

Summary
The human genome contains hundreds of thousands of regions exhibiting copy number variation (CNV). However, the phenotypic effects of most such polymorphisms are unknown because only larger CNVs have been ascertainable from SNP-array data generated by large biobanks. We developed a computational approach leveraging haplotype-sharing in biobank cohorts to more sensitively detect CNVs. Applied to UK Biobank, this approach accounted for approximately half of all rare gene inactivation events produced by genomic structural variation. This CNV call set enabled a detailed analysis of associations between CNVs and 56 quantitative traits, identifying 269 independent associations (P < 5 × 10−8) likely to be causally driven by CNVs. Putative target genes were identifiable for nearly half of the loci, enabling insights into dosage-sensitivity of these genes and uncovering several gene-trait relationships. These results demonstrate the ability of haplotype-informed analysis to provide insights into the genetic basis of human complex traits.
Graphical Abstract

In Brief:
Use of haplotype sharing across a biobank enables identification of copy number variants at a finer scale than previously possible and links the genotypes to a range of reported phenotypes.
Introduction
Copy number variants (CNVs), which duplicate and delete 50 base pair to megabase-scale genomic segments throughout the human genome (Abel et al., 2020; Collins et al., 2020; Sudmant et al., 2015), are known to contribute to numerous genomic disorders including neuropsychiatric diseases (Marshall et al., 2017; Sanders et al., 2011; Sebat et al., 2007) and have been estimated to account for a considerable fraction of all rare loss-of-function (LoF) events affecting protein-coding genes (Collins et al., 2020). Beyond disrupting coding sequences of genes, CNVs can also have unique functional consequences not producible by SNPs: for example, duplications can increase gene dosage, and deletions can eliminate regulatory elements. Investigating the broader phenotypic impacts of CNVs thus has the potential to uncover new large-effect variants and further our understanding of the genetic architecture of complex traits.
However, well-powered, phenome-wide CNV association analyses to date have been limited to considering large CNVs (tens of kilobases or longer) detectable from low-cost SNP-array data (Wang et al., 2007) available for biobank-scale cohorts. Moreover, CNV association studies have encountered analytical challenges such as how to harmonize imprecise breakpoints of CNV calls, how to group CNVs for association testing, and how to filter associations that only reflect linkage disequilibrium (LD) with nearby SNPs. Despite these difficulties, previous studies have made many important discoveries both by investigating the role of known pathogenic CNVs on various phenotypes (Crawford et al., 2019; Kendall et al., 2017; Owen et al., 2018) and by conducting association analysis on all CNVs detected in large cohorts (Macé et al., 2017; Aguirre et al., 2019; Li et al., 2020; Sinnott-Armstrong et al., 2021; Beyter et al., 2021; Chen et al., 2021; Auwerx et al., 2022; Collins et al., 2022), including UK Biobank (Bycroft et al., 2018). Here we developed a more sensitive CNV-detection method leveraging haplotype-sharing within biobank cohorts and applied it to UK Biobank, empowering exploration of the phenotypic effects of CNVs at much higher resolution than previously possible.
Results
Haplotype-informed copy-number variant detection
We developed a computational approach to CNV detection, called HI-CNV (Haplotype-Informed Copy-Number-Variation), that substantially increases CNV detection power in large cohorts by pooling information across individuals who share extended SNP haplotypes. The intuition behind this approach is that in large biobank cohorts, population-polymorphic CNVs are usually carried by multiple individuals who co-inherited a CNV on a shared haplotype originating from a common ancestor. As such, power to detect a CNV can be increased by sharing information about its presence (e.g., from genotyping array intensity data) across multiple carriers (Figure 1A).
Figure 1: Haplotype-informed CNV detection from SNP-array data in UK Biobank.

A The HI-CNV framework improves power to detect CNVs by analyzing SNP-array data from an individual together with corresponding data from individuals with long shared haplotypes (“haplotype neighbors”). In contrast, standard approaches analyze data from the individual alone. B SNP-specific genotype cluster priors map allele-specific (A and B allele) probe intensity measurements to probabilistic information about copy-number likelihoods. C Average number of CNVs called by PennCNV and HI-CNV per UK Biobank participant. D Distribution of total CNV length per individual in the HI-CNV call set. E Validation rate of CNV calls from PennCNV and HI-CNV on 43 UK Biobank participants with independent whole-genome sequencing data. Error bars, 95% CIs. F Distribution of CNV lengths in the HI-CNV call set. G Distributions (across increasingly constrained gene sets) of observed counts of whole-gene deletions and duplications and pLoF CNVs in n=452,500 UK Biobank participants. Centers, medians; box edges, 25th and 75th percentiles; whiskers, 5th and 95th percentiles.
To identify individuals who are likely to share a segment of genome inherited from a recent common ancestor (and therefore likely to have co-inherited any CNVs contained within the shared genomic tract), we adapted recent approaches that use the positional Burrows-Wheeler transform (PBWT) (Durbin, 2014) to rapidly identify identity-by-descent (IBD) segments (Zhou et al., 2020). Specifically, for each haplotype of each individual in a cohort, we use a PBWT-based algorithm to identify its closest “haplotype neighbors” – i.e., the longest IBD matches with other haplotypes in the cohort – spanning each genomic position (Figure 1A). Then, given quantitative information about the potential presence of a CNV in genetic data from the individual, as well as corresponding information from “haplotype neighbors,” we use a hidden Markov model (HMM) to detect CNVs co-inherited on shared haplotypes.
To apply our HI-CNV approach to SNP-array genotyping probe intensity data available for the UK Biobank cohort, we further developed methods to learn probabilistic models that map allele-specific probe intensity measurements to probabilistic information about copy-number likelihoods (Figure 1B). Intuitively, genotyping probes within CNVs produce distinctive intensity measurements compared to probes not within CNVs. While these deviations are difficult to detect given data from one SNP, the signal becomes clearer when consistent deviations are observed across multiple consecutive SNPs (Wang et al., 2007) – or, for HI-CNV, across multiple individuals co-inheriting a CNV. To optimize signal available from SNP-array probe intensities, we estimated SNP-specific genotype cluster priors corresponding to nine possible genotypes across copy-number states 1 (deletion), 2, and 3 (duplication) (Figure 1B), and we also denoised total intensities using principal component analysis. Full methodological details are provided in STAR Methods, and we have released a portable, open-source HI-CNV software implementation.
Modeling haplotype sharing increases CNV detection power in UK Biobank
We applied HI-CNV to detect CNVs across all UK Biobank participants with SNP-array genotyping, focusing our main analyses on CNVs called in 452,500 UK Biobank participants of European ancestry. HI-CNV detected >6 times as many CNVs per individual as the widely-used PennCNV method (Figure 1C), producing an average of 31.1 CNV calls per individual (18.4 deletions and 12.7 duplications spanning an average of 430kb and 899kb, respectively; Figures 1C and 1D; Table S1). In contrast, previous PennCNV-based analyses of UK Biobank SNP-array intensity data produced ~4–6 CNV calls per individual depending on quality-control filters applied (Aguirre et al., 2019; Kendall et al., 2017). Validation analyses using whole-genome sequencing (WGS) pilot data available for 43 participants estimated a validation rate of 91% for HI-CNV, similar to that of PennCNV (Figure 1E; Table S1; STAR Methods). This estimate was corroborated by further validation analyses using subsequently-released WGS for 500 participants (STAR Methods), with the validation rate increasing modestly with CNV length and with call confidence, as expected (Figure S1A; Table S1).
HI-CNV’s increased detection sensitivity was driven by improved ability to detect CNVs on the scale of 10kb or shorter (Figure 1F; Table S1), which account for the majority of all CNVs (Abel et al., 2020; Collins et al., 2020; Sudmant et al., 2015) but have traditionally been difficult to detect from SNP-array data. We designed HI-CNV with the goal of sensitively detecting low-frequency and rare CNVs of length >5kb (versus ~50kb for previous SNP-array-based analyses of UK Biobank), focusing on CNVs with minor allele frequency (MAF) < 5% because of their potential to be more deleterious and because SNP-array designs tend to avoid common CNV regions. Among such CNVs called from WGS pilot data and spanning at least two SNP-array probes (the minimum required by our approach), HI-CNV achieved a recall rate of 81% (Figure S2A; Table S1). Recall was unsurprisingly much lower (6%) when considering all MAF<5% CNVs called from WGS data (i.e., removing restrictions on size and array-overlap), consistent with most CNVs being shorter than the resolution of SNP-array probe spacing. However, recall of gene-overlapping CNVs was substantially higher (24%) because the UK Biobank SNP-array was designed to prioritize inclusion of coding variants (Bycroft et al., 2018). Moreover, the HI-CNV call set appeared to account for approximately half of the 10.2 genes per genome estimated to be altered by rare structural variants (Collins et al., 2020): restricting to rare (MAF < 1%) whole-gene duplications and CNVs predicted to cause loss-of-function (pLoF), a mean of 5.0 genes per individual were altered by such CNVs (2.8 pLoF and 2.2 gene duplications). Across 18,251 genes, whole-gene duplications and pLoF CNVs were called in a median of 6 and 8 individuals, respectively, with observed counts decreasing with increasing gene constraint (Figure 1G).
To explore the extensibility of HI-CNV to smaller cohorts and to other SNP-array data sets, we performed two additional analyses. First, we ran HI-CNV on subsamples of the UK Biobank data set, observing robust improvements in detection sensitivity even at ~100-fold smaller sample sizes (5,000 individuals; Figure 2). Second, we applied HI-CNV to 179,538 BioBank Japan participants (Nagai et al., 2017) (STAR Methods) and observed performance similar to UK Biobank: HI-CNV successfully leveraged haplotype-sharing within BioBank Japan to call an average of 28.4 calls per individual, with an estimated validation rate of 93% (Figure S1B; Table S1).
Figure 2: HI-CNV performance benchmarks on subsamples of the UK Biobank data set.
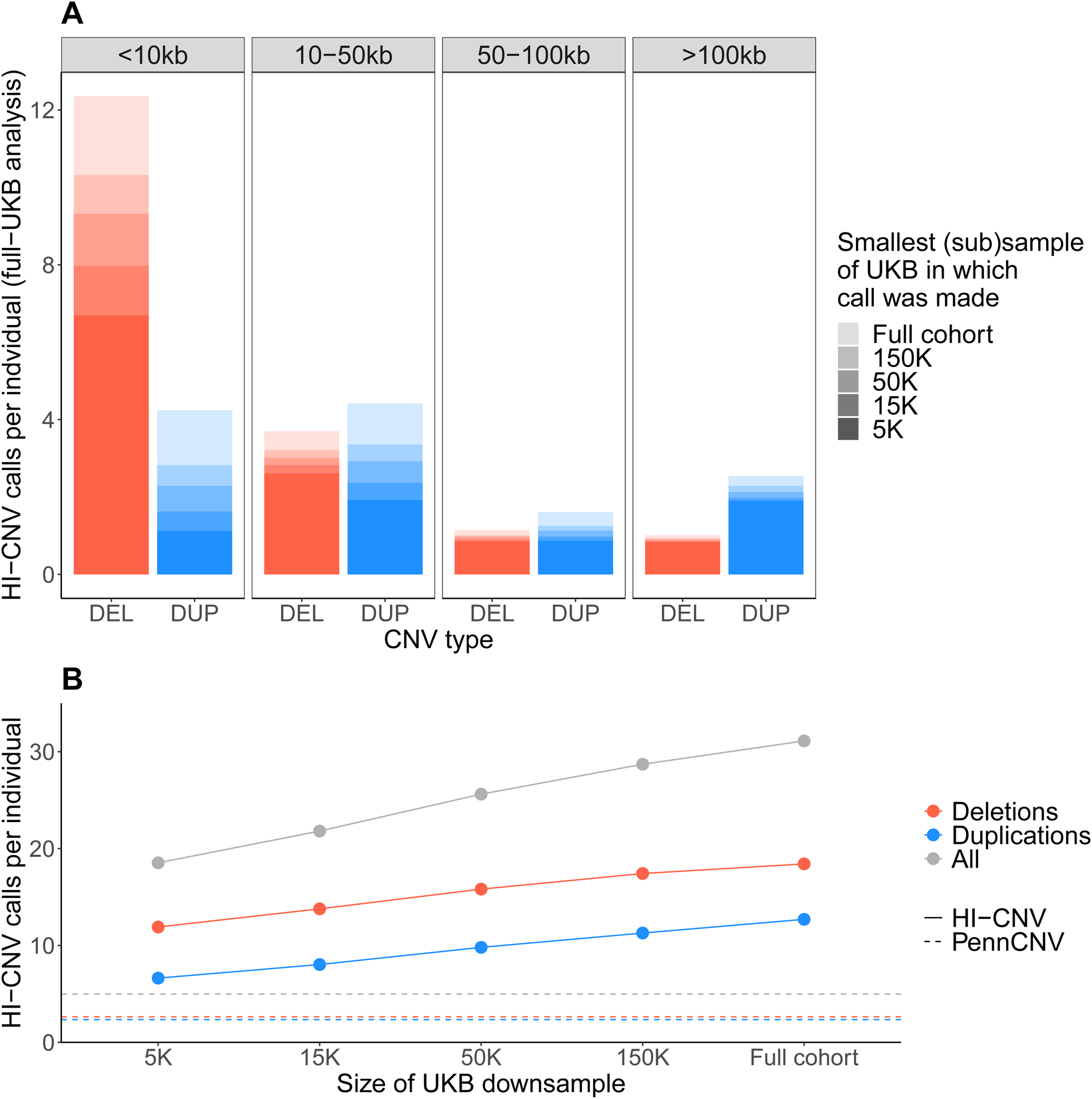
To evaluate the extent to which HI-CNV improves detection sensitivity in smaller sample sizes, we benchmarked the performance of HI-CNV across a range of subsamples of UK Biobank (N = 5K, 15K, 50K, and 150K). A For a subset of 500 individuals included in all subsamples, for each CNV call made in these individuals in the full N~500K analysis, we determined the minimal sample (N = 5K, 15K, 50K, 150K, or full cohort) in which the call was detected. Full bar heights indicate average numbers of calls across the 500 individuals (from the full N~500K analysis) stratified by event size and CNV type (deletion vs duplication). Shading reflects the subsample in which each call was first detected (defined as a call in the subsample overlapping or perfectly replicating the given call). These analyses showed that while detection sensitivity increased with sample size as expected (especially for small CNVs <10kb), most CNV calls made using the full UK Biobank cohort were already detectable by HI-CNV at a sample size of N=5K. B We compared the average number of calls per individual made by HI-CNV (on N = 5K, 15K, 50K, 150K, or all samples) to PennCNV. The average number of called CNVs per individual is plotted across the various subsamples, colored by CNV type. The horizontal lines reflect the average number of events detected by PennCNV across the entire UK Biobank cohort. (In each subsample, ~90% of calls (range: 89%–93%) replicated or overlapped calls made using the full cohort, indicating effective false-positive control in these downsampled analyses.)
Fine-mapping analyses reveal likely-causal CNV-trait associations
HI-CNV’s detection of many previously-undiscovered CNVs in UK Biobank suggested that CNV-phenotype association analyses might uncover new CNVs impacting human traits. We applied a combination of single-variant and burden-style analyses to test three categories of CNVs (gene-level, CNV-level, and probe-level; Figure 3A) for association with 56 heritable quantitative traits, including anthropometric traits, blood pressure, measures of lung function, bone mineral density, blood cell indices, and serum biomarkers (Table S2). We performed association analyses on up to 452,500 UK Biobank participants of European ancestry using linear mixed models implemented in BOLT-LMM (Loh et al., 2015, 2018a). We then removed associations that could potentially be explained by linkage disequilibrium with other variants by requiring each association to remain significant (P < 5 × 10−8) after conditioning on any other more-strongly-associated SNP, indel, or CNV within 3 megabases (STAR Methods). We previously observed that when fine-mapping associations involving rare variants (which comprised nearly all CNVs we detected), this pairwise LD filter effectively identifies variants likely to causally drive associations (Barton et al., 2021). This analysis pipeline resulted in 269 fine-mapped CNV-trait associations at 97 loci involving 252 likely-causal CNVs (Tables S3 and S4). The CNV calls involved in these associations exhibited an even higher WGS-based validation rate (94%) than the overall call set (Figure S1C; Table S1; STAR Methods), consistent with the idea that false-positive CNV calls are unlikely to confound association analyses.
Figure 3: Fine-mapping analyses reveal likely-causal CNV-trait associations.

A Association and fine-mapping pipeline; inset depicts the three categories of CNVs tested. B Effect size versus minor allele frequency for 269 likely-causal CNV-phenotype associations, colored by phenotype category. C Distributions of CNV length (left) and genic context (right) across all CNVs and across likely-causal CNVs. D Breakdown of 97 CNV loci according to prior literature status and whether a putative target gene was identified. E Candidate target genes, categorized according to whether (i) the CNV-phenotype association was previously reported, (ii) the target gene was previously implicated (either by a previously-reported coding variant association or by previous experimental work), or (iii) neither of the above. The rightmost column lists syndromic CNVs re-identified here. Colors indicate CNV type; bold font indicates noncoding CNVs potentially regulating the target gene. F Genic context of syndromic CNVs (bottom) and non-syndromic CNVs (top) stratified by the number of phenotype categories associated with the CNV.
Many of the 269 likely-causal CNV-phenotype associations had large effect sizes – including 59 associations with an absolute effect size greater than 1 standard deviation (s.d.) – and effect sizes generally increased with decreasing minor allele frequency (MAF) (Figure 3B). Only 10 of the 269 associations involved common (MAF > 5%) CNVs, whereas 186 associations involved CNVs with MAF < 0.1%. The associations affected most categories of phenotypes we considered, with blood cell phenotypes accounting for the majority of likely-causal associations (137 of 269 associations, involving 40 loci), reflecting their high heritability (average SNP-heritability of 0.31 (Barton et al., 2021)) and high representation among the quantitative traits we analyzed (19 of 56 phenotypes).
The likely-causal CNV-phenotype associations involved at least 252 unique CNVs (138 deletions, 114 duplications; Table S4; STAR Methods) which were enriched for multiple attributes correlated with functional impact (Figure 3C). Likely-causal CNVs tended to be longer than average (Li et al., 2020) and were much more likely to overlap coding sequences of genes (85.8% coding-overlapping vs. 22.1% expected for deletions; 94.7% vs. 43.4% expected for duplications; Figure 3C). For the small fraction of likely-causal deletions that did not overlap coding sequence (14.2%), roughly half overlapped enhancer annotations (42.1% vs. 8.4% expected; P = 7.76 × 10−5). The majority of likely-causal deletions affected either one gene (35%) or two genes (18%), facilitating further investigation of potential targets of trait-modifying CNVs.
CNV loci corroborate SNP associations and uncover gene-trait relationships
Of the 97 loci involved in the 269 fine-mapped CNV-trait associations, 72 loci had not been identified in previous large-scale CNV association studies (Aguirre et al., 2019; Auwerx et al., 2022; Beyter et al., 2021; Chen et al., 2021; Crawford et al., 2019; Li et al., 2020; Macé et al., 2017; Marshall et al., 2017; Sinnott-Armstrong et al., 2021). These previous studies included analyses of UK Biobank in which CNVs were genotyped using PennCNV (Aguirre et al., 2019; Auwerx et al., 2022; Crawford et al., 2019), which did not detect most likely-causal CNVs smaller than 20 kb (Figure S2B). For roughly half of the 72 previously unreported CNV loci (35 of 72 loci), we could identify a putative target gene (Figures 3D and 3E; Table S4). Among the 25 previously reported loci, half (13 loci) corresponded to syndromic CNVs known to cause genetic disorders (STAR Methods). These CNVs generally were longer, affected more phenotype categories, and overlapped more genes than CNVs at non-syndromic loci (Figure 3F), as expected. Many CNV associations corroborated target genes recently implicated by coding variant association studies (Barton et al., 2021; Marouli et al., 2017; Sinnott-Armstrong et al., 2021), including rare height-reducing deletions in CRISPLD2 and ADAMTS17, a rare sex hormone binding globulin (SHBG)-increasing deletion in HGFAC, and a rare IGF-1-decreasing partial deletion of MSR1 (Figure 3E). Several other CNV associations appeared to uncover genes contributing to the architecture of complex traits (Figure 3E).
To confirm the robustness of these associations, we performed two corroboratory analyses (STAR Methods). First, for associations involving CNVs predicted to cause loss-of-function (pLoF) of a putative target gene, we compared the effects of pLoF CNVs to the effects of ultra-rare pLoF SNP and indel variants in the same gene (Backman et al., 2021), which represent an independent class of genetic variants (and are guaranteed to be independent of overlapping deletion variants). We observed broadly consistent effect sizes between pLoF CNVs and pLoF SNP/indel variants (effect size correlation of 0.85, P = 8.0 × 10−21; Figure 4A). Among associations that we were well-powered to replicate (i.e., replication power >0.5 based on the effect size of the pLoF CNV and the combined allele frequency of ultra-rare (MAF<0.001%) pLoF SNPs and indels), we successfully replicated 35 of 36 associations (at nominal significance, P<0.05). Second, to obtain further confirmatory evidence supporting CNV associations implicating gene-trait relationships not previously identified (Figure 3E), we directly replicated CNV associations using HI-CNV calls in BioBank Japan. Among 14 associations (involving four genes) with suitable phenotyping and replication power in BioBank Japan, we observed broadly consistent effect sizes, with 13 out of 14 associations exhibiting the same effect direction as in UK Biobank (Figure 4B).
Figure 4: Corroboration and replication of CNV-phenotype associations.
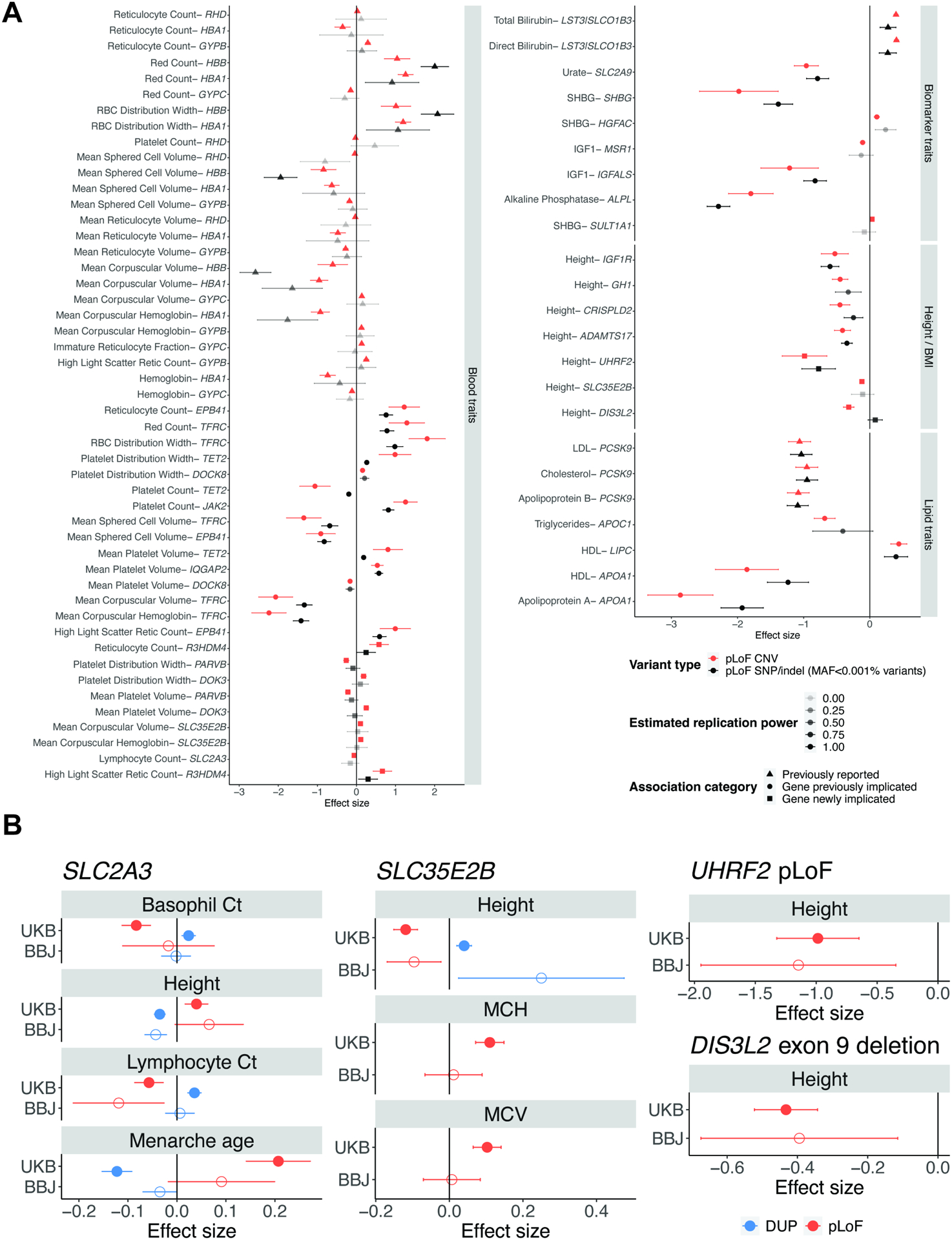
A Loss-of-function burden analyses in UK Biobank. For associations involving CNVs that we believed acted on a candidate target (focal) gene (Figure 3E), we compared the estimated effect of CNVs predicted to cause loss-of-function (pLoF) of the putative target gene to the estimated effect of ultra-rare pLoF SNP and indel variants in the same gene (recently reported in a whole-exome analysis of UK Biobank that performed SNP/indel pLoF burden tests (Backman et al., 2021)). Effect sizes and 95% confidence intervals are shown in red for the pLoF CNVs and in black for the pLoF SNP/indel burden; markers and error bars for the pLoF SNP/indel burden are shaded based on power to detect an association (assuming an effect size equal to the pLoF CNV and accounting for the combined allele frequency of the pLoF SNPs and indels). Previously reported associations are shown with a triangle, genes previously implicated are shown with a circle, and the remaining genes are shown with a square. B Replication of CNV-phenotype associations in BioBank Japan. We attempted to replicate 14 associations (selected based on available phenotyping and power in BioBank Japan) involved in gene-trait relationships putatively uncovered by our analysis of UK Biobank. Effect sizes and 95% confidence intervals are shown in red for pLoF CNVs and in blue for whole-gene duplications.
Given the large number of CNV loci identified here, we focus below on describing three classes of particularly interesting loci: (1) CNV associations stronger than any nearby SNP, (2) loci at which CNVs, together with nearby SNPs, created long allelic series, and (3) additional loci implicating putative target genes.
CNV associations stronger than nearby SNPs
Among 169 associations involving non-syndromic CNVs, a subset of 37 associations (22%) were stronger than associations of all SNPs within 500kb. Several of these associations appeared to uncover gene-trait relationships; here we highlight two loci with such associations. First, ultra-rare UHRF2 pLoF CNVs (carried by 19 UK Biobank participants) associated with a 1.11 (0.17) s.d. decrease in height (corresponding to 7.2 (1.1) cm shorter stature; P = 8.2 × 10−11; Figure 5A; Table S5). This association between UHRF2 and height was not visible from SNPs at the locus, none of which reached genome-wide significance (Figure 5A).
Figure 5: CNV-phenotype associations stronger than nearby SNPs. A UHRF2 locus.
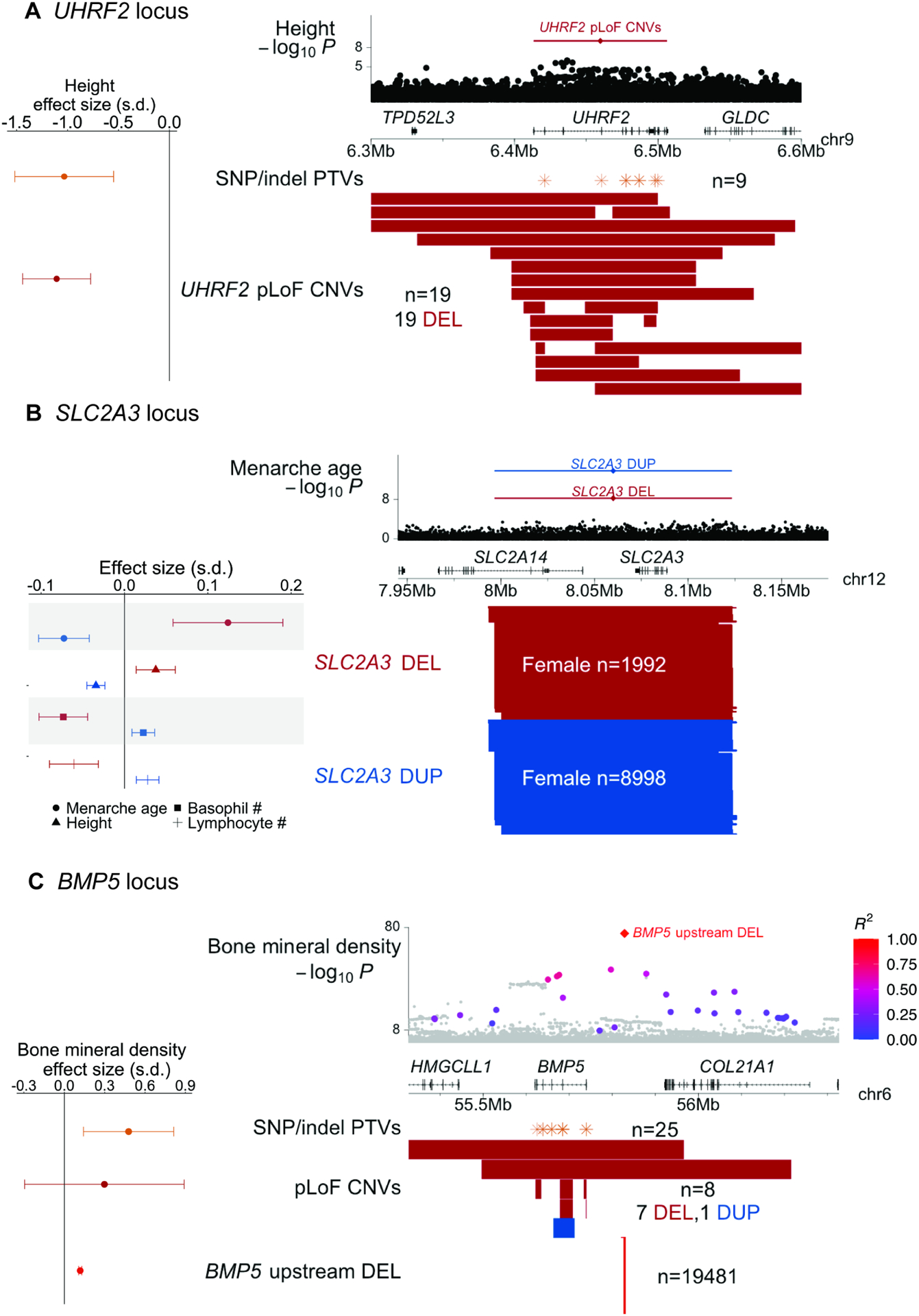
Top: height associations for UHRF2 pLoF CNVs and nearby SNPs. Bottom: locations of UHRF2 pLoF CNVs and SNP and indel PTVs; left: effect sizes for height. B SLC2A3 locus. Top: menarche age associations for SLC2A3 duplications and deletions and nearby SNPs. Bottom: locations of SLC2A3 deletions and duplications; left: effect sizes for menarche age, height, and basophil and lymphocyte counts. C BMP5 locus. Top: bone mineral density associations for a deletion upstream of BMP5 and nearby SNPs (colored according to linkage disequilibrium with the deletion, for SNPs with R2>0.1 to the deletion). Bottom: locations of the upstream deletion, BMP5 pLoF CNVs, and SNP and indel PTVs; left: effect sizes for bone mineral density. In all panels, deletions are colored red and duplications are colored blue. Error bars on effect sizes, 95% CIs. Numerical results are available in Table S5; example signal intensity plots are in Figure S3.
However, among 185,365 exome-sequenced UK Biobank participants (Szustakowski et al., 2021), nine carriers of UHRF2 protein-truncating SNP or indel variants (PTVs) exhibited 1.03 (0.25) s.d. decreased height (P = 3 × 10−5), corroborating the CNV association (Figure 5A; STAR Methods), which further replicated in BioBank Japan (Figure 4B). UHRF2 has not previously been implicated in large genome-wide association studies of height, demonstrating the utility of CNV association studies and motivating further study of how loss of one functional copy of UHRF2 (which encodes an E3 ubiquitin-protein ligase) impairs growth.
Another set of associations implicated copy-number variation of SLC2A3 as a modifier of age at menarche (P = 1.6 × 10−17), height (P = 7.7 × 10−12), and blood count phenotypes (Figure 5B; Table S3). SLC2A3 encodes GLUT3, a glucose transporter expressed in multiple tissues, and is prone to non-allelic homologous recombination that produces gene dosage-modifying ~130kb duplications and deletions (MAF = 1.9% and 0.4%, respectively, in our call set). SLC2A3 CNVs have been observed in many earlier studies, several of which have reported nominally significant associations with various clinical phenotypes; however, replication of these associations has been mixed (Ziegler et al., 2020). In UK Biobank, SLC2A3 deletions associated with delayed menarche (0.20 (0.03) years), increased height (0.25 (0.08) cm), and decreased basophil and lymphocyte counts, while duplications associated with reciprocal effects of roughly half the magnitude (Figure 5B; Table S5). Consistent effects were observed in BioBank Japan (Figure 4B). No individuals carried zero SLC2A3 copies (vs. 7.9 such individuals expected; P = 0.0009), consistent with previous literature suggesting that homozygous LoF mutations may be incompatible with life (Schmidt et al., 2009; Ziegler et al., 2020) (Figure S4A). These results support a dosage-sensitive role of GLUT3 in multiple organ systems.
Several other associations provided examples of loci at which SNP associations appeared to tag more-strongly-associated CNVs. Among the 37 associations for which a non-syndromic CNV attained the strongest association within 500kb, 21 involved loci at which a nearby SNP also reached significance. For six of those associations, the top SNP association became non-significant upon conditioning on the CNV. For example, a low-frequency (MAF = 2.2%) deletion upstream of BMP5, which encodes bone morphogenetic protein 5, associated strongly with increased bone mineral density (0.12 (0.01) s.d.; P = 9.2 × 10−82) and appeared to explain strong SNP associations nearby (P = 3.8 × 10−51, conditional P = 0.24; Figure 5C; Table S5), highlighting the importance of including structural variants in GWAS fine-mapping. BMP5 SNP and indel PTVs associated with stronger effects on bone mineral density (0.48 (0.17) s.d.; P = 0.005), suggesting that the deletion might affect an upstream regulatory region for BMP5, and motivating further exploration of allelic series including CNVs and SNPs.
Allelic series involving both regulatory and gene-altering CNVs
Several CNV-trait associations contributed to long allelic series involving both CNVs that appeared to modify regulatory elements as well as CNVs that directly affected genes, providing opportunities to explore the effects of such mutations relative to one another and to SNP and indel polymorphisms. At the α-globin locus, at which copy-number polymorphisms of HBA2 and HBA1 (both encoding α-globin) are known to cause thalassemias, an extended allelic series containing eight classes of CNVs enabled further insights into genetic control of alpha-globin expression (Figures 6A and S5; Table S5). α-globin and β-globin together compose hemoglobin, and both the production and balance of α- and β-globin are important for normal erythropoiesis (such that relatively too little α-globin can lead to α-thalassemia whereas α-globin duplication can increase the severity of β-thalassemia) (Piel and Weatherall, 2014; Taher et al., 2021). In UK Biobank, ultra-rare deletions that spanned either the α-globin gene pair, the upstream α-globin locus control region (HS-40), or the entire α-globin locus all associated with strongly decreased (~3 s.d.) mean corpuscular hemoglobin (MCH) and increased red blood cell (RBC) counts, consistent with such mutations causing α-thalassemia by inactivating the locus (Hatton et al., 1990; Hay et al., 2016; Liebhaber, 1990; Piel and Weatherall, 2014; Wilkie et al., 1990). “Silent” deletions of only HBA2 associated with a relatively milder 1.7 (0.2) s.d. decrease in MCH. Intriguingly, duplications of these genomic elements exhibited a further range of effects: while duplications that increased α-globin gene dosage by 1–2 copies appeared to have little or no impact on MCH, duplications of the entire α-globin locus appeared to have an effect similar to loss of one α-globin gene (1.9 (0.2) s.d. lower MCH). This allelic series suggests that increased and decreased α-globin expression result in similar hematological phenotypes (consistent with the importance of balance in α- and β-globin) and that enhancer function rather than α-globin gene dosage primarily limits increases in α-globin expression. These results illustrate the ability of biobank-scale CNV analyses to extend knowledge even at well-studied loci.
Figure 6: Allelic series involving both regulatory and gene-altering CNVs. A HBA locus.
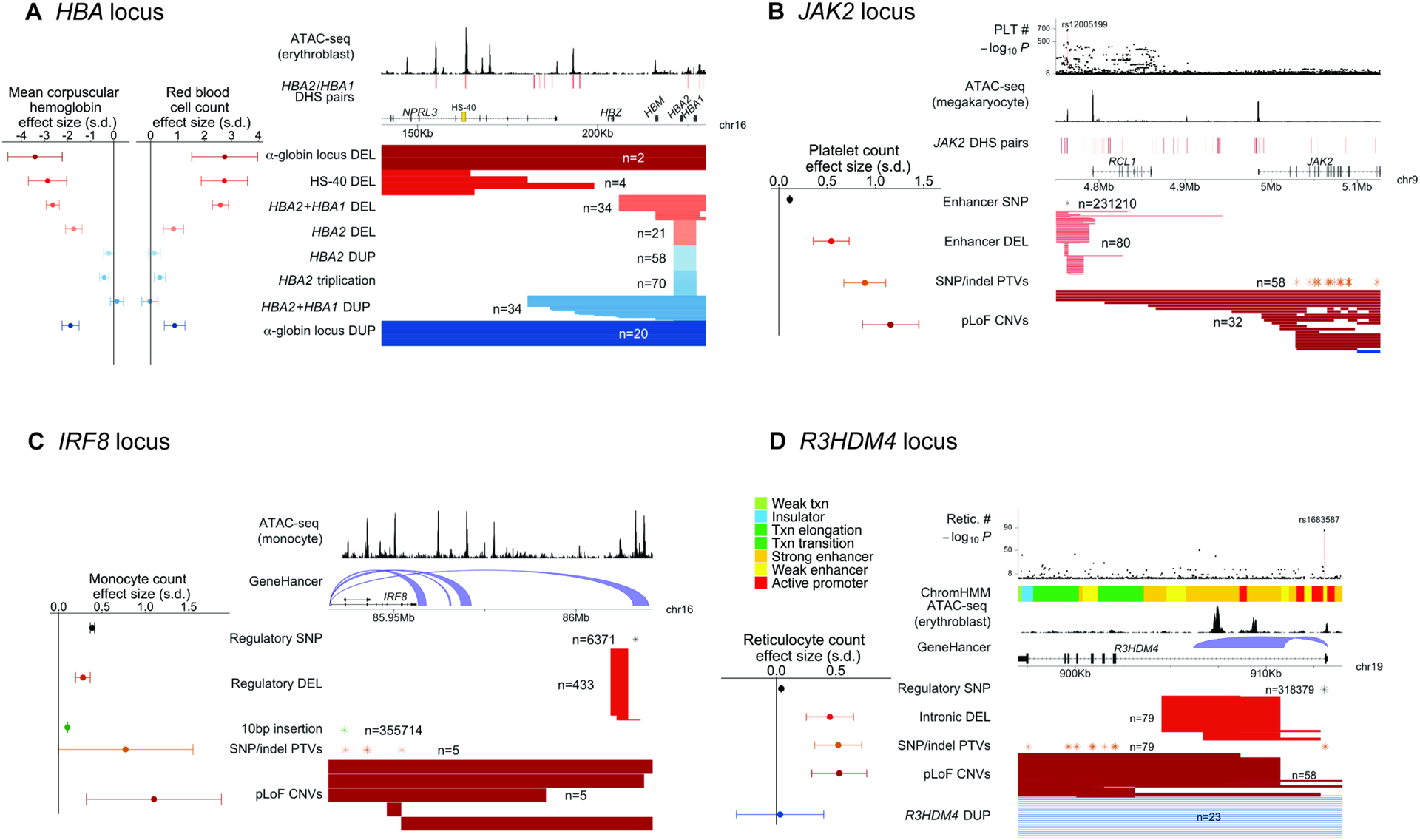
Eight classes of CNVs at the α-globin locus and their effect sizes for mean corpuscular hemoglobin and red blood cell counts. Genomic annotations indicate accessible chromatin regions in erythroblasts (Ulirsch et al., 2019) and distal DNase I hypersensitive sites (DHS) for HBA2/HBA1 (Thurman et al., 2012), highlighting the HS-40 super-enhancer. B JAK2 locus. Four classes of variants – JAK2 pLoF CNVs, JAK2 SNP and indel PTVs, a deletion of a distal enhancer, and the common SNP rs12005199 within the enhancer – and their effect sizes for platelet counts. Genomic annotations indicate accessible chromatin regions in megakaryocytes (Ulirsch et al., 2019) and JAK2 distal DHS pairs (Thurman et al., 2012), which colocalize with common-SNP platelet count associations (top) at the enhancer region ~220kb upstream of JAK2. C IRF8 locus. Fine-mapped common variants and rare pLoF variants at the IRF8 locus – including a putatively regulatory distal deletion, IRF8 pLoF CNVs, and IRF8 SNP and indel PTVs – and their effect sizes for monocyte counts. Genomic annotations indicate accessible chromatin regions in monocytes (Ulirsch et al., 2019) and GeneHancer connections (Fishilevich et al., 2017) between downstream regulatory regions and IRF8. D R3HDM4 locus. Rare CNVs, SNP and indel PTVs, and a common intronic SNP at R3HDM4 and their effect sizes for reticulocyte counts. Genomic annotations indicate ChromHMM (Ernst and Kellis, 2017) annotations, accessible chromatin regions in erythroblasts (Ulirsch et al., 2019), and GeneHancer connections (Fishilevich et al., 2017), all indicating regulatory function in the first intron of R3HDM4. The lead-associated SNP rs1683587 (top) also lies within this intron, suggesting regulatory function. In a and b, DHS pairs are colored by their correlation value, from light red (correlation < 0.8) to dark red (correlation >0.95). Error bars on effect sizes, 95% CIs. Numerical results are available in Table S5; example signal intensity plots are in Figure S3.
Some allelic series involved known gene-trait relationships but appeared to reveal CNV effects with no SNP analogues. At JAK2, ultra-rare CNVs predicted to cause loss of JAK2 function associated with a 1.16 (0.15) s.d. increase in platelet counts (P = 9.9 × 10−15; Figure 6B; Table S5). This association, which replicated in an analysis of SNP and indel PTVs (β = 0.89 (0.11) s.d., P = 1.1 × 10−15; Figure 6B), corroborated previous reports of an unexpected negative regulatory role for Jak2 in thrombopoiesis (Meyer et al., 2014). Interestingly, a distinct set of ultra-rare deletions centered ~220kb upstream of JAK2 associated with a 0.54 (0.09) s.d. increase in platelet counts (P = 9.5 × 10−9; Figure 6B; Table S5), roughly half the effect size of pLoF variants. The focal <4kb region shared by these deletions matched a strong megakaryocyte-specific accessible chromatin region previously implicated by common-SNP association and fine-mapping studies (Ulirsch et al., 2019) (Figure 6B) that appeared likely to regulate JAK2 (Table S5). However, deletion of the entire enhancer element associated with a five-fold larger effect on platelet counts than the single-base pair modifications produced by SNPs within the enhancer (Figure 6B; Table S5), highlighting the ability of CNVs to enable further insights into complex trait genetics by altering the genome in ways that SNPs cannot.
Copy-number variants also contributed to an extended allelic series at IRF8, which encodes a transcription factor critical to monocyte differentiation (Kurotaki et al., 2013). Strong SNP associations with monocyte counts have previously been observed at the IRF8 locus, led by a common noncoding 10bp insertion in IRF8 with a mild effect size (0.102 (0.002) s.d.; P = 7.8 × 10−587; Figure 6C; Table S5). Multiple SNPs downstream of IRF8 also associated independently with monocyte counts (consistent with the presence of multiple distal enhancers (Durai et al., 2019; Schönheit et al., 2013)), including a low-frequency SNP (rs11642657; MAF=0.8%) with a larger effect size (0.39 (0.01) s.d.; Figure 6C; Table S5). CNVs provided further insights into complex genetics at this locus: loss of one functional copy of IRF8 (identified in 10 carriers of either pLoF CNVs or PTVs) appeared to produce a larger increase in monocyte count (0.94 (0.28) s.d.; P = 0.0009), while a downstream deletion near rs11642657 had a moderate effect size similar to this SNP (0.28 (0.04) s.d.; P = 4.7 × 10−11), suggesting the presence of an important regulatory region (Figure 6C).
Some allelic series appeared to uncover gene-trait associations. Ultra-rare deletions at R3HDM4, a gene with unknown function, associated with 0.54 (0.08) s.d. higher reticulocyte counts (P = 3.5 × 10−11; Figure 6D; Table S3). This association was corroborated by R3HDM4 PTVs (β = 0.52 (0.10) s.d., P = 2.7 × 10−7), and a common intronic SNP also exhibited a mild-effect but strongly significant association with reticulocyte counts (β = 0.041 (0.002) s.d., P = 6.6 × 10−86; Figure 6D; Table S5). Interestingly, closer inspection of the deletions showed that they consisted of both exon-overlapping, pLoF deletions as well as intronic deletions falling fully within the first intron of R3HDM4, yet associating with a similar increase in reticulocyte counts (0.45 (0.10) s.d.; Figure 6D). These results suggest a key regulatory role of the intronic region spanned by the deletions, which contains an accessible chromatin region (in erythroblasts) with predicted R3HDM4 enhancer function (Ernst and Kellis, 2017; Fishilevich et al., 2017). Despite their associations with reticulocyte counts, neither type of deletion appeared to affect red blood cell counts (P = 0.17). These observations, which will require further understanding of R3HDM4 function to explain, again show the ability of regulatory CNVs to have significant phenotypic impacts, sometimes as strong as gene-dosage altering CNVs.
Diverse potential functional impacts of CNVs
The remaining likely-causal CNVs that appeared to uncover gene-trait associations (Figure 3E) seemed to alter gene dosage or function via a diversity of genomic modifications. Four rare deletions appeared to reduce or abolish gene function in a variety of ways. Two deletions associated with height: an inframe deletion spanning DIS3L2 exon 9 previously reported to reduce ribonuclease activity and cause Perlman syndrome (an autosomal recessive disease characterized by congenital overgrowth) (Astuti et al., 2012) surprisingly appeared to decrease height by 0.44 (0.04) s.d. in heterozygous carriers (P = 3.9 × 10−22), and a whole-gene deletion of SLC35E2B associated with modestly decreased height and increased MCH (Table S3). Interestingly, while both associations with height replicated in BioBank Japan and reciprocal duplications associated with increased height (Figures 4B, S4B, and S4C), pLoF variants in DIS3L2 appeared not to affect height (Figure 4A), such that further work will be necessary to decipher whether DIS3L2 exon 9 CNVs act through altering function of DIS3L2 or via a regulatory effect on a nearby gene (e.g., NPPC; STAR Methods). Two other deletions associated with ~0.2–0.3 s.d. effects on platelet traits: an inframe deletion spanning DOK3 exon 3 and a deletion spanning the final exon of PARVB (encoding 26 of 364 amino acids) (Table S3).
Another gene-trait association involved ultra-rare (MAF=0.003%), large (>700 kb) duplications that appeared to target a single gene, CXCR4, and associated with a 0.99 (0.17) s.d. decrease in monocyte counts (P = 5.5 × 10−9, Table S3). Gain-of-function mutations within CXCR4 (chemokine receptor 4) cause autosomal dominant WHIM syndrome, an immunodeficiency disease (Hernandez et al., 2003). Here, duplication of CXCR4 appeared to produce relatively milder decreases in leukocyte counts (including 0.5 (0.2) s.d. reduced neutrophil and lymphocyte counts) with no apparent disease phenotypes.
A final association with platelet distribution width involved a low-frequency (MAF=0.7%) variant that initially appeared to be a duplication at MTMR2 (Table S3) but was surprisingly absent from CNV reference data sets (Byrska-Bishop et al., 2021; Collins et al., 2020). Closer examination of sequencing reads from exome-sequenced carriers revealed that the structural variant actually constitutes a retroposition of the spliced MTMR2 transcript into an intron of LRCH1 (STAR Methods). A common SNP haplotype in a different intron of LRCH1 strongly and independently associated with increased platelet distribution width (P = 2.5 × 10−172), and both the SNP association and the insertion variant association (P = 3.5 × 10−17) appeared to be mediated by reduced LRCH1 expression (based on analyses of GTEx data (Aguet et al., 2020); STAR Methods), with the insertion exhibiting four-fold larger effects (Figure S4D and Table S5). This unexpected finding from SNP-array analysis hints at further discoveries that will be enabled by sequencing technologies capable of comprehensively genotyping structural variants.
Associations of CNVs with disease traits
Analyses of CNVs for association with 757 disease phenotypes curated by UK Biobank (STAR Methods) recovered known associations. Among 68 significant associations (P < 1 × 10−9) that remained after LD-clumping, 64 associations involved syndromic CNVs, three associations involved other known loci (HBA and HBB for thalassemia and RHD for maternal-fetal problems), and the remaining association appeared on follow-up to be a false positive (Table S6). These results reflect the challenge of performing disease analyses in generally-healthy population cohorts; larger CNV call sets or case-control cohorts will be necessary to power discovery of new CNV-disease associations.
Contrasting effects of deletions and duplications
Total genomic deletion burden and duplication burden have each been shown to associate with deleterious effects on several human traits (Dauber et al., 2011; Macé et al., 2017; Wheeler et al., 2013). We similarly observed associations of deletion and duplication burden with decreased height and years of education (even after excluding syndromic CNVs), with deletions appearing to be roughly four-fold as deleterious as duplications (Figures 7A and 7B; Table S7). The consistent negative effect directions of deletion burden and duplication burden contrasted with the opposite effect directions that we observed at several loci involving focal reciprocal CNVs (Table S3).
Figure 7: Contrasting phenotypic effects of deletions and duplications.

A,B Mean height (a) and years of education (b) as a function of total genomic length affected by deletions and duplications. Individuals carrying a known syndromic CNV were excluded from analysis. Numerical results are presented in Table S7. C Associations between whole-gene deletions and quantitative traits in targeted analyses of 41 gene-trait pairs for which we previously identified likely trait-altering PTVs(Barton et al., 2021) and for which the HI-CNV call set contained at least two whole-gene deletions. Effect sizes and 95% confidence intervals are shown in red for 16 genes for which whole-gene deletions exhibited nominally significant associations (P < 0.05); effect sizes for SNP or indel PTVs (Barton et al., 2021) are shown in black. D Observing 16 nominally significant associations was consistent with whole-gene deletions having the same effects as PTVs. Probability distributions indicate numbers of significant associations in simulations in which whole-gene deletions have no effect (grey), half the effect magnitude as PTVs (light pink), or the same effect magnitude as PTVs (red). E,F Analogous results for whole-gene duplications in targeted analyses of 139 gene-trait pairs, which produced 27 significant associations (P < 0.05), consistent with whole-gene duplications having less than half the effect magnitude of PTVs. (The aberrant effect directions of DOCK8 deletions and duplications relative to the DOCK8 PTV rs192864327 may be explained by this variant only causing loss of function in one of several transcripts.)
To more thoroughly explore the relative effects of focal deletions and duplications, we examined gene-trait pairs for which we had previously identified PTVs likely to alter quantitative traits (Barton et al., 2021). For each gene, we compared the effects of likely-causal PTVs to those of whole-gene deletions and duplications (STAR Methods). As expected, gene deletions acted similarly to PTVs, with 16 of 41 genes exhibiting nominally significant deletion associations (Figure 7C), consistent with available power (Figure 7D). In contrast, gene duplications tended to act in the opposite direction as PTVs and with smaller effect magnitudes: 27 of 139 genes exhibited nominally significant duplication associations (Figure 7E), consistent with duplications tending to have less than half the effects of deletions (Figure 7F; Table S7). These results suggest a contrast between CNV burden, which may be driven by large CNVs that disrupt many genes and tend to be deleterious regardless of deletion or duplication status, versus focal CNVs, which may tend to change the dosage of a specific key gene, resulting in reciprocal effects of deletions and duplications.
Discussion
These results demonstrate the power of haplotype-informed structural variant analysis that leverages pervasive distant relatedness within large biobank cohorts to pool information about variants co-inherited by individuals who share extended SNP haplotypes. Applied to explore CNV-phenotype associations in UK Biobank, this approach revealed many ways in which genetic variation influences complex traits. At several loci, large-effect CNVs uncovered putative target genes, and at several other loci, CNVs, together with nearby SNPs, created long allelic series illustrating the ability to CNVs to produce functional effects with no SNP analogues (e.g., gene copy-gain and regulatory element deletion or duplication).
Beyond the specific biological findings reported here, our study also provides a careful analytical approach for handling the statistical subtleties of performing association and fine-mapping analyses on difficult-to-call structural variants that can span large genomic regions. Additionally, the observation of several CNVs that represented lead associations at loci underscores the importance of considering structural variation even when performing statistical fine-mapping of SNP associations (Beyter et al., 2021; Mukamel et al., 2021).
These results also motivate further exploration of the far-larger set of CNVs that were not accessible to our analyses. While our approach enabled detection of 6-fold more CNVs than previous analyses of UK Biobank, and these CNVs appeared to account for roughly half of the rare LoFs estimated to arise from structural variation (Collins et al., 2020), the CNVs we detected from SNP-array data still represent only a small fraction of the thousands of CNVs typically present in each human genome (Abel et al., 2020; Collins et al., 2020). We anticipate that future studies analyzing short- and long-read sequencing data will provide many more insights into the phenotypic consequences of copy-number variation.
Limitations of the Study
The primary limitations of our study arose from inherent technical limitations of SNP-array probe intensity data. We were unable to ascertain CNVs smaller than the resolution of the SNP array, and we were also unable to genotype most common CNVs (MAF > 5%) due to inadequate SNP-array coverage and breakdown of modeling assumptions. Similarly, we were unable to genotype multi-copy CNVs due to limited differentiation of copy-number states in probe intensity data (Figure S6 and STAR Methods). These limitations could potentially be overcome by extending the HI-CNV framework to whole-exome or whole-genome sequencing data, which is a promising direction for future research, especially at loci that are challenging to genotype. A separate limitation of our study is that while we successfully replicated many of the CNV-phenotype associations we reported, other associations have yet to be externally validated, and in all cases experimental work will be necessary to conclusively demonstrate causality and determine mechanism.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Po-Ru Loh (poruloh@broadinstitute.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Summary statistics have been deposited at Zenodo and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
METHOD DETAILS
UK Biobank genetic and phenotypic data
Genome-wide SNP-array data, including allelic dosages of pairs of alleles (labeled A and B) for 805,426 biallelic variants (784,256 autosomal), was previously generated for 488,377 UK Biobank participants (Bycroft et al., 2018). For CNV-calling, these allelic intensities are typically transformed to measures of total intensity (LRR) and relative intensity (B-allele frequency, BAF). We analyzed the LRR values provided by UK Biobank after first applying two de-noising steps: (i) GC-correction of total allelic intensities and (ii) principal component (PC)-correction of LRR (Dennis et al., 2021); and we directly computed relative allelic intensities (see Transforming and denoising SNP-array genotyping intensities). We also analyzed pilot whole genome sequencing (WGS) data available for 48 individuals (for validation analyses) and whole exome sequencing (WES) data available for 200,643 individuals (Szustakowski et al., 2021) (for follow-up analyses) as well as subsequently-released WGS data for 500 individuals (for further validation).
We performed CNV analyses on the subset of 487,409 participants included in the UK Biobank imputed data set (Bycroft et al., 2018). We focused our primary analyses on individuals of self-reported European ancestry, excluding individuals with trisomy 21, blood cancer, or those who had withdrawn at the time of our study (see Quality control filtering in UK Biobank), resulting in 454,759 participants with array data, 43 individuals with WGS data, and 186,105 individuals with WES data.
We analyzed 56 heritable quantitative traits measured on the majority of UK Biobank participants. These traits included anthropometric traits, blood pressure, measures of lung function, bone mineral density, blood cell indices, and serum biomarkers (Table S2). Quality control and normalization of the quantitative traits was previously described (Barton et al., 2021; Loh et al., 2018a).
Overview of HI-CNV method for haplotype-informed CNV detection
We reasoned that CNV detection sensitivity from SNP-array data could be considerably increased via two orthogonal strategies: (a) estimating SNP-specific priors for allele combinations corresponding to CNV states (to enable more accurate assessment of probabilistic information about copy-number variation provided by probe intensities); and (b) integrating probe intensity data across individuals likely to have co-inherited a large genomic tract. To estimate SNP-specific priors for allele combinations corresponding to CNV states, we (i) directly estimated SNP-specific genotype cluster priors at a subset of SNPs covered by large, easily-called CNVs; and then (ii) used these SNPs as a reference set from which SNP-specific priors for other SNPs could be predicted (based on which SNPs in the reference set exhibited most-similar probe intensity patterns). To incorporate probe intensity data across individuals likely to have co-inherited a large genomic tract, for each individual and genomic position on the SNP-array, we used a PBWT-based algorithm to find the 10 longest identical-by-descent (IBD) matches (per haplotype of the individual) spanning the position under consideration.
We note that at loci containing multiple different types of copy-number polymorphism (e.g., CNVs with different sizes or breakpoints), haplotype-sharing information is still helpful even though different CNVs are expected to reside on different haplotypes (as they arose from distinct mutational events): as long as the shared haplotype postdates the mutational event that gave rise to a specific CNV, the individuals sharing that haplotype will still all carry the exact same CNV. In this respect, the different CNVs at a locus all behave like independent variants from the point of view of haplotype-sharing analysis.
We used a hidden Markov model to call CNVs, integrating probabilistic information about copy-number state across an individual and their “haplotype neighbors” by weighting each neighbor’s information according to length of IBD-sharing. In more detail, at each SNP, for the individual and for each haplotype neighbor, we computed Bayes factors for deletion and duplication states based on genotyping intensities from the corresponding sample. We then created a weighted sum of log Bayes factors at each SNP, giving higher weights to haplotype neighbors with longer IBD. We ran this analysis using several different weighting schemes (trading off sensitivity to more recent vs. older mutations) and compiled calls made across these weighting schemes.
We filtered CNV calls to deletions larger than 75bp and duplications larger than 500bp and removed individuals with more than 100 CNV calls. Many UK Biobank samples with aberrantly many CNV calls appeared to share rare technical artifacts in LRR that had escaped denoising. We therefore computed the first 10 principal components of LRR in these aberrant individuals, ranked all individuals by the amount of LRR variance explained by these artifact PCs, and further removed individuals in the top 0.5%. Finally, for all downstream analyses, we removed calls on any chromosome in which we had previously detected a mosaic CNV (Loh et al., 2020) as well as calls in regions with frequent somatic events. After these quality control filters, we had called CNVs in 452,500 UK Biobank participants (including 43 individuals with WGS data and 185,365 individuals with WES data). Further methodological details are available below (see Transforming and denoising SNP-array genotyping intensities; Estimating genotype cluster parameters; Finding longest identical-by-descent (IBD) matches per haplotype; Calling CNVs using intensity data across haplotype neighbors; Filtering, merging, and genotyping CNVs; and Quality control filtering in UK Biobank).
PennCNV call set in UK Biobank
We compared HI-CNV calls to previously-generated PennCNV (Wang et al., 2007) calls made by analyzing Affymetrix CEL files (UK Biobank Return 1701) (Crawford et al., 2019). Following suggested quality control procedures (Kendall et al., 2017), we filtered individuals with 30 or more calls, a genotype call rate less than 96%, or an absolute waviness factor greater than 0.3 and filtered individual CNV calls covered by <10 probes or with low probe density (< 1 probe per 20kb). To facilitate comparison to our HI-CNV call set, we then applied the same additional filtering of calls on chromosomes containing mosaic CNVs and in regions with frequent somatic events.
Precision and recall of HI-CNV and PennCNV call sets
To benchmark performance of HI-CNV and PennCNV, we analyzed independent WGS data available for 43 individuals using CNVnator (Abyzov et al., 2011) and DELLY (Rausch et al., 2012). To assess the precision, or validation rate, of array-based calls we computed the proportion of HI-CNV (respectively, PennCNV) calls that were either (1) replicated by CNVnator calls or (2) exhibited enrichment or depletion of read-depth (computed by CNVnator) consistent with the CNV call. To assess recall, or sensitivity, of HI-CNV and PennCNV, we analyzed calls from DELLY, which produced a merged call set across WGS samples that was helpful for computing recall of CNVs within allele frequency ranges. For each DELLY call, we annotated whether HI-CNV (respectively, PennCNV) called an overlapping event. Further details on computing precision and recall are provided below (see Summary measures of UK Biobank HI-CNV callset).
Stratifying carrier counts of gene dosage-modifying CNVs by LOEUF score
For each protein-coding gene, we computed the number of UK Biobank participants of European ancestry carrying whole-gene deletions, whole-gene duplications, and CNVs predicted to cause loss of function (pLoF; see Creating CNV genotypes for association tests). We then annotated each gene with its LOEUF sextile bin (‘oe_lof_upper_bin_6’ from the pLoF Metrics by Gene TSV file downloaded from https://gnomad.broadinstitute.org/downloads), which estimates strength of selection against protein-truncating mutations (Karczewski et al., 2020). We restricted to genes with a non-missing LOEUF sextile bin and genes with only one annotated canonical transcript. In Figure 1G, we reversed the order of LOEUF sextile bins such that higher-numbered bins correspond to more-constrained genes.
Association testing and statistical fine-mapping
We performed CNV-phenotype association analyses on three distinct classes of CNVs defined based on 1) SNP-array probe overlap, 2) gene overlap, and 3) specific CNVs. Analyses on the SNP probe level tested the hypothesis that a change in copy number (deletion or duplication, respectively) at the genomic location of the SNP alters the phenotype. Analyses on the gene level tested the hypothesis that a change in copy number affecting the gene in question (whole-gene deletion, whole-gene duplication, and pLoF, respectively) alters the phenotype. Analyses on the CNV level tested whether a specific CNV (allowing for slightly differing endpoints in calls from different samples) alters the phenotype. These tests comprised both burden-style analyses (the probe- and gene-level tests) and single-variant analyses (the CNV-level tests), for a total of ~1.7 million tests. Given that these tests contained a high degree of redundancy (e.g., because probe-level tests at consecutive SNPs tended to be very strongly correlated), we used the standard genome-wide significance threshold (P < 5 × 10−8) to determine significant associations.
We conducted association tests using BOLT-LMM (Loh et al., 2015, 2018a) (--lmmForceNonInf) with assessment center, genotyping array, sex, age, age squared and 20 genetic principal components included as covariates. We fit the mixed model on directly genotyped autosomal variants with MAF > 10−4 and missingness < 0.1 and computed association test statistics for CNVs in the three categories defined above; a similar pipeline produced association test statistics for SNP and indel variants imputed by UK Biobank (the imp_v3 release) and variants we previously imputed from the first tranche of exome-sequencing of 49,960 participants (Barton et al., 2021). We included all participants with non-missing phenotypes in the QC-ed European-ancestry HI-CNV call set described above.
To filter significant associations to a set of likely-causal associations, we used a pipeline we previously developed (Barton et al., 2021) to eliminate associations that could be explained by linkage disequilibrium (LD) with nearby variants (here, either SNP or indel variants from the UK Biobank imp_v3 release or variants we had imputed from WES (Barton et al., 2021)). This filter required CNVs to remain significant after conditioning on any other more strongly associated variant nearby. More explicitly, for every CNV i significantly associated with a given phenotype, we calculated its correlation rij with each more strongly associated variant j (including other CNVs and imputed SNPs and indels) within 3Mb using plink ‘--r’ (Chang et al., 2015). We then computed the approximate chi-square association statistic for CNV i conditioned on variant j as:
We defined likely-causal associations as those with the property that (P < 5 × 10−8) for all variants j more strongly associated with the trait than CNV i. We previously observed that this pairwise LD-based filter was effective for fine-mapping rare variant associations (Barton et al., 2021).
Defining and annotating CNV loci
To group phenotype-associated CNVs into genomic loci, we first identified a set of unique CNVs contributing to likely-causal associations (accounting for uncertainty in CNV breakpoints and for probe-level and gene-level tests aggregating signal across multiple CNVs; see CNVs contributing to likely-causal phenotype associations). We then ordered this set of likely-causal CNVs from smallest to largest, and if a CNV fell within 100kb of a previous CNV, we considered it to be part of the same locus. We annotated a likely-causal CNV as syndromic if it overlapped a previously-curated pathogenic CNV (Crawford et al., 2019) by more than 50%. We identified putative target genes of non-syndromic, likely-causal CNVs either by observing that a focal CNV association only overlapped a single gene or by finding independent supporting evidence for a particular gene within or near the CNV region (specifically, a coding variant association or experimental literature). Further details on defining and annotating loci are provided below (see Association testing and statistical fine-mapping).
Follow-up analyses at highlighted loci
At a subset of loci we investigated in greater detail (Figures 5 and 6), we identified carriers of high-confidence loss-of-function SNP and indel variants (annotated using LOFTEE (Karczewski et al., 2020)) among the 185,365 individuals with whole-exome sequencing data (Szustakowski et al., 2021) in our analysis set. To increase power to assess phenotypic impacts of SNP and indel PTVs, we residualized phenotypes for polygenic predictions of the phenotype using array-typed SNPs (omitting those within 2Mb of the gene of interest) that we generated using BOLT-LMM ‘—predBetasFile’ in 10-fold cross-validation (emulating linear mixed model association analysis) (Mefford et al., 2020). Residualized phenotypes could then be modeled as a function of SNP and indel PTV carrier status, as well as carrier status for other CNVs or SNPs of interest. We performed these analyses after our initial association analyses, such that numbers of carriers of CNVs differ slightly between Table S3 and the locus plots in Figures 5 and 6 (generated using karyoploteR (Gel and Serra, 2017)) due to participant withdrawals.
Binary association testing
We restricted disease association analyses to an unrelated subset of N=409,234 UK Biobank participants (within our primary European-ancestry sample set that passed quality control filters). Out of 1,126 “first-occurrence” binary disease phenotypes curated by UK Biobank, we tested 757 disease phenotypes which had at least 100 cases at the time of our study. We tested variants for association with binary traits using the BinomiRare test (Sofer, 2017) to obtain P-values robust to case-control imbalance while adjusting for age, sex, and 20 PCs. As previously described (Barton et al., 2022), for computational efficiency, we reimplemented the BinomiRare test and applied a binomial approximation when the number of observed cases among carriers exceeded 100. We identified approximately-independent CNV-disease associations using LD-clumping implemented in plink (Chang et al., 2015) (setting the LD threshold to 0.25 and the physical distance threshold to 250kb) and restricted results to CNV associations not within the MHC region that reached a significance threshold of P < 1 × 10−9.
HI-CNV analysis of BioBank Japan
We analyzed genotyping data previously generated for N=179,538 BioBank Japan participants using Illumina BeadChip platforms (either OmniExpressExome or a combination of OmniExpress and HumanExome) (Akiyama et al., 2017; Nagai et al., 2017). We analyzed genotyping probe intensities for 751,621 autosomal variants that passed quality control filters, extracting LRR values from Illumina GenomeStudio Final Report files and directly computing relative allelic intensities. We ran HI-CNV using haplotypes phased as previously described (Terao et al., 2020).
Transforming and denoising SNP-array genotyping intensities
UK Biobank provided genotyping intensity data generated by Affymetrix in two formats:
int files containing intensity values for the A and B alleles of each genotyped variant
baf and l2r files containing B allele frequency (BAF) and log2 R ratio (LRR) transformed intensity values (measuring relative and total genotyping intensities across the two alleles) used by typical CNV-calling pipelines.
Affymetrix’s genotype-calling algorithm modeled relative and total genotyping intensities by estimating bivariate normal distributions corresponding to “SNP clusters” for the three possible diploid (copy number 2; CN=2) genotypes (AA, AB, BB). We wished to extend this genotyping framework by additionally estimating bivariate normal SNP clusters for each possible genotype cluster corresponding to heterozygous CNVs, i.e., deletions (CN=1: A, B) and duplications (CN=3; AAA, AAB, ABB, BBB).
To do so, we required relative and total genotyping intensity measurements that were reasonably well-modeled by normal distributions. For relative genotyping intensities, the BAF values provided by UK Biobank did not meet this criterion because they had been truncated to fall between 0 and 1 (such that many individuals with homozygous genotypes had BAF of either 0 or 1). We therefore computed relative genotyping intensities from the int data for the A and B alleles by applying a polar-like transformation (Peiffer et al., 2006):
| (1) |
For total genotyping intensities, we analyzed the LRR (l2r) values provided by UK Biobank after first applying two denoising steps described below.
GC-correction of total allelic intensities (LRR)
We first corrected LRR values for “GC waves” (Diskin et al., 2008) using a simplified version of a previously-described pipeline (Jacobs et al., 2012; Loh et al., 2018b). Specifically, for each sample, we regressed LRR on proportions of GC and CpG content in 9 windows centered around each variant (spanning 50, 100, 500, 1k, 10k, 50k, 100k, 250k, and 1M bp) and analyzed the residuals. We computed GC content using bedtools (Quinlan and Hall, 2010) on the human reference (hg19), and we computed CpG content using the EpiGRAPH CpG annotation (Bock et al., 2007).
Principal component (PC)-correction of LRR
Even after GC-correction, top principal components of the LRR matrix explained large fractions of variance, indicating that the LRR data could be further-denoised by projecting out top PCs capturing unmodeled technical noise (Cooper et al., 2015). We took two precautions to guard against top PCs inadvertently capturing real signal from common CNVs:
We computed principal components on genome-wide LRR values for all autosomal variants at once (separately for each genotyping batch), reasoning that technical artifacts should behave similarly genome-wide (whereas inter-sample correlations in LRR driven by copy number variation would be locus-specific)—such that genome-wide PCs are more likely to pick up technical artifacts and less likely to “overfit” to local features.
We computed LRR PCs using only white British samples in order to reduce the potential for PCs to capture ancestry effects. (We then projected top PCs out of all samples in the genotyping batch: i.e., we regressed each sample’s LRR on top PCs and took the residuals).
We applied the above PC-correction procedure independently to each of the 106 genotyping batches, projecting the top 50 PCs out of LRR for each batch. We observed that these top 50 PCs explained an average of 58.5% of LRR variance. Additional PCs provided little marginal increase in variance explained (e.g., 100 PCs explained 61.6% of variance on average across batches).
Estimating genotype cluster parameters
SNP-array genotyping platforms use allele-specific oligonucleotide probes to quantify the abundance of each of two alleles (A and B) in a DNA sample. Genotyping of biallelic variants in regions of the genome that do not vary in copy number can then be performed by clustering measured probe intensities (across a batch of samples) into clusters corresponding to the three possible diploid genotypes (AA, AB, BB). Such clustering is usually performed using SNP-specific priors on the expected distribution of bivariate probe intensities assuming each possible genotype (AA, AB, BB), which for technical reasons can vary substantially among SNPs. Genotyping in this manner typically produces highly accurate genotype calls: e.g., ~99.9% accuracy with <1% missingness at most SNPs in UK Biobank (Bycroft et al., 2018).
SNP-array probe intensities are also informative of copy-number variants that overlap SNPs on an array, resulting in measured intensities that deviate from the clusters corresponding to the usual three diploid genotypes (AA, AB, BB) (Colella et al., 2007; Wang et al., 2007). Because these deviations are less dramatic than the differences in probe intensities that separate diploid genotypes, CNV-calling from the Affymetrix SNP-arrays used by UK Biobank (which produced relatively noisy probe intensity measurements) has tended to require combining signal across at least ~10 SNPs, resulting in detection of only an average of ~4–6 CNVs per sample (Aguirre et al., 2019; Kendall et al., 2019).
We reasoned that CNV detection sensitivity from SNP-array data could be considerably increased via two orthogonal strategies: (a) estimating SNP-specific priors for allele combinations corresponding to CNV states, thereby enabling more accurate assessment of probabilistic information about copy-number variation provided by probe intensities; and (b) incorporating probe intensity data from individuals likely to have co-inherited a large genomic tract. In this section we describe strategy (a), which was previously employed by the Birdsuite software (Korn et al., 2008); here, we leverage large-scale genotyping of thousands of samples to learn more information about SNP-specific priors from the data, requiring less extrapolation. The basic idea of our approach was to (i) directly estimate SNP-specific genotype cluster priors at a subset of SNPs covered by large, easily-called CNVs; and then (ii) use these SNPs as a reference set from which SNP-specific priors for other SNPs could be predicted (based on which SNPs in the reference set exhibited most-similar probe intensity patterns).
Partitioning samples into LRR-noise deciles
We first estimated a per-sample parameter reflecting overall amount of technical noise in probe intensities, which varied among samples. We computed this per-sample parameter as the RMSE (in standardized units) of LRR across autosomal variants on the SNP-array. That is, for each genotyped variant, we standardized LRR to have mean 0 and variance 1 across samples, and then for each sample, we computed the sample’s “noise scale factor” as the root-mean-square of standardized LRR across all autosomal variants.
We used these estimated noise scale factors to partition UK Biobank samples into noise deciles for downstream modeling of probe intensities, reasoning that the shapes and positions of probe intensity distributions might change somewhat depending on the amount of technical noise present in a sample. We also further adjusted for within-decile variation in noise scale factors when estimating Bayes factors for copy-number states given observed probe intensities (both in our initial LRR-based model and our final HI-CNV model; see the descriptions of these computations below for details).
Generating reference data via LRR-based calling of large CNVs
To obtain examples of probe intensities corresponding to copy-loss and copy-gain genotypes (loss = {A, B}; gain = {AAA, AAB, ABB, BBB}), we implemented a simple hidden Markov model (HMM) that called loss and gain events in each sample independently using only LRR values together with heterozygous SNP calls (used as evidence against deletions). This approach was designed to efficiently generate a high-confidence callset of large CNVs, providing data about probe intensity distributions for SNPs within these CNVs.
Specifically, the HMM contained three copy-number states (CN = 1, 2, 3), with transition and emission parameters defined as follows:
Transition penalties of 10−3 were assessed for jumping between adjacent states and 10−6 for jumping between CN=1 and CN=3.
- Emission probabilities were computed assuming that LRR was generated from a Gaussian distribution with:
- Mean equal to 0 for CN=2; mean equal to the empirical mean LRR in large deletions and duplications (estimated by iteratively running this HMM algorithm) for CN=1 and CN=3, respectively.
- Standard deviation estimated per-SNP as the empirical standard deviation of LRR across samples in a noise decile, multiplied by the relative noise scale factor (relative to the median-noise sample in the decile) for the sample being analyzed.
To limit the influence of outliers, relative emission probabilities for CN=1 vs. CN=2 and CN=3 vs. CN=2 were cropped to the range [10−4, 104].
An additional (multiplicative) emission penalty was assessed for the CN=1 state if a SNP had been called as heterozygous (since all SNPs within a deletion should be hemizygous). This penalty factor ranged from 5 × 10−6 (for the highest-confidence SNP calls) to 1 (for zero-confidence calls) according to genotype call confidence values provided by Affymetrix.
We used the Viterbi algorithm to identify putative CNVs (as segments of CN=1 and CN=3 states in the most likely path through the HMM). We then created a stringent set of (sample, SNP) pairs very likely to provide examples of probe intensity measurements arising from copy-loss or copy-gain states by restricting to:
SNPs well within deletion calls spanning 15+ SNPs (>3 SNPs from either end).
SNPs well within duplication calls spanning 50+ SNPs (>10 SNPs from either end).
We required deletions and duplications to be large both to minimize false positives in our reference data and to avoid ascertainment bias (which could occur for shorter CNVs if calls were only made in carriers for which LRR was especially large due to measurement noise). More precisely, because short CNVs are difficult to detect (especially from LRR alone in a single sample), including such CNV calls when creating reference CNV genotype clusters could bias the clusters to be too low (for DELs) or too high (for DUPs), similar to how “winner’s curse” biases effect size estimates to be too large in GWAS.
We also stringently trimmed the ends of CNV calls to avoid uncertainty in breakpoints (which was larger for duplications than for deletions), prioritizing data quality over quantity because reference data is sufficiently abundant in data sets containing thousands of samples. We note that because we did not attempt to model CN=0 or CN=4+ states, the reference data set we generated included a small fraction of homozygous CNVs; however, most of the large CNVs that we considered in this analysis were rare (MAF<0.01), such that the vast majority of DEL or DUP calls made by our preliminary LRR-based CNV caller could be assumed to be CN=1 or CN=3 (rather than CN=0 or CN=4). We could therefore simply ensure that our subsequent estimation of cluster priors (described below) was robust to outliers, circumventing the need to explicitly distinguish heterozygous vs. homozygous CNV genotypes.
Estimating parameters for clusters with available reference data
After identifying high-confidence within-CNV SNPs, we next needed to assign probe intensities from such SNPs (transformed to the (, LRR) scale) to genotype clusters for CN=1 (A, B) and CN=3 (AAA, AAB, ABB, BBB). We did so by dividing the (θ, LRR) plane into zones designed to typically contain most data points from each possible cluster (Figure S7A). We defined these zones in a SNP-specific, noise-decile-specific manner based on the locations and orientations of CN=2 clusters (i.e., distributions of AA, AB, BB genotype calls from standard SNP-array genotyping):
For CN=1, we split the plane left/right at the θ value of the CN=2 het (AB) cluster center.
For CN=3, we additionally split each of the above half-planes at a line passing through the point 2/3 of the way from the CN=2 het (AB) cluster center to the CN=2 hom (AA or BB) cluster center. We drew these lines parallel to regression lines indicating the relationship between LRR (treated as the independent variable) and θ (treated as the dependent variable) among points in the respective CN=2 hom clusters: e.g., we approximately separated AAA and AAB clusters by drawing a line “parallel to the AA cluster” located 2/3 of the way from the AB cluster to the AA cluster. (If one of the CN=2 hom clusters was very rare (n<25), we did not perform the additional split, assuming that the corresponding AAA or BBB cluster would have negligibly low frequency.)
After provisionally assigning within-CNV probe intensity data points to clusters according to the above zones, we next removed outliers farther from the median (in either coordinate, θ or LRR) than twice the interquartile range.
The above partitioning and outlier removal strategy worked well for most clusters, but visual inspection of the data showed that a sizable minority of provisional clusters still contained data points that should have been assigned to other clusters. We therefore applied a few post-processing filters to flag questionable-quality clusters for exclusion from our reference set:
Exclude all CN=1 minor-allele clusters for SNPs with MAF<0.05. Some of these provisional clusters contained a nontrivial fraction of data points that actually corresponded to CN=0, so we just excluded all such clusters (as we had no shortage of reference data from more-robust CN=1 clusters).
- Exclude any CN=3 cluster that overlaps with a neighboring CN=3 cluster with higher frequency (i.e., more data points). The rationale for this filter was that higher-frequency clusters tend to be only mildly affected by mis-assigned points that actually belong in lower-frequency clusters, but not vice versa. We defined “overlap” as follows:
- For the two CN=3 heterozygous clusters (AAB, ABB), we required the θ-distance between the center of the cluster and the center of each of each neighboring CN=3 cluster to be at least the sum of the cluster width and the neighboring cluster width: 2 · (s.d.(θ)cluster + s.d.(θ)neighbor).
- For the two CN=3 homozygous clusters (AAA, BBB) (which tended to be more affected by this problem), we required separation from the neighboring (het) CN=3 cluster center to be at least 2.5 · (s.d.(θ)cluster + s.d.(θ)neighbor).
Exclude any cluster with aberrantly large variance in either coordinate (θ or LRR): i.e., variance greater than 1.5 times the sum of variance (of the same coordinate) in each of the three CN=2 clusters (AA, AB, BB). This filter tended to catch remaining CN=1 clusters containing CN=0 data points.
Exclude all clusters from ultra-rare SNPs (n<25 het calls among samples in the noise decile).
For each noise decile, for each SNP, for each of the possible genotypes corresponding to CN=1 (A, B) and CN=3 (AAA, AAB, ABB, BBB), we considered the genotype cluster to be a suitable reference cluster if it contained at least 10 data points (after outlier removal) and had not been excluded by any of the above filters. Approximately 1% of all clusters satisfied this criterion. For each such cluster, we then estimated its five bivariate normal parameters—mean(θ), mean(LRR), var(θ), var(LRR), and cov(θ, LRR)—from its data points (Figure S7B).
Finally, we also estimated bivariate normal cluster parameters for CN=2 genotype clusters simply by assigning all samples with non-missing genotype calls (from standard SNP-array genotyping) to the corresponding cluster (and then removing outliers with either coordinate (θ or LRR) farther from the median than three times the interquartile range). As above, we required at least 10 data points to be assigned to a cluster in order to proceed with estimation of bivariate normal parameters; otherwise we set the cluster to missing. We did not attempt to identify and exclude data points corresponding to CNVs from this analysis given that (i) the vast majority of variants included on most SNP arrays are (by design) not in regions of the genome that harbor common copy-number variation; and (ii) our focus was on identifying rare, potentially-deleterious CNVs.
Predicting cluster parameters for all genotyped variants
Having determined approximate location and shape parameters for a small fraction of all CN=1 and CN=3 genotype clusters (~1% in UK Biobank) using the above procedure, we then sought to use this information to predict bivariate normal parameters at genotyped SNPs throughout the genome (most of which had insufficient or questionable-quality data from overlapping large CNVs). For each cluster to be predicted, the basic idea of our approach was to find the 20 reference SNPs with CN=2 clusters most similar to CN=2 clusters of the query SNP in question, and then predict the target cluster of the query SNP based on the location and shape of the corresponding cluster in the 20 reference SNPs. This approach is illustrated in Figure S7C and described below.
Explicitly, for each noise decile, for each SNP, for each side (left/right) of the cluster plot, for each CN=1 and CN=3 cluster on the side under consideration (i.e., A, AAA, AAB on the left side; B, ABB, BBB on the right side), we matched the SNP’s CN=2 clusters on the side under consideration (i.e., AA, AB on the left side; AB, BB on the right side) to the corresponding CN=2 clusters of reference SNPs at which the cluster had been estimated (typically ~10,000 SNPs in UK Biobank). We used squared Hellinger distance as a metric for assessing agreement between corresponding CN=2 clusters, summing across the two CN=2 clusters on the side under consideration. For example, for the left side:
| (2) |
where “query” denotes the SNP with clusters being predicted, “ref” denotes a reference SNP, and Hellinger distances are computed between the bivariate normal distributions at the “query” and “ref” SNPs for each of the two left-side CN=2 clusters (AA and AB). We computed Hellinger distances on bivariate normal distributions for CN=2 clusters that we estimated cohort-wide (instead of within noise deciles) to allow more-robust cluster-matching at rare SNPs, which had few data points in the het and hom-minor CN=2 clusters.
After ranking reference SNPs in this manner, we selected the top 20 reference SNPs that genotyped most similarly to the SNP whose cluster was being predicted. By design, such “ref” SNPs had CN=2 clusters that closely matched those of the “query” SNP; however, this alignment was not perfect. To adjust for small offsets between “query” SNP vs. “ref” SNP CN=2 cluster centers, we shifted each “ref” SNP’s CN=1/CN=3 clusters by the estimated offset (measuring the offset at the CN=2 cluster closest to the cluster being predicted, in the noise decile under consideration). Finally, we predicted bivariate normal parameters for the missing cluster of the “query” SNP by computing its mean and covariance assuming that it was an equal mixture of the 20 reference clusters.
This approach also allowed us to predict clusters for rare SNPs at which the hom-minor CN=2 cluster was missing (due to insufficient data points). For such SNPs, we predicted all clusters (CN = 1, 2, 3) on the missing (minor-allele) side using the same approach as above, but defining most-similar reference SNPs based on the opposite-side CN=2 clusters (hom-major and het).
We developed the above approach using cross-validation analyses (in which we attempted to predict held-out reference clusters using other reference SNPs), and visual inspection of predicted clusters corroborated good cross-validation performance as well as good containment of CNV data points in clusters for which coverage by large CNVs had been too low to estimate reference clusters.
Finding longest identical-by-descent (IBD) matches per haplotype
Beyond optimizing modeling of genotyping probe intensities, the main source of HI-CNV’s improved detection sensitivity is its use of IBD-sharing across distantly related individuals to amplify weak signals of CNV presence. This approach is inspired by the idea of validating variant calls in related samples (e.g., trios) by checking for Mendelian inheritance, a paradigm that is frequently used to benchmark variant callers or increase confidence in difficult-to-call variants. HI-CNV leverages the fact that population-scale cohorts such as UK Biobank contain extensive distant relatedness, such that any polymorphic variant present in at least a few individuals is likely to have been co-inherited on a long, readily-identifiable shared haplotype. In such scenarios, combining probabilistic information about CNV presence across individuals who share long IBD can dependably aid detection. This idea builds upon previous approaches that modeled linkage disequilibrium between CNVs and common SNPs by considering short ancestral haplotypes (Coin et al., 2010) and that performed SNP-haplotype-based refinement of CNV likelihoods (Handsaker et al., 2011, 2015).
In this section, we describe the algorithm we implemented to efficiently identify top IBD matches within very large cohorts such as UK Biobank: specifically, for each haplotype of each individual, and for each genomic position on the SNP-array, we wished to find the longest 10 IBD matches spanning the position under consideration. While several methods based on the positional Burrows-Wheeler transform (PBWT) (Durbin, 2014) have recently been developed for rapid IBD detection in large cohorts (Freyman et al., 2021; Naseri et al., 2019; Zhou et al., 2020), these methods aim to find all IBD segments above a fixed length (e.g., 2 or 3 cM) shared by pairs of haplotypes in a cohort—which could either result in too much output for our purposes (at loci containing very many IBD matches) or too little output (for haplotypes with only smaller lengths of IBD-sharing). We therefore implemented a simple PBWT-based algorithm (using a seed-and-extend approach similar to hap-IBD (Zhou et al., 2020) tailored to the specific task of finding longest-IBD matches. (Note that here we will be loose about the definition of “IBD”; a short, ~1-cM match might not arise from a recent-enough common ancestor to typically be considered “IBD” but might still be helpful for calling common CNVs contained within it that arose long ago, such that CNV genotypes segregate well with relatively short haplotypes.)
Identifying seed matches using the positional Burrows-Wheeler transform (PBWT)
The first step of our approach was to identify a set of long identical-by-state (IBS) segments among pairs of phased SNP-haplotypes, serving as seeds for extension into (potentially longer) IBD segments. We performed this search using the PBWT, which produces, at each genotyped SNP, a lexicographic sort of haplotype suffixes (when operating right-to-left) from which longest-IBS matches starting at each position can readily be obtained as bands of consecutive sorted haplotype suffixes (Durbin, 2014). Explicitly, every 32 SNPs processed by the PBWT, we augmented our set of IBS seed segments by selecting, for each haplotype, a band of adjacent haplotypes corresponding to K = 5 (first algorithmic iteration; see below) or K = 10 (second algorithmic iteration) longest IBS-suffix matches spanning at least 128 SNPs. For any IBS-suffix match that extended a sub-IBS-suffix previously selected, we eliminated the redundant, previously-selected sub-IBS-suffix from the seed set.
Extending IBD seeds
Most IBD segments do not consist of a single segment of perfect IBS (i.e., exact matching of a contiguous sequence of alleles along a pair of SNP-haplotypes); instead, IBD segments usually contain a sequence of IBS segments punctuated by mismatches (typically arising from genotyping errors or gene conversions). For each IBS seed identified by the PBWT-based algorithm above, we therefore attempted to extend the IBS segment into a longer IBD segment using an approach similar to hap-IBD (Zhou et al., 2020). (We did not attempt to model phase switch errors given that our phased haplotypes for UK Biobank had chromosome-scale accuracy (Loh et al., 2020).)
Explicitly, we attempted to extend each IBS seed to the left and right in an error-tolerant manner based on matching scores that we computed on blocks of 64 SNPs (using fast parallelization of bitwise operations):
| (3) |
where “soft” and “hard” errors were defined based on genotype call confidences (on a 0–1 scale) provided by Affymetrix and UK Biobank. Specifically:
We ignored errors at SNPs for which either sample in the pair had an estimated genotype error probability >0.002.
Otherwise, we considered a “soft” error to be a mismatch at a SNP for which at least one sample had an estimated genotype error probability in the range 0.0001–0.002.
The remaining errors (involving SNPs with very confident genotypes in both samples) were considered “hard” errors.
Under this scoring scheme, perfect matches of 64-SNP blocks incremented the score of a segment being extended by 1, while matches with non-ignored errors reduced the score by 1 or more (depending on the number and type of errors). Upon encountering a negatively-scored block, we required the total score to break even within the next 12 blocks; otherwise, we ended IBS seed extension at the first error encountered within the block. This approach effectively required that each “soft” error be counterbalanced by 127 matched SNPs and each “hard” error be counterbalanced by 255 matches.
Filtering to longest IBD matches per position per haplotype
From the list of IBD segments identified above, we wished to efficiently identify, for each haplotype and at each SNP-array position, a list of the top-K longest IBD segments spanning this position. To do so, we first post-processed the set of IBD segments by merging any duplicated or overlapping segments (involving the same pair of haplotypes). Then, for each haplotype, we identified top-K longest IBD matches at each SNP-array position using the following algorithm:
Sort all IBD matches involving the haplotype by start coordinate.
- Walk left to right across the chromosome, maintaining an “active set” of IBD matches spanning the current position, sorted in two ways: (i) by IBD length (longest to shortest); and (ii) by end coordinate (left to right). At each position:
- Update the active set if:
- Current position starts a new IBD match: add new match to active set.
- Current position ends an IBD match in the active set: delete ended match.
- Read off the top-K longest matches spanning the current position from the active set.
Correcting potential genotype errors
Our identification of top IBD matches for each haplotype at each genomic position provided an opportunity to correct some of the occasional SNP-allele mismatches that interrupted IBS within IBD tracts. Doing so could potentially improve the quality of IBS seeds identified by the PBWT, which is not robust to mismatches. We therefore implemented an “error-correction” strategy in which we used IBD information to identify haploid SNP-alleles that were inconsistent with haplotypes sharing longest IBD, and we subsequently ran a second iteration of the entire IBD-finding algorithm after modifying these SNP-alleles. We limited error-correction to SNP-alleles for which the genotype error probability estimated by Affymetrix was >0.002.
In more detail, for each haplotype, for each SNP-allele corresponding to a low-confidence genotype call, we identified the longest five IBD matches spanning the SNP-allele, as described above. We then examined the corresponding SNP-allele in each of the 5 IBD-neighbor haplotypes for which the SNP in question was located > 0.5 cM from the edge of the IBD segment. If at least four IBD-neighbors satisfied this requirement and only at most one of them agreed with the SNP-allele in the original haplotype, we recorded a likely error.
After analyzing all haplotypes in the above manner, we flipped the (haploid) SNP-allele genotypes at all recorded likely errors. We also used the information about potential errors to perform quality control on SNPs: for any SNP with likely errors in 0.25% or more haplotypes, we ignored this SNP in the next iteration of IBD-finding.
This algorithm produced long IBD calls for most haplotypes in UK Biobank at most genomic locations. For example, the longest match (“closest haplotype neighbor”) was >1cM 98% of the time, >5cM 80% of the time, and >10cM 58% of the time; the 5th longest haplotype match was >1cM 94% of the time, >5cM 51% of the time, and >10cM 18.5% of the time; and the 10th longest haplotype match was >1cM 90.5% of the time, >5cM 35% of the time, and >10cM 8% of the time.
Calling CNVs using intensity data across haplotype neighbors
The methods described in the previous sections provided the two key ingredients of the HI-CNV algorithm: (i) detailed, SNP-specific (and sample noise decile-specific) priors on probe intensities produced by different genotypes; and (ii) information about longest IBD matches for each haplotype at each genomic location. Here we describe the algorithm that we used to convert probe intensity data into probabilistic information about copy-number state and robustly integrate such information from individuals and their “haplotype neighbors” to call CNVs.
Estimating per-SNP Bayes factors for copy-number states
Our first task was to quantify the extent to which a given SNP-array measurement—i.e., observed relative intensity (θ) and total intensity (denoised LRR) for a given sample—supported the presence of a copy-gain, copy-loss, or no CNV spanning the SNP. We performed this quantification by estimating approximate Bayes factors for copy-gain vs. no CNV and for copy-loss vs. no CNV. To do so, we computed the probability density at the observed intensity data point (θ, LRR) for each of the bivariate normal genotype clusters we estimated above: two probability density values for the CN=1 clusters (A, B), three for the CN=2 clusters (AA, AB, BB), and four for the CN=3 clusters (AAA, AAB, ABB, BBB). We also included a cluster that accounted for occasional CN=0 data points; we situated this cluster at a constant offset below the CN=2 het (AB) cluster, with twice its variance parameters. We then computed maximum probability densities among the values obtained from copy-gain clusters (AAA, AAB, ABB, BBB), copy-loss clusters (A, B, and CN=0), and noCNV clusters (AA, AB, BB) and set the approximate Bayes factors for copy-gain vs. no CNV and copy-loss vs. no CNV to equal the ratios of the relevant maxima. Finally, we cropped these ratios to the range [3 × 10−3, 103/3] to limit the influence of potential outlier values.
We note that our use of maximum probability density values across genotype clusters within a copy-number state (e.g., AAA, AAB, ABB, BBB for CN=3) does not result in true Bayes factors: a formal Bayesian analysis would require a generative model that, for a given CN state, first defines a probability distribution over the genotype clusters corresponding to the CN state. We did not attempt to model the relative frequencies of genotype clusters because in practice, such modeling only becomes relevant for rare SNPs (with highly unbalanced cluster probabilities); but for such SNPs, almost all observations come from major-allele clusters, such that detailed modeling of cluster frequencies is rarely relevant. We found that in practice, CNV detection using the approximate Bayes factors we computed already increased detection sensitivity relative to previous PennCNV analyses of UK Biobank (Table S1; HI-CNV0 denotes analysis using our approximate Bayes factors without incorporating information from haplotype neighbors).
An additional detail regarding our computation of bivariate normal probability density values is that we applied individual-specific scale factors to the per-noise-decile bivariate normal clusters we had estimated. We did so because even though we estimated cluster parameters separately for each LRR-noise decile of samples, the samples within a decile still exhibited varying levels of noise. To account for this remaining variation in noise, we scaled all genotype cluster standard deviation parameters for a given sample by the ratio of s.d.(LRR) in the sample to the median s.d.(LRR) in the sample’s decile.
Masking genotyping intensities potentially influenced by nearby SNPs
We found that for some variants on the UK Biobank SNP-array, the presence of nearby SNPs (within ±30 bp) resulted in genotyping intensities similar to deletions, presumably because the additional nearby variant caused the local sequence no longer to hybridize to either of the oligonucleotide probes for the A or B allele of the variant being genotyped. To prevent such scenarios from potentially producing spurious deletion calls, we attempted to mask all genotyping intensity measurements that might be influenced by nearby SNPs. We did so by masking, in each individual, intensity data from all variants for which a nearby SNP (within ±30 bp) had been imputed (in the UKB imp_v3 release) with an imputed dosage >0.1 for the minor allele. This filter removed only a small fraction of the available data: at a typical heterozygosity rate of ~1 heterozygote per 1,000 basepairs, filtering when observing a SNP in the 60 bases within ±30 bp of a genotyped variant results in a variant being filtered ~6% of the time.
We also applied a similar mask to multi-allelic SNPs. Intuitively, a probe designed to look for the two most common alleles at a site may make carriers of a third allele look like carriers of deletions (no signal for either of the two common alleles). As such, we masked genotype intensities for imputed carriers of a third allele at a given SNP.
Hidden Markov model (HMM) using IBD-based weights
As in previous CNV-calling methods such as PennCNV (Wang et al., 2007), we used a hidden Markov model to identify sequences of consecutive SNPs at which genotyping intensity measurements consistently indicated the presence of a CNV (based on the Viterbi path passing through copy-gain or copy-loss states). Here, we needed to adapt this approach to incorporate probabilistic information not only from an individual but also from haplotype neighbors sharing IBD tracts. This task was nontrivial because fully modeling genotyping intensity data from all of these samples would require considering a combinatorial state space including the copy-number states of all haplotype neighbors (which might or might not match that of the individual in question, depending on recentness of IBD-sharing).
To retain computational tractability, we therefore incorporated information from haplotype neighbors using a simple heuristic approach somewhat analogous to a variational approximation. Specifically, at each genotyped SNP, we simply augmented the approximate Bayes factors for the individual (for copy-gain vs. no CNV and copy-loss vs. no CNV) with the corresponding Bayes factors from each haplotype neighbor, downweighted in such a way as to reflect the possibility that haplotype neighbors with shorter IBD-sharing might be too distantly related to the individual to have co-inherited a CNV. We ran this analysis using several different weighting schemes (trading off sensitivity to more recent vs. older CNV mutations, as described below) and compiled calls made across these weighting schemes (as described in the next section).
HMM states. We used a three-state HMM with copy-gain, copy-loss and no-CNV states. We did not attempt to have the HMM distinguish between CN=1 and CN=0 or between CN=3 and higher copy numbers given that our focus was on detecting rare biallelic CNVs.
-
Emission probabilities. Given that we ultimately wanted to perform inference based on the Viterbi path through the HMM, we could perform all computations in log space and work only with relative emission probabilities (i.e., log Bayes factors). As described above, at each SNP, our genotype cluster models allowed us to compute approximate log Bayes factors for copy-gain vs. no CNV and for copy-loss vs. no CNV from the genotyping intensities of the individual and likewise for each of the individual’s haplotype neighbors. To aggregate this information into a single log Bayes factor for copy-gain (respectively, copy-loss) vs. no CNV, we computed a weighted sum in which the individual’s log Bayes factor received a weight of 1 (corresponding to fully utilizing probabilistic information about CNV status from the individual’s genotyping intensities) and the haplotype neighbors’ log Bayes factors received weights between 0 and 1 depending on their lengths of IBD-sharing (so as to downweight information from individuals with shorter, less-confident IBD with the individual).
Explicitly, we considered a 1-parameter family of weighting functions that map a given IBD length to the probability that the time to the most recent common ancestor (TMCRA) is within T generations. Intuitively, this weighting scheme optimizes for detecting CNVs that arose roughly T generations ago (by incorporating information from haplotype neighbors who share more recent IBD— and thus have genotyping intensities informative of the co-inherited CNV—while discarding information from haplotype neighbors with TMRCA predating the CNV mutation). To power detection of CNVs of different ages, we ran HMM inference using six different values of T ∈ {0,5,10,25,50,100} generations, where T = 0 corresponds to ignoring haplotype neighbors entirely (i.e., performing single-sample analysis). For each T > 0, we performed two HMM runs, incorporating information from neighbors of each of the individual’s two haplotypes in turn.
To calculate the approximate probability that an IBD segment of length l Morgans has TMRCA (denoted t) less than T generations, we used the following derivation. For a population of constant size 2N haplotypes (N diploid individuals), we have (from page 117 of (Palamara, 2014)):
Letting N → ∞, we obtain:
Integrating from T to infinity,
Thus, the probability that an IBD segment of length l Morgans has TMRCA within T generations is approximately given by: -
Transition probabilities. We specified a transition probability matrix similar to PennCNV (Wang et al., 2007) in which the probabilities of changes in copy-number state between two consecutive probes depended on the distance between the probes (corresponding to the idea that copy-number state changes between nearby probes should be less likely than between distant probes).
Explicitly, we used the transition matrix:
where di is the distance between probes, and are the average number of deletions and duplications called using SNP-array data (set to 15 and 5, respectively), Ddel, Ddup are the average lengths of deletions and duplications (both set to 100kb), D is the genome size divided by the number of copy number variants (set to 3 × 109/20 = 150 Mb) and finally # probes is the number of SNPs on the array (set to 784,256 autosomal variants for UK Biobank).
Filtering, merging, and genotyping CNVs
In the previous sections, we described how we set up HMMs to call CNVs using information from haplotype neighbors. We incorporated such information via a set of IBD length-based weighting schemes (parameterized by a TMRCA parameter T ∈ {0,5,10,25,50,100} generations). Here we describe how we post-processed CNV calls from these HMMs to obtain a high-confidence set of CNVs (that merged calls across different values of T) and how we subsequently genotyped CNVs across samples. We note that the merging approach described below does not prioritize calls made using lower vs. higher values of T; the motivation for running the HMM using different values of T was not that some TMRCA parameters are inherently better than others, but rather that depending on the age of a CNV mutation, higher or lower values of the TMRCA parameter might offer more detection sensitivity.
Filtering and post-processing CNV calls from each HMM run
For each individual, for each run of the HMM (parameterized by T and by the haplotype of the individual used to identify neighbors), we extracted potential deletions (respectively, duplications) as consecutive sequences of copy-loss (respectively, copy-gain) states in the Viterbi path through the HMM. For each such sequence of states, we computed the log10 Bayes factor (BF) supporting the putative CNV event (as the sum of log10 BFs across the sequence of SNPs within the segment, including information from the focal individual as well as haplotype neighbors as in the HMM). We then applied an initial set of filters to these potential CNV segments: we required putative deletions to span at least 50 bp, and we required duplications to span at least 500 bp and have log10BF > 9 support.
We further post-processed the segments that survived filtering by bridging short gaps between consecutive segments of the same copy-number state (because the Viterbi path through long CNVs was sometimes interrupted by short sequences of no-CNV states). Specifically, we bridged gaps between nearby CNV segments if either (i) they included ≤4 probes and spanned <20 kb; or (ii) they spanned ≤20% of the combined length after bridging.
Merging CNV calls across HMM runs
To synthesize post-processed CNV calls across HMM runs from different values of the TMRCA parameter T (which had differing sensitivity to CNVs of different mutational ages and also exhibited stochastic variation in endpoints), we next performed a deduplication step to identify a nonredundant set of CNVs discovered in each individual. We performed this deduplication procedure on the aggregate set of CNV calls made across values of T and across which of the individual’s haplotypes had been used to identify neighbors. (Homozygous CNVs present on both haplotypes were collapsed into a single call during this step but handled later in a separate genotyping step described below.)
Specifically, we considered two CNV calls of the same type (DUP or DEL) to be duplicates if their endpoints matched within 4 SNP-array probes (i.e., Δ start ≤4 and Δ end ≤4). For each such duplicate pair, we retained the call with higher log10BF. We refer to the set of CNV calls remaining after this procedure as the “deduped” callset.
Because the deduped callset could still contain overlapping CNV calls (that were unwieldy for some downstream analyses), we also created a “unioned” callset in which we merged overlapping CNV calls of the same type (DUP or DEL). Lastly, we applied a final set of length filters on the CNV calls, requiring deletions to be >75 bp and duplications to be >500 bp (based on empirical validation analyses).
Creating CNV genotypes for association tests
We used the deduped and unioned callsets described above to create genotypes for single-variant and burden-style association tests on various classes of CNVs (grouping CNVs at the probe, gene, or CNV level). In more detail:
Probe-level tests: For each probe on the SNP-array, we used the unioned CNV callset to determine which individuals had a deletion or duplication spanning the given probe. This procedure created two 0/1 genotypes (for DEL and DUP) at each probe. (We did not distinguish homozygous from heterozygous genotypes for these tests.)
-
Gene-level tests: Similarly, for all protein-coding genes, we used the unioned CNV callset to construct three gene-level 0/1 genotypes (for DEL, DUP, and pLoF):
- Deletion (DEL): 1 if a deletion spans the entire gene (CNV boundaries 1 probe beyond first and last probe within coding sequence of gene);
- Duplication (DUP): 1 if a duplication spans the entire gene (CNV boundaries 1 probe beyond first and last probe within coding sequence of gene);
- Predicted loss of function (pLoF): 1 if a deletion spans any part of the coding sequence or a duplication is contained within coding sequence (i.e., CNV starting probe is at or after the first probe in coding sequence and last probe is at or before the last probe in coding sequence).
We created these genotypes using canonical transcripts for 20,091 genes (downloaded from https://github.com/im3sanger/dndscv/blob/master/data/refcds_hg19.rda).
CNV-level tests: For each CNV in the deduped callset with ≥ 5 carriers within the entire cohort, we constructed four versions of 0/1/2-genotypes for the CNV (parameterized by δ = {0,1,2,3}), reflecting four levels of tolerance to noise in breakpoints of CNV calls. Specifically, for a given CNV to be genotyped and a given value of δ, we considered an individual to be a carrier if the individual had a deduped CNV call with breakpoints that matched to within probes (i.e., Δ start ≤ δ and Δ end ≤ δ). We considered an individual to be homozygous for a CNV if for some T > 0, both HMM runs (using neighbors from the individual’s haplotype 1 and haplotype 2, respectively) had produced an approximately-matching CNV call with strong support from haplotype neighbors (i.e., neighbor-only log10BF > 6).
HI-CNV software implementation
To enable haplotype-informed CNV detection on data sets beyond UK Biobank, we have developed a portable, open-source HI-CNV software implementation designed to be readily applicable to other SNP-array-genotyped cohorts (10.5281/zenodo.7034987). This software package follows the same series of steps as our analysis of UK Biobank (described in the previous sections) with a few minor modifications to improve usability and generalizability:
LRR-denoising is performed using only principal component analysis, skipping the GC correction step (which appears to be obviated by PCA).
LRR principal components are computed using at most 5,000 randomly sampled individuals per genotyping platform (which is sufficient to estimate top PCs and capture technical noise).
Samples are partitioned into <10 LRR noise quantiles in data sets of <200,000 samples (to prevent sample noise quantiles from becoming too small to accurately estimate genotype clusters).
For LRR-based calling of large CNVs (used to generate reference genotype cluster data), expected LRR for DELs and DUPs are estimated via an iterative expectation-maximization (EM) approach (to allow for platform-dependent effects of CNVs on LRR).
Genotype call confidences are not considered when identifying haplotype matches (because genotyping confidence data is not always available and accounting for confidences in UK Biobank was only slightly beneficial).
Quality control filtering in UK Biobank
To further improve robustness of the UK Biobank HI-CNV callset, we performed several stages of filtering at the sample-level, chromosome-level, and CNV-level.
Individuals with trisomy 21 or blood cancer
To identify individuals with trisomy 21 we computed each individual’s mean denoised LRR across probes on chromosome 21. We identified 15 individuals (in the full UK Biobank cohort) with outlier values of chromosome 21 mean LRR consistent with potential trisomy 21 and removed these individuals from analysis.
To filter individuals whose DNA samples might be affected by blood cancers or premalignant conditions, we removed all individuals who self-reported any blood cancer at assessment or had a recorded date of first occurrence of blood cancer <5 years after assessment.
Technical artifacts producing aberrantly many CNV calls
We found that a small subset of samples with very low lymphocyte counts and red blood cell counts had aberrantly many duplication calls, apparently due to a technical artifact in LRR that had escaped denoising. We therefore filtered all samples with >100 CNV calls. Additionally, to identify individuals potentially affected more subtly by this type of artifact, we computed the first 10 principal components of LRR in these aberrant individuals, ranked all individuals by the amount of LRR variance explained by these artifact PCs, and removed individuals in the top 0.5% (corresponding to >1.1% of LRR variance explained by the 10 PCs).
Chromosomes with mosaic chromosomal alterations
For individuals with a mosaic chromosomal alteration call with cell fraction greater than 20% (Loh et al., 2020), we set all probe-level, gene-level, and CNV-level genotypes on the affected chromosome(s) to missing.
Somatic CNVs
We filtered calls intersecting the following regions (in hg19) frequently affected by somatic CNVs:
Immunoglobulin genes (IGK: chromosome 2; 89000000 – 90274235, IGH: chromosome 14; 106032614 – 107288051, IGL: chromosome 22; 22380474 – 23265085)
T cell receptor genes (TRG: chromosome 7; 38279625 – 38407656; TRB: chromosome 7; 141998851 – 142510972; TRA: chromosome 14; 22090057 – 23021075; TRD: chromosome 14; 22891537 – 22935569)
DLEU1 / DLEU2 locus (chromosome 13; 50556688 – 51297372)
Summary measures of UK Biobank HI-CNV callset
Validation rate of HI-CNV and PennCNV callsets
To assess the precision (i.e., validation rate) of CNVs called by HI-CNV (or PennCNV), we computed the proportion of HI-CNV (respectively, PennCNV) calls that were either (i) replicated by WGS-based CNV calls or (ii) exhibited enrichment or depletion of WGS read-depth consistent with the CNV call. We performed these analyses using whole-genome sequencing pilot data available for 43 individuals in our primary analysis set. For both the HI-CNV and PennCNV callsets, we removed calls that intersected regions that commonly contain somatic CNVs as well as all calls on chromosomes containing high-cell-fraction mosaic chromosomal alterations (see above). We note that the WGS data was aligned to hg38, whereas the SNP-array data analyzed by HI-CNV and PennCNV used hg19, so we lifted over the start and end of each HI-CNV and PennCNV call to hg38 and removed events which had an unmapped start or end.
We used CNVnator (Abyzov et al., 2011) to call CNVs following a standard pipeline (https://github.com/abyzovlab/CNVnator), using the -unique flag when extracting read mapping data from bam files and a binsize of 100 bp for computing WGS read-depth. We restricted to calls with a q0 (fraction of reads mapped with q0 quality) ≤ 0.5 (non-redundant) and a q0 not equal to −1 (couldn’t be calculated). We then used the python module pytools.io to extract CNVnator read depth data from the root file.
For all CNVs called by HI-CNV (or PennCNV), we annotated whether CNVnator called an overlapping CNV containing at least 50% of the probes in the SNP-array-based call. We also computed mean normalized read depth across the 100 bp windows spanning the CNV being validated (normalized by read depth across entire chromosome). We then compared this mean normalized read depth to the distribution of mean normalized read depth across the same CNV region among individuals with no CNV call in the region. We used the mean and standard deviation from this background distribution to compute a z-score and determine if there was a significant excess or depletion of read depth (P < 0.05).
The above computations allowed us to classify CNV calls into three categories containing CNVs (1) replicated by CNVnator, (2) supported by read-depth signal in the correct direction (e.g., depletion of read-depth for a deletion), (3) with read-depth signal in the incorrect direction. We estimated validation rate as the sum of the proportion of CNVs replicated by CNVnator and the excess of CNVs with read-depth signal in the correct versus incorrect direction: (1) + (2) – (3).
Additional validation of HI-CNV callset
We additionally validated HI-CNV calls within a randomly selected subset of 500 individuals with subsequently-released whole-genome sequencing data (Halldorsson et al., 2022). For each of these 500 individuals, we lifted the individual’s HI-CNV calls (post-QC) from hg19 to hg38 and assessed whether or not WGS read depth was higher (respectively, lower) than expected within the putative duplications (respectively, deletions) called by HI-CNV.
To calibrate WGS read-depth measurements, which we computed using mosdepth (Pedersen and Quinlan, 2018), we adjusted for genome-wide sequencing depth and also for local variation in GC content. Specifically, we trained sample-specific GC models by regressing observed read depth on GC-content and GC-content squared across a set of “well-behaved” 1kb bins (defined simply as bins having read depth between 15 and 75 in the 43 WGS pilot samples). We then used this model to compute GC-corrected read depth within CNV regions (dividing observed read depth by expected read depth based on the GC model), and we compared GC-corrected read depth in individuals putatively carrying a CNV to GC-corrected read depth in individuals with no CNV call overlapping the putative event (after calibrating GC-corrected read depth to have the same mean in each sample across all “well-behaved” 1kb bins).
Recall of HI-CNV and PennCNV callsets
We assessed the recall, defined as the proportion of CNVs called by WGS-based analysis for which overlapping calls were made by HI-CNV (or PennCNV). We removed WGS-based calls that intersected regions that could correspond to somatic events as well as all calls on chromosomes containing high-cell-fraction mosaic chromosomal alterations. We restricted analyses to rare or low-frequency CNVs, i.e., those with AC ≤ 5 in the full set of 48 UK Biobank participants with available pilot WGS data. As above, we lifted HI-CNV and PennCNV calls from hg19 to hg38 and removed events which had an unmapped start or end.
We used Delly (Rausch et al., 2012) to call CNVs following a standard pipeline for germline SV calling (https://github.com/dellytools/delly). We used Delly to assess recall because Delly performs joint-calling across a batch of samples and outputs allele frequencies for all called events, facilitating assessment of recall of CNVs within allele frequency ranges. We considered a Delly call to have been re-identified if a CNV of the same type (DEL/DUP) was called overlapping the CNV called by Delly. We assessed the recall of a variety of different subsets of CNVs (Figure S2A; Table S1).
Unique CNVs
For some downstream analyses, we wished to analyze the set of unique CNVs identified by HICNV. This task was nontrivial because a CNV mutation co-inherited by multiple individuals could be called with slightly different breakpoints in different carriers. Consequently, the set of unique CNV calls—i.e., unique pairs of (start, end) breakpoints for DELs and for DUPs—overcounted the actual number of unique mutational events identified.
To obtain a more accurate set of unique CNVs, we performed analyses to assess which CNV calls with similar breakpoints were likely to represent the same underlying CNV. Specifically, we started with CNV-level genotypes we had created for each unique CNV call (using the δ= 2 version of genotyping, in which individuals with CNV call endpoints matching within δ= 2 probes were considered to be carriers) and then pruned this set of CNV genotype vectors to an approximately independent subset.
Explicitly, given the complete set of δ = 2 CNV-level genotypes, we computed allele frequencies and pairwise D’ in unrelated self-reported Europeans using PLINK (Chang et al., 2015) and clumped CNVs (with frequency ≥ 5 × 10−6, corresponding to ≥5 carriers per CNV) with D’ ≥ 0.5, retaining higher-frequency CNVs. (While CNVs in LD can be distinct, we applied this clumping to try to be conservative in reporting numbers of unique CNVs.) We then removed all remaining CNVs that overlapped somatic event loci (see Somatic CNVs).
Finally, we refined the boundaries of the remaining, independent, δ= 2 CNV-level genotypes (because the breakpoints of the CNV calls used to “seed” these CNV-level genotypes could be off by 1–2 probes). To perform this refinement, for each remaining δ= 2 CNV genotype vector, we identified the most common (start, end) breakpoint pair among the CNV calls that contributed to this CNV-level genotype, and we took this most-common breakpoint pair to be our best guess of the breakpoints of the underlying CNV.
Association testing and statistical fine-mapping
We ran BOLT-LMM (Loh et al., 2015, 2018a) to compute association statistics between CNV genotypes—at the probe, gene, and CNV level (see Creating CNV genotypes for association tests)—and 56 quantitative traits (Table S2). We then used a pairwise linkage disequilibrium (LD)-based filter (that we previously developed for identifying likely-causal rare variant associations (Barton et al., 2021)) to remove CNV associations that could be explained by LD with a more strongly associated variant—either another CNV or an imputed SNP or indel (Barton et al., 2021; Bycroft et al., 2018)—within 3 Mb of the start of the CNV.
Filtering and annotating fine-mapped associations
We annotated all CNV-phenotype associations that passed our LD-based fine-mapping filter (involving either probe-, gene-, or CNV-level tests) with the following information (Table S3):
Trait associated with the CNV (trait)
Lead CNV (i.e., the tested CNV genotype vector with highest χ2 association statistic) and tied CNVs (all tested CNVs that had identical χ2 value as lead CNV; leadCNV,tiedCNVs)
Number of carriers and allele frequency among self-reported European UK Biobank participants (nCarriers,A1FREQ)
Genomic location: the chromosome, the median start and end location among CNV calls considered in the test, size (in kb) using the median start and end location; the median location of the probe before and after the CNV calls (Chr, medianStart, medianEnd, size_kb, median_loc_before_Start, median_loc_after_End)
Genic context: all genes intersecting the interval between the median start and end (either considering full genes or only exons; genes_exon_or_intron,gene_exons), genes that intersect an expanded interval ± 100 kb (genes_100kb).
Effect size and association strength: beta (effect size), standard error, χ2 and P from BOLT-LMM (BETA,SE,CHISQ_BOLT_LMM,P_BOLT_LMM)
-
Nearby SNP associations: most associated SNP (imputed in the UK Biobank imp_v3 release or from WES [30]) within 1Mb. For each such SNP, we annotated the ID of the SNP, the genomic location, the effect size, the P-value, and the 2 statistic from BOLT-LMM (mostAssocimpv3_SNP_1Mb,
mostAssocimpv3_SNP_1Mb_BP,
mostAssocimpv3_SNP_1Mb_beta,mostAssocimpv3_SNP_1Mb_P,
mostAssocimpv3_SNP_1Mb_CHISQ, mostAssocWES_SNP_1Mb,
mostAssocWES_SNP_1Mb_BP, mostAssocWES_SNP_1Mbbeta,
mostAssocWES_SNP_1Mb_P, mostAssocWES_SNP_1Mb_CHISQ)
We filtered all associations that involved CNVs that overlapped regions prone to somatic CNVs (see Somatic CNVs). We also filtered associations in the MHC region that had escaped our pairwise LD-based fine-mapping filter due to subtle differences in the genetic principal components we used as covariates in these analyses vs. the PCs that we had previously used as covariates when computing association test statistics for SNPs and indels (Barton et al., 2021). We verified (using linear regression analyses) that the difference in PCs only affected a small number of associations in the MHC region, at which long-range LD influenced one set of PCs more than the other.
Validation of fine-mapped HI-CNV calls
We validated fine-mapped HI-CNV calls using WGS read depth in a manner similar to how we validated HI-CNV calls for a subset of 500 individuals (see Additional validation of HI-CNV callset). In more detail, for each fine-mapped CNV-trait pair, we randomly selected a carrier of the involved CNV for whom whole-genome sequencing data available (which was possible for 268 of the 269 fine-mapped CNV-trait associations). We then lifted these CNV calls from hg19 to hg38, which succeeded for 259 of the 268 selected carriers of fine-mapped CNVs. After removing duplicates (which existed because some CNVs associated with multiple traits), we were left with a set of 250 distinct CNV calls, which we validated using the same WGS read-depth sign test described in Additional validation of HI-CNV callset.
CNVs contributing to likely-causal phenotype associations
Most of the CNV-phenotype associations that passed our fine-mapping filters (and were thus deemed likely-causal) involved burden-style tests: probe-level tests that considered all DELs or DUPs spanning a genomic position, and gene-level tests that considered all CNVs with a particular effect on a gene. CNV-level tests could also potentially include multiple distinct CNVs with slightly different breakpoints. We therefore undertook further analyses to roughly identify which unique CNVs underlay each association.
For each trait, we identified all δ = 2 CNV-level genotype vectors associated at nominal significance (P < 0.05). We then subsetted to genotype vectors that appeared to contribute to the association of interest, based on satisfying three additional criteria: (1) D’ ≥ 0.75 with the CNV genotype of interest (be it a probe, gene, or CNV level test), (2) MAF < 2 × MAF of the CNV genotype of interest, and (3) size of the CNV genotype of interest. Finally, among the remaining δ= 2 CNV-level genotypes, we pruned to an independent set following the same approach we used to identify unique CNVs (see Unique CNVs).
The above procedure produced a satisfactory set of unique CNVs underlying most phenotype associations, but for a few associated CNV genotypes that were very rare or combined deletions and duplications (specifically, pLoF gene-level tests), no δ= 2 CNV-level genotype was both in high D’ with the CNV genotype of interest and nominally associated with the trait. In these instances, we did not attempt to further identify specific unique CNVs contributing to the association.
Defining CNV loci
The above approach identified a set of CNVs likely to contribute to causal phenotype associations. To group these CNVs into loci, we sorted the CNVs by increasing size. For each chromosome, we denoted the smallest CNV on the chromosome as belonging to “locus1” and then iterated through other CNVs on the chromosome in order of size. For each CNV in turn, if it overlapped or fell within ±100 kb of one or more previously-defined loci, we annotated it as belonging to those loci, and otherwise we considered it to create a new locus.
Syndromic and non-syndromic loci, CNVs, and associations
We annotated a likely-causal CNV as syndromic if it overlapped a previously-curated pathogenic CNV (from the set of 92 pathogenic CNVs curated by (Crawford et al., 2019)) by more than 50%. We annotated a locus as syndromic if any CNV assigned to only that locus was annotated as syndromic. To annotate a CNV-phenotype association as being syndromic or non-syndromic, we examined all likely-causal CNVs that belonged to a single locus and contributed to the association and annotated the association as syndromic if at least one such CNV was syndromic.
Replication of phenotype associations
Loss-of-function SNP/indel burden analyses in UK Biobank
For associations involving CNVs that we believed acted on a candidate target (focal) gene (Figure 3E), we compared the estimated effect of CNVs predicted to cause loss-of-function (pLoF) of the putative target gene to the estimated effect of ultra-rare pLoF SNP and indel variants in the same gene (recently reported in a whole-exome analysis of UK Biobank that performed SNP/indel pLoF burden tests (Backman et al., 2021)). Specifically, we began by compiling a list of all gene-trait pairs implicated by CNV-phenotype associations (involving CNVs of any type) for which we had identified a putative target gene. For each such gene-trait pair, we then examined whether pLoF CNVs associated with the trait. We took forward for replication 74 such gene-trait pairs that exhibited Bonferroni-significant associations (P < 0.05/89).
For each gene-trait pair, we compared the effect size of pLoF CNVs to the effect size previously estimated for ultra-rare (MAF<0.001%) pLoF SNP and indel variants (Backman et al., 2021), excluding two genetrait pairs for which association statistics were unavailable for the trait (basophil count and age at menarche). In a few instances, we had to make a choice of which gene within a gene family should be used for replication: for the HBA locus we used HBA1, for the GYPB/GYPA locus we used GYPB, and for the FCGR2A–FCGR3A–FCGR3B–FCGR2B locus we did not attempt replication because it was unclear whether LoF of one of these genes would be expected to have the same effect as a CNV. Additionally, for LST3, we compared to results reported for SLCO1B3 (an alternative gene symbol).
Replicating CNV-phenotype associations in BioBank Japan
For fine-mapped associations that uncovered gene-trait relationships (rightmost column of Figure 3E), we performed additional replication analyses in BioBank Japan (N=180K; data set details above, see HI-CNV analysis of BioBank Japan). We first assessed which associations were suitable for replication based on available phenotyping and reasonable power in BBJ:
CXCR4 DUP: only 1 carrier with monocyte count available; excluded due to low power
DOK3 pLoF: phenotype not available; excluded
PARVB pLoF: phenotype not available; excluded
R3HDM4 pLoF: phenotype not available; excluded
SLC2A3 DUP/pLoF: included
SLC35E2B pLoF: included
FCGR3B DUP/pLoF: maximum of 14 carriers available with relevant phenotype (probably due to undercalling of CNVs at this locus); excluded due to low power
SULT1A1 pLoF: phenotype not available; excluded
DIS3L2 pLoF: included
UHRF2 pLoF: included
This investigation left us with four CNV loci (SLC2A3, SLC35E2B, DIS3L2, and UHRF2) to take forward for replication. Examination of CNVs and associated phenotypes at these loci led us to the following list of 14 associations to attempt to replicate:
SLC2A3: associations of both DUP and pLoF CNVs with basophil count, lymphocyte count, height, and menarche age.
SLC35E2B: associations of pLoF CNVs with mean corpuscular hemoglobin (MCH), mean corpuscular volume (MCV), and height. Upon closer inspection of this locus, we also noticed a strong association in UK Biobank between SLC35E2B DUPs and height (β =0.04 (0.01), P = 3.2 × 10−5), so we attempted to replicate this association in BioBank Japan as well.
DIS3L2: association of DEL spanning exon 9 (see Deletion spanning DIS3L2 exon 9) with height.
UHRF2: association of pLoF CNVs with height.
For the above events and phenotypes, we performed association analyses run on a 179,420-sample uniform-ancestry subset of BioBank Japan. For all phenotypes, residuals from a model regressing phenotype on age, sex, genotyping arrays (as factors) and 10 PCs as covariates were computed. These residuals were then inverse-normal transformed and used as outcome variables with the dependent variable being carrier status of events of interest.
Follow-up analyses at loci of interest
Here we provide details of additional analyses we performed at loci of interest, including refined analyses of specific phenotypes, corroborating analyses of SNP and indel PTVs, and further characterization of specific CNV events.
Extreme blood phenotypes
The CNV-phenotype association tests we ran using BOLT-LMM analyzed blood cell traits that we had previously normalized using an approach that included removal of outliers, defined as deviating from the median by >7x the interquartile range (IQR). However, we subsequently found that certain CNVs had large enough effect sizes that a substantial fraction of carriers had been removed as outliers. As such, when further investigating loci related to blood traits, we renormalized blood phenotypes without outlier removal (using covariate adjustment and inverse normal transforms as previously described) (Barton et al., 2021).
Residualization of phenotypes to emulate mixed model analysis
In follow-up analyses (e.g., of SNP and indel PTVs genotyped in a subset of individuals, or for categories of CNVs we did not initially genotype), we performed linear regression on phenotypes that we residualized for polygenic predictions using array-typed SNPs (omitting those within 2Mb of the gene of interest) that we generated using BOLT-LMM (--predBetasFile) in 10-fold cross-validation (to emulate the power of linear mixed model association analysis) (Mefford et al., 2020). We normalized residualized phenotypes to have a mean of zero across all non-removed individuals with non-missing phenotype.
PTVs in UK Biobank exome sequencing data
We identified carriers of high-confidence loss-of-function SNP and indel variants (on canonical transcripts annotated using LOFTEE (Karczewski et al., 2020)) from the 185,365 UK Biobank participants in our analysis set with whole-exome sequencing data available (Szustakowski et al., 2021). However, for R3HDM4 we analyzed carriers of high-confidence loss-of-function SNP and indels in any transcript as there were no high-confidence loss-of-function SNP and indels on the canonical transcript.
α-globin locus
Exons of HBA2 are located at 16:222911–223006; 16:223123–223328; and 16:223470–223599 whereas for HBA1 they are at 16:226715–226810, 16:226927–227132, and 16:227281–227410 (hg19 coordinates). The UK Biobank SNP-array contained 3 probes within either HBA2 or HBA1, with genomic coordinates listed as 227306, 227333, and 227365 (all within the last exon of HBA1, and all with extremely rare minor alleles). Due to the sequence similarity of HBA2 and HBA1 these 3 probes effectively measured copy number of both HBA2 and HBA1. The probe before these probes was at 221057 and the one after them was at 228306. HS-40 is located 40 kb upstream of the zeta-globin gene, around 162686.
Given the above information, we categorized CNV calls at the α-globin locus as follows:
Alpha-globin locus DEL: a deletion call with a start ≤ 140000 and end ≥230000.
HS-40 DEL: a deletion call with a start ≤ 162240 and end ≥162240 and < 226715.
HBA2+HBA1 DEL: a deletion call with a start of 205897 and end of 231021, or a deletion with a start of 216041 and end of 228306 or 231021.
HBA2 DEL: a deletion call with a start of 221057 and end of 227306 (indicating an −α4.2 deletion; Figure S5B).
HBA2 DUP: a duplication call with a start of 221057 and end of 227306 or 227333 (indicating an αααanti 4.2 duplication; Figure S5B).
HBA2 triplication: a duplication call with a start of 221057 and end of 227365 (suggesting an αααanti 4.2 triplication; Figure S5B). Whole-exome sequencing read-depth for carriers of such events confirmed triplication of HBA2 (Figure S5C).
HBA2+HBA1 DUP: a duplication call with a start ≥176743 and ≤ 221057 and end ≥ 230000.
Alpha-globin locus DUP: a duplication call with a start ≤140000 and end ≥ 230000.
Retroposition of spliced MTMR2 transcript into an intron of LRCH1
Our callset included a duplication call in MTMR2 on chromosome 11 with length ~10–20kb that was called in 2,522 UK Biobank participants (MAF=0.003). This variant associated with an increase in platelet distribution width of +0.12 (0.02) s.d. (P = 1.7 × 10−10) and passed our LD-based fine-mapping filter, with no nearby SNP on chromosome 11 reaching genome-wide significance. Surprisingly, this event was not called in gnomAD-SV (Collins et al., 2020) or the 1000 Genomes 30x SV callset (Byrska-Bishop et al., 2021), prompting further investigation.
Examination of sequencing reads from exome-sequenced carriers showed that the event was actually a retroposed pseudogene insertion of the MTMR2 processed transcript into an intron of LRCH1 on chromosome 13. We observed increases in read coverage only in exons of MTMR2 and split reads corresponding to splice junctions (usually seen in RNA-seq data rather than DNA sequencing). Split reads that partially aligned to the 5’ UTR of MTMR2 and partially aligned to chromosome 13 showed that that the MTMR2 transcript had been inserted into an intron of LRCH1.
Closer examination of UK Biobank SNP-array probes at MTMR2 contributing to the initial signal showed that an “indel” probe (Affx-52351109) actually directly genotyped the retroposed pseudogene insertion. Carriers of the duplication calls exhibited increased LRR at seven probes (not usually enough to sensitively call a duplication event—suggesting that MAF=0.003 was an underestimate, representing calls in only a subset of carriers). Six of the probes with increased LRR fell within coding exons or UTRs, as expected; the remaining probe (Affx-52351109, intended to genotype an indel 11:95595151:TTTA>T) fell just within intron 7–8 of MTMR2, 2bp beyond the end of exon 7. Inspection of the MTMR2 transcript showed that the minus-strand sequence ending in this “indel” actually corresponds to the splice junction created by joining exon 7 to exon 8. Further analysis of LRR at the seven probes confirmed that Affx-52351109 directly genotyped the retroposed insertion (identifying an expanded set of carriers; MAF=0.007). The 3bp indel that the probe was designed to genotype does not actually exist according to gnomAD (Karczewski et al., 2020).
Analyses of population allele frequencies and linkage disequilibrium of the MTMR2 retroposed insertion showed that the variant sits on a European haplotype (MAF=0.7%) containing rs145057384, a good tag SNP (R=0.87, MAF=1%). Allele frequencies in UK Biobank (based on Affx-52351109) were 0.69% in Europeans and 0.02–0.05% in non-Europeans (SAS, AFR, EAS). The insertion was also called in the 1000 Genomes 30x SV callset (Byrska-Bishop et al., 2021), which contains a 2,529 bp insertion consisting of most of the processed transcript of MTMR2 (excluding some 3’ UTR sequence typically present in transcripts according to GTEx v8 data (Aguet et al., 2020)), plus a poly-A tail, followed by another 15bp; the 1000 Genomes data set contained 11 carriers among N=3,202 individuals (10 EUR + 1 Colombian).
Our next question was whether the retroposed insertion affected platelet traits by disrupting LRCH1 in some way. LRCH1 LoFs were too rare to evaluate the effect of LoF on platelet distribution width (PDW), so we focused on investigating potential effects of LRCH1 variants on gene expression or splicing.
LRCH1 is broadly expressed in many tissues, and 11 carriers of the insertion in GTEx v8 (Aguet et al., 2020) appeared to have reduced LRCH1 expression. Carriers were identified based on chimeric sequence that we detected in 11 of 13 carriers of the tag SNP rs145057384. RNA-seq data was available for 0–8 carriers per GTEx tissue. Among the 25 tissues with RNA-seq data available for 4+ carriers (providing reasonable power), 22 of 25 tissues exhibited negative mean normalized expression of LRCH1 in carriers (P = 1.6 × 10−4; two-sided sign test). We were unable to determine a mechanism by which this ~2.5kb insertion might reduce expression: the inserted MTMR2 processed transcript does not appear to be transcribed (based on no evidence of expression of the truncated 3’ UTR), consistent with it lacking a promoter, and the insertion does not appear to affect splicing.
A common-SNP association with PDW (in a different intron of LRCH1) also appeared to be mediated by LRCH1 expression (Figure S4D). Interestingly, the association of the retroposed pseudogene insertion with PDW—which our GTEx analyses suggested was likewise LRCH1 expression-mediated—exhibited ~4-fold larger effect sizes on LRCH1 expression and PDW than the common SNPs (Table S5).
Deletion spanning DIS3L2 exon 9
Our fine-mapped CNV-phenotype associations included an association of deletions spanning a probe at chr2:233,022,511 (hg19) with a decrease in height of 0.44 (0.04) s.d. (P = 3.9 × 10−22). Our HI-CNV callset contained 271 such deletion calls among UK Biobank participants of European ancestry (MAF=0.0003). Further investigation revealed that these calls predominantly reflected a deletion spanning exon 9 in DIS3L2. This ~22kb deletion has previously been implicated in Perlman syndrome (an autosomal recessive disease characterized by congenital overgrowth), with previous work suggesting that exon 9 deletion abolishes the RNA-binding domain of DIS3L2, reducing ribonuclease activity (Astuti et al., 2012).
The presence of several different types of SNP and CNV polymorphisms at this locus offered the opportunity to search for further lines of evidence that might point to a potential mechanism underlying the association we observed with adult height in heterozygous carriers. In particular, beyond the rare deletion spanning exon 9 of DIS3L2, we also observed rare reciprocal duplications of the same region spanning DIS3L2 exon 9, consistent with this region being flanked by LINE1 elements and therefore being a hotspot of L1-mediated non-allelic homologous recombination [40]. We therefore examined these CNVs as well as common and rare SNPs for association with height:
Deletion spanning DIS3L2 exon 9. As noted above, in UK Biobank this deletion associated with a decrease in height of nearly half a standard deviation. We replicated the association in BioBank Japan: HI-CNV identified 49 deletion carriers with available height phenotypes, and the deletion associated with a decrease in height of 0.39 (0.14) s.d. (P = 0.006; Figure 4B).
Duplication spanning DIS3L2 exon 9. In UK Biobank, this reciprocal duplication associated with an increase in height of 0.85 (0.09) s.d. (P = 3.2 × 10−22; Figure S4B). Because the duplication was difficult to call even for HI-CNV (which undercalled the event due to its spanning only three genotyping probes and its even rarer frequency), we re-genotyped the duplication by performing a combined analysis of SNP-array probe intensity data at the three affected probes together with DIS3L2 exon 9 read-depth measured from whole-exome sequencing of N=454K UK Biobank participants (Backman et al., 2021) (Figure S4C).
Rare SNP/indel pLoF variants in DIS3L2. Despite ample power in UK Biobank, pLoF SNP and indel variants within DIS3L2 did not associate with height in published burden analyses of exome sequencing data (Backman et al., 2021) (Figure 4A).
Common SNPs. Common SNPs at the locus associate strongly with height, such that this locus was among the earliest height loci to be discovered (Estrada et al., 2009). However, these common SNPs associated with much smaller effects on height than the exon 9 deletion and duplication CNVs (Figure S4B).
Collectively, the associations of the reciprocal deletion and duplication with strong, opposite deviations in height suggest that these CNVs do causally influence height—but the apparent lack of an effect of DIS3L2 pLoF SNPs and indels leaves the mechanism unclear. Whereas the direct protein-coding consequence of exon 9 deletion and duplication would at first glance suggest a mechanism involving DIS3L2 function, this hypothesis is not supported by the pLoF SNP/indel burden analysis. Additionally, while exon 9 deletion could plausibly decrease DIS3L2 function (as previously suggested (Astuti et al., 2012)), it is unclear why exon 9 duplication would increase function.
An alternative possibility is that the deletion and duplication could have regulatory effects on a nearby gene. A plausible candidate is the gene NPPC (natriuretic peptide precursor C) ~35kb upstream of DIS3L2, which has previously been suggested as the potential target of height-associated variation at the locus (Estrada et al., 2009; Tassano et al., 2013). Examination of Hi-C data (Kerpedjiev, Abdennur, et al., 2018; Rao et al., 2014) does suggest that NPPC has long-range interactions with ~500kb worth of sequence extending through DIS3L2. We attempted to explore the possibility of a regulatory effect on NPPC using RNA-seq data but ultimately concluded that available data was insufficient: no carriers of DIS3L2 exon 9 deletions or duplications could be found within GTEx v8 (Aguet et al., 2020) and only one deletion carrier could be found within 1000 Genomes (using WGS read-depth) (Byrska-Bishop et al., 2021). Lymphoblastoid cell lines from this individual had been RNA-sequenced in the GEUVADIS project (Lappalainen et al., 2013), and the RNA-seq data exhibited the lowest DIS3L2 exon 9 expression among all GEUVADIS samples (consistent with exon 9 deletion); however, we were unable to assess any potential regulatory effect on NPPC due to insufficient expression in LCLs (i.e., most samples had no detectable NPPC expression).
Contrasting effect sizes of deletions and duplications
Selection of gene-trait pairs with likely-causal rare coding variants
To explore the relative effects of focal deletions and duplications, we examined 199 gene-trait pairs for which we had previously identified PTVs likely to alter quantitative traits (Supplementary Table 3 of (Barton et al., 2021)). For each gene on this list, we then compared the effects of likely-causal PTVs to those of whole-gene deletions and duplications.
At the level of individual loci, gene deletions acted similarly to PTVs; of the 41 genes for which there were at least 2 carriers of gene-deletions, 16 deletions were nominally significant for the given trait and 6 were Bonferroni significant (Figure 7). At the level of individual loci, gene duplications tended to act in the opposite direction as PTVs and with a smaller magnitude of effect; of the 139 genes for which there were at least 2 carriers of gene-duplications, 27 duplications were nominally significant for the given trait and 3 were Bonferroni significant (Figure 7).
Comparison of deletion and duplication effect sizes: power analysis
Consistent with the idea that duplications tend to have a weaker effect, there were far more examples of gene duplications than gene deletions with at least 2 carriers (139 vs. 41; Figure 7). We next wished to quantify the difference in effect sizes. For each of the 199 gene-trait pairs we could assess whether at least two individuals in UK Biobank carried a gene deletion or duplication, and for these events compare the effect sizes of likely-causal PTVs to the gene deletions and duplications.
More concretely, for a given trait t, and gene g, we are given:
, for CNV = {DEL,DUP}
Number of carriers of CNV = {DEL,DUP} (≥2)
Sample size (N)
Increase in effective sample size from using BOLT-LMM (equivalently, residualizing on genome-wide SNPs reduces σtrait to < 1); in BOLT-LMM output files the line ”Absolute prediction MSE, fold-best” contains an estimate of BOLT-LMM’s (after conditioning on genome-wide SNPs); denoted boltlmmboost
- Given multiple PTVs indexed by i, we compute the inverse variance weighted mean effect:
For each trait-gene pair, we can compute the power (powerg−t,CNV) for two sample (different sizes) t-test of means assuming the effect size , significance level 0.05, and the number of carriers and non-carriers for a given CNV = {DEL,DUP}.
For a given trait-gene pair, assuming independence across gene-trait pairs, we can consider the random indicator variable of whether a significant effect was seen for the . Across all trait-gene pairs we can then consider the observed number of significant CNV effects:
We can then compare the expected number of significant CNV effects for f = {0,0.5,1} to the number of observed significant CNV effects. We note that this approach ignores the sign of effect size (e.g., whether duplications have opposite vs. same effect directions as PTVs). Results were consistent with deletions having similar effect sizes as PTVs; assuming deletions had the same effect size as PTVs resulted in 18.5 expected nominally associated associations whereas assuming half the magnitude of PTVs resulted in 8.3 expected associations (Figure 7). Similar power analysis results for gene duplications show results are consistent with duplications having the opposite direction, and a smaller magnitude compared to the PTV-effect (Figure 7).
An extension of this approach is to search across the space 0 ≤ 𝑓 ≤ 1, and for each value compute the expected value of number of significant associations and find the value for which f results in the observed number of significant associations (Table S7).
As a sensitivity analysis we further performed likelihood-based analyses. We can compute the likelihood of observing assuming it came from ; assuming independence across gene-trait pairs, we can then compute the maximum likelihood estimate for c. We note that this approach incorporates the sign of effect size; however, one can also ignore the sign and quantify the absolute effect (agnostic to effect direction) by computing the likelihood of observing assuming it came from . We note that this approach ignores the standard error of however these PTVs come from a published set of significant (p < 5 × 10−8) variants and therefore the standard error can be considered to be much smaller than that of . Results can be found in Table S7.
HI-CNV at multi-copy regions: an investigation of SULT1A1
We examined the behavior of the HI-CNV method and underlying probe intensities at multi-copy regions by an analysis of the SULT1A1 locus, a known multiallelic, highly polymorphic CNV region. We estimated SULT1A1 copy number based on exome-sequencing read depth (in the N=200K UK Biobank WES release (Szustakowski et al., 2021)) followed by phasing and imputation using a computational pipeline we recently described (Mukamel et al., 2021), which provided precise copy number estimates for most individuals (Figure S6A). Copy number was estimated based on read depth within chr16:28,616,321–28,622,321 (hg19).
We compared these sequencing-derived estimates to copy numbers estimated by HI-CNV (which ranged from 0 to 4; HI-CNV can only call up to a single-copy increase per haplotype) for CNV calls starting within one probe of chr16:28,606,960 and ending within one probe of chr16:28,619,696 (the two probes closest to the ends of the CNV region). We observed that while HI-CNV did correctly call some carriers of SULT1A1 CNVs, many CNVs were missed, and higher-copy states were usually misclassified (Figure S6B). Consistent with this behavior, examination of probe intensity cluster plots showed that higher-copy states (CN≥4) provided very little differentiating probe intensity signal (Figure S6C).
Given these challenges observed at SULT1A1, we concluded that there is no straightforward way to considerably improve HI-CNV’s performance on SNP-array-based CNV calling at complex regions. However, we note that this reflects a limitation of SNP-array data rather than our haplotype-informed CNV detection framework, which we anticipate will be applicable for genotyping complex multi-copy regions from sequencing read-depth data.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details of exact analyses, statistical tests, and tools can be found in the main text and STAR Methods.
Supplementary Material
Supplemental Figure 1: Validation of HI-CNV; related to Figure 1 and STAR Methods. A Validation rate of HI-CNV calls assessed using WGS data for 500 individuals. The left and right plots indicate proportions (left) and numbers (right) of SNP-array-based HI-CNV calls that exhibited the correct WGS read-depth direction (below the mean for deletions and above the mean for duplications) as a function of various CNV properties: CNV length (top), and call confidence (middle, based on the Bayes factor estimated by the hidden Markov model; “log10BF”), and GC content (bottom). These sign-tests result in an estimated 90.6% and 86.9% validation rate for deletions and duplications, respectively, and 89.1% overall. B Average number of HI-CNV calls and proportion of HI-CNV calls with correct read-depth signal (sign test) assessed using WGS data for 100 BioBank Japan participants. C Validation rate of fine-mapped HI-CNV calls assessed using WGS data. The proportion and number of calls with the correct WGS read-depth direction (below the mean for deletions and above the mean for duplications) are shown for: (i) 250 randomly selected fine-mapped CNV calls (“Fine-mapped”; STAR Methods); and (ii) all HI-CNV calls made in 500 randomly selected individuals with WGS available (“All”; STAR Methods). These sign tests resulted in an estimated 93.7% and 95.7% validation rate for fine-mapped deletions and duplications, respectively (as compared to 90.6% and 86.9% for all events), and 94.4% overall (as compared to 89.1% for all events). Numerical results are available in Table S1.
Supplemental Figure 2: HI-CNV vs PennCNV; related to Figure 1 and STAR Methods. A Detection sensitivity (recall) of SNP-array based methods on a benchmark set of CNVs called from WGS data. We assessed recall of low-frequency (MAF≤5%) CNVs called by Delly in 43 individuals with whole-genome-sequence data available. Sensitivity increased with CNV size and probe overlap (left-to-right columns) and for gene-overlapping CNVs (bottom vs. top row). HI-CNV0 denotes analysis without incorporating information from haplotype neighbors (but still using our SNP-specific probabilistic models of genotyping intensities). Numerical results are available in Table S1. B Sensitivity of PennCNV to detect CNVs contributing to likely-causal phenotype associations identified by HI-CNV analysis. For each of the 269 likely-causal CNV-phenotype associations found via analysis of our HI-CNV callset, the number of carriers found using PennCNV versus HI-CNV is plotted. The four panels stratify the data by CNV size (median among the CNV calls that were considered in each association test). The dashed line shows the y=x line.
Supplemental Figure 3: Signal intensity plots for CNV-phenotype associations; related to Figure 5 and 6. Denoised LRR is plotted for four randomly selected CNV carriers at each locus shown in Figures 5 and 6. Dots indicating LRR at probes within a CNV call are colored in red (deletion called) or blue (duplication called).
Supplemental Figure 4: Investigation into various allelic series; related to Figure 5 and STAR Methods. A LRR signal for individuals with SLC2A3 deletion calls shows no evidence of homozygous deletions. Distributions of mean LRR (denoised and rescaled) across 60 probes spanned by reciprocal SLC2A3 DEL/DUP CNVs are shown for carriers of SLC2A3 deletion calls (3,768 individuals), SLC2A3 duplication calls (16,821 individuals), and 1,000 randomly selected controls. The mean LRR histogram for duplications has a heavy right tail suggesting the presence of individuals with CN=4, whereas the mean LRR histogram for deletions shows no evidence of homozygous deletions (binomial P =0.0009 assuming Hardy-Weinberg equilibrium). B,C Deletions and duplications of DIS3L2 exon 9 and associations with height. B Height associations (top) and effect sizes (bottom) for variants at the DIS3L2 locus. The reciprocal deletion and duplication spanning DIS3L2 exon 9 are shown in red and blue, respectively, and SNPs are shown in grey. SNP effect sizes are plotted only for genome-wide significant SNPs (P < 5 × 10−8). C Carriers of the DIS3L2 exon 9 deletion and duplication could be more accurately identified by analyzing average SNP-array probe intensity together with WES read depth within the event; carriers of deletions and duplications called in this way are colored in red and blue, respectively. The distribution of height (in units of standard deviations) is shown separately for carriers of the DIS3L2 exon 9 deletion, the duplication, and noncarriers. Mean and 95% CI are shown below the histograms. D A common-SNP association with platelet distribution width appears to be mediated by LRCH1 expression. Top: Manhattan plot of associations between SNPs and platelet distribution width (linear regression P -values computed using UK Biobank participants of self-reported European ancestry). Bottom: LRCH1 eQTL P-values from GTEx for the tissues with the top three signals. The red points toward the right of each plot correspond to the retroposed pseudogene insertion (directly genotyped by Affx-52351109 in the top plot) and the tag SNP rs145057384 in the bottom plots; the insertion has ~4-fold larger effect sizes than the top SNPs but has weaker associations because it is much rarer.
Supplemental Figure 5: α-globin locus; related to Figure 6 and STAR Methods. A α-globin locus CNVs at the α-globin locus and their effects on blood traits. For carriers of each category of α-globin CNVs shown in Figure 6A, we plot the average mean corpuscular hemoglobin (MCH; both raw and units of standard deviation; s.d.), red blood cell count (RBC; both raw and units of s.d.), mean corpuscular volume (MCV; both raw and units of s.d.), and the Mentzer index (MCV/RBC). Vertical lines denote population averages. B Schematic of non-allelic homologous recombination (NAHR) at the α-globin locus. The HBA2 and HBA1 genes lie within ~4 kb regions containing three distinct regions of high homology (X, Y, and Z boxes; Fig. 1 of (Wang et al., 2003)). The spacing between the X and Y boxes differs in the sequence left of HBA2 vs. HBA1, such that NAHR within the X boxes creates 4.2-kb DEL/DUP events (−α4.2 and αααanti 4.2) while NAHR within the Z boxes creates 3.7-kb DEL/DUP events (−α3.7 and αααanti 3.7). The UK Biobank SNP-array contained a probe in unique sequence at 16:221057 between X2 and Y2 (pink star). Because intensity at this probe is affected by 4.2-type events but unaffected by 3.7-type events, we concluded that duplication calls starting at the probe 16:221057 and extending through the three probes in HBA exons were probably 4.2-type events that involved full deletion or duplication of HBA2. C Mean LRR and normalized WES read depth for CNVs at the α-globin locus. Mean LRR across four probes (three within HBA1 and 16:221057) and mean normalized read depth for exome-sequencing reads mapping uniquely to HBA2 or HBA1 are plotted for each category of α−globin CNV we considered. Error bars, 95% CIs (computed as mean ±1.96× s.e.m. across carriers). WES read depth restricted to reads aligning uniquely to HBA2 and HBA1 (i.e., with nonzero mapping quality) was computed using mosdepth (Pedersen and Quinlan, 2018) and normalized by exome-wide coverage.
Supplemental Figure 6: Behavior of HI-CNV and SNP-array probe intensities at SULT1A1: a multiallelic, highly polymorphic CNV region; related to STAR Methods. A Estimated SULT1A1 copy number based on exome-sequencing read depth followed by phasing and imputation. B Comparison of sequencing-derived estimates to copy numbers estimated by HI-CNV (which ranged from 0 to 4; HI-CNV can only call up to a single-copy increase per haplotype). C Probe intensity cluster plots for a probe within SULT1A1. The probe examined was rs150459557 (hg19 chr16:28,617,472); intensity values are shown for 10 randomly selected individuals for each WES-derived copy-number state.
Supplemental Figure 7: Steps of SNP-specific genotype cluster prediction for deletion and duplication genotypes; related to STAR Methods. A Assignment of reference CNV probe intensity data to CN=1 genotype clusters (A, B) and CN=3 genotype clusters (AAA, AAB, ABB, BBB) by dividing the total vs. relative intensity plane into zones based on CN=2 cluster parameters. B Estimation of bivariate normal parameters corresponding to cluster assignments (for clusters with 10 or more assigned data points). C Prediction of CN=1 and CN=3 genotype clusters for other (“query”) SNPs. The basic idea was to perform prediction independently for left-half clusters (A, AAA, AAB) and right-half clusters (B, ABB, BBB) by identifying (for each half) reference SNPs with best-matching CN=2 clusters in the relevant half. The axes of each plot are (x-axis, analogous to BAF), and denoised LRR (y-axis), scaled to [−127, 127].
Table S1: Benchmarking and validation of HI-CNV; related to STAR Methods.
Table S2: Quantitative traits analyzed; related to Figure 3 and STAR Methods.
Table S3: Likely-causal CNV-phenotype associations; related to Figure 3 and STAR Methods.
Table S4: Annotation of CNV loci; related to Figure 3 and STAR Methods.
Table S5: Follow-up on allelic series; related to Figure 5, Figure 6, and STAR Methods.
Table S6: Binary associations; related to STAR Methods.
Table S7: Deletion and duplication burden; related to Figure 7 and STAR Methods.
Leveraging haplotype-sharing in biobank cohorts increases CNV detection sensitivity
HI-CNV software implementation enables haplotype-informed analysis of SNP-array data
Fine-mapped CNV-trait associations implicate regulatory and gene-altering CNVs
CNV loci corroborate SNP associations and uncover gene-trait relationships
Acknowledgements
We thank R. Handsaker and P. Palamara for helpful discussions and S. Stankovic for providing PTV annotations on UK Biobank exome-sequencing variants. We thank Dr. Yukihide Momozawa in RIKEN Center for Integrative Medical Sciences and the members of the BioBank Japan Project, headquartered in the University of Tokyo Institute of Medical Science, for supporting this project. This research was conducted using the UK Biobank Resource under application number 40709. M.L.A.H. was supported by US NIH Fellowship F32 HL160061. M.A.S. was supported by US NIH Fellowship F31 MH124393. A.R.B. was supported by US NIH grant fellowship F31 HL154537. R.E.M. was supported by US NIH grant K25 HL150334. V.G.S. received support from NIH grants R01 DK103794 and R01 HL146500, as well as the New York Stem Cell Foundation. C.T. was supported by Japan Agency for Medical Research and Development (AMED) grants JP21kk0305013, JP21tm0424220 and JP21ck0106642, and Japan Society for the Promotion of Science (JSPS) KAKENHI grant JP20H00462. P.-R.L. was supported by US NIH grant DP2 ES030554, a Burroughs Wellcome Fund Career Award at the Scientific Interfaces, the Next Generation Fund at the Broad Institute of MIT and Harvard, and a Sloan Research Fellowship. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. Computational analyses were performed on the O2 High Performance Compute Cluster, supported by the Research Computing Group, at Harvard Medical School (http://rc.hms.harvard.edu).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
V.G.S. serves as an advisor to and/or has equity in Branch Biosciences, Ensoma, Novartis, Forma, and Cellarity, all unrelated to the present work.
References
- Abel HJ, Larson DE, Regier AA, Chiang C, Das I, Kanchi KL, Layer RM, Neale BM, Salerno WJ, Reeves C, et al. (2020). Mapping and characterization of structural variation in 17,795 human genomes. Nature 583, 83–89. 10.1038/s41586-020-2371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abyzov A, Urban AE, Snyder M, and Gerstein M (2011). CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984. 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguet F, Barbeira AN, Bonazzola R, Brown A, Castel SE, Jo B, and Kasela S (2020). The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre M, Rivas MA, and Priest J (2019). Phenome-wide Burden of Copy-Number Variation in the UK Biobank. Am. J. Hum. Genet 105, 373–383. 10.1016/j.ajhg.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama M, Okada Y, Kanai M, Takahashi A, Momozawa Y, Ikeda M, Iwata N, Ikegawa S, Hirata M, Matsuda K, et al. (2017). Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat. Genet 49, 1458–1467. 10.1038/ng.3951. [DOI] [PubMed] [Google Scholar]
- Astuti D, Morris MR, Cooper WN, Staals RHJ, Wake NC, Fews GA, Gill H, Gentle D, Shuib S, Ricketts CJ, et al. (2012). Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat. Genet 44, 277–284. 10.1038/ng.1071. [DOI] [PubMed] [Google Scholar]
- Auwerx C, Lepamets M, Sadler MC, Patxot M, Stojanov M, Baud D, Mägi R, Porcu E, Reymond A, Kutalik Z, et al. (2022). The individual and global impact of copy-number variants on complex human traits. Am. J. Hum. Genet 109, 647–668. 10.1016/j.ajhg.2022.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman JD, Li AH, Marcketta A, Sun D, Mbatchou J, Kessler MD, Benner C, Liu D, Locke AE, Balasubramanian S, et al. (2021). Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 599, 628–634. 10.1038/s41586-021-04103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton AR, Sherman MA, Mukamel RE, and Loh P-R (2021). Whole-exome imputation within UK Biobank powers rare coding variant association and fine-mapping analyses. Nat. Genet 53, 1260–1269. 10.1038/s41588-021-00892-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton AR, Hujoel MLA, Mukamel RE, Sherman MA, and Loh P-R (2022). A spectrum of recessiveness among Mendelian disease variants in UK Biobank. Am. J. Hum. Genet 109, 1298–1307. 10.1016/j.ajhg.2022.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyter D, Ingimundardottir H, Oddsson A, Eggertsson HP, Bjornsson E, Jonsson H, Atlason BA, Kristmundsdottir S, Mehringer S, Hardarson MT, et al. (2021). Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat. Genet 53, 779–786. 10.1038/s41588-021-00865-4. [DOI] [PubMed] [Google Scholar]
- Bock C, Walter J, Paulsen M, and Lengauer T (2007). CpG Island Mapping by Epigenome Prediction. PLoS Comput. Biol 3. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209. 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, Corvelo A, Clarke WE, Musunuri R, Nagulapalli K, et al. (2021). High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv 10.1101/2021.02.06.430068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, and Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7. 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Abel HJ, Das I, Larson DE, Ganel L, Kanchi KL, Regier AA, Young EP, Kang CJ, Scott AJ, et al. (2021). Association of structural variation with cardiometabolic traits in Finns. Am. J. Hum. Genet 108, 583–596. 10.1016/j.ajhg.2021.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin LJM, Asher JE, Walters RG, El-Sayed Moustafa JS, de Smith AJ, Sladek R, Balding DJ, Froguel P, and Blakemore AIF (2010). cnvHap: an integrative population and haplotype–based multiplatform model of SNPs and CNVs. Nat. Methods 7, 541–546. 10.1038/nmeth.1466. [DOI] [PubMed] [Google Scholar]
- Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, and Ragoussis J (2007). QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 35, 2013–2025. 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, Khera AV, Lowther C, Gauthier LD, Wang H, et al. (2020). A structural variation reference for medical and population genetics. Nature 581, 444–451. 10.1038/s41586-020-2287-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Glessner JT, Porcu E, Lepamets M, Brandon R, Lauricella C, Han L, Morley T, Niestroj L-M, Ulirsch J, et al. (2022). A cross-disorder dosage sensitivity map of the human genome. Cell 185, 3041–3055.e25. 10.1016/j.cell.2022.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper NJ, Shtir CJ, Smyth DJ, Guo H, Swafford AD, Zanda M, Hurles ME, Walker NM, Plagnol V, Cooper JD, et al. (2015). Detection and correction of artefacts in estimation of rare copy number variants and analysis of rare deletions in type 1 diabetes. Hum. Mol. Genet 24, 1774–1790. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford K, Bracher-Smith M, Owen D, Kendall KM, Rees E, Pardiñas AF, Einon M, Escott-Price V, Walters JTR, O’Donovan MC, et al. (2019). Medical consequences of pathogenic CNVs in adults: analysis of the UK Biobank. J. Med. Genet 56, 131–138. 10.1136/jmedgenet-2018-105477. [DOI] [PubMed] [Google Scholar]
- Dauber A, Yu Y, Turchin MC, Chiang CW, Meng YA, Demerath EW, Patel SR, Rich SS, Rotter JI, Schreiner PJ, et al. (2011). Genome-wide Association of Copy-Number Variation Reveals an Association between Short Stature and the Presence of Low-Frequency Genomic Deletions. Am. J. Hum. Genet 89, 751–759. 10.1016/j.ajhg.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis J, Walker L, Tyrer J, Michailidou K, and Easton DF (2021). Detecting rare copy number variants from Illumina genotyping arrays with the CamCNV pipeline: Segmentation of z -scores improves detection and reliability. Genet. Epidemiol 45, 237–248. 10.1002/gepi.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin SJ, Li M, Hou C, Yang S, Glessner J, Hakonarson H, Bucan M, Maris JM, and Wang K (2008). Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 36, e126–e126. 10.1093/nar/gkn556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durai V, Bagadia P, Granja JM, Satpathy AT, Kulkarni DH, Davidson JT, Wu R, Patel SJ, Iwata A, Liu T-T, et al. (2019). Cryptic activation of an Irf8 enhancer governs cDC1 fate specification. Nat. Immunol 20, 1161–1173. 10.1038/s41590-019-0450-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R (2014). Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics 30, 1266–1272. 10.1093/bioinformatics/btu014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2017). Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc 12, 2478–2492. 10.1038/nprot.2017.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada K, Krawczak M, Schreiber S, van Duijn K, and Stolk L (2009). A genome-wide association study of northwestern Europeans involves the C-type natriuretic peptide signaling pathway in the etiology of human height variation. Hum. Mol. Genet 18, 3516–3524. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, Rosen N, Kohn A, Twik M, Safran M, et al. (2017). GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database 2017. 10.1093/database/bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyman WA, McManus KF, Shringarpure SS, Jewett EM, Bryc K, The 23 and Me Research Team, and Auton A (2021). Fast and Robust Identity-by-Descent Inference with the Templated Positional Burrows–Wheeler Transform. Mol. Biol. Evol 38, 2131–2151. 10.1093/molbev/msaa328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gel B, and Serra E (2017). karyoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 33, 3088–3090. 10.1093/bioinformatics/btx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halldorsson BV, Eggertsson HP, Moore KHS, Hauswedell H, Eiriksson O, Ulfarsson MO, Palsson G, Hardarson MT, Oddsson A, Jensson BO, et al. (2022). The sequences of 150,119 genomes in the UK Biobank. Nature 607, 732–740. 10.1038/s41586-022-04965-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker RE, Korn JM, Nemesh J, and McCarroll SA (2011). Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet 43, 269–276. 10.1038/ng.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, and McCarroll SA (2015). Large multiallelic copy number variations in humans. Nat. Genet 47, 296–303. 10.1038/ng.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton C, Wilkie A, Drysdale H, Wood W, Vickers M, Sharpe J, Ayyub H, Pretorius I, Buckle V, and Higgs D (1990). Alpha-thalassemia caused by a large (62 kb) deletion upstream of the human alpha globin gene cluster. Blood 76, 221–227. 10.1182/blood.V76.1.221.221. [DOI] [PubMed] [Google Scholar]
- Hay D, Hughes JR, Babbs C, Davies JOJ, Graham BJ, Hanssen LLP, Kassouf MT, Oudelaar AM, Sharpe JA, Suciu MC, et al. (2016). Genetic dissection of the α-globin super-enhancer in vivo. Nat. Genet 48, 895–903. 10.1038/ng.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Klotman ME, and Diaz GA (2003). Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat. Genet 34, 5. . [DOI] [PubMed] [Google Scholar]
- Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, Hutchinson A, Deng X, Liu C, Horner M-J, et al. (2012). Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet 44, 651–658. 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall KM, Rees E, Escott-Price V, Einon M, Thomas R, Hewitt J, O’Donovan MC, Owen MJ, Walters JTR, and Kirov G (2017). Cognitive Performance Among Carriers of Pathogenic Copy Number Variants: Analysis of 152,000 UK Biobank Subjects. Biol. Psychiatry 82, 103–110. 10.1016/j.biopsych.2016.08.014. [DOI] [PubMed] [Google Scholar]
- Kendall KM, Bracher-Smith M, Fitzpatrick H, Lynham A, Rees E, Escott-Price V, Owen MJ, O’Donovan MC, Walters JTR, and Kirov G (2019). Cognitive performance and functional outcomes of carriers of pathogenic copy number variants: analysis of the UK Biobank. Br. J. Psychiatry 214, 297–304. 10.1192/bjp.2018.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerpedjiev P, Abdennur N, Lekschas F, McCallum C, Dinkla K, Strobelt H, Luber JM, Ouellette SB, Azhir A, Kumar N, et al. (2018). HiGlass: web-based visual exploration and analysis of genome interaction maps. Genome Biol. 19. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, et al. (2008). Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet 40, 1253–1260. 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotaki D, Osato N, Nishiyama A, Yamamoto M, Ban T, Sato H, Nakabayashi J, Umehara M, Miyake N, Matsumoto N, et al. (2013). Essential role of the IRF8-KLF4 transcription factor cascade in murine monocyte differentiation. Blood 121, 1839–1849. 10.1182/blood-2012-06-437863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, t Hoen PAC, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. (2013). Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, Glessner JT, Coe BP, Li J, Mohebnasab M, Chang X, Connolly J, Kao C, Wei Z, Bradfield J, et al. (2020). Rare copy number variants in over 100,000 European ancestry subjects reveal multiple disease associations. Nat. Commun 11, 255. 10.1038/s41467-019-13624-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebhaber SA (1990). Inactivation of human a-globin gene expression by a de novo deletion located upstream of the a-globin gene cluster. Proc Natl Acad Sci USA 5. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P-R, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, Chasman DI, Ridker PM, Neale BM, Berger B, et al. (2015). Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet 47, 284–290. 10.1038/ng.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P-R, Kichaev G, Gazal S, Schoech AP, and Price AL (2018a). Mixed-model association for biobank-scale datasets. Nat. Genet 50, 906–908. 10.1038/s41588-018-0144-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P-R, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF, Birmann BM, Talkowski ME, Bakhoum SF, McCarroll SA, et al. (2018b). Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 559, 350–355. 10.1038/s41586-018-0321-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P-R, Genovese G, and McCarroll SA (2020). Monogenic and polygenic inheritance become instruments for clonal selection. Nature 584, 136–141. 10.1038/s41586-020-2430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macé A, Tuke MA, Deelen P, Kristiansson K, Mattsson H, Nõukas M, Sapkota Y, Schick U, Porcu E, Rüeger S, et al. (2017). CNV-association meta-analysis in 191,161 European adults reveals new loci associated with anthropometric traits. Nat. Commun 8, 744. 10.1038/s41467-017-00556-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR, Fine RS, Lu Y, Schurmann C, Highland HM, et al. (2017). Rare and low-frequency coding variants alter human adult height. Nature 542, 186–190. 10.1038/nature21039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, Antaki D, Shetty A, Holmans PA, Pinto D, et al. (2017). Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet 49, 27–35. 10.1038/ng.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford J, Park D, Zheng Z, Ko A, Ala-Korpela M, Laakso M, Pajukanta P, Yang J, Witte J, and Zaitlen N (2020). Efficient Estimation and Applications of Cross-Validated Genetic Predictions to Polygenic Risk Scores and Linear Mixed Models. J. Comput. Biol 27, 599–612. 10.1089/cmb.2019.0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer SC, Keller MD, Woods BA, LaFave LM, Bastian L, Kleppe M, Bhagwat N, Marubayashi S, and Levine RL (2014). Genetic studies reveal an unexpected negative regulatory role for Jak2 in thrombopoiesis. Blood 124, 2280–2284. 10.1182/blood-2014-03-560441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamel RE, Handsaker RE, Sherman MA, Barton AR, Zheng Y, McCarroll SA, and Loh P-R (2021). Protein-coding repeat polymorphisms strongly shape diverse human phenotypes. Science 373, 1499–1505. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai A, Hirata M, Kamatani Y, Muto K, Matsuda K, Kiyohara Y, Ninomiya T, Tamakoshi A, Yamagata Z, Mushiroda T, et al. (2017). Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol 27, S2–S8. 10.1016/j.je.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseri A, Liu X, Tang K, Zhang S, and Zhi D (2019). RaPID: ultra-fast, powerful, and accurate detection of segments identical by descent (IBD) in biobank-scale cohorts. Genome Biol. 20, 143. 10.1186/s13059-019-1754-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D, Bracher-Smith M, Kendall KM, Rees E, Einon M, Escott-Price V, Owen MJ, O’Donovan MC, and Kirov G (2018). Effects of pathogenic CNVs on physical traits in participants of the UK Biobank. BMC Genomics 19, 867. 10.1186/s12864-018-5292-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamara PF (2014). Population Genetics of Identity By Descent. Columbia University. [Google Scholar]
- Pedersen BS, and Quinlan AR (2018). Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics 34, 867–868. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw CA, Belmont J, et al. (2006). High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 16, 1136–1148. 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel FB, and Weatherall DJ (2014). The α-Thalassemias. N. Engl. J. Med 371, 1908–1916. 10.1056/NEJMra1404415. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn A, Machol I, Omer AD, Lander ES, et al. (2014). A three-dimensional map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, and Korbel JO (2012). DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339. 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. (2011). Multiple Recurrent De Novo CNVs, Including Duplications of the 7q11.23 Williams Syndrome Region, Are Strongly Associated with Autism. Neuron 70, 863–885. 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S, Hommel A, Gawlik V, Augustin R, Junicke N, Florian S, Richter M, Walther DJ, Montag D, Joost H-G, et al. (2009). Essential role of glucose transporter GLUT3 for post-implantation embryonic development. J. Endocrinol 200, 23–33. 10.1677/JOE-08-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönheit J, Kuhl C, Gebhardt ML, Klett FF, Riemke P, Scheller M, Huang G, Naumann R, Leutz A, Stocking C, et al. (2013). PU.1 Level-Directed Chromatin Structure Remodeling at the Irf8 Gene Drives Dendritic Cell Commitment. Cell Rep. 3, 1617–1628. 10.1016/j.celrep.2013.04.007. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. (2007). Strong Association of De Novo Copy Number Mutations with Autism. Science 316, 445–449. 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnott-Armstrong N, Tanigawa Y, Amar D, Mars N, Benner C, Aguirre M, Venkataraman GR, Wainberg M, Ollila HM, Kiiskinen T, et al. (2021). Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat. Genet 53, 185–194. 10.1038/s41588-020-00757-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofer T (2017). BinomiRare: A robust test of the association of a rare variant with a disease for pooled analysis and meta-analysis, with application to the HCHS/SOL: Sofer. Genet. Epidemiol 41, 388–395. 10.1002/gepi.22044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, et al. (2015). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szustakowski JD, Balasubramanian S, Kvikstad E, Khalid S, Bronson PG, Sasson A, Wong E, Liu D, Wade Davis J, Haefliger C, et al. (2021). Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nat. Genet 53, 942–948. 10.1038/s41588-021-00885-0. [DOI] [PubMed] [Google Scholar]
- Taher AT, Musallam KM, and Cappellini MD (2021). β-Thalassemias. N. Engl. J. Med 384, 727–743. 10.1056/NEJMra2021838. [DOI] [PubMed] [Google Scholar]
- Tassano E, Buttgereit J, Bader M, Lerone M, Divizia MT, Bocciardi R, Napoli F, Pala G, and Gimelli G (2013). Genotype-Phenotype Correlation of 2q37 Deletions Including NPPC Gene Associated with Skeletal Malformations. PLOS ONE 8, 7. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao C, Suzuki A, Momozawa Y, Akiyama M, Ishigaki K, Yamamoto K, Matsuda K, Murakami Y, McCarroll SA, Kubo M, et al. (2020). Chromosomal alterations among age-related haematopoietic clones in Japan. Nature 584, 130–135. 10.1038/s41586-020-2426-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. (2012). The accessible chromatin landscape of the human genome. Nature 489, 75–82. 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulirsch JC, Lareau CA, Bao EL, Ludwig LS, Guo MH, Benner C, Satpathy AT, Kartha VK, Salem RM, Hirschhorn JN, et al. (2019). Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet 51, 683–693. 10.1038/s41588-019-0362-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SFA, Hakonarson H, and Bucan M (2007). PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 17, 1665–1674. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Ma ESK, Chan AYY, Prior J, Erber WN, Chan LC, Chui DHK, and Chong SS (2003). Single-Tube Multiplex-PCR Screen for Anti-3.7 and Anti-4.2 α-Globin Gene Triplications. Clin. Chem 49, 1679–1682. 10.1373/49.10.1679. [DOI] [PubMed] [Google Scholar]
- Wheeler E, Huang N, Bochukova EG, Keogh JM, Lindsay S, Garg S, Henning E, Blackburn H, Loos RJF, Wareham NJ, et al. (2013). Genome-wide SNP and CNV analysis identifies common and low-frequency variants associated with severe early-onset obesity. Nat. Genet 45, 513–517. 10.1038/ng.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie M, Lamb J, Harris PC, Finneyt RD, and Higgs DR (1990). A truncated human chromosome 16 associated with a thalassaemia is stabilized by addition of telomeric repeat (TTAGGG)n. 346, 4. . [DOI] [PubMed] [Google Scholar]
- Zhou Y, Browning SR, and Browning BL (2020). A Fast and Simple Method for Detecting Identity-by-Descent Segments in Large-Scale Data. Am. J. Hum. Genet 106, 426–437. 10.1016/j.ajhg.2020.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler GC, Almos P, McNeill RV, Jansch C, and Lesch K (2020). Cellular effects and clinical implications of SLC2A3 copy number variation. J. Cell. Physiol 235, 9021–9036. 10.1002/jcp.29753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Validation of HI-CNV; related to Figure 1 and STAR Methods. A Validation rate of HI-CNV calls assessed using WGS data for 500 individuals. The left and right plots indicate proportions (left) and numbers (right) of SNP-array-based HI-CNV calls that exhibited the correct WGS read-depth direction (below the mean for deletions and above the mean for duplications) as a function of various CNV properties: CNV length (top), and call confidence (middle, based on the Bayes factor estimated by the hidden Markov model; “log10BF”), and GC content (bottom). These sign-tests result in an estimated 90.6% and 86.9% validation rate for deletions and duplications, respectively, and 89.1% overall. B Average number of HI-CNV calls and proportion of HI-CNV calls with correct read-depth signal (sign test) assessed using WGS data for 100 BioBank Japan participants. C Validation rate of fine-mapped HI-CNV calls assessed using WGS data. The proportion and number of calls with the correct WGS read-depth direction (below the mean for deletions and above the mean for duplications) are shown for: (i) 250 randomly selected fine-mapped CNV calls (“Fine-mapped”; STAR Methods); and (ii) all HI-CNV calls made in 500 randomly selected individuals with WGS available (“All”; STAR Methods). These sign tests resulted in an estimated 93.7% and 95.7% validation rate for fine-mapped deletions and duplications, respectively (as compared to 90.6% and 86.9% for all events), and 94.4% overall (as compared to 89.1% for all events). Numerical results are available in Table S1.
Supplemental Figure 2: HI-CNV vs PennCNV; related to Figure 1 and STAR Methods. A Detection sensitivity (recall) of SNP-array based methods on a benchmark set of CNVs called from WGS data. We assessed recall of low-frequency (MAF≤5%) CNVs called by Delly in 43 individuals with whole-genome-sequence data available. Sensitivity increased with CNV size and probe overlap (left-to-right columns) and for gene-overlapping CNVs (bottom vs. top row). HI-CNV0 denotes analysis without incorporating information from haplotype neighbors (but still using our SNP-specific probabilistic models of genotyping intensities). Numerical results are available in Table S1. B Sensitivity of PennCNV to detect CNVs contributing to likely-causal phenotype associations identified by HI-CNV analysis. For each of the 269 likely-causal CNV-phenotype associations found via analysis of our HI-CNV callset, the number of carriers found using PennCNV versus HI-CNV is plotted. The four panels stratify the data by CNV size (median among the CNV calls that were considered in each association test). The dashed line shows the y=x line.
Supplemental Figure 3: Signal intensity plots for CNV-phenotype associations; related to Figure 5 and 6. Denoised LRR is plotted for four randomly selected CNV carriers at each locus shown in Figures 5 and 6. Dots indicating LRR at probes within a CNV call are colored in red (deletion called) or blue (duplication called).
Supplemental Figure 4: Investigation into various allelic series; related to Figure 5 and STAR Methods. A LRR signal for individuals with SLC2A3 deletion calls shows no evidence of homozygous deletions. Distributions of mean LRR (denoised and rescaled) across 60 probes spanned by reciprocal SLC2A3 DEL/DUP CNVs are shown for carriers of SLC2A3 deletion calls (3,768 individuals), SLC2A3 duplication calls (16,821 individuals), and 1,000 randomly selected controls. The mean LRR histogram for duplications has a heavy right tail suggesting the presence of individuals with CN=4, whereas the mean LRR histogram for deletions shows no evidence of homozygous deletions (binomial P =0.0009 assuming Hardy-Weinberg equilibrium). B,C Deletions and duplications of DIS3L2 exon 9 and associations with height. B Height associations (top) and effect sizes (bottom) for variants at the DIS3L2 locus. The reciprocal deletion and duplication spanning DIS3L2 exon 9 are shown in red and blue, respectively, and SNPs are shown in grey. SNP effect sizes are plotted only for genome-wide significant SNPs (P < 5 × 10−8). C Carriers of the DIS3L2 exon 9 deletion and duplication could be more accurately identified by analyzing average SNP-array probe intensity together with WES read depth within the event; carriers of deletions and duplications called in this way are colored in red and blue, respectively. The distribution of height (in units of standard deviations) is shown separately for carriers of the DIS3L2 exon 9 deletion, the duplication, and noncarriers. Mean and 95% CI are shown below the histograms. D A common-SNP association with platelet distribution width appears to be mediated by LRCH1 expression. Top: Manhattan plot of associations between SNPs and platelet distribution width (linear regression P -values computed using UK Biobank participants of self-reported European ancestry). Bottom: LRCH1 eQTL P-values from GTEx for the tissues with the top three signals. The red points toward the right of each plot correspond to the retroposed pseudogene insertion (directly genotyped by Affx-52351109 in the top plot) and the tag SNP rs145057384 in the bottom plots; the insertion has ~4-fold larger effect sizes than the top SNPs but has weaker associations because it is much rarer.
Supplemental Figure 5: α-globin locus; related to Figure 6 and STAR Methods. A α-globin locus CNVs at the α-globin locus and their effects on blood traits. For carriers of each category of α-globin CNVs shown in Figure 6A, we plot the average mean corpuscular hemoglobin (MCH; both raw and units of standard deviation; s.d.), red blood cell count (RBC; both raw and units of s.d.), mean corpuscular volume (MCV; both raw and units of s.d.), and the Mentzer index (MCV/RBC). Vertical lines denote population averages. B Schematic of non-allelic homologous recombination (NAHR) at the α-globin locus. The HBA2 and HBA1 genes lie within ~4 kb regions containing three distinct regions of high homology (X, Y, and Z boxes; Fig. 1 of (Wang et al., 2003)). The spacing between the X and Y boxes differs in the sequence left of HBA2 vs. HBA1, such that NAHR within the X boxes creates 4.2-kb DEL/DUP events (−α4.2 and αααanti 4.2) while NAHR within the Z boxes creates 3.7-kb DEL/DUP events (−α3.7 and αααanti 3.7). The UK Biobank SNP-array contained a probe in unique sequence at 16:221057 between X2 and Y2 (pink star). Because intensity at this probe is affected by 4.2-type events but unaffected by 3.7-type events, we concluded that duplication calls starting at the probe 16:221057 and extending through the three probes in HBA exons were probably 4.2-type events that involved full deletion or duplication of HBA2. C Mean LRR and normalized WES read depth for CNVs at the α-globin locus. Mean LRR across four probes (three within HBA1 and 16:221057) and mean normalized read depth for exome-sequencing reads mapping uniquely to HBA2 or HBA1 are plotted for each category of α−globin CNV we considered. Error bars, 95% CIs (computed as mean ±1.96× s.e.m. across carriers). WES read depth restricted to reads aligning uniquely to HBA2 and HBA1 (i.e., with nonzero mapping quality) was computed using mosdepth (Pedersen and Quinlan, 2018) and normalized by exome-wide coverage.
Supplemental Figure 6: Behavior of HI-CNV and SNP-array probe intensities at SULT1A1: a multiallelic, highly polymorphic CNV region; related to STAR Methods. A Estimated SULT1A1 copy number based on exome-sequencing read depth followed by phasing and imputation. B Comparison of sequencing-derived estimates to copy numbers estimated by HI-CNV (which ranged from 0 to 4; HI-CNV can only call up to a single-copy increase per haplotype). C Probe intensity cluster plots for a probe within SULT1A1. The probe examined was rs150459557 (hg19 chr16:28,617,472); intensity values are shown for 10 randomly selected individuals for each WES-derived copy-number state.
Supplemental Figure 7: Steps of SNP-specific genotype cluster prediction for deletion and duplication genotypes; related to STAR Methods. A Assignment of reference CNV probe intensity data to CN=1 genotype clusters (A, B) and CN=3 genotype clusters (AAA, AAB, ABB, BBB) by dividing the total vs. relative intensity plane into zones based on CN=2 cluster parameters. B Estimation of bivariate normal parameters corresponding to cluster assignments (for clusters with 10 or more assigned data points). C Prediction of CN=1 and CN=3 genotype clusters for other (“query”) SNPs. The basic idea was to perform prediction independently for left-half clusters (A, AAA, AAB) and right-half clusters (B, ABB, BBB) by identifying (for each half) reference SNPs with best-matching CN=2 clusters in the relevant half. The axes of each plot are (x-axis, analogous to BAF), and denoised LRR (y-axis), scaled to [−127, 127].
Table S1: Benchmarking and validation of HI-CNV; related to STAR Methods.
Table S2: Quantitative traits analyzed; related to Figure 3 and STAR Methods.
Table S3: Likely-causal CNV-phenotype associations; related to Figure 3 and STAR Methods.
Table S4: Annotation of CNV loci; related to Figure 3 and STAR Methods.
Table S5: Follow-up on allelic series; related to Figure 5, Figure 6, and STAR Methods.
Table S6: Binary associations; related to STAR Methods.
Table S7: Deletion and duplication burden; related to Figure 7 and STAR Methods.
Data Availability Statement
Summary statistics have been deposited at Zenodo and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table