IgG Fc, BioLegend), IgMs (Alexa Fluor® 647-conjugated AffiniPure Goat Anti-Human IgM, Fc5μ Fragment Specific, Jackson ImmunoResearch), IgAs (Alexa Fluor® 647-conjugated AffiniPure Goat Anti-Human Serum IgA, α Chain Specific, Jackson ImmunoResearch) or Total Igs (Alexa Fluor® 647-conjugated AffiniPure Goat Anti-Human IgA + IgG + IgM (H + L), Jackson ImmunoResearch) for 20 minutes at 25°C. The cells were then washed and fixed in a 2% PBS-Formaldehyde solution. The percentage of transduced cells (GFP + cells) was determined by gating on the living cell population based on the viability dye staining (Aqua fluorescent reactive dye, Invitrogen). Non-specific staining was evaluated using CEM.NKr CCR5+ parental cells and subtracted from the staining on the live GFP + cells in the CEM.NKr.Spike cells. Samples were acquired on an LSRII cytometer (BD Biosciences) and data analysis was performed using FlowJo v10.7.1 (Tree Star).
Pseudovirus neutralization assay
To produce the pseudoviruses, 293T cells were transfected with the lentiviral vector pNL4.3 R-E− Luc (NIH AIDS Reagent Program) and a plasmid encoding for the indicated S glycoprotein (D614G) at a ratio of 10:1. Two days post-transfection, cell supernatants were harvested and stored at −80°C until use. For the neutralization assay, 293T-ACE2 target cells were seeded at a density of 1 × 104 cells/well in 96-well luminometer-compatible tissue culture plates (Perkin Elmer) 24h before infection. Pseudoviral particles were incubated with several plasma dilutions (1/50; 1/250; 1/1250; 1/6250; 1/31250) for 1h at 37°C and were then added to the target cells followed by incubation for 48 h at 37°C. Then, cells were lysed by the addition of 30 μL of passive lysis buffer (Promega) followed by one freeze-thaw cycle. An LB942 TriStar luminometer (Berthold Technologies) was used to measure the luciferase activity of each well after the addition of 100 μL of luciferin buffer (15mM MgSO4, 15mM KPO4 [pH 7.8], 1mM ATP, and 1mM dithiothreitol) and 50 μL of 1mM d-luciferin potassium salt (Prolume). The neutralization half-maximal inhibitory dilution (ID50) represents the plasma dilution to inhibit 50% of the infection of 293T-ACE2 cells by pseudoviruses.
SARS-CoV-2 neutralization assay
Serial two-fold dilutions of heat inactivated (56°C for 30 min) CCPs (1:4, 1:16, 1:64, 1:256, 1:1024) were prepared in triplicates in a volume of 50 μL. 50 μL of WA1, Delta and Beta VOCs (a virus concentration to generate 30–50 plaques per well in six well plate) was mixed with diluted plasma and incubated for 1 h at 37°C. The virus-plasma mixes were then added to Vero E6 cells (7.5 × 105 cells/well) seeded 24 h earlier, in 6-well tissue culture plates and allowed to interact with cells for 1 h. The cells were then overlayed with 1 mL of pre-warmed 0.6% Avicel (RC-581 FMC BioPolymer) made in complete RPMI medium. Plaques were resolved after 72 h by fixing cells in 10% paraformaldehyde for 15 min followed by staining for 15 minutes with 0.2 % crystal violet made in 20% ethanol. Plates were rinsed in water to visualize FFU. The FFU counts from virus samples without antibody incubation were set to 100% (30–50 FFU/well). IC50 was calculated by plotting the log (plasma dilution) vs normalized FFUs and using non-linear fit option in GraphPad Prism.
Quantification and statistical analysis
Data were analyzed and plotted using GraphPad Prism software (La Jolla, CA, USA). Statistical significance for pairwise comparisons were derived by applying non-parametric Mann-Whitney test (two-tailed). To obtain statistical significance for survival curves, grouped data were compared by log-rank (Mantel-Cox) test. To obtain statistical significance for grouped data we employed 2-way ANOVA followed by Tukey’s multiple comparison tests. p values lower than 0.05 were considered statistically significant. p values were indicated as ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001.
Acknowledgments
This work was supported by NIH grant R01AI163395 to W.M., le Ministère de l’Économie et de l’Innovation du Québec, Programme de soutien aux organismes de recherche et d’innovation, Foundation du CHUM, CIHR grant nos. 352417 and 177958, a Canada Research Chair on Retroviral Entry no. RCHS0235 950-232424 to A.F., and Canada Foundation for Innovation (CFI) #41027 to A.F. M.W.G. is a recipient of the Gruber Science Fellowship and supported by T32AI055403, K.D. was funded by a PREMIER fellowship, CIHR and FRQS fellowships to G.B.B., le Ministère de l’Économie et de l’Innovation du Québec, Fondation du CHU Sainte-Justine Fonds de recherche du Québec – Santé #281662 to P.B.
Author contributions
Conceptualization, P.D.U., A.F., R.B., and PB; Methodology, P.D.U., I.U., M.C., A.F., and R.B.; Investigation, I.U., K.S., P.D.U., G.B.B., E.D., A.T., A.L., and M.W.G.; Writing – Original Draft, P.D.U.; Writing – Review & Editing, P.D.U., A.F., W.M., P.K., R.B., P.B., M.C., I.U., and M.W.G.; Funding Acquisition, W.M., A.F., P.B., and R.B.; Resources, W.M., P.K., A.F., and R.B.; Supervision, P.D.U., A.F., and R.B.
Declaration of interests
The authors declare no competing interests.
Published: December 16, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100893.
Contributor Information
Priti Kumar, Email: priti.kumar@yale.edu.
Renée Bazin, Email: renee.bazin@hema-quebec.qc.ca.
Andrés Finzi, Email: andres.finzi@umontreal.ca.
Pradeep D. Uchil, Email: pradeep.uchil@yale.edu.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request. Additional Supplemental Items are available from Mendeley Data, https://doi.org/10.17632/xx7j82dv97.1.
-
•
This paper does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from lead contact upon request.
References
- 1.Casadevall A., Scharff M.D. Return to the past: the case for antibody-based therapies in infectious diseases. Clin. Infect. Dis. 1995;21:150–161. doi: 10.1093/clinids/21.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mair-Jenkins J., Saavedra-Campos M., Baillie J.K., Cleary P., Khaw F.M., Lim W.S., Makki S., Rooney K.D., Nguyen-Van-Tam J.S., Beck C.R., Convalescent Plasma Study Group The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: a systematic review and exploratory meta-analysis. J. Infect. Dis. 2015;211:80–90. doi: 10.1093/infdis/jiu396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luke T.C., Kilbane E.M., Jackson J.L., Hoffman S.L. Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Ann. Intern. Med. 2006;145:599–609. doi: 10.7326/0003-4819-145-8-200610170-00139. [DOI] [PubMed] [Google Scholar]
- 4.Cao H., Shi Y. Convalescent plasma: possible therapy for novel coronavirus disease 2019. Transfusion. 2020;60:1078–1083. doi: 10.1111/trf.15797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beraud M., Goodhue Meyer E., Lozano M., Bah A., Vassallo R., Brown B.L. Lessons learned from the use of convalescent plasma for the treatment of COVID-19 and specific considerations for immunocompromised patients. Transfus. Apher. Sci. 2022;61:103355. doi: 10.1016/j.transci.2022.103355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ljungquist O., Lundgren M., Iliachenko E., Månsson F., Böttiger B., Landin-Olsson M., Wikén C., Rosendal E., Överby A.K., Wigren B.J., et al. Convalescent plasma treatment in severely immunosuppressed patients hospitalized with COVID-19: an observational study of 28 cases. Inf. Disp. 2022;54:283–291. doi: 10.1080/23744235.2021.2013528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinkus G.S., Said J.W. Leu-M1 immunoreactivity in nonhematopoietic neoplasms and myeloproliferative disorders. An immunoperoxidase study of paraffin sections. Am. J. Clin. Pathol. 1986;85:278–282. doi: 10.1093/ajcp/85.3.278. [DOI] [PubMed] [Google Scholar]
- 8.Tauzin A., Beaudoin-Bussières G., Gong S.Y., Chatterjee D., Gendron-Lepage G., Bourassa C., Goyette G., Racine N., Khrifi Z., Turgeon J., et al. Humoral immune responses against SARS-CoV-2 Spike variants after mRNA vaccination in solid organ transplant recipients. iScience. 2022;25 doi: 10.1016/j.isci.2022.104990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ledford H. Antibody therapies could be a bridge to a coronavirus vaccine - but will the world benefit? Nature. 2020;584:333–334. doi: 10.1038/d41586-020-02360-y. [DOI] [PubMed] [Google Scholar]
- 10.Greaney A.J., Starr T.N., Bloom J.D. An antibody-escape estimator for mutations to the SARS-CoV-2 receptor-binding domain. Virus Evol. 2022;8:veac021. doi: 10.1093/ve/veac021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tada T., Zhou H., Dcosta B.M., Samanovic M.I., Chivukula V., Herati R.S., Hubbard S.R., Mulligan M.J., Landau N.R. Increased resistance of SARS-CoV-2 Omicron variant to neutralization by vaccine-elicited and therapeutic antibodies. EBioMedicine. 2022;78 doi: 10.1016/j.ebiom.2022.103944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tartof S.Y., Slezak J.M., Puzniak L., Hong V., Frankland T.B., Xie F., Ackerson B.K., Valluri S.R., Jodar L., McLaughlin J.M. Durability of BNT162b2 vaccine against hospital and emergency department admissions due to the omicron and delta variants in a large health system in the USA: a test-negative case-control study. Lancet Respir. Med. 2022 doi: 10.1016/S2213-2600(22)00101-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Planas D., Saunders N., Maes P., Guivel-Benhassine F., Planchais C., Buchrieser J., Bolland W.H., Porrot F., Staropoli I., Lemoine F., et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2022;602:671–675. doi: 10.1038/s41586-021-04389-z. [DOI] [PubMed] [Google Scholar]
- 14.Liu L., Iketani S., Guo Y., Chan J.F.W., Wang M., Liu L., Luo Y., Chu H., Huang Y., Nair M.S., et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature. 2022;602:676–681. doi: 10.1038/s41586-021-04388-0. [DOI] [PubMed] [Google Scholar]
- 15.Cao Y., Wang J., Jian F., Xiao T., Song W., Yisimayi A., Huang W., Li Q., Wang P., An R., et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature. 2022;602:657–663. doi: 10.1038/s41586-021-04385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villa C.H. 2021. Clinical Memorandum for Use of COVID-19 Convalescent Plasma EUA 26382.https://www.fda.gov/media/141480/download [Google Scholar]
- 17.Basheer M., Saad E., Shlezinger D., Assy N. Convalescent plasma reduces mortality and decreases hospitalization stay in patients with moderate COVID-19 pneumonia. Metabolites. 2021;11:761. doi: 10.3390/metabo11110761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dispinseri S., Secchi M., Pirillo M.F., Tolazzi M., Borghi M., Brigatti C., De Angelis M.L., Baratella M., Bazzigaluppi E., Venturi G., et al. Neutralizing antibody responses to SARS-CoV-2 in symptomatic COVID-19 is persistent and critical for survival. Nat. Commun. 2021;12:2670. doi: 10.1038/s41467-021-22958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Writing Committee for the R.-C.A.P.I., Estcourt L.J., Turgeon A.F., McQuilten Z.K., McVerry B.J., Al-Beidh F., et al. Effect of Convalescent Plasma on Organ Support-Free Days in Critically Ill Patients With COVID-19: A Randomized Clinical Trial. JAMA. 2021;326:1690–1702. doi: 10.1001/jama.2021.18178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korley F.K., Durkalski-Mauldin V., Yeatts S.D., Schulman K., Davenport R.D., Dumont L.J., El Kassar N., Foster L.D., Hah J.M., Jaiswal S., et al. Early convalescent plasma for high-risk outpatients with covid-19. N. Engl. J. Med. 2021;385:1951–1960. doi: 10.1056/NEJMoa2103784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bégin P., Callum J., Jamula E., Cook R., Heddle N.M., Tinmouth A., Zeller M.P., Beaudoin-Bussières G., Amorim L., Bazin R., et al. Convalescent plasma for hospitalized patients with COVID-19: an open-label, randomized controlled trial. Nat. Med. 2021;27:2012–2024. doi: 10.1038/s41591-021-01488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Natarajan H., Crowley A.R., Butler S.E., Xu S., Weiner J.A., Bloch E.M., Littlefield K., Wieland-Alter W., Connor R.I., Wright P.F., et al. SARS-CoV-2 antibody signatures robustly predict diverse antiviral functions relevant for convalescent plasma therapy. medRxiv. 2020 doi: 10.1101/2020.09.16.20196154. Preprint at. [DOI] [Google Scholar]
- 23.Beaudoin-Bussières G., Chen Y., Ullah I., Prévost J., Tolbert W.D., Symmes K., Ding S., Benlarbi M., Gong S.Y., Tauzin A., et al. A Fc-enhanced NTD-binding non-neutralizing antibody delays virus spread and synergizes with a nAb to protect mice from lethal SARS-CoV-2 infection. Cell Rep. 2022;38 doi: 10.1016/j.celrep.2022.110368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ullah I., Prévost J., Ladinsky M.S., Stone H., Lu M., Anand S.P., Beaudoin-Bussières G., Symmes K., Benlarbi M., Ding S., et al. Live imaging of SARS-CoV-2 infection in mice reveals that neutralizing antibodies require Fc function for optimal efficacy. Immunity. 2021;54:2143–2158.e15. doi: 10.1016/j.immuni.2021.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Erp E.A., Luytjes W., Ferwerda G., van Kasteren P.B. Fc-mediated antibody effector functions during respiratory syncytial virus infection and disease. Front. Immunol. 2019;10:548. doi: 10.3389/fimmu.2019.00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winkler E.S., Gilchuk P., Yu J., Bailey A.L., Chen R.E., Chong Z., Zost S.J., Jang H., Huang Y., Allen J.D., et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell. 2021;184:1804–1820.e16. doi: 10.1016/j.cell.2021.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tay M.Z., Wiehe K., Pollara J. Antibody-dependent cellular phagocytosis in antiviral immune responses. Front. Immunol. 2019;10:332. doi: 10.3389/fimmu.2019.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel K.R., Roberts J.T., Barb A.W. Multiple variables at the leukocyte cell surface impact Fc gamma receptor-dependent mechanisms. Front. Immunol. 2019;10:223. doi: 10.3389/fimmu.2019.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pereira N.A., Chan K.F., Lin P.C., Song Z. The "less-is-more" in therapeutic antibodies: afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. mAbs. 2018;10:693–711. doi: 10.1080/19420862.2018.1466767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu L.L., Suscovich T.J., Fortune S.M., Alter G. Beyond binding: antibody effector functions in infectious diseases. Nat. Rev. Immunol. 2018;18:46–61. doi: 10.1038/nri.2017.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamin R., Jones A.T., Hoffmann H.H., Schäfer A., Kao K.S., Francis R.L., Sheahan T.P., Baric R.S., Rice C.M., Ravetch J.V., Bournazos S. Fc-engineered antibody therapeutics with improved anti-SARS-CoV-2 efficacy. Nature. 2021;599:465–470. doi: 10.1038/s41586-021-04017-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schäfer A., Muecksch F., Lorenzi J.C.C., Leist S.R., Cipolla M., Bournazos S., Schmidt F., Maison R.M., Gazumyan A., Martinez D.R., et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J. Exp. Med. 2021;218:e20201993. doi: 10.1084/jem.20201993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W., Chen Y., Prévost J., Ullah I., Lu M., Gong S.Y., Tauzin A., Gasser R., Vézina D., Anand S.P., et al. Structural basis and mode of action for two broadly neutralizing antibodies against SARS-CoV-2 emerging variants of concern. Cell Rep. 2022;38 doi: 10.1016/j.celrep.2021.110210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jennewein M.F., MacCamy A.J., Akins N.R., Feng J., Homad L.J., Hurlburt N.K., Seydoux E., Wan Y.H., Stuart A.B., Edara V.V., et al. Isolation and characterization of cross-neutralizing coronavirus antibodies from COVID-19+ subjects. Cell Rep. 2021;36 doi: 10.1016/j.celrep.2021.109353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tauzin A., Nayrac M., Benlarbi M., Gong S.Y., Gasser R., Beaudoin-Bussières G., Brassard N., Laumaea A., Vézina D., Prévost J., et al. A single dose of the SARS-CoV-2 vaccine BNT162b2 elicits Fc-mediated antibody effector functions and T cell responses. Cell Host Microbe. 2021;29:1137–1150.e6. doi: 10.1016/j.chom.2021.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anand S.P., Prévost J., Nayrac M., Beaudoin-Bussières G., Benlarbi M., Gasser R., Brassard N., Laumaea A., Gong S.Y., Bourassa C., et al. Longitudinal analysis of humoral immunity against SARS-CoV-2 Spike in convalescent individuals up to 8 months post-symptom onset. Cell Rep. Med. 2021;2 doi: 10.1016/j.xcrm.2021.100290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richardson S.I., Manamela N.P., Motsoeneng B.M., Kaldine H., Ayres F., Makhado Z., Mennen M., Skelem S., Williams N., Sullivan N.J., et al. SARS-CoV-2 Beta and Delta variants trigger Fc effector function with increased cross-reactivity. Cell Rep. Med. 2022;3 doi: 10.1016/j.xcrm.2022.100510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaplonek P., Fischinger S., Cizmeci D., Bartsch Y.C., Kang J., Burke J.S., Shin S.A., Dayal D., Martin P., Mann C., et al. mRNA-1273 vaccine-induced antibodies maintain Fc effector functions across SARS-CoV-2 variants of concern. Immunity. 2022;55:355–365.e4. doi: 10.1016/j.immuni.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gundlapalli A.V., Salerno R.M., Brooks J.T., Averhoff F., Petersen L.R., McDonald L.C., Iademarco M.F., CDC COVID-19 Response SARS-CoV-2 serologic assay needs for the next phase of the US COVID-19 pandemic response. Open Forum Infect. Dis. 2021;8:ofaa555. doi: 10.1093/ofid/ofaa555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Russell M.W., Moldoveanu Z., Ogra P.L., Mestecky J. Mucosal immunity in COVID-19: a neglected but critical aspect of SARS-CoV-2 infection. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.611337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ku Z., Xie X., Hinton P.R., Liu X., Ye X., Muruato A.E., Ng D.C., Biswas S., Zou J., Liu Y., et al. Nasal delivery of an IgM offers broad protection from SARS-CoV-2 variants. Nature. 2021;595:718–723. doi: 10.1038/s41586-021-03673-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gasser R., Cloutier M., Prévost J., Fink C., Ducas É., Ding S., Dussault N., Landry P., Tremblay T., Laforce-Lavoie A., et al. Major role of IgM in the neutralizing activity of convalescent plasma against SARS-CoV-2. Cell Rep. 2021;34 doi: 10.1016/j.celrep.2021.108790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Z., Lorenzi J.C.C., Muecksch F., Finkin S., Viant C., Gaebler C., Cipolla M., Hoffmann H.H., Oliveira T.Y., Oren D.A., et al. Enhanced SARS-CoV-2 neutralization by dimeric IgA. Sci. Transl. Med. 2021;13:eabf1555. doi: 10.1126/scitranslmed.abf1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaplonek P., Cizmeci D., Fischinger S., Collier A.R., Suscovich T., Linde C., Broge T., Mann C., Amanat F., Dayal D., et al. mRNA-1273 and BNT162b2 COVID-19 vaccines elicit antibodies with differences in Fc-mediated effector functions. Sci. Transl. Med. 2022;14 doi: 10.1126/scitranslmed.abm2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brunet-Ratnasingham E., Anand S.P., Gantner P., Dyachenko A., Moquin-Beaudry G., Brassard N., Beaudoin-Bussières G., Pagliuzza A., Gasser R., Benlarbi M., et al. Integrated immunovirological profiling validates plasma SARS-CoV-2 RNA as an early predictor of COVID-19 mortality. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abj5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gunn B.M., Lu R., Slein M.D., Ilinykh P.A., Huang K., Atyeo C., Schendel S.L., Kim J., Cain C., Roy V., et al. A Fc engineering approach to define functional humoral correlates of immunity against Ebola virus. Immunity. 2021;54:815–828.e5. doi: 10.1016/j.immuni.2021.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seehusen F., Clark J.J., Sharma P., Bentley E.G., Kirby A., Subramaniam K., Wunderlin-Giuliani S., Hughes G.L., Patterson E.I., Michael B.D., et al. Neuroinvasion and neurotropism by SARS-CoV-2 variants in the K18-hACE2 mouse. Viruses. 2022;14 doi: 10.3390/v14051020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park J.G., Pino P.A., Akhter A., Alvarez X., Torrelles J.B., Martinez-Sobrido L. Animal models of COVID-19: transgenic mouse model. Methods Mol. Biol. 2022;2452:259–289. doi: 10.1007/978-1-0716-2111-0_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Halfmann P.J., Iida S., Iwatsuki-Horimoto K., Maemura T., Kiso M., Scheaffer S.M., Darling T.L., Joshi A., Loeber S., Singh G., et al. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature. 2022;603:687–692. doi: 10.1038/s41586-022-04441-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng J., Wong L.Y.R., Li K., Verma A.K., Ortiz M.E., Wohlford-Lenane C., Leidinger M.R., Knudson C.M., Meyerholz D.K., McCray P.B., Jr., Perlman S. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature. 2021;589:603–607. doi: 10.1038/s41586-020-2943-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCray P.B., Jr., Pewe L., Wohlford-Lenane C., Hickey M., Manzel L., Shi L., Netland J., Jia H.P., Halabi C., Sigmund C.D., et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 2007;81:813–821. doi: 10.1128/JVI.02012-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dekkers G., Bentlage A.E.H., Stegmann T.C., Howie H.L., Lissenberg-Thunnissen S., Zimring J., Rispens T., Vidarsson G. Affinity of human IgG subclasses to mouse Fc gamma receptors. mAbs. 2017;9:767–773. doi: 10.1080/19420862.2017.1323159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kerntke C., Nimmerjahn F., Biburger M. There is (scientific) strength in numbers: a comprehensive quantitation of Fc gamma receptor numbers on human and murine peripheral blood leukocytes. Front. Immunol. 2020;11:118. doi: 10.3389/fimmu.2020.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith P., DiLillo D.J., Bournazos S., Li F., Ravetch J.V. Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc. Natl. Acad. Sci. USA. 2012;109:6181–6186. doi: 10.1073/pnas.1203954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seidel U.J.E., Schlegel P., Lang P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front. Immunol. 2013;4:76. doi: 10.3389/fimmu.2013.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Honko A.N., Storm N., Bean D.J., Henao Vasquez J., Downs S.N., Griffiths A. Rapid Quantification and Neutralization Assays for Novel Coronavirus SARS-CoV-2 Using Avicel RC-591 Semi-solid Overlay. Preprints. 2020;2020050264 doi: 10.20944/preprints202005.0264.v1. [DOI] [Google Scholar]
- 57.Lodge R., Lalonde J.P., Lemay G., Cohen E.A. The membrane-proximal intracytoplasmic tyrosine residue of HIV-1 envelope glycoprotein is critical for basolateral targeting of viral budding in MDCK cells. EMBO J. 1997;16:695–705. doi: 10.1093/emboj/16.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Z., Pan Q., Ding S., Qian J., Xu F., Zhou J., Cen S., Guo F., Liang C. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe. 2013;14:398–410. doi: 10.1016/j.chom.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 59.Finzi A., Xiang S.H., Pacheco B., Wang L., Haight J., Kassa A., Danek B., Pancera M., Kwong P.D., Sodroski J. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol. Cell. 2010;37:656–667. doi: 10.1016/j.molcel.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xie X., Muruato A., Lokugamage K.G., Narayanan K., Zhang X., Zou J., Liu J., Schindewolf C., Bopp N.E., Aguilar P.V., et al. An infectious cDNA clone of SARS-CoV-2. Cell Host Microbe. 2020;27:841–848.e3. doi: 10.1016/j.chom.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prévost J., Gasser R., Beaudoin-Bussières G., Richard J., Duerr R., Laumaea A., Anand S.P., Goyette G., Benlarbi M., Ding S., et al. Cross-sectional evaluation of humoral responses against SARS-CoV-2 Spike. Cell Rep. Med. 2020;1 doi: 10.1016/j.xcrm.2020.100126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beaudoin-Bussières G., Richard J., Prévost J., Goyette G., Finzi A. A new flow cytometry assay to measure antibody-dependent cellular cytotoxicity against SARS-CoV-2 Spike-expressing cells. STAR Protoc. 2021;2 doi: 10.1016/j.xpro.2021.100851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perreault J., Tremblay T., Fournier M.J., Drouin M., Beaudoin-Bussières G., Prévost J., Lewin A., Bégin P., Finzi A., Bazin R. Waning of SARS-CoV-2 RBD antibodies in longitudinal convalescent plasma samples within 4 months after symptom onset. Blood. 2020;136:2588–2591. doi: 10.1182/blood.2020008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Y., Sun L., Ullah I., Beaudoin-Bussières G., Anand S.P., Hederman A.P., Tolbert W.D., Sherburn R., Nguyen D.N., Marchitto L., et al. Engineered ACE2-Fc counters murine lethal SARS-CoV-2 infection through direct neutralization and Fc-effector activities. Sci. Adv. 2022;8 doi: 10.1126/sciadv.abn4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bauché D., Joyce-Shaikh B., Jain R., Grein J., Ku K.S., Blumenschein W.M., Ganal-Vonarburg S.C., Wilson D.C., McClanahan T.K., Malefyt R.d.W., et al. LAG3(+) regulatory T cells restrain interleukin-23-producing CX3CR1(+) gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity. 2018;49:342–352.e5. doi: 10.1016/j.immuni.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 66.Moynihan K.D., Opel C.F., Szeto G.L., Tzeng A., Zhu E.F., Engreitz J.M., Williams R.T., Rakhra K., Zhang M.H., Rothschilds A.M., et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med. 2016;22:1402–1410. doi: 10.1038/nm.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request. Additional Supplemental Items are available from Mendeley Data, https://doi.org/10.17632/xx7j82dv97.1.
-
•
This paper does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from lead contact upon request.