

Abstract
Mucus forms the first line of defence of the intestinal mucosa barrier, and mucin is its core component. Glutamine is a vital energy substance for goblet cells; it can promote mucus synthesis and alleviate damage to the intestinal mucus barrier after burn injury, but its mechanism is not fully understood. This study focused on the molecular mechanisms underlying the effects of glutamine on the synthesis and modification of mucin 2 (MUC2) by using animal and cellular models of burn sepsis. We found that anterior gradient-2 (AGR2) plays a key role in the posttranslational modification of MUC2. Oxidative stress induced by burn sepsis enhanced the S-glutathionylation of AGR2, interfered with the processing and modification of MUC2 precursors by AGR2 and blocked the synthesis of mature MUC2. Further studies revealed that NADPH, catalysed by glucose-6-phosphate dehydrogenase (G6PD), is a key molecule in inhibiting oxidative stress and regulating AGR2 activity. Glutamine promotes O-linked N-acetylglucosamine (O-GlcNAc) modification of G6PD via the hexosamine pathway, which facilitates G6PD homodimer formation and increases NADPH synthesis, thereby inhibiting AGR2 S-glutathionylation and promoting MUC2 maturation, ultimately reducing damage to the intestinal mucus barrier after burn sepsis. Overall, we have demonstrated that the central mechanisms of glutamine in promoting MUC2 maturation and maintaining the intestinal mucus barrier are the enhancement of G6PD glycosylation and inhibition of AGR2 S-glutathionylation.
Keywords: Glutamine, MUC2, AGR2, G6PD, Intestinal mucosa barrier, S-glutathionylation
Graphical abstract
Highlights
-
•
Oxidative stress triggered by burn sepsis can enhance AGR2 S-glutathionylation and decrease its enzyme activity.
-
•
NADPH is a core molecule in maintaining redox homeostasis in AGR2; G6PD is the rate-limiting enzyme of NADPH synthesis.
-
•
Glutamine administration can inhibit AGR2 S-glutathionylation by promoting G6PD glycosylation.
1. Introduction
Patients with severe burns have extensive skin destruction combined with intense postinjury stress, tissue ischaemia/hypoxia, hypermetabolism and immune dysfunction, which puts them at high risk of developing sepsis [1,2]. Currently, multiple organ dysfunction syndrome (MODS) caused by sepsis has become the leading factor in burn lethality, accounting for 50–60% of total mortality [[3], [4], [5]]. Therefore, burn sepsis has become a common concern in the fields of burns and critical care medicine. In recent years, the role of an impaired intestinal mucosal barrier in the development and progression of sepsis has attracted increasing attention, and maintenance of intestinal function has become an important aspect in sepsis therapy [[6], [7], [8], [9]]. The intestinal mucosal barrier is a core tissue structure for maintaining the stability of the body's internal environment. It consists of mechanical, immune, biological and mucus barriers, of which the mucus barrier is the first line of defence against the invasion of pathogenic microorganisms [10,11]. The intestinal mucus plays an important role in maintaining the stability of the internal environment by coordinating with the other three barriers [[12], [13], [14]].
Intestinal mucus is a multicomponent gel complex whose core component is mucin [15]. There are several subtypes of mucins in the intestine, with mucin2 (MUC2) being the major subtype in the colon [14]. The posttranslational MUC2 precursor is first folded and glycosylated in the endoplasmic reticulum (ER) to form mature MUC2, which is subsequently modified with salivary acidification in the Golgi apparatus to form large gel-like mucus that is secreted into the intestinal lumen [[15], [16], [17], [18]]. Thus, the processing of mucin precursors into mature mucins in the ER is the first and critical step in mucus synthesis. Anterior gradient-2 (AGR2) in the ER is regarded as a key enzyme for the synthesis and modification of mucins [19,20]. It is a member of the protein disulfide isomerase (PDI) family with redox and molecular chaperone functions that process and modify immature mucins into mature mucins [21,22]. The activity of AGR2 depends on its redox balance, and oxidative stress can lead to a decrease in AGR2 enzyme activity, hence impeding the synthesis and modification of mucins [23].
In pathological states such as burns, trauma, and sepsis, large amounts of reactive oxygen species (ROS) are generated due to tissue ischaemia‒reperfusion and excessive inflammatory responses [24,25]. It will continuously consume reduced glutathione (GSH), leading to an increase in oxidized glutathione (GSSG), triggering cellular redox imbalance [25,26]. Excessive ROS not only leads to cellular damage directly, but also triggers ER stress (ERS) due the imbalance of the ratio of GSH/GSSG, leading to impaired mucin synthesis and disruption of the intestinal mucus barrier [26,27]. Further research has found that a decrease in the intracellular GSH/GSSG ratio when exposed to ROS can lead to glutathionylation modification of a variety of sulfhydryl-containing proteins, which is called protein S-glutathionylation [27,28]. In mammalian cells, S-glutathionylation functions as a protective mechanism against irreversible oxidation of the sulfhydryl group of cysteine [29]. However, it inevitably causes changes in the charge and spatial structure of the protein, interferes with protein function, and leads to enhanced or suppressed activity [30,31]. More evidence shows that redox regulation by S-glutathionylation contributes to physiological processes, and aberrant S-glutathionylation is associated with various diseases, from diabetes, cancer, neurodegeneration, and pulmonary fibrosis to liver and cardiovascular diseases [27,[31], [32], [33]]. Whether reduced AGR2 activity and impaired mucin synthesis due to oxidative stress are associated with enhanced S-glutathionylation of AGR2 is unclear, and is one of the focuses of this study.
The GSH and GSSG equilibrium constitutes the most critical cellular redox system, and its ratio affects the glutathionylation status of a protein [30]. Changes in GSH/GSSG ratios could influence many proteins by causing glutathionylation modification at specific protein cysteinyl residues [27,34]. The mutual transformation of GSH and GSSG depends on the involvement of reduced nicotinamide adenine dinucleotide phosphate (NADPH), which is considered a key reducing transmitter in the body and can provide electron donor or acceptor for glutathione conversion through the NADPH/NADP + cycle [27,35]. There are several NADPH synthesis pathways in vivo. In addition to the predominant pentose glucose phosphate pathway (PPP), other generation pathways of NADPH include reactions catalysed by isocitrate dehydrogenase 1 (IDH1), malic enzyme 1 (ME1) and methylene tetrahydrofolate dehydrogenase 1 (MTHFD1). Of these, the PPP contributes over 60% to the synthesis of NADPH [36,37]. Glucose-6-phosphate dehydrogenase (G6PD) is the rate-limiting enzyme in the PPP. It catalyses the conversion of glucose-6-phosphate to ribose 5-phosphate and generates bimolecular NADPH in a two-step oxidation reaction [38,39]. Therefore, research on G6PD may contribute to elucidating the deeper mechanisms of mucin synthesis and modification.
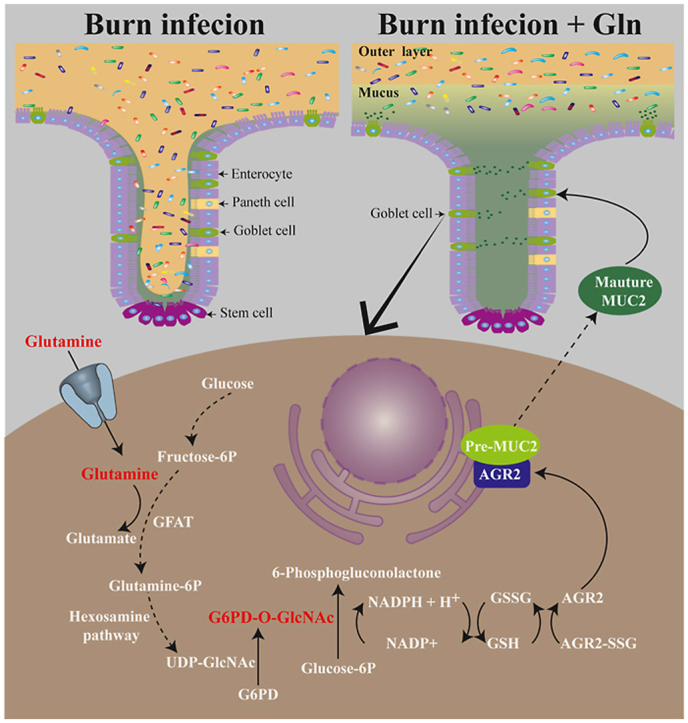
Glutamine (Gln) is the most important energy substance for intestinal epithelial cells, and it plays a critical role in reducing intestinal mucosa damage after burns [[40], [41], [42]]. Our previous studies have shown that Gln administration apparently reduces postburn intestinal goblet cell damage, promotes mucus synthesis and secretion, and maintains the intestinal mucus barrier. The mechanism is related to promoting NADPH synthesis and increasing GSH content [26,43], but past research has not considered G6PD. It has been reported in the literature that Gln promotes G6PD synthesis through the Nrf2 signaling pathway, but whether it affects enzymatic activity has not been determined [44]. We hypothesized that Gln might potentially have a regulatory effect on G6PD activity. To test this hypothesis, in the current study, we used a burn sepsis mouse model and a cellular model of LPS-stimulated HT29 Cl.16E cells to explore the mechanism of Gln maintenance of the intestinal mucus barrier. Our experiments revealed that Gln can enhance the glycosylation of G6PD, induce homodimer formation to enhance enzyme activity, and promote NADPH synthesis, thereby reducing burn sepsis-induced oxidative stress, maintaining AGR2 redox homeostasis, promoting the synthesis of mature MUC2, and maintaining the intestinal mucus barrier.
2. Materials and methods
2.1. Animals
Healthy male BALB/c mice (22–25 g, 8–10 weeks old) were purchased from the Laboratory Animal Center of the Third Military Medical University. The animals were housed in individually ventilated cages under specific pathogen-free conditions with a 12-h light/dark cycle and free access to food and water. The mice were fed standardized diets for one week before the experiment. All procedures were in agreement with institutional guidelines and regulations, and the experimental protocols were approved before implementation by the Laboratory Animal Welfare and Ethics Committee of the Third Military Medical University (Permit Number: AMUWEC20210636).
2.2. Experimental models
In this study, we used the classic model of burn sepsis, i.e., the burn subscab bacterial injection model, which is also referred to as the burn invasive wound infection model (abbreviated “burn infection model”). The specific groups and operations were as follows: sixty mice were randomly divided into three groups, the sham group (n = 20), burn infection group (BI group, n = 20) and burn infection with Gln supplementation group (BI + Gln group, n = 20), according to descriptions of previous burn sepsis models with slight modifications [6,[45], [46], [47]]. Under general anaesthesia (i.p. injection of 80 mg/kg ketamine and 10 mg/kg xylazine), the dorsal surface of each mouse was shaved. The anaesthetized mice were placed in a template exposing ∼20% of the total body surface area (TBSA) as calculated by the Meeh formula [48]. The shaved dorsum was exposed to water at 90 ± 0.2 °C for 8 s to induce scalding of approximately 20% of the overall surface area, after which the mouse was intraperitoneally injected with 1.5 mL kg−1%TBSA−1 lactated Ringer's solution for fluid resuscitation and 100 μL of buprenorphine (0.3 mg/mL) for analgesia. The burned mice were housed individually in sterile cages and provided sterile water and food. Sham mice underwent the same experimental procedure, except that the mice were exposed to 37 °C water. For burn wound infection, 100 μL of 10 mM MgSO4 containing 5 × 105 colony-forming units (cfu) of Pseudomonas aeruginosa clinical isolate PA14 was intradermally injected at the burn eschar site immediately after the burn insult. An equal injection of 100 μL of MgSO4 was used for the sham mice. Then, gavage was started twice daily for 7 days with i) 0.3 ml saline (sham group); ii) 0.3 ml saline (BI group); or iii) 1 g/kg body weight Gln in 0.3 ml saline (BI + Gln group). After burn infection for 1, 3, 5, and 7 days, the mice were euthanized by cervical dislocation. Then, the abdomen was opened aseptically through a midline laparotomy, and the distal colon was aseptically harvested. The experimental design is shown in Fig. S1.
2.3. HT-29 Cl.16E cell culture
HT-29 Cl.16E cells were generated by culturing the human colonic cancer cell line HT29 (ATCC, HTB-38) in the presence of 5 mmol/L sodium butyrate for 9 days and then subculturing the cells into medium containing sodium butyrate for an additional 14 days. The cells were then cultured in standard medium without sodium butyrate. During this time, the cells differentiated into a goblet-like phenotype with the ability to produce large amounts of mucus [49]. HT-29 Cl.16E cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) heat-inactivated foetal bovine serum, penicillin/streptomycin (100 U/mL/100 mg/mL), and 1 mmol/L sodium pyruvate at 37 °C in 5% CO2.
2.4. Stable knockout (KO) and overexpressing cell lines
HT-29 CL.16E cells with AGR2 KO and G6PD KO were obtained by CRISPR/Cas9 technology. Briefly, the guide RNA oligonucleotide (5′- ATGTCTGAGTCCAGATGAGT-3′) that targets exon 3 of the human AGR2 gene and (5′- ATATGTGTGTATCCG ACTGA-3′) that targets exon 2 of the human G6PD gene were designed using Tools for Guide Design (zlab.bio/guide-design-resources). The control sequence was 5′- CGCTTCCGCGGCCCGTTCAA-3'. Both sequences were cloned into a LentiCRISPR vector (Genechem Technology) using BsmBI restriction cloning.
HT-29 CL.16E cells (1 × 105 cells) were transfected with pLenti- U6-AGR2-EF1a-Cas9-FLAG-P2A-puromycin or pLenti-U6-G6PD-EF1a-Cas9-FLAG-P2A-puromycin. After 3 days, the cells were exposed to puromycin (5 μg/ml), and pools of resistant cells were sorted out and seeded as individual colonies in 96-well plates. Western blotting was used to detect AGR2 or G6PD expression in KOAGR2 and KOG6PD cells. Two clones with undetectable AGR2 or G6PD expression were selected separately for further experiments.
Rescue of the wild-type (WT G6PD) and mutant (S84V G6PD) genes in KO cells was successful only when cDNAs, where gRNAs bind, were modified by three nucleotides, while the amino acid sequences remained unchanged. The cDNAs of the target genes were cloned into the pLenti-Ubi-MCS-3FLAG-SV40-mCherry lentivector (Genechem Technology). HT-29 CL.16E cells were infected with the lentiviruses and selected for monoclonal cells with red fluorescence.
2.5. Histological scoring and morphometric analysis of mucin
Samples of distal colon were fixed in tubes with 5 mL methanol-Carnoy's fixative. The tissue was fixed for a minimum of 3 h at room temperature and processed routinely for embedding in paraffin. Tissue sections (5 μm) were stained with haematoxylin and eosin (H&E) or with Alcian blue (AB)/Periodic Acid–Schiff (PAS). The histological examination was performed in a blinded fashion using a scoring system previously validated and described [50,51] as follows: crypt architecture, 0 (normal) to 5 (crypt abscesses); tissue damage, 0 (none) to 3 (extensive mucosal damage); goblet cell depletion, 0 (normal) to 3 (>50% depletion); and inflammatory cell infiltration, 0 (occasional infiltration) to 3 (transmural infiltration). Crypt length (0–4) was measured using CellSens software (Olympus). This scoring system determined a combined score of histological colitis (maximum of 18) via assessment of the abovementioned parameters to reflect tissue damage and colitis severity.
Goblet cells were counted in the distal colon from the basal half of the crypts. Quantification of goblet cells containing mature mucins was performed in AB- and PAS-stained paraffin sections of intestine. Goblet cells stained with light blue and magenta/purple were counted as AB+ and PAS+, respectively. Goblet cells fitting neither category were counted as AB-PAS+. The morphometric analysis was performed by ImagePro Plus 7.0 software.
2.6. Mucin isolation and detection
The colon was flushed slowly 5 times with 3 M urea (2 ml each flush). The insoluble mucus was extracted from flushed colon or HT29 CL.16E cells by using guanidium chloride extraction buffer (6 M GuCl, 0.1 M Tris pH 8.0, 1 mM EDTA). Following extraction, the mucus was analysed using composite AgPAGE. The gel consisted of a gradient of 0.5–1.0% agarose, 0–10% glycerol and 0–6% acrylamide. Before gel electrophoresis, mucus samples were reduced with 1 mM DTT for 3 h at 37 °C to break up the disulfide bonds. After reduction, 0.125 M iodoacetamide was added to the mucus sample, which was incubated for 30 min at room temperature in the dark so that iodoacetamide could carboxymethylate the mucus to prevent reformation of the disulfide bonds. The isolated samples in GuCl were then dialyzed in 3–6 M urea using Tube™ dialyzers (with a molecular weight cut-off of 14 kDa or below). The protein samples were immunoblotted using a MUC2-N antibody (Abcam, ab90007) to show the amount of mature MUC2 in colonic tissue and HT29 CL.16E cells.
HT29 Cl.16E cells or colon tissue were lysed with lysis buffer (1 mmol/L EDTA, 10 mmol/L Tris-HCl, 10% SDS, 5 mmol/L NaCl, 20% Triton X-100, phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail; Sigma‒Aldrich). The samples were separated by composite AgPAGE, Western blotted onto a nitrocellulose membrane, and probed with two MUC2 precursor antibodies (MUC2-VNTR, Invitrogen, MA5-12345; MUC2-VNTR, Novus, NBP2-25221), which detect sequences in the tandem repeat of MUC2, to detect immature MUC2.
2.7. Western blotting and coimmunoprecipitation
For Western blot analysis, proteins were electrophoresed in 10% sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene fluoride membranes using standard electroblotting procedures. The membranes were blocked with 5% BSA in TBST (TBS; 50 mM Tris-Cl, 150 mM NaCl, pH 7.5) containing 0.1% Tween 20 and immunolabelled overnight at 4 °C with primary antibodies against G6PD (Abcam, ab993), CHOP (Abcam, ab11419), AGR2 (Abcam, ab76473), O-GlcNAC (Abcam, ab2739), glutathione (Virogen, 101-A), GRP78 (Cell Signaling, 3177S), or GAPDH (Aksomics, KC-5G4). The appropriate HRP-conjugated secondary antibodies (Cell Signaling, 7074S & 7076S) were applied, and immunolabeling was detected by an enhanced chemiluminescence kit (Thermo Fisher Scientific, A38556).
For coimmunoprecipitation experiments, tissue or cell lysates were incubated with antibodies against AGR2 (Abcam, ab76473), MUC2-VNTR (Invitrogen, MA5-12345), or control immunoglobulin (Proteintech, B900620 & B900610) overnight. The antibody-bound proteins were precipitated with Protein A/G magnetic beads (Thermo Scientific Pierce, 88805) and washed three times with lysis buffer. The samples were prepared for further analysis by Western blotting.
2.8. Immunofluorescence staining
The cells were cultured on small round cover glasses, fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, blocked with goat serum, and treated with antibodies against MUC2-VNTR (Novus, NBP2-25221), MUC2-N (Abcam, ab90007), AGR2 (Abcam, ab76473) and glutathione (Virogen, 101-A) overnight at 4 °C. The samples were then incubated with fluorescently labelled secondary antibodies, which were anti-rabbit Alexa Fluor 594 (Invitrogen, A-11012) and anti-mouse Alexa Fluor 488 (Invitrogen, A-10680). Colocalization was assessed by confocal laser scanning microscopy (Zeiss LSM 880).
The same method was employed for immunofluorescence staining of colon tissue samples. Sections were deparaffinized and then subjected to antigen repair with 10 μmol/L citrate buffer at pH 6.0. After serum closure, sections were incubated with a MUC2-C antibody (Abcam, ab272692) and an AGR2 antibody (Santa Cruz, sc-101211) to observe MUC2 and AGR2 colocalization or with a MUC2-VNTR antibody (Invitrogen, MA5-12345) to observe immature MUC2 expression after burn injury. The cells were washed and incubated with anti-rabbit Alexa Fluor 488 (Invitrogen, A-32731), anti-mouse Alexa Fluor 594 (Invitrogen, A-11005) and anti-mouse Alexa Fluor 488 antibodies at room temperature for 1 h. The cell nuclei were stained with DAPI, and immunofluorescence-stained images were visualized and quantified under a confocal laser scanning microscope (Zeiss LSM 880) and analysed using ZEN Blue software (Zeiss).
2.9. Fluorescence in situ hybridization
Fluorescence in situ hybridization was performed as previously described. The tissue sections were incubated with 250 μg of Cy3–conjugated universal bacterial probe EUB338 (5′-GCTGCCTCCCGTAGGAGT-3′, bp 337–354 within bacteria EU622773, Invitrogen) in 50 μl of hybridization buffer (20 mM Tris·HCl [pH 7.4], 0.9 M NaCl, 0.1% SDS) at 50 °C overnight. The slices were rinsed in washing buffer (20 mM Tris HCl [pH 7.4], 0.9 M NaCl), washed at 50 °C for 20 min, and re-stained with DAPI. Coimmunostaining with anti-MUC2C (Abcam, ab272692) was performed at 4 °C without antigen retrieval, and the sections were mounted in ProLong Gold antifade (Invitrogen).
2.10. Determination of ROS levels and related redox couples
Cellular ROS and cytosolic superoxide were measured by H2DCFDA (Sigma‒Aldrich,D6883) and DHE (Sigma‒Aldrich, D7008), respectively. In brief, H2DCFDA or DHE was dissolved in DMSO to obtain a 10 mM stock solution and further diluted before use. The adherent cells were incubated with PBS containing 5 μM staining solution at 37 °C in the dark for 30 min, harvested with 0.05% trypsin-EDTA solution, suspended in fresh medium, and immediately analysed with a flow cytometer (NovoCyte; 488 nm laser). The ROS-Glo™ H2O2 assay (Progema, G8820) was used to measure intracellular H2O2. In brief, HT29 CL.16E cells were grown adherent on opaque white 96-well plates. After different treatments, H2O2 substrate solution was added to bring the final volume to 100 μl in each well. The plate was incubated for 6 h in a 5% CO2 incubator at 37 °C. Then 100 μl of the ROS-Glo detection solution was added to each well, and the plate was further incubated for 30 min at room temperature. The luminescence was recorded using a Varioskan flash multimode reader (Thermo Fisher Scientific). H2O2 concentrations were calculated from the standard curve obtained by the luminescence value of H2O2 at different concentrations.
The NADPH/NADP + ratios in colon tissues and cultured cells were determined with commercial kits according to the manufacturer's protocols (Beyotime). Total glutathione and oxidized glutathione contents were measured by a GSSG/GSH Quantification Kit (Dojindo, G263) according to the manufacturer's instructions. The concentrations of GSH and GSSG were calculated using a standard curve. The results were expressed as nanomoles of GSH per mg of cellular proteins.
2.11. AGR2 redox state monitoring
To study AGR2 redox states in colon tissues and cultured cells, samples were prepared through protein precipitation using trichloroacetic acid (Macklin, T885181) and labelled with Protein-SHifter Plus in accordance with the manufacturer's instructions for the -SulfoBiotics- Protein Redox State Monitoring Kit Plus (Dojindo, SB12). After cell and colon tissue extracts were subjected to SDS-PAGE, the gels were exposed to UV light for 10 min on a transilluminator to remove the protein-shifter. The proteins in the gel were then electrotransferred to a polyvinylidene fluoride. The membrane was blocked with 5% nonfat dry milk and incubated with anti-AGR2 antibody (Abcam, ab76473) or anti-AGR2 antibody (Santa Cruz, sc-101211). Signals were obtained by using a horseradish peroxidase-linked secondary antibody and an enhanced chemiluminescence system, and the Western blot band intensities were computed.
2.12. Detection of S-glutathionylation of AGR2
To detect glutathione protein in colon tissue extracts, the EZ link Sulfo–NHS–Biotin (100 μL, 64 mM; Thermo Fisher Scientific, 21217) was added to 100 mM potassium phosphate buffer (pH 8.0) containing GSSG (100 μL, 32 mM). The mixture (200 μL) was derivatized for 1 h at room temperature. Untreated biotin was quenched by ammonium bicarbonate buffer (70 μL, 0.6 M). The colon tissue extract was incubated with freshly synthesized BioGSSG (5 mM) for 1 h at room temperature. Free BioGSSG and excess reagents were removed by Bio-Gel P10. The level of total biotin-GSS-protein conjugates in colon tissue was determined using nonreducing Western blot analysis with streptavidin-HRP. After the GSS-protein was pulled down with streptavidin-agarose, the glutathione level of AGR2 was analysed by Western blotting with an antibody against AGR2.
To detect GSS-AGR2 adducts in HT29 CL.16E cells, HT29 CL.16E cells were incubated with BioGEE (250 μM; Invitrogen, G36000) for 1.5 h and then stimulated with H2O2 or LPS. The cells were lysed with a protein extract containing N-ethyl maleimide (20 mM). The level of GSS-protein adducts in cells was determined using nonreducing Western blot analysis with streptavidin-HRP. AGR2 glutathione levels in cells were detected by the same method as in colon tissues.
For immunoprecipitation experiments, cells were lysed in NP-40 lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA) containing 20 mM N-ethylmaleimide (Sigma‒Aldrich, E1271) and protease inhibitor cocktail (Roche, 04693159001). The lysates were incubated overnight at 4 °C with anti-glutathione (Virogen, 101-A) antibody-conjugated SureBeads Protein A/G magnetic beads (Thermo Fisher Scientific, 88805). Coimmunoprecipitation (Co-IP) was performed on a magnetic rack (Bio-Rad). Immunoblotting analysis was performed with anti-AGR2 antibody (Abcam, ab76473) or anti-AGR2 antibody (Santa Cruz, sc-101211).
2.13. Analysis of G6PD glycosylation
Chemoenzymatic labeling and biotinylation of proteins in colon tissues or cell lysates were carried out as described previously [52]. In short, colon tissues and cell lysates were labelled according to the Click-IT™ O-GlcNAc Enzymatic Labeling System (Invitrogen, C33368). The samples were subsequently labelled with the alkyne-biotin dye per the Click-IT™ Biotin Protein Analysis Detection Kit protocol (Invitrogen, C33372). The biotinylated lysates were resolubilized in 1% SDS and quenched with an equal volume of neutralization buffer (6% NP-40, 100 mM Na2HPO4, 150 mM NaCl). The biotinylated proteins were then incubated with Streptavidin Agarose (Sigma‒Aldrich, 85881) at 4 °C overnight. The agarose beads were then washed three times with 1 ml of low-salt buffer (100 mM Na2HPO4, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate) and three times with 1 ml of high-salt buffer (100 mM Na2HPO4, 500 mM NaCl, 0.2% Triton X-100). After washing, the beads were boiled in elution buffer (50 mM Tris-HCl pH 6.8, 2.5% SDS, 100 mM DTT, 10% glycerol, 20 mM biotin) for 10 min. Western blot analysis was carried out with anti-G6PD antibody (Abcam, ab993). For immunoprecipitation experiments, cell lysates were incubated with Protein A/G beads coupled to anti-G6PD (Abcam, ab993) or anti-Flag antibody (Abcam, ab205606) overnight at 4 °C. Samples were prepared for further analysis by Western blotting. The ratio of glycosylated G6PD protein to total G6PD protein was taken as the level of glycosylation.
2.14. Determination of G6PD activity and the G6PD dimer
G6PD activity was measured using a G6PD Activity Assay Kit (Beyotime). Briefly, 1 × 106 cells or 20 mg tissues were harvested and lysed with G6PD extraction solution at 4 °C. The protein concentration was determined by a BCA protein quantification kit (Beyotime, P0010) and normalized. Fifty microlitres of protein sample and 50 μL of the G6PD test solution were added to each well of the 96-well plate. After incubating at 37 °C for 10 min in the dark, the OD (optical density) value was tested at 450 nm by a microplate reader.
DSS (Thermo Fisher Scientific, 21655) cross-linking was used to detect G6PD dimerization. Briefly, the culture medium in the cells was removed, and the cells were washed twice with ice-cold PBS. Then, the cells were scraped with PBS supplemented with an EDTA-free protease inhibitor mixture (Roche, 04693159001). Samples with the DSS cross-linker were incubated under rotation for 30 min at 37 °C. The reaction was quenched by the addition of 1 M Tris, pH 7.5, to a final concentration of 20 mM in the reaction mixture and incubated further for 15 min at RT. The cells were lysed in M-PER (Thermo Fisher Scientific, 78501) buffer and then centrifuged at 16,000×g for 10 min at 4 °C. The supernatant was saved for Western blot analysis.
2.15. Statistical analysis
Statistical analysis was performed using GraphPad Prism v.8.0.1 (GraphPad, La Jolla, CA). The Shapiro‒Wilk normality test was performed to determine the data distribution. Normally distributed data are reported as the mean ± standard deviation (SD), and nonnormally distributed data are reported as the median with interquartile range (IQR). The two groups were compared using an independent-samples t-test or Mann‒Whitney U test. One-way ANOVA with Bonferroni correction was performed for comparisons among multiple groups. P values < 0.05 were considered to indicate statistical significance. For all statistical analyses: *P < 0.05; **P < 0.01; ***P < 0.001.
3. Results
3.1. Supplementation with Gln lessened burn infection–induced loss of mucins and colonic injury
To investigate the effect of Gln on intestinal barrier repair in burns, a mouse model of burn infection was employed. We used the modified histopathologic grading system to analyse the distal colons from mice subjected to different treatments.
Major changes in the BI group compared with the sham group included shortening and atrophic collapse of the crypt, infiltration of inflammatory cells, and loss of goblet cells from H&E staining (Fig. 1A1-A2, B1-B2 and E). These changes were attenuated in Gln-supplemented mice (Fig. 1C1-C2 and E), and significantly less colonic injury was observed in Gln-supplemented mice than in burn-infected mice (Fig. 1D).
Fig. 1.

Gln lessened burn infection–induced loss of mucins and colonic injury
H&E and AB/PAS staining of longitudinal sections of distal colons from the (A) sham, (B) burn infection (BI) group and (C) burn infection with Gln supplementation (BI + Gln) group at 5 days post-injury. In the sham group, the mucosa had a uniform, fenestrated, regular surface epithelium with long and straight crypt foci in the colon (A2, yellow lines). Goblet cells (A2, white arrows) were abundant in the surface epithelium and within the proximal third of the crypt but less so at the base of the crypt (A2). The surface epithelium and goblet cells in the proximal part of the crypt contain PAS + neutral mucin in a dark purple colour (A4, red arrow), and the goblet cells at the base of the crypt contain mainly AB + acidic fluid in a light blue colour (A4, yellow arrow). In burn-infected mice, the mucosal surface of the colon had an uneven appearance, the colonic crypt (B2, yellow line) was atrophied and lost, epithelial cell growth was disorganized, basal lymphocytes were aggregated (B2, black arrow), goblet cell numbers (B2, white arrow) were reduced, and AB + mucin was transformed to PAS+ (B4, red arrow). Compared with the burn-infected mice, the mice with Gln supplementation had a regular surface epithelium of the colonic mucosa, longer colonic crypts (C2, yellow lines), increased numbers of goblet cells (C2, white arrows), and increased AB + mucin (C4, yellow arrows). Scale bar, 50 μm or 20 μm.(D) Histological score of the colon calculated from H&E-stained colon sections (maximum score = 14, N = 5 per group). (E) Length of colonic crypts in the mice from the sham, BI and BI + Gln groups (N = 5 per group). (F) Quantification of goblet cells in the mice from the sham, BI and BI + Gln groups (N = 5 per group). (G) Counts of AB + goblet cells (N = 5 per group). . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
AB/PAS, which stains heavily glycosylated proteins, including mucins, revealed mucus-containing goblet cells within the colons of sham mice, but the staining was markedly reduced in burn-infected mice (Fig. 1A3-A4 and B3-B4). The mucin transition from acidic to neutral mucin in the colonic goblet cells of burn-infected mice was further enhanced, an early event prevalent in burn-infected mice. However, compared with burn-infected mice, Gln supplementation for 5 days exhibited increased goblet cells and the proportion of acidic mucin in crypt, suggesting that mucin components are involved in repair (Fig. 1C3-C4 and F–G). These results indicated that Gln supplementation preserved the colonic mucus barrier in burn-infected mice.
3.2. Gln supplementation promoted MUC2 maturation and attenuated colonic mucus barrier damage after burn injury
Gln supplementation can repair burn infection-induced loss of mucins, and MUC2 is a core component of colonic mucus, we hypothesized that Gln could assist the synthesis and modification of MUC2. First, we examined the level of mature MUC2 protein in colon tissue from mice subjected to different treatments. Compared with sham mice, burn-infected mice exhibited markedly reduced mature MUC2 protein levels at 3, 5, and 7 days post-injury. These changes were attenuated after 5 and 7 days of Gln supplementation (Fig. 2A–H). The results suggest that Gln supplementation promotes MUC2 protein synthesis in burn-infected mice. The same positive effect of Gln was found in both HT-29 CL.16E cells and LPS-treated cells in vitro (Fig. 2I–L).
Fig. 2.

Gln supplementation promoted MUC2 maturation and attenuated colonic mucus barrier damage after burn injury
(A, B, C, D) Exploration of the expression of mature MUC2 in distal colon tissue at 3, 5, and 7 days post-injury from the sham, BI and BI + Gln groups. (E, F, G, H) Quantification of the expression of mature MUC2 in distal colon tissue at 3, 5, and 7 days post-injury. (N = 5 per group). (I, K) The expression of mature MUC2 was explored and quantified in HT-29 CL.16E cells with or without 2 mM Gln for 12 h (N = 6 per group). (J, L) The expression of mature MUC2 was explored and quantified in HT-29 CL.16E cells. The cells were exposed for 12 h to LPS (100 ng/ml), and cultured in the absence or presence of 2 mM Gln (N = 6 per group). (M) Immunostaining of colon sections using antibodies against MUC2-C (green) and FISH (with bacterial 16S rRNA gene probe) at 5 days post-injury. The red arrow represents bacterial staining. The inner stratified mucus layer (s) and outer mucus layer (o) are marked with a white segmented line. Blue indicates nuclear (DAPI) staining (scale bar: 50 μm). . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To evaluate the effect of Gln supplementation on the function of the colonic mucus layer, immunostaining of the Carnoy-fixed mouse distal colon with an MUC2-C antibody (green) was performed, which identified two primary mucus layers in addition to MUC2-positive goblet cells. Next, we analysed the tissue sections for bacterial presence by in situ hybridization using a general 16S rRNA probe. Bacteria were detected (red) in the outer mucus layer (o) and were excluded from the inner stratified layer (s) in sham mice. However, the inner firm mucus layer was penetrated by bacteria in the burn-infected mice. This meant that the inner mucus layer could not act as a physical barrier impenetrable to bacteria after burn sepsis. Gln supplementation alleviated this condition and maintained the function of the inner mucus layer (Fig. 2M).
These data indicated that Gln treatment promoted MUC2 protein synthesis and maintained the function of the inner mucus layer in burn-infected mice.
3.3. S-glutathionylation of AGR2 restricted the processing of MUC2
AGR2 plays a direct role in the processing of MUC2 by forming mixed disulfide bonds [22]. To probe the role of AGR2 in MUC2 maturation, we used three types of anti-MUC2 antibodies: MUC2-N antibody (mature MUC2), MUC2-VNTR antibody (MUC2 precursor) and MUC2-C antibody (MUC2 precursor and mature MUC2). To determine whether AGR2 physically associates with immature MUC2, we immunoprecipitated AGR2-containing complexes from mucus-producing HT-29 CL.16E cells. Immunoblotting with MUC2-VNTR antibody demonstrated that the MUC2 precursor was physically associated with AGR2 (Fig. 3A). Immunofluorescence localization showed that immature MUC2 was located within AGR2-containing areas (Fig. 3B). Increased levels of MUC2 precursor and decreased levels of mature MUC2 were observed in the cells with AGR2 KO (Fig. 3C). In addition, immunofluorescence revealed a significant decrease in the amount of intracellular mature MUC2 (red) and a significant increase in the amount of its precursor (green), resulting in a large accumulation of immature MUC2 in the cells with AGR2 KO (Fig. 3D). We concluded that AGR2 plays a critical role in the processing of MUC2.
Fig. 3.

S-glutathionylation of AGR2 restricted the processing of MUC2.
(A) AGR2 was immunoprecipitated from HT-29 CL.16E cells, and associated MUC2 was detected by immunoblotting. Rabbit or mouse IgG was used as a control. A total of 5% of the volume of lysate used for the immunoprecipitation was run on the gel for comparison (input). (B) Immunofluorescent localization of the MUC2 precursor (green) and AGR2 (red) in HT-29 CL.16E cells. Blue indicates nuclear (DAPI) staining. Scale bar: 10 μm. (C) The expression levels of the MUC2 precursor, mature MUC2 and AGR2 were assessed in AGR2 Knockout HT-29 CL.16E cell line. (D) Immunofluorescence staining for the MUC2 precursor (green) and mature MUC2(red) in AGR2 Knockout HT-29 CL.16E cell line. Blue indicates nuclear (DAPI) staining. Scale bar: 10 μm. (E) Expression of mature MUC2, immature MUC2, AGR2, GRP78, and CHOP in the distal colons of sham, burn–infected mice at 5 days post-injury (N = 5 per group). (F) Staining for ER-localized MUC2 precursor (green) in mouse goblet cells to assess MUC2 biosynthesis at 5 days post-injury. Scale bar: 50 μm. (G) Representative confocal microscopy images of distal colon sections for analysis of AGR2 (red) and MUC2 (green) at 5 days post-injury, with yellow arrows representing immature MUC2 and red arrows indicating mature MUC2. Scale bar: 50 μm. (H) Cysteine thiol levels of AGR2 in the distal colons of sham and burn-infected mice at 5 days post-injury. Tissue lysates were prepared, incubated with Protein-SHifter Plus, and subjected to SDS‒PAGE without BME (N = 3 per group). (I) Immunoblotting for S-glutathionylation of protein in colon tissues (DTT, negative control; GAPDH, loading control; N = 4 per group). (J) Co-IP showing S-glutathionylated AGR2 in colon tissue lysates from sham or burn-infected mice at 5 days post-injury (IP, GSH; IB, AGR2). (K) Tissue lysates were incubated with a MUC2-VTNR antibody coupled to Thermo Scientific Pierce Protein A/G magnetic beads for 24 h. The precipitates were subjected to Western blotting with anti-MUC2-VTNR and anti-AGR2 antibodies. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To investigate the regulatory effect of AGR2 on MUC2 in burn infection, we evaluated the expression changes in AGR2, immature MUC2, mature MUC2 and ER stress-responsive proteins. Compared with sham, burn-infected mice had no changes in AGR2 levels, but mature MUC2 levels were decreased, while immature precursor protein levels were significantly increased, along with the levels of CHOP and GRP78 (Fig. 3E). Immunofluorescence revealed a significant increase in the location overlap of the MUC2 precursor (green) and AGR2 (red) in burn-infected mice (Fig. 3F–G). We hypothesized that the function of AGR2 is inhibited in burn-infected mice, limiting the processing of immature MUC2. However, the precise molecular function of AGR2 remains to be defined. It is known that the activity of AGR2 depends on its redox balance, and oxidative stress can lead to a decrease in AGR2 enzyme activity [23]. We therefore suspect that the peroxidation state of AGR2 is responsible for its reduced efficiency in promoting immature MUC2 processing in burn-infected mice.
Protein cysteine thiol status is a major determinant of oxidative stress and oxidant signaling. A -SulfoBiotics- Protein Redox State Monitoring Kit was used to investigate protein thiol states. The cysteine thiol level of AGR2 in burn-infected mice was decreased compared with that in sham mice, suggesting that posttranslational modifications (PTMs) occurred on the only cysteine of AGR2 (Fig. 3H). To determine which redox-dependent PTMs occurred on cysteine, we first examined the S-nitrosylation level of AGR2 and found no S-nitrosylated AGR2 in burn-infected mice. Subsequently, S-glutathiolation modification of AGR2 was found in BiogSSG-treated colonic tissue. (Figs. S2A–B). BioGSSG offers a useful tool for the study of protein S-glutathiolation. However, BioGSSG cannot be used to assess protein S-glutathiolation in cell or tissue samples with intrinsic oxidative stress [53]. Therefore, in our study, GSH antibody were used to detect protein S-glutathiolation in the colonic tissues of burn-infected mice. We observed that S-glutathionylation of both total protein and AGR2 was enhanced after burn infection (Fig. 3I–J). In addition, S-glutathiolation of AGR2 interfered with the molecular chaperone activity of AGR2 and eliminated the ability of AGR2 to form complexes with the MUC2 precursor (Fig. 3K). These results suggested that the S-glutathionylation of AGR2 restricted the processing of immature MUC2 after burn infection.
3.4. Gln supplementation dampened the S-glutathionylation of AGR2 caused by LPS-induced oxidative stress
Gln has been shown to inhibit oxidative stress [54]. Therefore, we hypothesized that Gln can promote MUC2 maturation during oxidative stress because it attenuates oxidative stress and S-glutathionylation of AGR2. First, we used LPS to induce inflammatory and oxidative responses, and then measured intracellular ROS production using H2DCFA probes. The ROS level was significantly increased in HT-29 CL.16E cells stimulated with LPS and significantly decreased in the Gln treatment group (Fig. 4A). To further confirm the source of intracellular ROS, we quantified intracellular hydrogen peroxide (H2O2) and superoxide anion radical (O2•(−)). The intracellular concentrations of O2•(−) and H2O2 were enhanced under LPS stimulation, and Gln treatment could inhibit the effect (Fig. 4B–C). Further studies conclusively showed that the effect of Gln was related to its ability to promote GSH synthesis. The GSH/GSSG ratio was significantly reduced in HT-29 CL.16E cells after LPS exposure (36.8 ± 2.8 to 8.2 ± 1.4, p < 0.001), and the ratio was increased after Gln administration (8.2 ± 1.4 to 28.2 ± 2.3, p < 0.001) (Fig. 4D–E).
Fig. 4.

Gln mitigated LPS-induced oxidative stress and S-glutathionylation of AGR2, which regulates MUC2 processing.
HT-29 CL.16E cells were cultured in the absence or presence of Gln (2 mM) and LPS (100 ng/ml) for 8 h. (A) The fluorescence intensity in HT-29 CL.16E cells was measured by flow cytometry using H2DCFDA dye (N = 6 per group). (B) The intracytoplasmic superoxide anion concentration in HT-29 CL.16E cells was assessed by flow cytometry using a dihydroethidium (DHE) probe (N = 6 per group). (C) Quantitative estimation of LPS-induced H2O2. Hydrogen peroxide released by cells was detected with the ROS-Glo™ Hydrogen Peroxide Kit, which was calculated by comparing the hydrogen peroxide standard curve. (D) GSH levels in cell lysates (N = 5 per group). (E) GSH/GSSG ratio in cell lysates (N = 5 per group). (F) Immunoblotting analysis of the cysteine thiol levels of AGR2 under different cellular treatments. HT-29 CL.16E cells were exposed for 30 min to H2O2 (200 μM), then stimulated with or without 5 mM sodium pyruvate (SP), (DTT, positive control). (G) Nonreducing electrophoresis and Western blot analysis with streptavidin-HRP to determine the S-glutathionylation level of the protein. HT-29 CL.16E cells were preloaded with BioGEE (250 μM) for 1.5 h and stimulated with H2O2 (200 μM) for 30 min, then cultured in the presence and absence of 5 mM sodium pyruvate (DTT, negative control). (H) Reduction electrophoresis and Western blot to determine the S-glutathionylation level of AGR2 after pulling down the biotin-GSS-protein adducts. (I) Immunoblots of AGR2 glutathionylation under different cellular treatments. HT-29 CL.16E cells were preloaded with BioGEE (250 μM) for 1.5 h, stimulated with H2O2 (200 μM) for 30 min, and then incubated with or without 2 mM N-acetylcysteine (DTT, negative control). (J) Western blot analysis with streptavidin-HRP to determine the S-glutathionylation level of protein. HT-29 CL.16E cells were preloaded with BioGEE (250 μM) for 1.5 h and exposed for 8 h to LPS (100 ng/ml), next cultured in the absence or presence of Gln (2 mM) and 2 mM N-acetylcysteine (DTT, negative control). (K) Immunoblotting to determine the S-glutathionylation level of AGR2 after pulling down the biotin-GSS-protein adducts. (L) Immunoblotting analysis of the cysteine thiol levels of AGR2 under different cellular treatments. HT-29 CL.16E cells were exposed for 8 h to LPS (100 ng/ml), and cultured in the absence or presence of 2 mM Gln (DTT, positive control; H2O2, negative control; N = 3 per group). (M) Confocal imaging of glutathionylated proteins. HT-29 CL.16E cells were cultured in the absence or presence of LPS and Gln 8 h. Green fluorescent labeling was performed using an anti-glutathionylated antibody. Blue indicates nuclear (DAPI) staining. Scale bar, 10 μm. (N) The expression of mature MUC2, immature MUC2, GRP78 and CHOP was analysed by immunoblotting. HT-29 CL.16E cells were cultured in the absence or presence of LPS and Gln (N = 3 per group). * Compared with control group, # Compared with LPS group, #P < 0.05; # #P < 0.01; # # #P < 0.001. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Oxidative stress has been shown to induce the S-glutathionylation of free thiol groups (-SH) on the cysteine residues of proteins to form protein-glutathione mixed disulfide adducts (Pr-SSG) [55]. Therefore, we first examined the cysteine thiol level of AGR2. The thiol level of AGR2 in H2O2- treated HT-29 CL.16E cells was obviously lower than in the control, while the thiol level increased remarkably after adding DTT or sodium pyruvate. The results suggested that the cysteine thiol level of AGR2 was regulated by reducing agents and oxidants (Fig. 4F). To explore the variation of S-glutathionylation levels of AGR2 under different REDOX state, the use of GSH antibody and biotin labeling were chosen in the assay. We found that the S-glutathionylation levels of total protein and AGR2 in HT29CL.16E cells were both increased by H2O2 stimulation, and the changes were inhibited by DTT or sodium pyruvate treatment (Fig. 4G–H, Fig. S3A). In addition, N-acetylcysteine, as a precursor of GSH, alleviated the S-glutathionylation level of AGR2 induced by H2O2 (Fig. 4I). Thus, these results suggested that S-glutathionylation was a reversible post-translational modification at cysteine residues that was regulated by intracellular GSH.
Further studies conclusively showed that Gln administration could inhibit LPS-induced cellular oxidative stress. The S-glutathionylation levels of total protein (Fig. 4J, M) and AGR2 (Fig. 4K, Fig. S3B) decreased appreciably, and the cysteine thiol level of AGR2 obviously increased through the administration of Gln (Fig. 4L). In addition, the levels of mature MUC2 were increased, while the levels of the immature precursor proteins CHOP and GRP78 were significantly decreased by Gln treatment (Fig. 4N). As expected, Gln dampened the S-glutathionylation of AGR2 caused by LPS-induced oxidative stress.
3.5. G6PD attenuated oxidative stress and assisted MUC2 maturation
GSH is an important reductant against oxidative stress, the levels of which can be maintained by NADPH generated mainly via the PPP and its rate-limiting enzyme, glucose-6-phosphate dehydrogenase (G6PD) [56]. The role of G6PD in the control of protein glutathionylation has been demonstrated [57]. Given that we showed that the processing of immature MUC2 was limited by S-glutathionylation of AGR2 during oxidative stress, we hypothesized that G6PD activation might induce an increase in NADPH that would promote MUC2 maturation.
To test this hypothesis, we first wanted to confirm whether NADPH is a key product of G6PD against oxidative stress. The ratios of NADPH/NADP+ and GSH/GSSG were significantly diminished by G6PDi-1 (an inhibitor of G6PD) treatment, while the levels of total ROS and H2O2 were enhanced. However, the effects were alleviated by exogenous NADPH supplementation (Fig. 5A–C, Fig. S4A). At the same time, G6PDi-1 or NADPH treatment seemed to have no significant effect on intracellular superoxide anion radicals (Fig. S4B). These results indicated that G6PD may reduce LPS-induced H2O2 by promoting GSH synthesis through NADPH. The cysteine thiol level of AGR2 was significantly decreased, S-glutathionylation level of AGR2 was increased by G6PDi-1 treatment, and the inhibitory effect was alleviated by exogenous NADPH supplementation (Fig. 5D–E). Correspondingly, mature MUC2 levels were increased by exogenous NADPH supplementation in LPS and G6PDi-1-treated cells (Fig. 5F). To further confirm the effect of G6PD on the S-glutathionylation of AGR2, we generated G6PD-KO cells using CRISPR‒Cas9 and showed results similar to those obtained by using G6PD inhibitors (Fig. 5G–K, Figs. S4C–D). Finally, as predicted, G6PD activated by AG1 (a small-molecule activator of G6PD [58]) promoted the levels of the G6PD dimer and cysteine thiol of AGR2. At same time, the treatment dampened the levels of the G6PD monomer and S-glutathionylated AGR2 (Fig. 5L–N). Correspondingly, a significant increase in mature MUC2 and a decrease in immature MUC2 were observed after AG1 treatment (Fig. 5O).
Fig. 5.

G6PD activation induced an increase in NADPH that promoted MUC2 maturation.
HT-29 CL.16E cells were exposed for 6 h to LPS (100 ng/ml), and cultured in the absence or presence of G6PDi-1 (50 μM), then stimulated with or without 20 mM NADPH. (A) NADPH/NADP + ratio. (B) GSH/GSSG ratio. (C) Quantitative estimation of the dose-dependent H2O2 generation. (N = 5 per group). (D) Immunoblotting analysis of the cysteine thiol levels of AGR2 under different cellular treatments. HT-29 CL.16E cells were exposed for 6 h to LPS (100 ng/ml), cultured in the absence or presence of G6PDi-1 (50 μM), and then stimulated with or without 20 mM NADPH (DTT, positive control; H2O2, negative control) (N = 3 per group). (E) S-glutathionylated AGR2/AGR2 measured by immunoblot analysis (N = 3 per group). (F) Expression of mature MUC2, immature MUC2, and G6PD as determined by immunoblot analysis (N = 5 per group). * Compared with control, # Compared with the G6PDi-1 treated group, #P < 0.05; # #P < 0.01; # # #P < 0.001. After exposure to LPS (100 ng/mL) for 6 h, G6PD-KO cells were treated with or without exogenous NADPH. (G) GSH/GSSG ratio. (H) Quantitative estimation of the dose-dependent H2O2 generation. (N = 5 per group). (I) S-glutathionylated AGR2/AGR2 as measured by immunoblot analysis (N = 3 per group). (J) Cell lysates were incubated with Protein-Shifter Plus and immunoblotted with an AGR2 antibody (N = 3 per group). (K) Expression of mature MUC2 and immature MUC2 as determined by immunoblot analysis (N = 5 per group). * Compared with control, # Compared with G6PD-KO cells, #P < 0.05; # #P < 0.01; # # #P < 0.001.
Taken together, these data suggest that modulation of G6PD activity can affect NADPH levels, which can regulate MUC2 maturation.
HT-29 CL.16E cells were exposed for 6 h to LPS (100 ng/ml), then stimulated with or without 10 μM AG1. (L) 1 mM DSS cross-linked protein with monomeric and dimeric changes detected by immunoblotting using a G6PD antibody. (M) A -SulfoBiotics- Protein Redox State Monitoring Kit was used to determine the thiol redox state of AGR2 (N = 3 per group). (N) S-glutathionylated AGR2/AGR2 as measured by immunoblot analysis (N = 3 per group). (O) Immunoblotting for the expression of mature MUC2, immature MUC2, and G6PD (N = 3 per group).
3.6. O-GlcNAcylation of G6PD promoted G6PD dimer formation and increased NADPH synthesis
Activation of G6PD activity via O-GlcNAcylation upregulates glucose flux through the PPP, providing reducing power in the form of NADPH and GSH to combat oxidative stress [59]. Given that we showed that G6PD activation by AG1 treatment induced an increase in NADPH and GSH to combat oxidative stress, we hypothesized that O-GlcNAcylation of G6PD might induce an increase in NADPH and GSH that would promote MUC2 maturation.
To test this hypothesis, we first examined whether O-GlcNAcylation of G6PD is dynamically regulated in the colonic tissue of burn-infected mice. We confirmed that the levels of O-GlcNAcylation in the colonic tissue of sham mice were higher than those in burn-infected mice (Fig. 6A). Then, we employed a well-established chemoenzymatic labeling approach and performed capture with streptavidin-agarose beads to probe the O-GlcNAcylation levels of G6PD. Subsequent immunoblotting of the captured proteins with an antibody against G6PD showed reduced O-GlcNAcylation of G6PD in burn-infected mice (Fig. 6B). Consistently, the G6PD activity was diminished with low O-GlcNAcylation levels (Fig. 6C).
Fig. 6.

O-GlcNAcylation of G6PD regulates its activity through dimerization to promote MUC2 maturation.
(A) Immunoblot analysis of protein O-GlcNAc modification in the distal colons of sham and burn-infected mice at 5 days post-injury (N = 5 per group). (B) Detection of O-GlcNAcylation of G6PD in the distal colons of sham and burn-infected mice by using chemical enzyme labeling and biotinylation (N = 5 per group). (C) G6PD enzyme activity assay (N = 5 per group). (D) Labeling of G6PD glycosylation levels in WT and S84V G6PD cells. (E) Immunoblot for O-GlcNAcylation of G6PD. Cells labelled WT and S84V were treated with DON (20 μM) and TMG (50 μM). (F) G6PD enzyme activity assay of cells under different treatments (N = 5 per group). (G) Oligomerization status in WT and S84V G6PD cells measured by immunoblotting. Cross-linking with 1 mM DSS. (H) Immunoblot for O-GlcNAcylation of G6PD. WT and S84V G6PD cells were exposed to LPS (100 ng/mL) for 6 h and treated with or without NADPH. (I) G6PD enzyme activity assay under different treatments (N = 5 per group). (J) GSH levels in cell lysates (N = 5 per group). (K) GSH/GSSG ratio in cell lysates (N = 5 per group). (L) Co-IP showing S-glutathionylated AGR2 in cell lysates (N = 3 per group). (M) Immunoblot detection of mature MUC2 and immature MUC2 (N = 3 per group). * Compared with the WT group, # Compared with the S84V group without NADPH, #P < 0.05; # #P < 0.01; # # #P < 0.001.
Following a previous report on the O-GlcNAcylation of G6PD [59], we created a system to study the effect of G6PD glycosylation on antioxidation. Mutation of Ser-84 to valine resulted in the elimination of glycosylation signaling, supporting Ser-84 as an O-GlcNAcylation site for G6PD (Fig. 6D). To further understand the effect of O-GlcNAcylation on G6PD activity, we first examined G6PD enzyme activity in WT G6PD and S84V G6PD cells treated with the two inhibitors. Pharmacological inhibition of O-GlcNAc hydrolase with the specific inhibitor thiamet-G (TMG) significantly increased G6PD enzyme activity. 6-Diazo-5-oxo-L-nor-leucine (DON) inhibits the production of glucosamine 6-phosphate by the rate-limiting enzyme of the hexosamine pathway, glutamine: fructose 6-phosphate aminotransferase (GFAT). Compared with WT G6PD cells, S84V G6PD cells showed a negligible response to TMG or DON treatment (Fig. 6E–F). The dimeric forms of G6PD are catalytically active [58]. To examine whether S84 glycosylation affects the oligomerization state of G6PD, we performed protein cross-linking experiments using DSS. Inhibition of OGA activity to enhance O-GlcNAcylation levels resulted in a significant increase in the G6PD dimer level, but no change in G6PD dimers was detected in the S84V mutant (Fig. 6G). The results indicated that the O-GlcNAcylation of G6PD regulates its activity through dimerization.
To further confirm that O-GlcNAcylation of G6PD plays an important role in antioxidant defence, we compared the redox status between WT G6PD and S84V G6PD cells treated with LPS. O-GlcNAc levels were suppressed in S84V G6PD cells compared with WT G6PD cells (Fig. 6H). Consistently, G6PD activity was diminished with low O-GlcNAc levels (Fig. 6I). Blocking glycosylation of G6PD significantly suppressed GSH and GSH/GSSG in S84V G6PD cells compared with WT G6PD cells, but this inhibitory effect was alleviated by exogenous NADPH supplementation (Fig. 6J–K). Consistently, compared with WT G6PD cells, S84V G6PD cells exhibited higher S-glutathionylation levels of AGR2. Correspondingly, a significant increase in immature MUC2 and a decrease in mature MUC2 were observed. These changes were alleviated by exogenous NADPH supplementation (Fig. 6L–M). These results indicated that G6PD O-GlcNAcylation plays an important role in regulating redox homoeostasis to promote MUC2 maturation.
3.7. Gln promoted MUC2 maturation by stabilizing AGR2 redox homeostasis induced by O-GlcNAcylation of G6PD
O-GlcNAcylation is the product of nutrient flux through the hexosamine biosynthetic pathway (HBP). GFAT is the rate-limiting step of the HBP, and Gln is an indispensable substrate for GFAT [60]. Given that we showed that G6PD O-GlcNAcylation plays an important role in regulating redox homoeostasis to promote MUC2 maturation, we hypothesized that Gln might induce an increase in G6PD O-GlcNAcylation that would promote MUC2 maturation in burn-infected mice.
We first confirmed whether G6PD O-GlcNAcylation is regulated through HBP by Gln. We measured the O-GlcNAcylation of G6PD in HT-29 CL.16E cells by IP and Western blotting. The O-GlcNAcylation levels of G6PD were higher in the Gln- or glucosamine-supplemented group than in the control group (Fig. 7B). The increased G6PD O-GlcNAcylation by Gln supplementation was inhibited by DON (a GFAT inhibitor). However, the increased G6PD O-GlcNAcylation induced by glucosamine did not respond to the addition of DON (Fig. 7C). We next investigated the capacity to Gln enhance the O-GlcNAcylation of G6PD under LPS-induced oxidative stress. The O-GlcNAcylation levels of total proteins and G6PD were both higher in the Gln-treated group than in the LPS group. However, this increase was also inhibited by DON (Fig. 7D–E). Correspondingly, a similar trend of change was observed at the G6PD dimer level (Fig. 7F). Then we further confirmed that G6PD activated by Gln treatment can promote cysteine thiol levels and dampen the S-glutathionylation of AGR2 (Fig. 7G–H), thereby promoting MUC2 maturation (Fig. 7I).
Fig. 7.

Gln activated G6PD activity and stabilized AGR2 redox homeostasis via the HBP
(A) Hexosamine biosynthetic pathway. (B, C) Effects of Gln and glucosamine on G6PD protein and O-GlcNAcylation levels as determined by immunoblotting of G6PD protein with antibodies against the G6PD protein and O-GlcNAc. HT-29 CL.16E cells were treated with 2 mM Gln or 10 mM glucosamine for 12 h, and cultured in the absence or presence of DON (20 μM). (D) Immunoblot detection of cellular protein O-GlcNAcylation. HT-29 CL.16E cells were exposed for 12 h to LPS (100 ng/ml), cultured in the absence or presence of Gln (2 mM), and then stimulated with or without Don (N = 3 per group). (E) Chemical enzyme labeling and biotinylation to detect the O-GlcNAcylation of G6PD (N = 3 per group). (F) Immunoblot detection of G6PD dimers and monomers. Cells were cross-linked with 1 mM DSS (N = 3 per group). (G) A -SulfoBiotics- Protein Redox State Monitoring Kit was used to determine the thiol redox state of recombinant proteins (N = 3 per group). (H) S-glutathionylated AGR2/AGR2 was measured by immunoblot analysis (N = 3 per group).(I) Immunoblot detection of mature MUC2 and immature MUC2 protein expression (N = 3 per group). * Compared with control, & Compared with the LPS group, & P < 0.05; &&P < 0.01; &&& P < 0.001. # Compared with LPS + Gln group, #P < 0.05; # #P < 0.01; # # #P < 0.001.
To further confirm the mechanism by which Gln promotes MUC2 maturation in burn infection, we examined the effect of Gln in burn-infected mice. Treatment with Gln promoted O-GlcNAcylation levels of total proteins and G6PD, GSH levels, GSH/GSSG ratio, AGR2 cysteine thiol level and mature MUC2 level in the colonic tissues of burn-infected mice, while S-glutathionylation levels of total proteins and AGR2, immature MUC2, CHOP and GRP78 were decreased significantly (Fig. 8A–I). These results suggested that Gln plays an important role in regulating AGR2 redox homoeostasis to promote MUC2 maturation by G6PD O-GlcNAcylation via the HBP. Thus, we proposed a mechanism for the effect of Gln on promoting MUC2 maturation in burn-infected mice (Fig. 8J).
Fig. 8.

Gln promoted MUC2 maturation by stabilizing AGR2 redox homeostasis induced by O-GlcNAcylation of G6PD in burn infection. (A) O-GlcNAcylation levels in the distal colons after 5 days of glutamine supplementation in burn-infected mice (N = 3 per group). (B) Chemical enzyme labeling and biotinylation to detect G6PD O-GlcNAcylation (N = 3 per group). (C) GSH levels in distal colon tissue lysates (N = 3 per group). (D) GSH/GSSG in distal colon tissue lysates (N = 3 per group). (E) Immunoblot analysis of the cysteine thiol level of AGR2. Tissue lysates were incubated with Protein-Shifter Plus and subjected to SDS‒PAGE in the absence of BME (N = 3 per group). (F) Immunoblot analysis of the S-glutathionylation levels of total protein (N = 3 per group). (G) Co-IP showing S-glutathionylated AGR2 in tissue lysates (N = 3 per group). * Compared with sham group, # Compared with BI group, #P < 0.05; # #P < 0.01; # # #P < 0.001. (H) Immunoblotting for the expression of mature MUC2, immature MUC2, GRP78 and CHOP in the distal colon (N = 5 per group). (I) Representative AGR2 and MUC2 protein staining in colon sections. The yellow arrows indicate immature MUC2, and the red arrows indicate mature MUC2. (J) Proposed mechanism for the effect of Gln on promoting MUC2 maturation in burn-infected mice. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
As the core component of colonic mucus, MUC2 needs to be processed and modified in the ER after translation to complete the transformation from the precursor to the mature protein, and AGR2 plays a key role in the process [22,23,61]. In this study, we demonstrated that the oxidative stress triggered by burn sepsis led to enhanced AGR2 glutathionylation modification, which hindered the processing of MUC2 by AGR2, resulting in restrained MUC2 maturation. Further studies revealed that NADPH synthesized by G6PD is the core molecule that inhibits oxidative stress and regulates AGR2 activity. By promoting G6PD glycosylation, Gln facilitated G6PD homodimer formation, enhanced NADPH synthesis, maintained AGR2 redox homeostasis and increased mature MUC2 synthesis, thereby lessening the intestinal mucus barrier damage triggered by burn sepsis.
AGR2 is a member of the PDI family and has two key attributes: ER localization and a functional thioredoxin-like domain with a CXXS motif. The former makes it an ER-resident protein, and the latter makes it function as an oxidoreductase and molecular chaperone [[21], [22], [23]]. In the presence of AGR2, immature mucins undergo folding, and coiling and eventually transform into mature mucins [20,22]. Genetic defects or knockdown of AGR2 can lead to impaired synthesis of MUC2 and exacerbate intestinal damage, and it is also an important factor in triggering inflammatory bowel disease [[19], [20], [21], [22],62]. In this study, we found that knockdown of AGR2 resulted in a significant decrease in the level of mature MUC2 and a remarkable increase in the level of immature MUC2 (Fig. 3C–D).
Structural biology shows that ARG2 and MUC2 can form a heterodimer via a mixed disulfide bond that is formed by condensation of two cysteine residues located on AGR2 and MUC2 [21,22]. This reaction is a prerequisite for posttranslational modification of MUC2 by AGR2. This study confirmed that AGR2 can bind to MUC2, which is consistent with the literature [22]. Intriguingly, we found that AGR2 only bound to immature MUC2 (Fig. 3A–B), which is consistent with the function of AGR2, and this finding provides new and strong evidence that AGR2 modifies MUC2. It is now known that there is only one cysteine (Cys-81) in the amino acid sequence of AGR2, which is a key target for AGR2 to form a heterodimer with the modified protein, or two AGR2 monomers to form a homodimer [21,63]. During the processing and modification of MUC2 precursors, it is important to maintain the dynamic balance between AGR2 monomer and its homodimer. It is now known that AGR2 in the monomeric state facilitates its modification of the target protein, while in the dimeric state facilitates the maintenance of AGR2 stability and alleviates ER stress. Therefore, AGR2 dimers act as sensors of ER homeostasis [64,65]. The unfolded protein response resulted from ER stress induces the conversion of AGR2 from a dimer to a monomer to exert its molecular chaperone effects and manage misfolded proteins.
Under physiological conditions, the cysteine of AGR2 is in a reduced state, and its thiol can condense with those of other cysteines to form disulfide bonds [23]. Oxidative stress leads to disruption of the thiol on the cysteine of AGR2, upon which the ability to form covalent bonds with mucin is lost [66,67]. We found that the oxidative stress triggered by burn sepsis led to the accumulation of a large amount of ROS, which significantly reduced the cysteine thiol level of AGR2, while glutathione modification was obviously enhanced (Fig. 3H–J). The essence of this change was that the –SH group on Cys-81 of AGR2 was replaced by a -SSG group, which made it difficult to form disulfide bonds between AGR2 and MUC2, and inhibited the ability of AGR2 to process and modify MUC2. Large amounts of MUC2 precursor protein accumulated in the ER, further exacerbating burn sepsis-induced ER stress, with obvious increases in the levels of the corresponding marker proteins GRP78 and CHOP (Fig. 3E).
S-glutathionylation is a common post-translational modification of protein cysteines and is usually activated during oxidative stress [27]. It can protect against the irreversible oxidation of protein thiol groups, and is involved in protein function regulation and oxidative stress signal transduction [29]. A large number of proteins have been identified as potentially regulated by reversible S-glutathionylation, such as c-Jun, NF-κB, Ras, and MEKK1 [29,55,68]. S-glutathionylation has been shown to regulate the structure/function of proteins in a complex manner, either positively or negatively (enhanced or suppressed activity) [30]. It has been reported that oxidative stress can promote the Cys118 on p21ras S-glutathionylation in both endothelial and smooth muscle cells, trigger downstream signaling through phosphorylation of ERK and AKT, and mediate hypertrophy of vascular smooth muscle cells [29]. Similar results could be found in S-glutathionylation of the Cys188 residue of IL- 1β, which positively regulates its bioactivity [55]. However, more studies have shown that most proteins are inhibited in their activity by S-glutathionization occurred on cysteines, which is particularly evident in the case of enzymes [29,68,69]. AGR2 is a key enzyme regulating the synthesis and modification of MUC2. If AGR2 S-glutathionylation enhanced due to oxidative stress after burn sepsis, it will inevitably interfere with the formation of the disulfide bond between AGR2 and MUC2 and hinder the modification of MUC2 by AGR2. In addition, excessive S-glutathionylation might also prevent AGR2 from forming homodimers, thus exacerbating ER stress. In this study, the results confirm the above hypothesis (Fig. 3 E; 3 K). S-glutathionylation is regulated by the glutathione cycle; GSSG promotes this reaction, while GSH inhibits it [34]. We found that administration of Gln enhanced GSH synthesis, inhibited oxidative stress, decreased AGR2 S-glutathionylation, protected vulnerable thiols from over-oxidation, increased the synthesis of mature MUC2, and reduced ERS (Fig. 4D–N). Similarly, Tang's group demonstrated that glutamine could inhibit the glutathione modification of mitochondrial complex I by promoting GSH synthesis, but the author did not explore the deeper mechanism [70]. Our previous studies have shown that Gln inhibits oxidative stress by promoting NADPH synthesis and accelerating the conversion of GSSG to GSH [26,43]. NADPH is the most important reducing equivalent in the body, and it acts as a transmitter of hydrogen to maintain redox homeostasis and regulate the GSH cycle through the NADPH/NADP + cycle [57]. Therefore, we hypothesized that NADPH may play a key role in the inhibition of AGR2 S-glutathionylation.
There are several known NADPH synthesis pathways in the body, including the PPP, the ME1 pathway and the IDH1 pathway. The PPP is the main process for NADPH synthesis, accounting for more than 60% of the total [36,37]. Therefore, in this study we chose to explore the mechanism regulating NADPH synthesis with the PPP. G6PD is the rate-limiting enzyme in this pathway and controls carbon flux and NADPH production [57,69,71]. The two molecules of NADPH generated by the PPP are obtained by two-step oxidation reactions catalysed by G6PD [36,37,39]; the importance of G6PD is therefore self-evident. In this study, the G6PD inhibitor treatment or knockdown of G6PD resulted in a significant reduction in NADPH synthesis and enhanced S-glutathionylated AGR2, leading to a reduction in the mature MUC2 level (Fig. 5A–K). Conversely, supplementation with exogenous NADPH or G6PD agonists markedly increased the GSH/GSSG ratio, maintained the redox balance of AGR2, and increased the mature MUC2 content. Thus, G6PD activity is closely related to AGR2 redox homeostasis and the maturation of MUC2.
Rao et al. [59] reported that compared with the G6PD monomer, the G6PD dimer has better bioactivity and stability, and glycosylation modification is key to promoting the formation of homodimers of G6PD. Furthermore, the serine at N-terminal position 84 of G6PD (Ser-84) is the only glycosylation site. In this study, we mutated the site and found that the glycosylation of G6PD was markedly reduced, resulting in a decrease in the number of G6PD dimers (Fig. 6G). This result confirmed that glycosylation modification of G6PD is a core factor in the formation of G6PD homodimers. Gln can promote the synthesis of glucosamine, a glycosylated substrate involved in the glycosylation modification of some proteins, such as HSP70 and Sp1 [[72], [73], [74]]. However, whether it promotes the glycosylation modification of G6PD has not been reported. Our experimental results revealed that the G6PD glycosylation was significantly reduced and the NADPH synthesis was obviously inhibited in septic mice (Fig. 6A–C). Gln administration notably increased the G6PD glycosylation level and dimer content, thereby promoting NADPH synthesis, inhibiting the glutathionylated modification of AGR2, and maintaining redox homeostasis (Figs. 7 and 8).
There are two main types of protein glycosylation modifications, namely, O-glycosylation and N-glycosylation [75]. Gln is involved in protein glycosylation through the HBP, a subtype of O-glycosylation [76,77]. The rate-limiting enzyme of HBP is GFAT, under its catalysis, an acylamino from Gln is added to the sugar chain of fructose 6-phosphate to form the substrate of O-glycosylation, i.e., uracil diphosphate-N-acetylglucosamine (UDP-N-GlcNAc) [73,76,77]. In this study, we used DON, a specific inhibitor of GFAT, and found that the effect of Gln on promoting G6PD glycosylation was significantly inhibited. Administration of glucosamine reversed the effect of DON, thus confirming that Gln promotes G6PD glycosylation via the HBP (Fig. 7C–E).
In recent years, the regulatory role of Gln in cellular metabolism has received increasing attention, including its role in the metabolic reprogramming of the TCA cycle [79,80], regulation of the urea cycle and redox homeostasis [[78], [79], [80]], and protein glycosylation modification [81,82]. In this study, we used G6PD glycosylation modification as an entry point and NADPH as an effector to investigate the molecular mechanism by which Gln maintains AGR2 redox homeostasis and reduces intestinal mucus barrier injury due to burn sepsis. The most important findings are twofold: first, we demonstrated that the S-glutathionylation of AGR2 is key to inhibiting its activity; second, we found that the core mechanism by which Gln maintains the intestinal mucus barrier involves promotion of G6PD glycosylation, an increase in NADPH synthesis, attenuation of AGR2 glutathionylation, and upregulation of mature MUC2 synthesis. These results help to deepen the understanding of Gln-mediated regulation of cellular metabolism, and they provide a theoretical basis for the rational use of Gln in burn clinics. Nevertheless, some details are not entirely clear, such as how is the formation of AGR2 dimer versus monomer precisely regulated, and what are the biological functional of AGR2 dimerization, and the mechanism of regulating the balance between glutathionylation versus deglutathionylation of AGR2? These issues will be investigated in our future studies.
Authors’ contributions
XP designed the trial and controlled the process. DW, YW, LX and SF participated in relevant animal experiments and obtained specimens and data. DW, SS, GY, QH and YY obtained the test data. SS, DW and XZ participated in the statistical analysis and drafted the manuscript. All authors discussed the results and commented on the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81971838) and Innovative Leading Talents Project of Chongqing, China (NO. cstc2022ycjh-bgzxm0148).
Declaration of competing interest
All authors declare that they have no competing interests.
Acknowledgements
We sincerely thank Dr. Min-Dian Li (Dept. Cardiovas. Med. & C4CM) and Dr. Lei Cai (Department of Hepatobiliary Surgery, Chongqing People's Hospital) for their help in experimental design and manuscript writing.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102581.
Contributor Information
Dan Wu, Email: hao20111985@163.com.
Sen Su, Email: 1441suse@163.com.
Xule Zha, Email: zhaxl1005@126.com.
Yan Wei, Email: 56333849@qq.com.
Gang Yang, Email: ygyg11a@163.com.
Qianying Huang, Email: huangqianying0@126.com.
Yongjun Yang, Email: yyj85@tmmu.edu.cn.
Lin Xia, Email: 375441866@qq.com.
Shijun Fan, Email: fanshijun1211@hotmail.com.
Xi Peng, Email: pxlrmm@tmmu.edu.cn, pxlrmm@163.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Dvorak J.E., Ladhani H.A., Claridge J.A. Review of sepsis in burn patients in 2020. Surg. Infect. 2021;22(1):37–43. doi: 10.1089/sur.2020.367. [DOI] [PubMed] [Google Scholar]
- 2.Rech M.A., Mosier M.J., McConkey K., Zelisko S., Netzer G., Kovacs E.J., et al. Outcomes in burn-injured patients who develop sepsis. J. Burn Care Res. 2019;40(3):269–273. doi: 10.1093/jbcr/irz017. official publication of the American Burn Association. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manning J. Sepsis in the burn patient. Crit. Care Nurs. Clin. 2018;30(3):423–430. doi: 10.1016/j.cnc.2018.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Zhang P., Zou B., Liou Y.C., Huang C. The pathogenesis and diagnosis of sepsis post burn injury. Burns & trauma. 2021;9 doi: 10.1093/burnst/tkaa047. tkaa047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenhalgh D.G. Sepsis in the burn patient: a different problem than sepsis in the general population. Burns & trauma. 2017;5:23. doi: 10.1186/s41038-017-0089-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adiliaghdam F., Cavallaro P., Mohad V., Almpani M., Kühn F., Gharedaghi M.H., et al. Targeting the gut to prevent sepsis from a cutaneous burn. JCI insight. 2020;5(19) doi: 10.1172/jci.insight.137128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haussner F., Chakraborty S., Halbgebauer R., Huber-Lang M. Challenge to the intestinal mucosa during sepsis. Front. Immunol. 2019;10:891. doi: 10.3389/fimmu.2019.00891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niu M., Chen P. vol. 9. Burns & trauma; 2021. (Crosstalk between Gut Microbiota and Sepsis). tkab036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fay K.T., Ford M.L., Coopersmith C.M. The intestinal microenvironment in sepsis. Biochim. Biophys. Acta, Mol. Basis Dis. 2017;1863(10 Pt B):2574–2583. doi: 10.1016/j.bbadis.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lei X., Teng W., Fan Y., Zhu Y., Yao L., Li Y., et al. The protective effects of HIF-1α activation on sepsis induced intestinal mucosal barrier injury in rats model of sepsis. PLoS One. 2022;17(5) doi: 10.1371/journal.pone.0268445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capaldo C.T., Powell D.N., Kalman D. Layered defense: how mucus and tight junctions seal the intestinal barrier. J. Mol. Med. (Berl.) 2017;95(9):927–934. doi: 10.1007/s00109-017-1557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang B.X., Wu C.M., Ribbeck K. Home, sweet home: how mucus accommodates our microbiota. FEBS J. 2021;288(6):1789–1799. doi: 10.1111/febs.15504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelaseyed T., Bergström J.H., Gustafsson J.K., Ermund A., Birchenough G.M., Schütte A., et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014;260(1):8–20. doi: 10.1111/imr.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paone P., Cani P.D. Mucus barrier, mucins and gut microbiota: the expected slimy partners? Gut. 2020;69(12):2232–2243. doi: 10.1136/gutjnl-2020-322260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bansil R., Turner B.S. The biology of mucus: composition, synthesis and organization. Adv. Drug Deliv. Rev. 2018;124:3–15. doi: 10.1016/j.addr.2017.09.023. [DOI] [PubMed] [Google Scholar]
- 16.Hansson G.C. Mucins and the microbiome. Annu. Rev. Biochem. 2020;89:769–793. doi: 10.1146/annurev-biochem-011520-105053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergstrom K., Shan X., Casero D., Batushansky A., Lagishetty V., Jacobs J.P., et al. Proximal colon-derived O-glycosylated mucus encapsulates and modulates the microbiota. Science. 2020;370(6515):467–472. doi: 10.1126/science.aay7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao Y., Kim G., Shafer S., Chen Z., Kubo S., Ji Y., et al. Mucus sialylation determines intestinal host-commensal homeostasis. Cell. 2022;185(7):1172–1188. doi: 10.1016/j.cell.2022.02.013. e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y.C., Lu Y.F., Li I.C., Hwang S.P. Zebrafish Agr2 is required for terminal differentiation of intestinal goblet cells. PLoS One. 2012;7(4) doi: 10.1371/journal.pone.0034408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye X., Wu J., Li J., Wang H. Anterior gradient protein 2 promotes mucosal repair in pediatric ulcerative colitis. BioMed Res. Int. 2021;2021 doi: 10.1155/2021/6483860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergström J.H., Berg K.A., Rodríguez-Piñeiro A.M., Stecher B., Johansson M.E., Hansson G.C. AGR2, an endoplasmic reticulum protein, is secreted into the gastrointestinal mucus. PLoS One. 2014;9(8) doi: 10.1371/journal.pone.0104186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park S.W., Zhen G., Verhaeghe C., Nakagami Y., Nguyenvu L.T., Barczak A.J., et al. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc. Natl. Acad. Sci. U.S.A. 2009;106(17):6950–6955. doi: 10.1073/pnas.0808722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shishkin S.S., Eremina L.S., Kovalev L.I., Kovaleva M.A. AGR2, ERp57/GRP58, and some other human protein disulfide isomerases. Biochemistry. Biokhimiia. 2013;78(13):1415–1430. doi: 10.1134/S000629791313004X. [DOI] [PubMed] [Google Scholar]
- 24.El Ayadi A., Salsbury J.R., Enkhbaatar P., Herndon D.N., Ansari N.H. Metal chelation attenuates oxidative stress, inflammation, and vertical burn progression in a porcine brass comb burn model. Redox Biol. 2021;45 doi: 10.1016/j.redox.2021.102034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong Z.Y., Yuan Z.Q., Dong Z.W., Peng Y.Z. Glutamine with probiotics attenuates intestinal inflammation and oxidative stress in a rat burn injury model through altered iNOS gene aberrant methylation. Am. J. Tourism Res. 2017;9(5):2535–2547. [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z.E., Wu D., Zheng L.W., Shi Y., Wang C., Chen Z.H., et al. Effects of glutamine on intestinal mucus barrier after burn injury. Am. J. Tourism Res. 2018;10(11):3833–3846. [PMC free article] [PubMed] [Google Scholar]
- 27.Musaogullari A., Chai Y.C. Redox regulation by protein S-glutathionylation: from molecular mechanisms to implications in health and disease. Int. J. Mol. Sci. 2020;21(21):8113. doi: 10.3390/ijms21218113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gallogly M.M., Mieyal J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007;7(4):381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Dalle-Donne I., Rossi R., Giustarini D., Colombo R., Milzani A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007;43(6):883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 30.Mieyal J.J., Gallogly M.M., Qanungo S., Sabens E.A., Shelton M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxidants Redox Signal. 2008;10(11):1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiong Y., Uys J.D., Tew K.D., Townsend D.M. S-glutathionylation: from molecular mechanisms to health outcomes. Antioxidants Redox Signal. 2011;15(1):233–270. doi: 10.1089/ars.2010.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rashdan N.A., Shrestha B., Pattillo C.B. S-glutathionylation, friend or foe in cardiovascular health and disease. Redox Biol. 2020;37 doi: 10.1016/j.redox.2020.101693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsui R., Ferran B., Oh A., Croteau D., Shao D., Han J., Pimentel D.R., et al. Redox regulation via glutaredoxin-1 and protein S-glutathionylation. Antioxidants Redox Signal. 2020;32(10):677–700. doi: 10.1089/ars.2019.7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalinina E., Novichkova M. Glutathione in protein redox modulation through S-glutathionylation and S-nitrosylation. Molecules. 2021;26(2):435. doi: 10.3390/molecules26020435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moreno-Sánchez R., Marín-Hernández Á., Gallardo-Pérez J.C., Vázquez C., Rodríguez-Enríquez S., Saavedra E. Control of the NADPH supply and GSH recycling for oxidative stress management in hepatoma and liver mitochondria. Biochim. Biophys. Acta Bioenerg. 2018;1859(10):1138–1150. doi: 10.1016/j.bbabio.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 36.Ying M., You D., Zhu X., Cai L., Zeng S., Hu X. Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol. 2021;46 doi: 10.1016/j.redox.2021.102065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen L., Zhang Z., Hoshino A., Zheng H.D., Morley M., Arany Z., et al. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nature metabolism. 2019;1:404–415. [PMC free article] [PubMed] [Google Scholar]
- 38.Yen W.C., Wu Y.H., Wu C.C., Lin H.R., Stern A., Chen S.H., et al. Impaired inflammasome activation and bacterial clearance in G6PD deficiency due to defective NOX/p38 MAPK/AP-1 redox signaling. Redox Biol. 2020;28 doi: 10.1016/j.redox.2019.101363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koju N., Qin Z.H., Sheng R. Reduced nicotinamide adenine dinucleotide phosphate in redox balance and diseases: a friend or foe? Acta Pharmacol. Sin. 2022;43(8):1889–1904. doi: 10.1038/s41401-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen S., Xia Y., Zhu G., Yan J., Tan C., Deng B., et al. Glutamine supplementation improves intestinal cell proliferation and stem cell differentiation in weanling mice. Food Nutr. Res. 2018;62 doi: 10.29219/fnr.v62.1439. 10.29219/fnr.v62.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alpers D.H. Glutamine: do the data support the cause for glutamine supplementation in humans? Gastroenterology. 2006;130(2 Suppl 1):S106–S116. doi: 10.1053/j.gastro.2005.11.049. [DOI] [PubMed] [Google Scholar]
- 42.Wischmeyer P.E. Glutamine in burn injury. Nutr. Clin. Pract. : Offi. Pub. Am.Soc. Parenter. Enteral Nutrit. 2019;34(5):681–687. doi: 10.1002/ncp.10362. [DOI] [PubMed] [Google Scholar]
- 43.Yang Y.J., Liu M.M., Zhang Y., Wang Z.E., Dan-Wu, Fan S.J., et al. Effectiveness and mechanism study of glutamine on alleviating hypermetabolism in burned rats. Nutrition. 2020;79–80 doi: 10.1016/j.nut.2020.110934. [DOI] [PubMed] [Google Scholar]
- 44.Polat I.H., Tarrado-Castellarnau M., Benito A., Hernandez-Carro C., Centelles J., Marin S., et al. Glutamine modulates expression and function of glucose 6-phosphate dehydrogenase via NRF2 in colon cancer cells. Antioxidants. 2021;10(9):1349. doi: 10.3390/antiox10091349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hernandez A., Patil N.K., Bohannon J.K. A murine model of full-thickness scald burn injury with subsequent wound and systemic bacterial infection. Walker W.E., editor. Sepsis. Method. Molecular Biol. 2021;2321:111–120. doi: 10.1007/978-1-0716-1488-4_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patil N.K., Bohannon J.K., Luan L., Guo Y., Fensterheim B., Hernandez A., et al. Flt3 ligand treatment attenuates T cell dysfunction and improves survival in a murine model of burn wound sepsis. Shock. 2017;47(1):40–51. doi: 10.1097/SHK.0000000000000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faezi S., Sattari M., Mahdavi M., Roudkenar M.H. Passive immunisation against Pseudomonas aeruginosa recombinant flagellin in an experimental model of burn wound sepsis. Burns. 2011;37(5):865–872. doi: 10.1016/j.burns.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 48.Gilpin D.A. Calculation of a new Meeh constant and experimental determination of burn size. Burns. 1996;22(8):607–611. doi: 10.1016/s0305-4179(96)00064-2. [DOI] [PubMed] [Google Scholar]
- 49.Nanni P., Mezzanotte L., Roda G., Caponi A., Levander F., James P., et al. Differential proteomic analysis of HT29 Cl.16E and intestinal epithelial cells by LC ESI/QTOF mass spectrometry. J. Proteonomics. 2009;72(5):865–873. doi: 10.1016/j.jprot.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 50.Cakir B., Cevik H., Contuk G., Ercan F., Ekşioğlu-Demiralp E., Yeğen B.C. Leptin ameliorates burn-induced multiple organ damage and modulates postburn immune response in rats. Regul. Pept. 2005;125(1–3):135–144. doi: 10.1016/j.regpep.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 51.Erben U., Loddenkemper C., Doerfel K., Spieckermann S., Haller D., Heimesaat M.M., et al. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol. 2014;7(8):4557–4576. [PMC free article] [PubMed] [Google Scholar]
- 52.Clark P.M., Dweck J.F., Mason D.E., Hart C.R., Buck S.B., Peters E.C., et al. Direct in-gel fluorescence detection and cellular imaging of O-GlcNAc-modified proteins. J. Am. Chem. Soc. 2008;130(35):11576–11577. doi: 10.1021/ja8030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brennan J.P., Miller J.I., Fuller W., Wait R., Begum S., Dunn M.J., et al. The utility of N, N-biotinyl glutathione disulfide in the study of protein S-glutathiolation. Mol. Cell. Proteomics: MCP. 2006;5(2):215–225. doi: 10.1074/mcp.M500212-MCP200. [DOI] [PubMed] [Google Scholar]
- 54.Durante W. The emerging role of l-glutamine in cardiovascular health and disease. Nutrients. 2019;11(9):2092. doi: 10.3390/nu11092092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo Y., Liu Y., Zhao S., Xu W., Li Y., Zhao P., et al. Oxidative stress-induced FABP5 S-glutathionylation protects against acute lung injury by suppressing inflammation in macrophages. Nat. Commun. 2021;12(1):7094. doi: 10.1038/s41467-021-27428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hwang S., Mruk K., Rahighi S., Raub A.G., Chen C.H., Dorn L.E., et al. Correcting glucose-6-phosphate dehydrogenase deficiency with a small-molecule activator. Nat. Commun. 2018;9(1):4045. doi: 10.1038/s41467-018-06447-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ayene I.S., Biaglow J.E., Kachur A.V., Stamato T.D., Koch C.J. Mutation in G6PD gene leads to loss of cellular control of protein glutathionylation: mechanism and implication. J. Cell. Biochem. 2008;103(1):123–135. doi: 10.1002/jcb.21394. [DOI] [PubMed] [Google Scholar]
- 58.Raub A.G., Hwang S., Horikoshi N., Cunningham A.D., Rahighi S., Wakatsuki S., et al. Small-molecule activators of glucose-6-phosphate dehydrogenase (G6PD) bridging the dimer interface. ChemMedChem. 2019;14(14):1321–1324. doi: 10.1002/cmdc.201900341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rao X., Duan X., Mao W., Li X., Li Z., Li Q., et al. O-GlcNAcylation of G6PD promotes the pentose phosphate pathway and tumor growth. Nat. Commun. 2015;6:8468. doi: 10.1038/ncomms9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang X., Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017;18(7):452–465. doi: 10.1038/nrm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brychtova V., Mohtar A., Vojtesek B., Hupp T.R. Mechanisms of anterior gradient-2 regulation and function in cancer. Semin. Cancer Biol. 2015;33:16–24. doi: 10.1016/j.semcancer.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 62.Al-Shaibi A.A., Abdel-Motal U.M., Hubrack S.Z., Bullock A.N., Al-Marri A.A., Agrebi N., et al. Human AGR2 deficiency causes mucus barrier dysfunction and infantile inflammatory bowel disease. Cellular Molecular gastroenterol. Hepatol. 2021;12(5):1809–1830. doi: 10.1016/j.jcmgh.2021.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Delom F., Nazaraliyev A., Fessart D. The role of protein disulphide isomerase AGR2 in the tumour niche. Biol. Cell. 2018;110(12):271–282. doi: 10.1111/boc.201800024. [DOI] [PubMed] [Google Scholar]
- 64.Maurel M., Obacz J., Avril T., Ding Y.P., Papadodima O., et al. Control of anterior GRadient 2 (AGR2) dimerization links endoplasmic reticulum proteostasis to inflammation. EMBO Mol. Med. 2019;11(6) doi: 10.15252/emmm.201810120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ryu J., Park S.G., Lee P.Y., Cho S., Lee D.H., et al. Dimerization of pro-oncogenic protein Anterior Gradient 2 is required for the interaction with BiP/GRP78. Biochem. Biophys. Res. Commun. 2013;430(2):610–615. doi: 10.1016/j.bbrc.2012.11.105. [DOI] [PubMed] [Google Scholar]
- 66.Ma Y., Zhou G., Li Y., Zhu Y., Yu X., Zhao F., et al. Intake of fish oil specifically modulates colonic Muc2 expression in middle-aged rats by suppressing the glycosylation process. Mol. Nutr. Food Res. 2018;62(4) doi: 10.1002/mnfr.201700661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Worfolk J.C., Bell S., Simpson L.D., Carne N.A., Francis S.L., Engelbertsen V., et al. Elucidation of the AGR2 interactome in esophageal adenocarcinoma cells identifies a redox-sensitive chaperone hub for the quality control of MUC-5AC. Antioxidants Redox Signal. 2019;31(15):1117–1132. doi: 10.1089/ars.2018.7647. [DOI] [PubMed] [Google Scholar]
- 68.Dalle-Donne I., Rossi R., Colombo G., Giustarini D., Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem. Sci. 2009;34(2):85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 69.Xiong Y., Manevich Y., Tew K.D., Townsend D.M. S-glutathionylation of protein disulfide isomerase regulates estrogen receptor α stability and function. Inter. J. Cell Biol. 2012 doi: 10.1155/2012/273549. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li K., Cui Y.C., Zhang H., Liu X.P., Zhang D., Wu A.L., et al. Glutamine reduces the apoptosis of H9C2 cells treated with high-glucose and reperfusion through an oxidation-related mechanism. PLoS One. 2015;10(7) doi: 10.1371/journal.pone.0132402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ou W., Liang Y., Qin Y., Wu W., Xie M., Zhang Y., et al. Hypoxic acclimation improves cardiac redox homeostasis and protects heart against ischemia-reperfusion injury through upregulation of O-GlcNAcylation. Redox Biol. 2021;43 doi: 10.1016/j.redox.2021.101994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singleton K.D., Wischmeyer P.E. Glutamine induces heat shock protein expression via O-glycosylation and phosphorylation of HSF-1 and Sp1. JPEN. J. Parenter. Enteral Nutr. 2008;32(4):371–376. doi: 10.1177/0148607108320661. [DOI] [PubMed] [Google Scholar]
- 73.Brasse-Lagnel C., Fairand A., Lavoinne A., Husson A. Glutamine stimulates argininosuccinate synthetase gene expression through cytosolic O-glycosylation of Sp1 in Caco-2 cells. J. Biol. Chem. 2003;278(52):52504–52510. doi: 10.1074/jbc.M306752200. [DOI] [PubMed] [Google Scholar]
- 74.Gong J., Jing L. Glutamine induces heat shock protein 70 expression via O-GlcNAc modification and subsequent increased expression and transcriptional activity of heat shock factor-1. Minerva Anestesiol. 2011;77(5):488–495. [PubMed] [Google Scholar]
- 75.Liu H.M., Ma L.L., Cao B., Lin J.Z., Han L., Li C.Y., et al. Progress in research into the role of abnormal glycosylation modification in tumor immunity. Immunol. Lett. 2021;229:8–17. doi: 10.1016/j.imlet.2020.11.003. [DOI] [PubMed] [Google Scholar]
- 76.Liu J., Marchase R.B., Chatham J.C. Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J. Mol. Cell. Cardiol. 2007;42(1):177–185. doi: 10.1016/j.yjmcc.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hamiel C.R., Pinto S., Hau A., Wischmeyer P.E. Glutamine enhances heat shock protein 70 expression via increased hexosamine biosynthetic pathway activity. Am. J. Physiol. Cell Physiol. 2009;297(6):C1509–C1519. doi: 10.1152/ajpcell.00240.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou Y., Eid T., Hassel B., Danbolt N.C. Novel aspects of glutamine synthetase in ammonia homeostasis. Neurochem. Int. 2020;140 doi: 10.1016/j.neuint.2020.104809. [DOI] [PubMed] [Google Scholar]
- 79.Jafri F., Seong G., Jang T., Cimpeanu E., Poplawska M., Dutta D., et al. L-glutamine for sickle cell disease: more than reducing redox. Ann. Hematol. 2022;101(8):1645–1654. doi: 10.1007/s00277-022-04867-y. [DOI] [PubMed] [Google Scholar]
- 80.Kim J.S., Martin M.J. Redox redux? Glutamine, catabolism, and the urea-to-creatinine ratio as a novel nutritional metric. Crit. Care Med. 2022;50(7):1156–1159. doi: 10.1097/CCM.0000000000005520. [DOI] [PubMed] [Google Scholar]
- 81.Oliveira I.A., Allonso D., Fernandes T., Lucena D., Ventura G.T., Dias W.B., et al. Enzymatic and structural properties of human glutamine: fructose-6-phosphate amidotransferase 2 (hGFAT2) J. Biol. Chem. 2021;296 doi: 10.1074/jbc.RA120.015189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Campbell S., Mesaros C., Izzo L., Affronti H., Noji M., Schaffer B.E., et al. Glutamine deprivation triggers NAGK-dependent hexosamine salvage. Elife. 2021;10 doi: 10.7554/eLife.62644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.