

Abstract
Patient: Male, 14-year-old
Final Diagnosis: Carney complex
Symptoms: Gynecomastia • testicular pain
Medication: —
Clinical Procedure: Invasive adrenal venous sampling
Specialty: Endocrinology and Metabolic
Objective:
Rare disease
Background:
Carney complex (CNC) is a rare multiple neoplasia syndrome with autosomal dominant inheritance. CNC is frequently misdiagnosed owing to its diverse clinical characteristics. We reported the case of a 14-year-old Saudi boy with a history of gynecomastia, Cushing syndrome, large-cell calcifying Sertoli cell tumor of the testis, and CNC.
Case Report:
The patient was referred to the pediatric endocrine clinic for evaluation of bilateral slow progressing gynecomastia for 1-year duration. His clinical examination revealed lentigenes, bilateral diffuse breast enlargement (consistent with Tanner stage III), and asymmetrical testicular enlargement, more on the left side. Other systemic examinations were unremarkable. The initial blood workup showed elevated estradiol level with unsup-pressed cortisol after an overnight 1-mg dexamethasone suppression test. Breast ultrasound (US) confirmed true gynecomastia. Testicular US revealed microcalcification and the testicular biopsy confirmed diagnoses of large-cell calcifying Sertoli cell tumor (LCCSCT). A 2-step dexamethasone suppression test showed a paradoxical rise in serum and urine cortisol levels, which are characteristic for PPNAD. LCCSCT and PPNAD are 2 major criteria fulfilling a diagnosis of CNC. The gene test showed heterozygous mutation in the PRKAR1A gene, which is diagnostic for CNC. The patient underwent bilateral mastoplasty and was planned for radical left orchiectomy.
Conclusions:
Gynecomastia and LCCSCT can be presenting features of CNC, which mandates careful, thorough clinical examination and tailored investigation to reach a diagnosis.
Keywords: Carney Complex, Sertoli Cell Tumor
Background
Carney complex (CNC) is a rare genetic disorder involving multiple neoplasia syndrome, first described in 1985 [1]. More than 50% of cases are familial owing to an autosomal dominant inheritance pattern, or appear occasionally as a result of a de novo genetic defect [2].
The pathogenesis of PPNAD is unclear, but it appears to be associated with pathogenic variants of the PRKAR1A gene [3]. CNC is inherited in an autosomal dominant pattern with high penetrance but a heterogeneous expression [4]. Approximately one-quarter of cases are due to de novo mutations. There are 2 genetic sites that have been associated with CNC. Inactivating mutations in the protein kinase A type I-alpha regulatory subunit (PRKAR1A) gene on chromosome 17q22-24 are found in most CNC patients [5]. A second locus on chromosome 2p16 is also associated with CNC [6]. Cushing syndrome is the most frequently observed endocrine tumor in CNC, occurring in approximately 25% of affected individuals. Large-cell calcifying Sertoli cell tumors (LCCSCTs) are observed in one-third of affected males within the first decade of life and in most adult males. Up to 75% of individuals with CNC have multiple thyroid nodules, most of which are nonfunctioning thyroid follicular adenomas. Clinically evident acromegaly from a growth hormone (GH)-producing adenoma occurs in approximately 10% of adult patients. Psammomatous melanotic schwannoma (PMS), a rare tumor of the nerve sheath, occurs in an estimated 10% of affected individuals. The median age at diagnosis is 20 years. The diagnosis of CNC is established in a proband with 2 or more major diagnostic criteria and/or by identification of a heterozygous germline pathogenic variant in PRKAR1A on molecular genetic testing [7,8]. It has been observed that most CNC patients (>80%) have spotty skin pigmentation or skin growths, which usually emerge early in life and can occur anywhere on the body, classically on the mucosa, lips, face, and genital region [2]. Nevertheless, the majority of non-cutaneous lesions found in CNC are cardiac myxomas, accounting for over 50% of CNC-related deaths [9–11].
Sertoli cell tumor is a rare subtype of sex cord-stromal tumors and it contributes to less than 1% of all testicular tumors [12,13]. However, there are 3 subtypes of Sertoli cell tumors: not otherwise specified, and 2 variants (sclerosing Sertoli cell tumors and LCCSCTs) [13]. LCCSCT is an uncommon variant of Sertoli cell tumor [14]. LCCSCT is typically described by distinguishing histology: an intratubular growth pattern, with hyalinization of the tubular basement membrane and cells arranged in circumscribed nodules. Large round Sertoli cells with abundant cytoplasm are characteristic, in addition to massive calcifications, which are essential for the diagnosis of LCCSCT [14]. To the best of our knowledge, our case report is the first to describe LCCSCT in a Saudi boy associated with CNC for which conservative treatment was initially provided along with regular follow-up. Conservative treatment is initially indicated if there is no sign of distressing malignancy. Physical and ultra-sonographic examinations are necessary once a year as part of the follow-up [15]. Aromatase inhibitors can be an efficacious treatment option for patients with LCCSCT [16], but data from clinical trials are needed. The present case is comparable to that of an adolescent who presented with a testicular mass at age 9 years followed by PPNAD diagnosed at age 22 years [1].
CNC is a rare condition, but over 750 patients have been described in different ethnic groups [7]. Given the significant variability in the clinical manifestations, even within a given family, careful clinical evaluation of first-degree family members is warranted in presumed “sporadic” cases [17]. Familial transmission has been reported more frequently via the affected mother, suggesting a non-Mendelian inheritance or impaired male fertility of affected individuals [17]. Early diagnosis allows vigilant continuing observation of tumors and complications, thus improving disease prognosis by early treatment. Herein, we report the case of a 14-year-old boy who had CNC with large-cell calcifying Sertoli cell tumor (LCCSCT) of the testis and primary pigmented nodular adrenocortical disease (PPNAD).
Case Report
A 14-year-old boy was referred to our Endocrinology Department at King Fahd Medical City (KFMC) in 2017 for evaluation of bilateral breast enlargement and hyperprolactinemia. The bilateral breast enlargement was noticed by the family 1 year prior to presentation. It was slowly progressive but not painful, with no discharge from nipples or galactorrhea, and no skin changes over the breast area. There was no report of headache, vomiting, visual impairment symptoms, or acne. He had no history of intracranial tumor or head irradiation. The parents of the patient were second-degree cousins. The patient was the eldest child among 3 healthy siblings and was developmentally normal, with average school performance.
The examination revealed a healthy-appearing patient who was not dysmorphic, pale, or jaundiced. His weight was 46 kg (90th percentile), height was 156 cm (90th percentile), and body mass index was 18.5 kg/m2 (80th percentile). Lentigines were found on the membrane lining the eyes and on the sclera (Figure 1). There was hyperpigmentation of the gingiva. Bilateral diffuse breast enlargement was consistent with Tanner stage III. Also, he had neither focal breast lesion nor palpable axillary lymph nodes. Pubic Hair Tanner stage was II. The physical examination also revealed enlargement of the left testis.
Figure 1.

(A, B) A 14-year-old Saudi boy with gynecomastia, Cushing syndrome, large-cell calcifying Sertoli cell tumor of the testis, and Carney complex. The eyes show lentigines in the conjunctival membranes (arrows).
The clinical laboratory findings revealed a serum prolactin level of 576 ng/mL (normal is less than 20 ng/ml) and serum estradiol 178 pg/mL (normal is 10–50 pg/ml). However, other laboratory findings, such as complete blood count, liver function test, and kidney function test, were normal. The serum sodium and potassium levels were 136 mEq/L and 3.9 mEq/L, respectively. Lactic acid dehydrogenase, luteinizing hormone, follicle-stimulating hormone, testosterone, and dehydroepiandrosterone sulfate were in normal ranges. The β-Human chorionic gonadotropin hormone and α-fetoprotein serum levels showed a normal profile.
An ultrasonography evaluation of the breast revealed prominent fibro-fatty tissue bilaterally, likely a manifestation of physiological gynecomastia. However, there was no evidence of suspicious solid or cystic mass lesions. An ultrasonography evaluation of the testis revealed multiple microcalcifications (Figure 2). The computed tomography (CT) scan of the pelvis revealed bilateral testicular calcification, larger on the left side (Figure 3).
Figure 2.
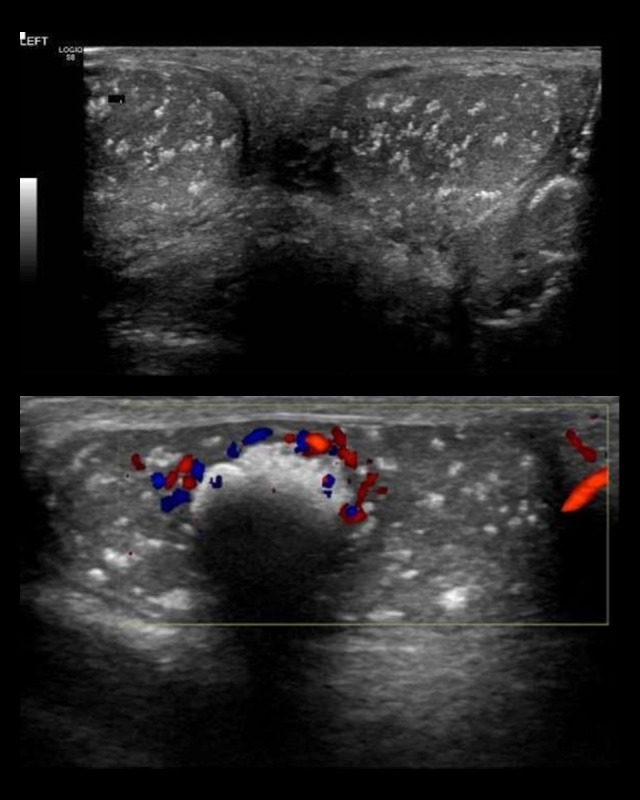
A 14-year-old Saudi boy with gynecomastia, Cushing syndrome, large-cell calcifying Sertoli cell tumor of the testis, and Carney complex. Testicular ultrasound imaging of the testes shows diffuse calcifications within the tumor tissue (arrows).
Figure 3.
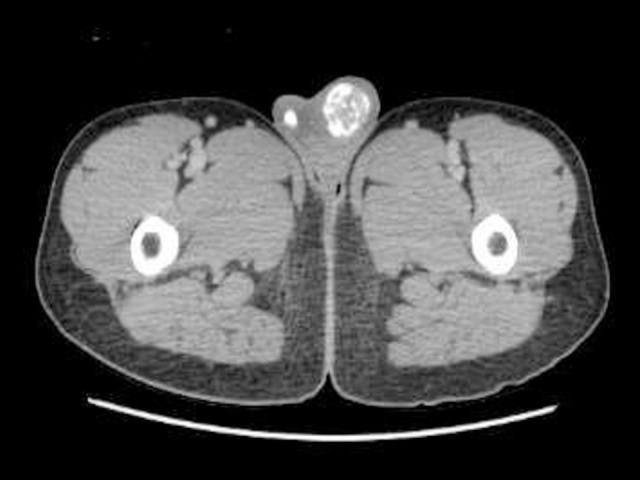
Computed tomography ( CT ) scan of the pelvis showed bilateral testicular calcification more on the left side.
Based on findings of ultrasonography, testicular biopsy was performed, which revealed a large-cell calcifying Sertoli cell tumor (Figure 4). The case was discussed in the Tumor Board meeting and conservative treatment with regular follow-up was initiated.
Figure 4.

Photomicrograph of the diagnostic histopathology of a large cell calcifying Sertoli cell tumor of the testis in a 14-year-old Saudi boy. The tumor consists of cords and nests of uniform pale eosinophilic cells with small regular nuclei. There is no necrosis, and no mitoses are present. A focus of microcalcification is seen (arrow), which is typical of large cell calcifying Sertoli cell tumor of the testis. Hematoxylin and eosin (H&E). Magnification ×40.
High-dose adrenocorticotropic hormone (ACTH) stimulation testing revealed a normal profile and did not suggest a diagnosis of adrenal insufficiency. Similarly, molecular genetics reports on congenital adrenal hyperplasia also were negative. The low-dose (1-mg) overnight dexamethasone suppression test showed high cortisol levels, at 139 nmol/L (a normal response to dexamethasone suppression test should be less than 50 nmol/l). The 6-day (2-step) standard dexamethasone suppression test (Table 1) showed a low serum ACTH level, at 0.8 (normal range: 5–50 pg/mL), a paradoxical rise in serum cortisol level to 503 nmol/l, and 24-h urine free cortisol more than 1000 nmol/L on day 6 (normally 15–224 nmol/l), which is characteristic of primary pigmented nodular adreno-cortical disease (PPNAD). Therefore, the diagnosis of ACTH-independent Cushing syndrome (CS) was confirmed. The left testicular mass biopsy report (Figure 4) described a sex cord-stromal tumor, favoring large-cell calcifying Sertoli cell tumor. Thus, because of large-cell calcifying Sertoli cell tumor of the testis and PPNAD, the diagnosis of CNC was suspected.
Table 1.
Six-day standard (2-step) dexamethasone suppression test.
| ACTH Pmol/l | Cortisol nmol/L | 24-h urine cortisol nmol/L | |
|---|---|---|---|
| Baseline | 1.4 (1.6–13.9) | 215 | 333+ (15–224) |
| 1.6 | 232 | ||
| 4th day | 1.8 | 270 | 805++ |
| 6th day | 0.8 | 503 | 1000++ |
ACTH – adrenocorticotropic hormone; Pmol/l – picomoles per liter; nmol/l – nanomoles per liter.
Molecular genetic analysis (Table 2).
Table 2.
Gene finding description.
| Gene | Variant coordinates | Amino acid change | SNP identifier | Zygosity | Type and classification |
|---|---|---|---|---|---|
| PRKAR1A | NM_001276289.1: c.682C >T | p.(Arg228*) | RS281864784 | Heterozygous | Nonsense Likely Pathogenic |
PRKAR1A – Protein Kinase CAMP-Dependent Type 1 Regulatory Subunit Alpha, SNP – single nucleotide polymorphism. Molecular analysis, using a next-generation sequencing (NGS) panel for LCCST, identified heterozygous pathogenic variant in the PRKAR1A gene (nonsense mutation at nucleotide number 682 with change of nucleobase cytosine to thiamine, resulting of amino acid change to arginine).
A blood sample was sent to Centogene Lab (Germany), at which a gene panel for LCCSCT using next-generation sequencing (NGS) was done and showed heterozygous, nonsense mutation in the PRKAR1A gene, likely a pathogenic variant, consistent with the genetic diagnoses of Carney complex type 1.
Regarding other possible comorbidities of CNC, a wide examination was performed to exclude the involvement of other organs. Ultrasonography for the thyroid and pelvis, as well as echocardiography, did not reveal abnormalities. Brain and pituitary gland MRI were normal.
Invasive bilateral adrenal venous sampling was done (Table 3), and showed almost double the normal amount of cortisol secretion from the left adrenal gland, which confirmed the presence of left adrenal lateralization (predominance) as a source of cortisol secretion. After confirming the diagnosis, the family members were offered genetic testing for the same mutation (target gene mutation test), the mother and siblings have not carried the mutation (wild-type gene). His father was unable to undergo genetic testing because of special social circumstances. The patient underwent bilateral mastoplasty because of distressing gynecomastia. For the Cushing syndrome, the conservative management vs laparoscopic adrenalectomy was initially discussed with the family, and as the patient was not showing any apparent clinical manifestation of Cushing syndrome (eg, hypertension or abnormal BMI), we agreed on conservative management with regular follow-up.
Table 3.
Invasive adrenal venous sampling for cortisol, aldosterone, and adrenaline levels.
| Right adrenal | Left adrenal | Inferior vena cava | |
|---|---|---|---|
| Cortisol | 3532 nmol/l | 6290 nmol/l | 180 nmol/l |
| Aldosterone | 960 ng/dl | 383 ng/dl | 8.6 ng/dl |
| Adrenaline | 5310 ng/l | 1020 ng/l | 80 ng/l |
nmol/l – nanomoles per liter; ng/dl – nanograms per deciliter.
The patient is 16 years old at present. During regular follow-up visits in the urology clinic, he has been reporting left testicular pain, for which testicular US was repeated and showed significant progression of left testicular calcification, and a radical left orchiectomy was planned. In the endocrine clinic, he started to show intermittent elevation in blood pressure, higher BMI (29–31) kg/m2, and new appearance of pink stria on the abdominal wall. His repeated Hba1c, thyroid function test, and IGF-1 levels were normal. For the Cushing features, 24-h urine collection for urinary free cortisol (UFC) excretion showed a level of 34.7 mcg/m2/day (normal level is less than 70 mcg/m2/day), with follow-up for reevaluation.
Discussion
This case report shows that gynecomastia can be the presenting feature of an underlying multisystem disorder, which mandates thorough examination and tailored investigation to reach a diagnosis. We presented a patient with classic clinical characteristics of CNC, including spotty skin pigmentation on the membrane lining the eyes and on the sclera, LCCSCT, and ACTH-independent Cushing syndrome – PPNAD. The molecular genetic analysis identified a known pathogenic mutation of PRKAR1A. Considering all this together, our patient met the diagnostic criteria of CNC (Table 4) [7]: (1) spotty skin pigmentation with the distinctive presence on the face, lip, and sclera; (2) LCCSCT, as confirmed by testicular biopsy result; and (3) ACTH-independent Cushing syndrome caused by PPNAD (as identified in our patient).
Table 4.
Diagnostic criteria of Carney complex.
Major criteria
|
Supplemental criteria
|
Adapted from Correa R, Salpea P, Stratakis CA. Carney complex: An update. Eur J Endocrinol. 2015;173:M85-97 [3].
The patient has also manifested primary pigmented nodular adrenal disease (PPNAD), in association with ACTH-independent Cushing syndrome (CS). The diagnosis of ACTH-independent CS related to PPNAD can be challenging and the best screening test for CS is measurement of urine free cortisol excretion [18]. The current case report found more than 1000 nmol/L free cortisol in a 24-h urine sample on day 6 of the dexamethasone suppression test. Moreover, a low serum ACTH level of 0.8 (normal range: 5–50 pg/mL) and serum cortisol level of 503 nmol/L were noted. Our patient had a less aggressive course of the CNC in comparison to a previous case report by Rosenblum et al in 2017 [1], in which the patient had LCCSCT, pancreatic cancer, and Cushing syndrome due to adrenocortical carcinoma.
The CT examination of the adrenal gland with PPNAD is often interpreted as normal and occasionally it represents the appearance of a string of beads due to the presence of multiple small nodules in the otherwise atrophic adrenal cortex [19]. However, it is worth emphasizing that, in cases like this, because of the patient’s low levels of ACTH and normal adrenal imaging, a hypothesis of exogenous glucocorticoid must also be persistently investigated and ruled out. Furthermore, it is important to make sure that the sampling and assaying of ACTH have been performed adequately; otherwise, the ACTH values might be falsely found to be low, thus inducing diagnostic error [20]. PPNAD is a rare form of ACTH-independent CS that can appear alone, but 90% of cases are linked to CNC [21]. The symptoms are often mild, and patients typically present long after the onset of symptoms. The disease commonly appears during the second decade of life, but an age range of 4–44 years has been recorded in the literature [17,22]. CS in PPNAD can present as overt CS, subclinical CS, cyclic CS, or atypical (asthenic) CS, of which overt CS is the most common [23]. Our patient was classified as having subclinical CS. The preferred treatment option for PPNAD with overt CS is bilateral total adrenalectomy [24]. A laparoscopic approach has a lower rate of morbidity in comparison to the open technique, and it involves less postoperative pain, shorter length of hospital stay, and low overall cost [19]. In case of surgical failure or before adrenalectomy, pharmacotherapy with ketoconazole, metyrapone, mitotane, and trilostane alone or in combination can control hypercortisolism by inhibiting steroidogenesis. Fluconazole has recently been suggested as a safer alternative to ketoconazole [25].
Conclusions
We presented a case of CNC with gynecomastia, large-cell calcifying Sertoli cell tumor (LCCSCT) of the testis and primary pigmented nodular adrenocortical disease. PPNAD is a rare cause of ACTH-independent CS in childhood and can indicate underlying CNC. Moreover, LCCSCTs are very rare tumors and most of them are benign. It is highly recommended that clinical assessments and genetic tests be conducted promptly in such patients. The account of clinical manifestations of this patient adds to the knowledge about CNC and PRKAR1A mutations. The ability to distinguish CNC is important for the prevention of severe complications. PRKAR1A mutation analysis must be conducted as soon as possible in suspected CNC patients.
Footnotes
Declaration of Figures’ Authenticity
All figures submitted have been created by the authors who confirm that the images are original with no duplication and have not been previously published in whole or in part.
References:
- 1.Rosenblum F, Koenig RG, Mikhail FM, et al. An adolescent with large cell calcifying Sertoli cell tumor of the testis and undiagnosed Carney Complex: A case report. Diagn Cytopathol. 2017;45(7):634–39. doi: 10.1002/dc.23700. [DOI] [PubMed] [Google Scholar]
- 2.Sandrini F, Stratakis C. Clinical and molecular genetics of Carney complex. Mol genet Metab. 2003;78(2):83–92. doi: 10.1016/s1096-7192(03)00006-4. [DOI] [PubMed] [Google Scholar]
- 3.Bertherat J. Carney complex (CNC) Orphanet J Rare Dis. 2006;1(1):21. doi: 10.1186/1750-1172-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papanastasiou L, Fountoulakis S, Voulgaris N, et al. Identification of a novel mutation of the PRKAR1A gene in a patient with Carney complex with significant osteoporosis and recurrent fractures. Hormones. 2016;15(1):129–35. doi: 10.14310/horm.2002.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirschner LS, Carney JA, Pack SD, et al. Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26(1):89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 6.Matyakhina L, Pack S, Kirschner LS, et al. Chromosome 2 (2p16) abnormalities in Carney complex tumours. J Med Genet. 2003;40(4):268–77. doi: 10.1136/jmg.40.4.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Correa R, Salpea P, Stratakis CA. Carney complex: An update. Eur J Endocrinol. 2015;173(4):M85–97. doi: 10.1530/EJE-15-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Q, Tong D, Liu G, et al. Carney complex with PRKAR1A gene mutation: A case report and literature review. Medicine (Baltimore) 2017;96(50):e8999. doi: 10.1097/MD.0000000000008999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Espiard S, Bertherat J. Carney complex. Front Hom Res. 2013;41:50–62. doi: 10.1159/000345669. [DOI] [PubMed] [Google Scholar]
- 10.Bertherat J, Horvath A, Groussin L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5’-monophosphate-dependent protein kinase (PRKAR1A): Phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab. 2009;94(6):2085–91. doi: 10.1210/jc.2008-2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boikos Sosipatros A, Stratakis Constantine A. Carney complex: The first 20 years. Curr Opin Oncol. 2007;19(1):24–29. doi: 10.1097/CCO.0b013e32801195eb. [DOI] [PubMed] [Google Scholar]
- 12.Sato K, Ueda Y, Sakurai A, et al. Large cell calcifying Sertoli cell tumor of the testis: Comparative immunohistochemical study with Leydig cell tumor. Pathol Int. 2005;55(6):366–71. doi: 10.1111/j.1440-1827.2005.01838.x. [DOI] [PubMed] [Google Scholar]
- 13.Moch H, Cubilla AL, Humphrey PA, et al. The 2016 WHO classification of tumours of the urinary system and male genital organs – part A: Renal, penile, and testicular tumours. Eur Urol. 2016;70(1):93–105. doi: 10.1016/j.eururo.2016.02.029. [DOI] [PubMed] [Google Scholar]
- 14.Proppe KH, Scully RE. Large-cell calcifying Sertoli cell tumor of the testis. Am J Clin Pathol. 1980;74(5):607–19. doi: 10.1093/ajcp/74.5.607. [DOI] [PubMed] [Google Scholar]
- 15.Gourgari E, Saloustros E, Stratakis CA. Large-cell calcifying Sertoli cell tumors of the testes in pediatrics. Curr Opin Pediatr. 2012;24(4):518–22. doi: 10.1097/MOP.0b013e328355a279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grandone A, del Giudice EM, Cirillo G, et al. Prepubertal gynecomastia in two monozygotic twins with Peutz-Jeghers syndrome: Two years’ treatment with anastrozole and genetic study. Horm Res Paediatr. 2011;75(5):374–79. doi: 10.1159/000324178. [DOI] [PubMed] [Google Scholar]
- 17.Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: Diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metabol. 2001;86(9):4041–46. doi: 10.1210/jcem.86.9.7903. [DOI] [PubMed] [Google Scholar]
- 18.Navarro Moreno C, Delestienne A, Marbaix E, et al. Familial forms of Cushing syndrome in primary pigmented nodular adrenocortical disease presenting with short stature and insidious symptoms: A clinical series. Horm Res Paediatr. 2018;89(6):423–33. doi: 10.1159/000488761. [DOI] [PubMed] [Google Scholar]
- 19.Bavadiya G, Roy A, Sarkar KK, et al. Primary pigmented nodular adrenocortical disease (PPNAD) presenting as Cushing syndrome in a child and review of literature. Acta Endocrinol (Buchar) 2020;16(3):362–65. doi: 10.4183/aeb.2020.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castro M, Moreira AC. Diagnóstico laboratorial da síndrome de Cushing. Arquivos Brasileiros de Endocrinologia & Metabologia. 2002;46(1):97–105. [in Portuguese] [Google Scholar]
- 21.Sandrini F, Stratakis C. Clinical and molecular genetics of primary pigmented nodular adrenocortical disease. Arq Bras Endocrinol Metab. 2004;48(5):637–41. doi: 10.1590/s0004-27302004000500007. [DOI] [PubMed] [Google Scholar]
- 22.Manipadam MT, Abraham R, Sen S, Simon A. Primary pigmented nodular adrenocortical disease. J Indian Assoc Pediatr Surg. 2011;16(4):160–62. doi: 10.4103/0971-9261.86881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Memon SS, Thakkar K, Patil V, et al. Primary pigmented nodular adrenocortical disease (PPNAD): single centre experience. J Pediatr Endocrinol Metab. 2019;32(4):391–97. doi: 10.1515/jpem-2018-0413. [DOI] [PubMed] [Google Scholar]
- 24.Powell AC, Stratakis CA, Patronas NJ, et al. Operative management of Cushing syndrome secondary to micronodular adrenal hyperplasia. Surgery. 2008;143(6):750–58. doi: 10.1016/j.surg.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riedl M, Maier C, Zettinig G, et al. Long term control of hypercortisolism with fluconazole: case report and in vitro studies. Eur J Endocrinol. 2006;154(4):519–24. doi: 10.1530/eje.1.02120. [DOI] [PubMed] [Google Scholar]