

Abstract
CD3-targeted lentiviral vectors (CD3-LVs) mediate selective transduction of human T lymphocytes in vitro and in vivo while simultaneously activating the targeted cells. Previously, we have demonstrated that CD3-LV leads to downmodulation of the CD3:T cell receptor (TCR) complex. We therefore hypothesized that inhibition of CD3 phosphorylation by Src/Abl tyrosine kinase inhibitors such as dasatinib results in enhancement of gene delivery by T cell-targeted LVs. Indeed, dasatinib treatment of T cells prior to incubation with CD3-LV increased reporter gene delivery by 3- to 10-fold. Moreover, the presence of dasatinib enhanced selective transduction into non-activated target cells present in whole blood. When combined with delivery of the CD19-chimeric antigen receptor (CAR) gene, dasatinib increased CAR T cell numbers by close to 10-fold. Importantly, the short-term exposure of T cells to dasatinib during vector incubation did not interfere with tumor cell killing by the resulting CAR T cells and rather came along with less upregulated exhaustion markers and a more naive phenotype. Our data suggest that dasatinib prevents CD3-LV-induced phosphorylation and CD3:TCR intake, thereby increasing the amount of CD3-LV bound to the cell surface. This is the first description of dasatinib as transduction enhancer, an activity particularly relevant for CAR T cell generation with CD3-LV.
Keywords: dasatinib, T cell targeting, CD3-LV, transduction enhancer, CAR T cells, exhaustion, T cell receptor
Graphical abstract
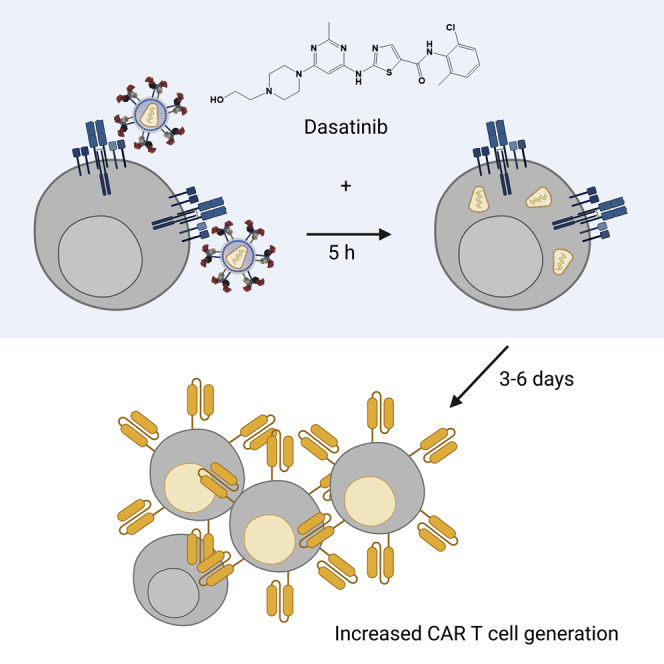
This paper describes the discovery of dasatinib as transduction enhancer for gene delivery into T lymphocytes by CD3-targeted lentiviral vectors. Dasatinib acts through preventing CD3:TCR intake, thereby increasing the amount of vector particles bound to the T cell surface. Improved generation of CAR T cells is illustrated as an immediate application.
Introduction
Chimeric antigen receptor (CAR) T cells have shown great success in the treatment of B cell lymphoma and recently also against multiple myeloma, with five approved products on the European market and several trials ongoing.1,2,3 Most CAR T cell products are generated by genetic modification of T cells using lentiviral vectors (LVs) pseudotyped with the vesicular stomatitis virus (VSV) glycoprotein G. Still, many challenges remain in the manufacturing process, which in its current form is highly laborious, time intensive, and expensive.4 T cells need to be isolated from the patient’s blood by leukapheresis, activated through the endogenous CD3:T cell receptor (TCR) using recombinant antibodies against CD3 and CD28, genetically modified by viral vectors, and expanded several days prior to reinfusion into the patient.5,6 The first step, being the T cell isolation from blood, needs to be performed very accurately as remaining non-target leukemic cells can cause a safety issue during gene transfer. Secondly, T cell activation is time consuming and causes progressive differentiation, shown to be associated with loss of anti-leukemic effect.7 During the step of gene delivery, the transduction of solely target cells is crucial as gene transfer to a single leukemic cell can lead to a CAR T cell-resistant clone followed by relapse and death of the patient.8 At last, expansion of CAR T cells prior to reinjection further prolongs ex vivo culturing time, leading to progressive differentiation and eventual exhaustion of the cells.
Some hurdles may be overcome by T cell-targeted vectors that deliver the CAR gene precisely into T lymphocytes. Such LVs are pseudotyped with engineered envelope glycoproteins derived from alphaviruses or paramyxoviruses.9 For the latter, attachment and membrane fusion proteins from measles virus (MV) or Nipah virus (NiV) are in use.10,11 The attachment glycoprotein is blinded toward its natural receptor and is then fused to a targeting ligand such as an antibody fragment (here: single-chain variable fragment [scFv]) that confers the desired specificity for the cell surface receptor of choice. Based thereupon, a CD8-targeted LV was generated and shown to deliver CAR genes with more than 99% precision into its targeted subpopulation of T lymphocytes.12,13,14,15 More recently, LVs have been engineered to specifically recognize the T cell surface antigens CD4 or CD3.16,17,18 CD3-LVs recognize both, CD4+ and CD8+ T cell subsets, equally well, while remaining highly selective for T cells. An additional feature of CD3-LVs are their agonistic activity: These vectors are able to stimulate resting T cells, consequently mediating gene transfer to unstimulated and cytokine-only-stimulated T cells at similar efficiencies as activated T cells, outperforming VSV-LV transduction under these conditions.18 Such vectors are important novel tools in gene therapy and immunotherapy, since they have the potential to increase safety and to simplify the complex manufacturing process of T cell therapy by shortening ex vivo time and yielding a better cellular product.
To implement targeted LVs as therapeutical vectors in gene therapy, their moderate gene delivery rates must be improved. The different cell entry mechanisms might affect gene delivery rates. Targeted LVs based on MV- and NiV-derived glycoproteins undergo pH-independent cell binding and fusion, whereas VSV-LV relies on acidification of the endosomes.19,9 Efficient cell contact necessary for pH-independent cell entry might not be sufficiently ensured and result in reduced gene transfer by T cell-targeted LVs. To circumvent this issue, transduction enhancers can be used to improve gene delivery. The most commonly used transduction enhancers, mainly polymers such as Vectofusin-1 (VF-1),20 polybrene,21 or LentiBOOST,22 are known to increase lentiviral transduction by receptor independent vector-to-cell association, e.g., by facilitating vector-to-cell binding through nanofibril-mediated cell contact and neutralization of charge repulsion or cell entry by permeabilization of the cell membrane. VF-1 enhanced transduction for all T cell-targeted LVs 2- to 3-fold, which for CD3-LV, however, resulted in only moderate transduction levels.20,18 This might be caused by CD3-LV’s strong agonistic effect on T cells mediated by CD3 cross-linking leading to T cell activation and downregulation of CD3:TCR.18,23
Dasatinib is a small molecule tyrosine kinase inhibitor approved for the treatment of Philadelphia chromosome-positive chronic myeloid leukemia and acute lymphoblastic leukemia.24 As it has less stringent binding requirements and the ability to recognize multiple states of the kinase, in its active as well as inactive form, dasatinib functions against many imatinib-resistant kinase domain mutations of BCR-ABL, resulting in enhanced therapeutic potential.25 As a member of the Src/Abl tyrosine kinase inhibitor family, dasatinib also interferes with Lck, an upstream kinase in the T cell activation cascade.23,26 By blocking Lck, phosphorylation of the CD3-zeta domain and continuously ZAP-70 are inhibited, preventing activation-induced downregulation of the TCR.23,26
Here we show that dasatinib increased CD3-LV-mediated gene transfer to resting and activated T cells by blocking T cell activation. Mechanistically, activation inhibition increased the amount of CD3:TCR-bound vector, resulting in increased numbers of functionally active CAR T cells.
Results
Enhanced transduction of activated and non-activated primary T lymphocytes by dasatinib
To test if dasatinib enhances gene transfer mediated by CD3-LV, a NiV glycoprotein Gmut pseudotyped LV11 displaying a scFv derived from the CD3-specific agonistic antibody TR66 (TR66.opt)18 was used. We started out with reporter gene transfer by delivering gfp under control of the SFFV promotor into activated human peripheral blood mononuclear cells (PBMCs) exposed to 50 nM dasatinib for 1 h prior to LV addition. Increasing amounts, starting at four particles per cell, were added and dasatinib was removed 4 h after LV addition. Treating the cells with dasatinib increased transduction efficiencies by 3- to 10-fold, causing a significantly higher increase when applying low vector amounts (Figure 1A). When determining gene transfer activities of different CD3-LV stocks on primary human lymphocytes, dasatinib increased their titers 8-fold on average (Figure S1A). Next, we investigated the optimal conditions for dasatinib treatment of PBMCs by applying different dasatinib concentrations and incubation times. When testing increasing dasatinib concentrations with constant vector amounts of 2.5 × 1010 particles, a dose dependency was observed: the lowest concentration of 0.39 nM dasatinib was not sufficient to enhance transduction, while a concentration of 6.25 nM increased transduction by 2-fold. A plateau was reached at concentrations ≥50 nM (Figure 1B). A dasatinib incubation time of 5 h was optimal, since a shorter period reduced the effect, and a longer incubation did not improve transduction efficiency further (Figures 1C and 1D). Importantly, none of the treatment conditions compromised T cell viability or T cell proliferation (Figures S1B and S1C). Thus, incubation of activated T cells for 5 h with 50 nM dasatinib was optimal for transduction enhancement. Interestingly, dasatinib was more potent as transduction enhancer for the CD3-targeted LV than VF-1, but the combination with VF-1 further boosted gene delivery (Figure S2A). Dasatinib had no effect on the pH-dependent cell entry of VSV-LV, and in line with previous data, VF-1 strongly inhibited VSV-LV-mediated transduction on T cells in absence or presence of dasatinib (Figure S2B).20
Figure 1.

Dasatinib enhances CD3-targeted gene transfer into activated PBMCs in a time- and dose-dependent manner
Freshly thawed primary human PBMCs were activated for 3 days with αCD3 and αCD28 antibodies in presence of IL-7/15 prior to transduction with CD3-LV. (A) Cells were incubated in 50 nM dasatinib-containing medium for 5 h with increasing particle numbers. Fold increase in transduction compared with transduction in absence of dasatinib is shown for four vector stocks. Each data point is an average value referring to a particular vector stock tested on one to four donors. ∗p < .05, by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. (B) Cells were incubated in 0.39–100 nM dasatinib-containing medium for 7 h in presence of vector particles. Dose-dependent increase in GFP-expression 3 days after transduction with 2.5 × 1010 CD3-LV particles is shown for four to five donors. (C and D) Cells were incubated without (w/o), 25 nM, 50 nM, or 100 nM dasatinib-containing medium for 3 h, 5 h, or 7 h in presence of vector particles. (C) Representative dot plots of GFP-expression 3 days after transduction. (D) Superimposed symbols for different concentrations connected by line show GFP-expression 3 days after transduction. Data are based on three donors, and values are shown as mean ± standard error of the mean (SEM).
To assess if dasatinib also enhances transduction into non-activated T cells in an in vivo-like setup, in which all components active in blood such as erythrocytes, thrombocytes, and plasma proteins are present, dasatinib was mixed into human whole blood followed by the addition of vector particles (Figure 2A). CD3-LV transduced 0.5% of T cells without and above 2% in presence of dasatinib exclusively within the CD3+ cell population (Figures 2B and 2C). In contrast, VSV-LV showed lower transduction rates into both CD3+ and CD3− cells (0.1%–0.4%) regardless of dasatinib exposure. Thus, the transduction-enhancing activity of dasatinib was confirmed for non-activated T cells and not affected by components and cells present in blood.
Figure 2.

Dasatinib increases transduction of non-activated human T cells in whole blood
Whole blood from healthy donors was incubated with 100 nM dasatinib or without (w/o) for 1 h prior to addition of 8.5 × 1010 particles CD3-LV or 3 × 1010 particles VSV-LV. PBMCs were subsequently isolated by density centrifugation and cultivated in T cell medium (TCM) or NutriT medium with IL-7/15 for 5–6 days. (A) Experimental design. (B) Representative dot plots showing GFP-expression 6 days post transduction. Numbers indicate GFP-positive cells of all viable cells. (C) Bar diagrams show fold increase of GFP-positive cells 5–6 days after transduction determined by flow cytometry. Data represent mean ± SEM values of three donors. Ns, non-significant; ∗p < .05, by ratio paired t test.
Enhanced vector particle cell association and reduced agonistic activity of CD3-LV in presence of dasatinib
To better understand the mechanism behind dasatinib’s transduction-enhancing effect, we investigated the effect of dasatinib treatment on CD3-LV’s agonistic activity. Freshly isolated PBMCs were exposed to IL-7/15 overnight and subsequently incubated with dasatinib 1 h prior to incubation with vector particles. Both, αCD3/αCD28- and CD3-LV-treated cells showed an upregulation of the activation marker CD25 to above 80% compared with background expression of around 20%. In presence of dasatinib, however, this effect was reduced: incubation with activating antibodies only led to a slight upregulation of CD25, while CD25 levels upon incubation with CD3-LVs remained close to background. As expected, CD8-and VSV-LV, which do not possess agonistic activity, did not increase CD25 expression neither in presence nor absence of dasatinib (Figure 3A). These results are in line with dasatinib’s interference in T cell activation through inhibition of the tyrosine kinase Lck.23,26
Figure 3.

Dasatinib prevents T cell activation causing increased availability of the CD3 receptor for CD3-LV binding and transduction
(A) PBMCs were cultured in IL-7/15 supplemented medium overnight without further stimulation. The next day, cells were treated with activating antibodies αCD3/αCD28, CD3-LV, VSV-LV, CD8-LV or remained untreated (ut). Cells were incubated in 100 nM dasatinib-containing medium or medium without (w/o) dasatinib for 7 h during vector incubation. Bar diagrams show expression of the activation marker CD25 4 days after transduction. Mean ± SEM of three to four donors is shown, of which one was measured in technical triplicates. ∗p < .05, ∗∗p < .01, by paired t test. (B) Activated PBMCs were incubated in 100 nM dasatinib-containing medium or without dasatinib for 7 h during incubation with CD3-LVs. Timelines show mean fluorescence intensity (MFI) of anti-His staining, normalized to 7 h HuM291-LV, over 72 h after treatment with dasatinib, allowing detection of LVs bound to PBMCs. Filled symbols and lines show the binding of LVs to cells treated with dasatinib (+dasat.), and blank dots connected by dashed lines show binding of LVs to cells in absence of dasatinib. Mean ± SEM of three donors measured in technical duplicates. (C) MFI of the CD3 receptor 3 days after transduction with CD3-LV in presence or absence of dasatinib. Each data point is an average value referring to a particular vector stock tested on one to three donors. Values are shown as mean ± SEM. ∗p < .05, ∗∗p < .01, by repeated measure one-way ANOVA followed by Tukey’s multiple comparison test. (D) Activated PBMCs were incubated in 0–20 nM bafilomycin A1 (BflA1)-containing medium for 5 h during transduction. Fold change in transduction compared with transduction in absence of BflA is shown for three donors as mean ± SEM.
Next, we wanted to know whether the reduction of CD3-LV’s agonistic activity led to reduced CD3 intake, which could increase the amount of CD3-LV bound to CD3:TCR on the cell surface. Therefore, binding of three different CD3-targeted vector particles to activated cells was monitored over 3 days. These LVs displayed either the modified TR66.opt scFv (TR66.opt-LV), a non-modified TR66 scFv (TR66-LV), or a scFv derived from an alternative CD3-specific antibody, HuM291 (HuM291-LV).18 Cells incubated with the CD3-LVs for 7 h in presence or absence of dasatinib were analyzed for bound vector particles by using an antibody recognizing the His-tag attached to the binding glycoprotein of the LVs. In presence of dasatinib, all CD3-LVs showed substantially increased binding to the cells. This was especially pronounced at the early time point after 7 h and remained detectable for 24 h (Figures 3B and S3). Additionally, the CD3 receptor density on the T cell surface was significantly higher after dasatinib treatment (Figure 3C). Inhibition of endocytosis using bafilomycin A1 (BflA1) on primary cells reduced VSV-LV mediated transduction, while it did not change CD3-LV transduction efficiency (Figure 3D). Therefore we hypothesize that dasatinib maintains higher levels of CD3 and prolongs its presence on the cell surface, thus allowing more efficient binding of CD3-LVs to target cells, which results in better transduction.
Dasatinib increases CAR gene transfer resulting in potent CAR T cells
Having demonstrated substantial increase of reporter gene delivery by CD3-LV through dasatinib, we wondered if CAR gene transfer can be enhanced as well and if dasatinib would compromise the activity of the generated CAR T cells. CD3-LV packaging the CD19-CAR gene reached titers of up to 2 × 107 TU/mL on primary T cells in presence of dasatinib, corresponding to a 9-fold increase compared with the vector alone (Figure S4A). When combined with dasatinib and VF-1, CD3-LV(CD19.CAR) generated up to 70% CAR T cells (Figures 4A, 4B, and S4B). In agreement with the data shown for reporter gene transfer, the effect of dasatinib on CAR gene delivery was concentration and incubation time dependent, with the maximal transduction enhancing effect at 50 nM and 5 h of incubation (Figures S4C and S4D).
Figure 4.

Dasatinib enhances CD3-targeted CAR gene transfer allowing long-term killing activity
Activated PBMCs were incubated in 50 nM dasatinib-containing medium or without (w/o) dasatinib for 5 h during transduction. (A) Exemplary dot plots show CAR-expression of PBMCs, transduced in presence or absence of dasatinib on day 3 after transduction. (B) Percent of CAR-expression without or in presence of dasatinib (+dasat.) determined 3 days after transduction by flow cytometry is shown as scatter bar diagrams. Mean ±95% CI of four donors. ∗∗p < .01 by paired t test. (C–E) For the repetitive killing assay, PBMCs were incubated in 50 nM dasatinib-containing medium (+dasta.) or without dasatinib for 5 h during transduction before adding CellTraceViolet (CTV)-labeled Nalm-6 cells in a 1:1 ratio. (C) Increasing numbers of freshly labeled Nalm-6 cells were added every alternate day. (D) Percent of lysed tumor cells, after subtraction of dead tumor cells in absence of T cells, is enumerated at the denoted time points. Data represent mean ±95% CI values of four donors, of which one was measured in technical triplicates. (E) CAR T cell levels of four donors are shown in separate plots over the time of the killing assay.
Next, activated PBMCs were incubated with vector particles in presence or absence of dasatinib and shortly thereafter mixed with increasing amounts of CD19+ Nalm-6 cells every other day, starting with a 1:1 effector to target ratio (Figure 4C). Thus, CAR T cells arose in presence of tumor cells precluding conclusions about dose-efficacy correlations in this assay. Untransduced T cells incubated with or without dasatinib were not able to lyse the increasing amount of tumor cells from day 4 on (Figure 4D). In contrast, CD3-LV(CD19.CAR) incubated T cells effectively controlled tumor cells throughout the complete assay duration of 8 days including four tumor cell challenges, showing CAR T cell-specific tumor cell elimination from day 4 onward. While tumor cell control did not vary substantially between donors, CAR T cell levels were donor dependent but in each case strongly increased in presence of dasatinib (Figure 4E).
To further compare the cytotoxic activity and exhaustion profile of CAR T cells that had been shortly exposed to dasatinib during incubation with vector particles, 5 × 103 CAR T cells generated either in presence or absence of dasatinib were co-cultured with 1 × 104 Nalm-6 cells (0.5:1 effector to target). Killing assays monitored for 4 or 24 h both revealed equal killing activities of CAR T cells generated in presence or absence of dasatinib (Figures 5A and 5B). Moreover, exhaustion markers such as LAG-3, TIM-3, and PD-1 were significantly higher in T cells that had never been exposed to dasatinib (Figures 5C–5E and S5). This effect was observed 3 days after vector and dasatinib exposure, prior to killing, as well as 4 h and 24 h after addition of target cells (Figures 5C–5E and S5). Additionally, T cells exposed to dasatinib showed a tendency for a less differentiated phenotype with more central memory cells (Tcm) compared with CAR T cells generated in absence of dasatinib (Figure S6).
Figure 5.

Dasatinib mediates generation of functional, less exhausted CAR T cells
(A and B) CAR T cells were generated in presence (+dasat.) or in absence (w/o) of 50 nM dasatinib. 3 days after transduction, untransduced or CD3-LV(CAR)-transduced T cells were co-cultured with CTV-labeled Nalm-6 cells at 0.5:1 effector to target ratio. Percent of dead CTV-labeled target cells were determined by flow cytometry after 4 h (A) and 24 h (B). Bar diagrams show killing as mean ±95% CI of four donors determined in technical triplicates. ∗p < .05, ∗∗∗p < .001, by repeated measure one-way ANOVA followed by Tukey’s multiple comparison test. (C–E) 3 days after transduction the level of exhaustion markers LAG-3 (C), TIM-3 (D), and PD-1 (E) were determined on T cells, prior to killing (= 0 h), after 4 h of killing, and after 24 h of killing. Data are shown as mean ± SEM of four to seven donors. ∗p < .05, ∗∗p < .01, ∗∗∗p < .001, by repeated measure one-way ANOVA followed by Tukey’s multiple comparison test.
Taken together, transduction of PBMCs with CD3-LV(CAR) in presence of dasatinib allowed the generation of an increased number of CAR T cells. These CAR T cells were able to efficiently kill increasing amounts of target cells for several days and were not compromised in their cytotoxic activity compared with untreated CAR T cells.
Discussion
Here, we discovered that the tyrosine kinase inhibitor dasatinib can function as transduction enhancer substantially increasing gene delivery by CD3-LVs. Dasatinib enhanced gene delivery rates especially well when delivering CARs with CD3-LV, with up to 10-fold increase without compromising selectivity for CD3+ cells. This is remarkable since commonly used transduction enhancers rather achieve improvements in the range of 2-fold.22,20 Dasatinib differs from other transduction enhancers as it particularly enhances CD3-LV-mediated gene transfer. Common transduction enhancers act by facilitating cell binding receptor independently, while dasatinib exerts its transduction-enhancing effect via its kinase inhibitor activity by preventing CD3-LV-mediated T cell activation. Due to these differences in the mode of action, dasatinib can be combined with available enhancers, potentially resulting in synergistic increase of gene delivery rates. Here, we combined dasatinib with VF-1, thus exploiting both improved vector-to-cell association and increased receptor availability, which resulted in up to 70% CAR T cells upon a single cycle of CD3-LV-mediated transduction, a value which is well within the range of what is usually achieved with VSV-LV.
Our data suggest that dasatinib enhances transduction by promoting receptor availability for CD3-LV. We have shown previously that the agonistic CD3-specific scFv displayed on CD3-LV does not only mediate selective transduction of CD3+ T cells, but also CD3:T cell receptor downmodulation.18 By blocking Lck activity, phosphorylation of the CD3-zeta domain and ZAP-70 are inhibited, preventing activation-induced downregulation of the TCR.23,26 Thus, inhibition of Lck by dasatinib is the most likely cause for increased transduction efficiency by CD3-LV, while we cannot exclude that other pathways might have impacted additionally. As a result, levels of cell surface bound particles increased about 2-fold upon dasatinib treatment. CD3 surface levels remained increased for at least 3 days compared with CD3-LV-only treated T cells. Interestingly, dasatinib was active also on non-activated cells. This was surprising since dasatinib counteracted CD3-LV’s agonistic characteristic, as determined by the CD25 expression level. Yet, this did not interfere with gene delivery. On the contrary, transduction of untouched T cells in human whole blood increased by up to 4-fold. This observation can be explained by dasatinib’s reversible activity. After removing dasatinib from the cells, signaling in T lymphocytes is rapidly and completely restored having no effect on viability and or proliferation.23,27 Thus, in presence of dasatinib, binding of vector particles is enhanced, while after its removal, T cell activation and transduction proceed.
The described mode of action of dasatinib also sheds light on the entry pathway taken by CD3-LV. The moderate transduction rates in absence of dasatinib are not only caused by internalization of the vector bound to the CD3:TCR complex,18 followed by acidification of the endosome and degradation of the vector by pH-dependent endosomal proteases, as for example described for EpCAM-LV.11 Indeed, endocytosis inhibitors showed a slight increase in CD3-LV-mediated transduction on cell lines.18 However, we could not find any benefit on primary T cells. Instead, it is likely that entry into primary T lymphocytes requires a threshold of CD3 receptor molecules exposed on the cell surface. Receptor availability is necessary for efficient transduction with CD3-LV, as NiV-G pseudotyped LVs rely on binding to the target receptor and subsequent fusion with the cell membrane at neutral pH.11,9 Upon treatment with dasatinib, a higher proportion of particles binds to the cells, which is potentially required not only for proper membrane fusion but also for the evasion of restriction factors while trafficking through the cytoplasm.28,29
Improving CAR T cell manufacturing has become a major challenge in CAR T cell therapy, as this could make the treatment more broadly available, cheaper, and ultimately safer. Several approaches for rapid manufacturing of CAR T cells are being tested and improved.7,30,31,32 Now, CD3-LV, which in combination with VF-1 and dasatinib produces similar CAR T cell numbers as VSV-LV, offers additional options. Due to its specificity for CD3+ T cells, the intense and sensitive purification during T cell isolation can be omitted, while the risk of unwanted, potentially fatal transduction of cancer cells as described previously is dramatically reduced.8
Dasatinib has been used in context of CAR T cells before, aiming at goals other than transduction enhancement. When present during tumor cell killing, dasatinib inhibits the cytolytic activity, cytokine secretion, and proliferation of CAR T cells by blocking Src/Lck phosphorylation. Accordingly, it has been suggested as a potent OFF-switch for CAR T cell activity to counteract cytokine release syndrome.33,27 Likewise, dasatinib has been used to improve CAR T cell fitness and cytotoxic function by inducing rest from tonic CAR signaling, thus reversing exhaustion of differentiated CAR T cells through prolonged dasatinib exposure of several days to weeks before assessing cytotoxic activity.34 Most recently, dasatinib was used to prevent fratricide during manufacturing of CAR T cells recognizing T cell surface antigens. The resulting CAR T cells showed improved effector function performing better in long-term and repetitive killing assays in vitro and in vivo.35 In our setting using dasatinib purely as transduction enhancer, it was present only for a few hours during exposure of T lymphocytes to vector stocks but not during further cultivation and killing assays. Even after this short exposure, we documented reduced presence of exhaustion markers. While repetitive killing assays will have to be in focus of further work to confirm reduced functional exhaustion, our observations are well in line with the studies mentioned above.34,35 The use of dasatinib as transduction enhancer during manufacturing is well compatible with its further use during CAR T cell manufacture or for pharmacological control after administration into patients.
Therefore, using dasatinib in combination with CD3-LV provides a viable alternative to current CAR T cell generation methods with VSV-LV. This might substantially refine specific and efficient LV gene delivery strategies in T cell-based therapies.
Materials and methods
Primary cells
Human PBMCs were isolated from blood of healthy and anonymous donors who had given their consent or from buffy coats acquired from DRK Blutspendedienst Baden-Württemberg-Hessen (Frankfurt, Germany) in agreement with the ethics committee of the Frankfurt University Hospital. PBMCs were isolated using Pancoll (PAN-Biotech, Aidenbach, Germany) density gradient centrifugation and cultivated in T cell medium (TCM; complete RPMI 1640 [Biowest, Nuaillé, France] containing 0.5% penicillin/streptomycin and 25 mM HEPES [Sigma-Aldrich, Munich, Germany]) or NutriT medium (4Cell Nutri-T media; Sartorius, Göttingen, Germany; containing 0.5% penicillin/streptomycin) supplemented with IL-2 (50 U/ml) or IL-7 (25 U/ml) and IL-15 (U/ml) (all Miltenyi Biotec, Bergisch Gladbach, Germany). For T cell activation prior to transduction, plates were coated with 1 μg/mL anti-human CD3 (αCD3; clone OKT3; Miltenyi Biotec), and freshly isolated or thawed PBMCs were cultivated with 3 μg/mL anti-human CD28 (αCD28; clone 15E8; Miltenyi Biotec)-containing medium for 3 days.
Cultivation of cell lines
HEK-293T, Lenti-X-293T, and β2Mk/oCD47high 293T cells were grown in DMEM (Biowest) supplemented with 10% fetal bovine serum (FBS; Biochrome, Berlin, Germany) and 2 mM L-glutamine (Sigma-Aldrich). Nalm-6 cells were cultured in RPMI 1640 (Biowest) supplemented with 10% FBS and 2 mM L-glutamine.
Vector production, titration, and characterization
LVs were produced in HEK-293T (ATCC CRL-11268), Lenti-X-293T, or β2Mk/oCD47high 293T cells via polyethyleneimine transfection as recently described in a detailed protocol.36 To determine particle numbers, nanoparticle tracking analysis was performed at the NanoSight NS300 (Malvern Panalytical, Malvern, United Kingdom) or p24 enzyme-linked immunosorbent assay (ZeptoMetrix, Buffalo, NY, USA) following the manufacturer’s instructions. Vector titers of purified LV stocks were determined in transducing units per milliliter (TU/mL) from 5-fold dilutions of LV on PBMC. Dilution steps showing linear correlation to the number of transgene-positive cells were included in the titer calculations.
Transduction of primary cells
Activated or IL-7/15-stimulated PBMCs were seeded at 8 × 104 cells/well in TCM or NutriT medium supplemented with IL-2 or IL-7 and IL-15 in a 96-well plate. For T cell transduction in presence of the tyrosine kinase inhibitor dasatinib (Selleck Chemicals, Houston, TX, USA, reconstituted in DMSO) or the endocytosis inhibitor bafilomycin A1 (BflA1; Sigma-Aldrich, reconstituted in DMSO), concentrations ranging from 0.39 to 100 nM dasatinib or 1–20 nM BflA1 (diluted in medium) were added to the culturing medium 1 h prior to transduction. The cells were transduced using 5 μL (particle numbers depicted in Table 1) of targeted vector stocks or serial dilutions for titer determination. For transduction with VSV-LV, 5 μL of a 1:10 dilution of the vector stock was used, to stay below the maximal tolerated dose of 1 × 109 particles.18 Cells were subsequently centrifuged for 90 min at 850 x g and 32°C. After spinfection, 100 μL of fresh medium either containing the different inhibitor concentrations or medium without inhibitors was added, and the cells were cultured at 37°C. The medium was changed to fresh TCM or NutriT medium with IL-2 or IL-7/15 without dasatinib 3 h, 5 h, or 7 h after start of incubation with dasatinib or 5 h after start of incubation with BflA1. Samples were used for analysis at indicated time points.
Table 1.
Particle numbers of vector stocks used for transduction of primary T cells
| Vector | Particles per 5 μL |
|---|---|
| CD3(TR66.opt)-LV(GFP) | 2–3 x 1010 |
| CD3(TR66)-LV(GFP) | 2 × 1010 |
| CD3(HuM291)-LV(GFP) | 1 × 1010 |
| VSV-LV(GFP) | 7–9 x 108 |
| CD8-LV(GFP) | 2 × 1010 |
| CD3(TR66.opt)-LV(CAR) | 1 × 1010 |
CAR T cells were generated by transduction of 8 × 104 PBMCs with CD3-LV(CD19.CAR) in presence or absence of 50 nM dasatinib as described above. A second-generation CD19-CAR expressed under the SFFV promotor was used, consisting of a CD19-directed scFv derived from FMC63 and an intracellular CD28 costimulatory domain. To further increase CAR gene transfer, transduction was optionally carried out in presence of Vectofusin-1 (VF-1) (Miltenyi Biotec) as described previously.20
Transduction in whole blood
Whole blood was taken from healthy anonymous donors who had given their consent or from buffy coats acquired from DRK Blutspendedienst Baden-Württemberg-Hessen. The blood was transferred to a 24-well plate at 1 mL/well; subsequently 100 nM dasatinib or vehicle (DMSO) was added to the blood and incubated on a tumbling table at 120 rpm and 37°C for 60 min. After 1 h of incubation with dasatinib, 1 × 1011 CD3-LV particles or 3 × 109 VSV-LV particles were added to the blood and incubated for 6 h at 120 rpm and 37°C. Subsequently, PBMCs were isolated using density gradient centrifugation and cultured in TCM or NutriT medium supplemented with IL-7/15.
Cytotoxicity assay
Cytotoxic activity of CAR T cells was determined by killing of CD19+ Nalm-6 target cells. CAR expression and cell count were determined and normalized to equal T cell numbers for each condition. Cells were washed twice and 5 × 103 CAR T cells were seeded in cytokine-free NutriT medium. Nalm-6 cells were labeled with CellTraceViolet (CTV; Thermo Fisher Scientific, Waltham, MA, USA) following manufacturer’s instructions. 1 × 104 labeled Nalm-6 cells were added to the CAR T cells (0.5:1 effector to target ratio). The amount of dead target cells was determined via flow cytometry 4 h and 24 h after start of killing.
Repetitive killing assay
Activated PBMCs were seeded at 1 × 104 cells/well in cytokine-free NutriT medium containing 50 nM dasatinib or without dasatinib. 1 h after incubation with dasatinib, 5 μL (6–9 x 109 particles) CD3-LV(CD19.CAR) was added to the wells, and spinfection was performed. 5 h after addition of the vector, medium was removed and fresh medium containing 1 × 104 CTV-labeled Nalm-6 cells was added. The co-culture was cultivated at 37°C for 8 days. Every alternate day, starting on day 2, fluorescent antibody staining and flow cytometry were performed, to determine target cell killing and CAR expression. Doubling amounts of freshly labeled Nalm-6 cells were added to the remaining wells.
Flow cytometry
Cells were washed twice with wash buffer (PBS, 2% FBS, 2 mM NaN3). Optionally, cells were treated with human FcR blocking reagent (Miltenyi Biotec) prior to antibody staining. The cells were stained for 30 min at 4°C with the appropriate antibody mix (all Miltenyi Biotec) for determination of CAR expression (anti-myc [FITC] clone SH1-26e7.1.3), particle binding (anti-His [PE] clone GG11-8F3.5.1), or T cell characterization (anti-CD4 [VioBlue] clone VIT4; anti-CD3 [PerCP] clone BW264/56; anti-CD8 [APC] clone BW135/80; anti-CD45RA [VioBlue] clone T6D11; anti-CD62L [PE-Vio770] clone 145/15; anti-CD45RO [APC] clone UCHL1; anti-CD223 [VioBlue] clone REA351; anti-CD279 [PE-Vio770] clone RPA-T8; anti-CD336 [APC] clone REA635). T cell viability was determined by addition of fixable viability dye eFluor780 (Thermo Fisher Scientific). After staining, cells were washed twice with wash buffer and fixed with PBS containing 1% paraformaldehyde. The samples were measured using MACSQuant X (Miltenyi Biotec), and the results were analyzed using FCS Express (De Novo Software).
To assess T cell proliferation, PBMCs were cultivated in NutriT medium supplemented with IL-7/15 overnight. Cells were stained with CTV (Thermo Fisher Scientific) prior to incubation with dasatinib and vector particles. CTV fluorescence was measured by flow cytometry prior to incubation, on day 1 and on day 3.
Statistical analysis
Statistical analysis of all data was performed using GraphPad Prism 8 software (GraphPad). Data were analyzed for statistical significance using the indicated tests and considered significant at p < .05.
Acknowledgments
The authors thank Gundula Braun and Julia Brynza for excellent technical assistance in production and characterization of vector particles; Alexander Michels for the support in data analysis; and Frederic B. Thalheimer and Filippos T. Charitidis for helpful discussions.
Author contributions
Conceptualization: C.J.B., A.H.B., and A.M.F. Methodology: A.H.B. and N.H. Investigation: A.H.B. Formal analysis: A.H.B. Supervision: C.J.B. Writing – original draft: A.H.B. and C.J.B. Writing – review & editing: A.H.B., C.J.B., A.M.F., and N.H. Funding acquisition: C.J.B.
Declaration of interests
C.J.B., A.M.F., and A.H.B. are inventors of a patent describing the use of dasatinib as a transduction enhancer.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.12.002.
Supplemental information
Data availability
All authors declare that data are available within the article or the supplemental information files.
References
- 1.Albinger N., Hartmann J., Ullrich E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021;28:513–527. doi: 10.1038/s41434-021-00246-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korell F., Berger T.R., Maus M.V. Understanding CAR T cell-tumor interactions: paving the way for successful clinical outcomes. Med. 2022;3:538–564. doi: 10.1016/j.medj.2022.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Holzinger A., Abken H. Treatment with living drugs: pharmaceutical aspects of CAR T cells. Pharmacology. 2022;107:446–463. doi: 10.1159/000525052. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe N., Mo F., McKenna M.K. Impact of manufacturing procedures on CAR T cell functionality. Front. Immunol. 2022;13:876339. doi: 10.3389/fimmu.2022.876339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X., Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol. Ther. Oncolytics. 2016;3:16015. doi: 10.1038/mto.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine B.L., Miskin J., Wonnacott K., Keir C. Global manufacturing of CAR T cell therapy. Mol. Ther. Methods Clin. Dev. 2017;4:92–101. doi: 10.1016/j.omtm.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghassemi S., Nunez-Cruz S., O'Connor R.S., Fraietta J.A., Patel P.R., Scholler J., Barrett D.M., Lundh S.M., Davis M.M., Bedoya F., et al. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol. Res. 2018;6:1100–1109. doi: 10.1158/2326-6066.CIR-17-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruella M., Xu J., Barrett D.M., Fraietta J.A., Reich T.J., Ambrose D.E., Klichinsky M., Shestova O., Patel P.R., Kulikovskaya I., et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018;24:1499–1503. doi: 10.1038/s41591-018-0201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frank A.M., Buchholz C.J. Surface-engineered lentiviral vectors for selective gene transfer into subtypes of lymphocytes. Mol. Ther. Methods Clin. Dev. 2019;12:19–31. doi: 10.1016/j.omtm.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Funke S., Maisner A., Mühlebach M.D., Koehl U., Grez M., Cattaneo R., Cichutek K., Buchholz C.J. Targeted cell entry of lentiviral vectors. Mol. Ther. 2008;16:1427–1436. doi: 10.1038/mt.2008.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bender R.R., Muth A., Schneider I.C., Friedel T., Hartmann J., Plückthun A., Maisner A., Buchholz C.J. Receptor-targeted Nipah virus glycoproteins improve cell-type selective gene delivery and reveal a preference for membrane-proximal cell attachment. PLoS Pathog. 2016;12:e1005641. doi: 10.1371/journal.ppat.1005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Q., Schneider I.C., Edes I., Honegger A., Bach P., Schönfeld K., Schambach A., Wels W.S., Kneissl S., Uckert W., Buchholz C.J. T-cell receptor gene transfer exclusively to human CD8(+) cells enhances tumor cell killing. Blood. 2012;120:4334–4342. doi: 10.1182/blood-2012-02-412973. [DOI] [PubMed] [Google Scholar]
- 13.Pfeiffer A., Thalheimer F.B., Hartmann S., Frank A.M., Bender R.R., Danisch S., Costa C., Wels W.S., Modlich U., Stripecke R., et al. In vivo generation of human CD19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol. Med. 2018;10:e9158. doi: 10.15252/emmm.201809158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frank A.M., Weidner T., Brynza J., Uckert W., Buchholz C.J., Hartmann J. CD8-specific DARPins improve selective gene delivery into human and primate T lymphocytes. Hum. Gene Ther. 2020;31:679–691. doi: 10.1089/hum.2019.248. [DOI] [PubMed] [Google Scholar]
- 15.Charitidis F.T., Adabi E., Thalheimer F.B., Clarke C., Buchholz C.J. Monitoring CAR T cell generation with a CD8-targeted lentiviral vector by single-cell transcriptomics. Mol. Ther. Methods Clin. Dev. 2021;23:359–369. doi: 10.1016/j.omtm.2021.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Q., Uhlig K.M., Muth A., Kimpel J., Lévy C., Münch R.C., Seifried J., Pfeiffer A., Trkola A., Coulibaly C., et al. Exclusive transduction of human CD4+ T cells upon Systemic delivery of CD4-targeted lentiviral vectors. J. Immunol. 2015;195:2493–2501. doi: 10.4049/jimmunol.1500956. [DOI] [PubMed] [Google Scholar]
- 17.Agarwal S., Hanauer J.D., Frank A.M., Riechert V., Thalheimer F.B., Buchholz C.J. In vivo generation of CAR T cells selectively in human CD4+ lymphocytes. Mol. Ther. 2020;20:30239–30242. doi: 10.1016/j.ymthe.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank A.M., Braun A.H., Scheib L., Agarwal S., Schneider I.C., Fusil F., Perian S., Sahin U., Thalheimer F.B., Verhoeyen E., Buchholz C.J. Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood Adv. 2020;4:5702–5715. doi: 10.1182/bloodadvances.2020002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun X., Roth S.L., Bialecki M.A., Whittaker G.R. Internalization and fusion mechanism of vesicular stomatitis virus and related rhabdoviruses. Future Virol. 2010;5:85–96. doi: 10.2217/FVL.09.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jamali A., Kapitza L., Schaser T., Johnston I.C.D., Buchholz C.J., Hartmann J. Highly efficient and selective CAR-gene transfer using CD4- and CD8-targeted lentiviral vectors. Mol. Ther. Methods Clin. Dev. 2019;13:371–379. doi: 10.1016/j.omtm.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toyoshima K., Vogt P.K. Enhancement and inhibition of avian sarcoma viruses by polycations and polyanions. Virology. 1969;38:414–426. doi: 10.1016/0042-6822(69)90154-8. [DOI] [PubMed] [Google Scholar]
- 22.Höfig I., Atkinson M.J., Mall S., Krackhardt A.M., Thirion C., Anastasov N. Poloxamer synperonic F108 improves cellular transduction with lentiviral vectors. Achtung, funktioniert bei mir nicht (Tobi) J. Gene Med. 2012;14:549–560. doi: 10.1002/jgm.2653. [DOI] [PubMed] [Google Scholar]
- 23.Schade A.E., Schieven G.L., Townsend R., Jankowska A.M., Susulic V., Zhang R., Szpurka H., Maciejewski J.P. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood. 2008;111:1366–1377. doi: 10.1182/blood-2007-04-084814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maiti A., Cortes J.E., Patel K.P., Masarova L., Borthakur G., Ravandi F., Verstovsek S., Ferrajoli A., Estrov Z., Garcia-Manero G., et al. Long-term results of frontline dasatinib in chronic myeloid leukemia. Cancer. 2020;126:1502–1511. doi: 10.1002/cncr.32627. [DOI] [PubMed] [Google Scholar]
- 25.Tokarski J.S., Newitt J.A., Chang C.Y.J., Cheng J.D., Wittekind M., Kiefer S.E., Kish K., Lee F.Y.F., Borzillerri R., Lombardo L.J., et al. The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006;66:5790–5797. doi: 10.1158/0008-5472.CAN-05-4187. [DOI] [PubMed] [Google Scholar]
- 26.Lissina A., Ladell K., Skowera A., Clement M., Edwards E., Seggewiss R., van den Berg H.A., Gostick E., Gallagher K., Jones E., et al. Protein kinase inhibitors substantially improve the physical detection of T-cells with peptide-MHC tetramers. J. Immunol. Methods. 2009;340:11–24. doi: 10.1016/j.jim.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mestermann K., Giavridis T., Weber J., Rydzek J., Frenz S., Nerreter T., Mades A., Sadelain M., Einsele H., Hudecek M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019;11:eaau5907. doi: 10.1126/scitranslmed.aau5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao X., Li J., Winkler C.A., An P., Guo J.-T. IFITM genes, variants, and their roles in the control and pathogenesis of viral infections. Front. Microbiol. 2018;9:3228. doi: 10.3389/fmicb.2018.03228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hornick A.L., Li N., Oakland M., McCray P.B., Sinn P.L. Human, pig, and mouse interferon-induced transmembrane proteins partially restrict pseudotyped lentiviral vectors. Hum. Gene Ther. 2016;27:354–362. doi: 10.1089/hum.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J., He J., Zhang X., Li J., Wang Z., Zhang Y., Qiu L., Wu Q., Sun Z., Ye X., et al. Next-day manufacture of a novel anti-CD19 CAR-T therapy for B-cell acute lymphoblastic leukemia: first-in-human clinical study. Blood Cancer J. 2022;12:104. doi: 10.1038/s41408-022-00694-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang C., He J., Liu L., Wang J., Wang S., Liu L., Ge J., Gao L., Gao L., Kong P., et al. Novel CD19 chimeric antigen receptor T cells manufactured next-day for acute lymphoblastic leukemia. Blood Cancer J. 2022;12:96. doi: 10.1038/s41408-022-00688-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghassemi S., Durgin J.S., Nunez-Cruz S., Patel J., Leferovich J., Pinzone M., Shen F., Cummins K.D., Plesa G., Cantu V.A., et al. Rapid manufacturing of non-activated potent CAR T cells. Nat. Biomed. Eng. 2022;6:118–128. doi: 10.1038/s41551-021-00842-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weber E.W., Lynn R.C., Sotillo E., Lattin J., Xu P., Mackall C.L. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 2019;3:711–717. doi: 10.1182/bloodadvances.2018028720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber E.W., Parker K.R., Sotillo E., Lynn R.C., Anbunathan H., Lattin J., Good Z., Belk J.A., Daniel B., Klysz D., et al. Science; 2021. Transient Rest Restores Functionality in Exhausted CAR-T Cells through Epigenetic Remodeling; p. 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hebbar N., Epperly R., Vaidya A., Thanekar U., Moore S.E., Umeda M., Ma J., Patil S.L., Langfitt D., Huang S., et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat. Commun. 2022;13:587. doi: 10.1038/s41467-022-28243-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weidner T., Agarwal S., Perian S., Fusil F., Braun G., Hartmann J., Verhoeyen E., Buchholz C.J. 2020. Genetic in Vivo Engineering of Human T Lymphocytes in Mouse Models. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All authors declare that data are available within the article or the supplemental information files.